

Abstract
A low-molecular weight, solid CO surrogate that only requires low-power LED for activation to release two equivalents of CO is reported. The surrogate is universally implementable in various palladium-catalyzed carbonylative transformations. It is also compatible with protocols employing blue-light to activate conventionally inaccessible substrates such as non-activated alkyl halides. Furthermore, we demonstrate that the photo-labile CO releasing scaffold can be installed into polymeric materials thereby creating new materials with CO releasing capabilities.
Graphical Abstract
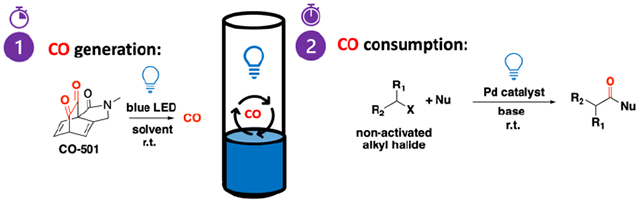
Transition metal-catalyzed carbonylation, pioneered by Heck and co-workers,1 is a one-step route to directly introduce molecular complexity in organic synthesis enabling access to a wide range of carbonyl containing intermediates.2 However, widespread application of carbonylation in small-scale laboratory setting is hindered by the inherent toxicity, storage, and handling issues associated with CO gas tanks. Thus, development of bench-stable, solid or liquid reagents that replace the direct use of CO gas is an active research area. CO surrogates developed thus far include a wide array of compounds ranging from low molecular weight compounds such as CO2,3 formic acid and its derivatives,4–7 dimethylformamide,8 chloroform,9, 10 oxalyl chloride,11 acyl chlorides,12 oxalic acid,13 paraformaldehyde and methanol14, 15 to more complex compounds such as metal carbonyls,16, 17 aldehydes,18–20 silacarboxylic acids,21 cyclopropenones,22 norbornenones,23 and 9-methyl-fluorene-9-carbonyl chloride,12, 24 among many others. Although, these surrogates provide advantages over the use of CO gas, additional reagents are required to initiate CO release. In most instances, spatial separation of the CO surrogate from the carbonylation reaction is necessary since reaction conditions for CO release are incompatible with the carbonylation chemistry. Current approaches involve thermal activation or transition metal-mediated CO release in one vessel, run in parallel with a secondary vessel where carbonylation takes place (Fig 1A). There are several CO surrogates that have been employed in one-pot systems.25, 26 These surrogates have decarbonylation chemistries that can occur concurrent with the carbonylation reaction. Another possible way to achieve orthogonal reaction conditions is by using light,27 instead of adding other chemicals or heating, for the activation of the CO surrogate.
Figure 1.

(A) For light-assisted carbonylation reactions using CO surrogates, a two-chamber reaction vessel is needed. (B) Light-activated CO donor simplifies set-up and protocol.
Another major hurdle in transition metal-catalyzed carbonylation is the inaccessibility of alkyl halides as substrates. This is due to the inherently slow oxidative addition step in alkyl halides coupled with their increased propensity to undergo isomerization via β-elimination.28 Recent reports successfully employ light to achieve alternative radical reaction pathways for these challenging substrates.29–32 With the aforementioned current barriers in the field, light seems to be a “reagent” that can fulfill two roles – as an activator for CO release from the surrogate, and as catalyst for non-activated alkyl halide substrates (Fig 1B).
We describe the design and synthesis of a high-content CO surrogate that utilizes low-power blue LED as remote trigger for CO release. This photo-activated CO donor performs as a versatile CO surrogate for various conventional Pd-catalyzed carbonylation reactions. Its utility is further exemplified in carbonylation reactions employing light to access usually restrictive, less-reactive alkyl halides (Fig 1B).29–31 In these examples, the same blue light that activates the CO donor also assists in the catalysis. Furthermore, we showcase an example wherein the photo-labile CO releasing unit is built into polymeric scaffolds affording new materials endowed with CO-releasing capabilities. The polymeric version was shown to be suitable in fully aqueous systems which could be an entry point for either green chemistry or biological CO applications.
We based our design on the photo-labile chemistry of bridged 1,2-diketones. Aromatic 1,2-diketone photoprecursors of polyacenes have been previously shown to quantitatively extrude the diketone moiety as two equivalents of CO.33, 34 We sought to synthesize an aliphatic version of the bridged 1,2-diketone and test if this will still result in extrusion of two equivalents of CO under visible light irradiation. To construct the aliphatic 1,2-diketone moiety, literature precedents describe an intermolecular Diels-Alder reaction between 1,2-quinones and strained alkyne partners,35 which requires tedious synthesis. A simplified synthesis (3-4 steps) via an intramolecular approach starting from the cheap and readily available catechol, 2,3-dihydroxybenzoic acid was carried out to prepare the photoactivated CO donor, CO-501 (Scheme 1). Amidation to a propargylic amine installs an alkyne intramolecular to the latent quinone group formed after oxidation of the catechol moiety. The diene of the benzoquinone reacts via a Diels-Alder reaction with the tethered alkyne. Echoing our earlier findings,36, 37 tethering a regular alkyne intramolecular to its dienone partner obviated the need for a strained alkyne to drive the Diels-Alder reaction.
Scheme 1.

Strategy to prepare CO-501.
The UV-Vis spectrum of CO-501 shows two absorption bands. The absorption between 420 to 500 nm with λmax of 441 nm corresponds to the n-π* transition of the carbonyl groups. Irradiation for 5 minutes with blue LED (440-445 nm) led to the gradual disappearance of the absorption at this wavelength (Fig 2A). Under ambient light, CO-501 remains stable (Fig 2B, Fig S2) for at least 2 h (stable in the dark over a few months). 1H-NMR study reveals that upon irradiation, all three alkene protons (a, b, d) and bridgehead proton (c) are converted to aromatic protons (g-j). The accompanying disappearance of complex splitting due to the diastereotopic methylene protons (e and e’) is consistent with the formation of photoproduct 3 (Fig 2C). Likewise, 13C NMR shows disappearance of two carbonyl carbons (α and β) and downfield shift of the two bridgehead carbons (γ and c) to the aromatic region. Furthermore, GC-TCD headspace analysis of CO-501 confirmed unloading of two equivalents of CO (27 mol wt% CO) upon exposure to blue LED. These studies indeed show that CO-501 undergoes bisdecarbonylation quantitatively under blue LED irradiation (Fig 2D). Next, we sought to validate the possible use of CO-501 as a CO surrogate for carbonylation reactions.
Figure 2.
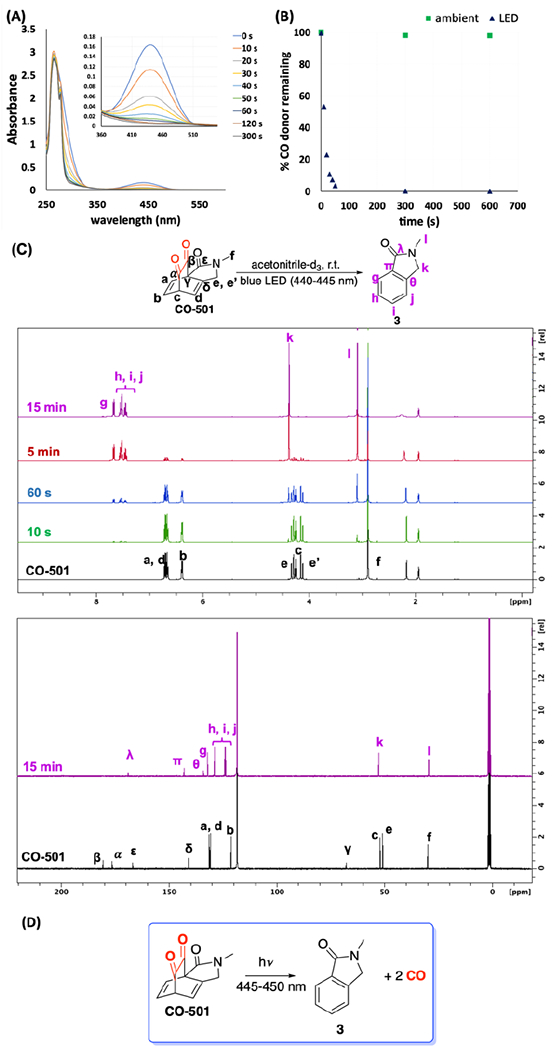
(A) UV-Vis spectrum CO-501 (1 mM in DMSO) upon exposure to blue LED light. (B) CO-501 is stable under ambient light for at least 2 h but releases CO when exposed to blue LED. (C) NMR studies confirming conversion of CO-501 to photoproduct 3. (D) CO-501 undergoes blue-light induced decarbonylation to release 2 eq of CO.
However, 1,2-diketones are known to readily and reversibly hydrate in aqueous systems.38 Liao and co-workers reported that hydration prevents 1,2-diketones from releasing CO, and that micellar structures34, 39 and hydrophobic moieties such as tert-butyl groups40 can circumvent this. CO-501 also undergoes hydration. However, instead of abolishing its CO releasing capability, hydration merely slows down the process. Fig S4 shows that the diketone n-π* transition at 441 nm present in DMSO also disappears in the presence of water while another peak appears at ~380 nm. This suggests that one of the carbonyl groups is hydrated, leaving one ketone group intact. NMR studies show that the hydration equilibrium favors the hydrated form in DMSO:water solutions (Fig S6–7). Upon extended irradiation with LED (440-445 nm) for around 65 min, the hydrated products were converted to compounds with spectra corresponding to that of by-product 3 (Fig S8). Reversibility of the hydration was further confirmed when resuspension in DMSO of a lyophilized 1:1 DMSO-d6:D2O solution gave an NMR spectrum corresponding to that of intact CO-501 (Fig S9). CO yield is also dependent on water ratio. At 50% DMSO, only 0.8 eq of CO was recovered after 5 minutes of irradiation. Increasing the irradiation time to 30 min increased CO recovery up to 1.5 eq. Further increase in water content up to 98% water in DMSO led to a yield of 1.2 eq of CO (Fig S3) after irradiation for 30 min, representing a slight decrease in CO production. These findings indicate that these 1,2-diketones are compatible with applications in aqueous systems. Nevertheless, irradiation time of CO-501 will need to be adjusted accordingly to achieve maximal CO yield.
Furthermore, due to the vicinal carbonyl moieties, 1,2-diketones such as CO-501 are rendered more susceptible to attack by nucleophiles. Control experiments indicate when all reaction components are added together with the CO donor, no carbonylated products are observed (Table S1). This could be explained by the reaction of CO-501 with the base and/or amine nucleophiles that are usually added in excess. In the presence of triethylamine and hexylamine, CO yield is decreased and completely abolished, respectively (Fig S10). Because of this, physical separation of the CO generating reaction from the carbonylation reaction in a two-chamber strategy is one of the possible workarounds to preventing CO-501 inactivation. Just like in most CO surrogates, the use of commercially available two-chamber gas reactors have been shown to be effective.41 But because CO generation is light-activated in CO-501, instead of spatial separation, we utilize light as an external trigger to temporally enrich the reaction vessel with CO before the addition of reaction components that may cause inactivation. Indeed, we find that chronological separation of the CO generation process and the carbonylation reaction allowed the use of CO-501 in various carbonylative reactions under a wide range of reaction conditions (Table 1). There is no temperature restriction or partitioning that needs to be factored in with the one-pot set-up. With other surrogates requiring thermal activation, the CO generating pot needs to be maintained at a different temperature than the carbonylation pot. Furthermore, in this system, no additional transition metal catalysts, additives, and/or bases are needed to activate CO release. But reaction vessel volume needs to be carefully selected to make sure it will be able to accommodate the accumulated CO. We carried out the reactions in various vessel volumes ranging from 6-20 mL vials and in 50-mL round-bottom flasks for larger scale reactions (Fig S12) and found CO-501 and this protocol to be widely applicable.
Table 1.
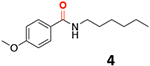
Proof-of-concept studies on the applicability of CO-501 as a CO surrogate for various carbonylation reactions (≈ 0.1 mmol scale) of 4-methoxyiodobenzene (0.1 mmol, # ≈ 1 mmol scale) under different reaction conditions.
| Carbonylative Reactions | Other Starting Material | Catalyst/Ligand | Base | Solvent | Temp (°C) | Product | %yield, isolated |
|---|---|---|---|---|---|---|---|
| Aminocarbonylation 12 | hexan-1-amine (2 eq) | Pd(dba)2 (6mol%), PPh3 (10mol%) | Et3N (2 eq) | dioxane | 80 |
|
89 (98)# |
| Aminocarbonylation 31 | hexan-1-amine (2 eq) | Pd(PPh3)4 (6mol%) | K2CO3 (1 eq) | MeTHF: H2O | r.t. |

|
82 |
| Carbonylative Suzuki-Miyaura 42 | phenylboronic acid (1 eq) | Pd2(dba)3 (1mol%) | K2CO3 (3 eq) | anisole | 100 |

|
80 |
| Double Carbonylation 43 | butan-1-amine (3 eq) | Pd(t-Bu3P)2 (3mol%) | DBU (3 eq) | THF | r.t. |

|
77 |
| Alkoxycarbonylation 44 | methanol (10 eq) | Pd(OAc)2 (5mol%), Xantphos (5mol%) | Et3N (18 eq) | acetonitrile | 80 |

|
56 |
| Carbonylative Sonogashira 45 | 4-ethynyltoluene (2 eq) | PdCl2 (3mol%), Xantphos (5 mol%) | Et3N (3 eq) | dioxane | 80 |

|
79 |
Because CO-501 releases two equivalents of CO, sub-stoichiometric amount of the donor is needed to provide more than one equivalent of CO to the reaction mixture. The low-power visible light needed for the reaction coupled with the fast kinetics (Fig 2) associated with the photo-bisdecarbonylation of CO-501 provides CO within 30 minutes of irradiation. Results also indicate that photoproduct 3 is a stable, benign by-stander that does not interfere with the carbonylation process (Table 1, S1).
Up to this point we showed that CO-501 is a CO donor that can be used in conventional Pd-catalyzed carbonylation reactions. Next, we demonstrate that the photolysis conditions to release CO from CO-501 is compatible with the conditions needed for light-assisted carbonylation of challenging substrates. In prior works utilizing CO surrogates for light-mediated carbonylation, a two-chamber set-up is required to accommodate the different reaction conditions employed for CO generation and light-assisted carbonylation reactions.30, 31 Physical separation of the CO generation and carbonylation reactions is necessary because of the incompatibility of the chemistries involved in the two processes. Here, we show that CO-501 can be used in a simplified, one-pot reaction set-up that is convenient for light-mediated carbonylation reactions. Cyclohexyl iodide and butyl iodide with either primary or secondary amine nucleophiles were carbonylated under these reaction conditions (Scheme 2)
Scheme 2.

Light-activated CO surrogate for light-assisted carbonylation reactions of non-activated alkyl iodides substrates.
We explored immobilization of the photo-labile 1,2-diketone core as a way around some of CO-501’s limitations. We prepared CO-502, a light-activated polymeric version of CO-501, as shown in Scheme S3. Using a six-carbon aminocarboxylic acid linker, we attached the protected propargylic catechol first on the aminomethyl polystyrene resin via an amide linkage. Then, subsequent deprotection and oxidation installed the light-activated 1,2-diketone moiety on the resin. CO-502 was assayed to have a CO loading degree of 1.2 mmol CO/g. We demonstrate its utility in two scenarios – (1) improved yield for carbonylation in a fully aqueous system indicating some protection against hydration, (2) simplified work-up and purification of a specific carbonylation reaction wherein the desired product co-elutes with by-product 2 (Scheme S4). While these are improvements of the surrogate under specific conditions, immobilization of this light-sensitive CO-releasing moiety on polymeric structures may potentially open other applications including biological applications.
In summary, the light-activated CO donors CO-501 and CO-502 deliver CO upon activation by low-power blue LED light. To the best of our knowledge, these are the first photo-activated, one-pot CO donors that have been demonstrated to be applicable under different carbonylation reaction conditions. More specifically, the use of this light-activated donor has been demonstrated to be compatible with blue light-assisted carbonylation reactions of non-activated alkyl halide substrates. Furthermore, with CO-502, we showed that the photo-labile CO releasing moiety can be installed into polymeric materials endowing them with CO-releasing capabilities. The convenience and practicality of these CO surrogates in various applications have been described herein. Future improvement to this system would be to bring down cost of production, especially for applications beyond research labs (N-methylprop-2-yn-1-amine is an expensive starting material) and to design a 1,2-diketone-based donor for C-13 and/or O-18 labelled CO.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful for financial support from the National Institutes of Health (R01DK119202), the Georgia Research Alliance through an Eminent Scholar fund, and internal resources at Georgia State University.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supporting Information. (Experimental details, photographs of experimental setup, compound characterization data, in PDF format)
REFERENCES
- 1.Schoenberg A; Bartoletti I; Heck RF, Palladium-catalyzed carboalkoxylation of aryl, benzyl, and vinylic halides. J. Org. Chem 1974, 39, 3318–3326. [Google Scholar]
- 2.Peng J-B; Geng H-Q; Wu X-F, The Chemistry of CO: Carbonylation. Chem 2019, 5, 526–552. [Google Scholar]
- 3.Ren X; Zheng Z; Zhang L; Wang Z; Xia C; Ding K, Rhodium-Complex-Catalyzed Hydroformylation of Olefins with CO2 and Hydrosilane. Angew. Chem. Int. Ed. Engl 2017, 56, 310–313. [DOI] [PubMed] [Google Scholar]
- 4.Morimoto T; Kakiuchi K, Evolution of Carbonylation Catalysis: No Need for Carbon Monoxide. Angew. Chem. Int. Ed. Engl 2004, 43, 5580–5588. [DOI] [PubMed] [Google Scholar]
- 5.Cao J; Zheng Z-J; Xu Z; Xu L-W, Transition-metal-catalyzed transfer carbonylation with HCOOH or HCHO as non-gaseous C1 source. Coordination Chemistry Reviews 2017, 336, 43–53. [Google Scholar]
- 6.Konishi H; Manabe K, Formic Acid Derivatives as Practical Carbon Monoxide Surrogates for Metal-Catalyzed Carbonylation Reactions. Synlett 2014, 25, 1971–1986. [Google Scholar]
- 7.Veryser C; Van Mileghem S; Egle B; Gilles P; De Borggraeve WM, Low-cost instant CO generation at room temperature using formic acid, mesyl chloride and triethylamine. Reaction Chemistry & Engineering 2016, 1, 142–146. [Google Scholar]
- 8.Panda B; Albano G, DMF as CO Surrogate in Carbonylation Reactions: Principles and Application to the Synthesis of Heterocycles. Catalysts 2021, 11, 1531. [Google Scholar]
- 9.Grushin VV; Alper H, Novel palladium-catalyzed carbonylation of organic halides by chloroform and alkali. Organometallics 1993, 12, 3846–3850. [Google Scholar]
- 10.Mondal K; Halder P; Gopalan G; Sasikumar P; Radhakrishnan KV; Das P, Chloroform as a CO surrogate: applications and recent developments. Org. Biomol. Chem 2019, 17, 5212–5222. [DOI] [PubMed] [Google Scholar]
- 11.Hansen SVF; Ulven T, Oxalyl Chloride as a Practical Carbon Monoxide Source for Carbonylation Reactions. Org. Lett 2015, 17, 2832–2835. [DOI] [PubMed] [Google Scholar]
- 12.Hermange P; Lindhardt AT; Taaning RH; Bjerglund K; Lupp D; Skrydstrup T, Ex Situ Generation of Stoichiometric and Substoichiometric 12CO and 13CO and Its Efficient Incorporation in Palladium Catalyzed Aminocarbonylations. J. Am. Chem. Soc 2011, 133, 6061–6071. [DOI] [PubMed] [Google Scholar]
- 13.Shao C; Lu A; Wang X; Zhou B; Guan X; Zhang Y, Oxalic acid as the in situ carbon monoxide generator in palladium-catalyzed hydroxycarbonylation of arylhalides. Org. Biomol. Chem 2017, 15, 5033–5040. [DOI] [PubMed] [Google Scholar]
- 14.Liu Q; Yuan K; Arockiam P-B; Franke R; Doucet H; Jackstell R; Beller M, Regioselective Pd-Catalyzed Methoxycarbonylation of Alkenes Using both Paraformaldehyde and Methanol as CO Surrogates. Angew. Chem. Int. Ed. Engl 2015, 54, 4493–4497. [DOI] [PubMed] [Google Scholar]
- 15.Natte K; Dumrath A; Neumann H; Beller M, Palladium-Catalyzed Carbonylations of Aryl Bromides using Paraformaldehyde: Synthesis of Aldehydes and Esters. Angew. Chem. Int. Ed. Engl 2014, 53, 10090–10094. [DOI] [PubMed] [Google Scholar]
- 16.Kaiser NF; Hallberg A; Larhed M, In situ generation of carbon monoxide from solid molybdenum hexacarbonyl. A convenient and fast route to palladium-catalyzed carbonylation reactions. J. Comb. Chem 2002, 4, 109–11. [DOI] [PubMed] [Google Scholar]
- 17.Åkerbladh L, Odell, Luke R; Larhed M, Palladium-Catalyzed Molybdenum Hexacarbonyl-Mediated Gas-Free Carbonylative Reactions. Synlett 2018. [Google Scholar]
- 18.Morimoto T; Fuji K; Tsutsumi K; Kakiuchi K, CO-Transfer Carbonylation Reactions. A Catalytic Pauson–Khand-Type Reaction of Enynes with Aldehydes as a Source of Carbon Monoxide. J. Am. Chem. Soc 2002, 124, 3806–3807. [DOI] [PubMed] [Google Scholar]
- 19.Ueda T; Konishi H; Manabe K, Palladium-Catalyzed Reductive Carbonylation of Aryl Halides with N-Formylsaccharin as a CO Source. Angew. Chem. Int. Ed. Engl 2013, 52, 8611–8615. [DOI] [PubMed] [Google Scholar]
- 20.Konishi H, Creation of Novel Toxic Gas Surrogates and the Development of Safe and Facile Catalytic Reactions. Chem Pharm Bull (Tokyo) 2018, 66, 1–19. [DOI] [PubMed] [Google Scholar]
- 21.Friis SD; Taaning RH; Lindhardt AT; Skrydstrup T, Silacarboxylic Acids as Efficient Carbon Monoxide Releasing Molecules: Synthesis and Application in Palladium-Catalyzed carbonylation Reactions. J. Am. Chem. Soc 2011, 133, 18114–18117. [DOI] [PubMed] [Google Scholar]
- 22.Poloukhtine A; Popik VV, Highly Efficient Photochemical Generation of a Triple Bond: Synthesis, Properties, and Photodecarbonylation of Cyclopropenones. J. Org. Chem 2003, 68, 7833–7840. [DOI] [PubMed] [Google Scholar]
- 23.Payne CM; Cho K; Larsen DS, 5-Bromo-norborn-2-en-7-one derivatives as a carbon monoxide source for palladium catalyzed carbonylation reactions. RSC Adv. 2019, 9, 30736–30740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zoller B; Zapp J; Huy PH, Rapid Organocatalytic Formation of Carbon Monoxide: Application towards Carbonylative Cross Couplings. Chemistry – A European Journal 2020, 26, 9632–9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Konishi H; Manabe K, Recent progress on catalytic Heck carbonylations using carbon monoxide surrogates. Tetrahedron Lett. 2019, 60, 151147. [Google Scholar]
- 26.Gautam P; Bhanage BM, Recent advances in the transition metal catalyzed carbonylation of alkynes, arenes and aryl halides using CO surrogates. Catal. Sci. Technol 2015, 5, 4663–4702. [Google Scholar]
- 27.Albini A; Fagnoni M In The Greenest Reagent in Organic Synthesis: Light, Dordrecht 2008. Springer Netherlands: Dordrecht; pp 173–189. [Google Scholar]
- 28.Sumino S; Fusano A; Fukuyama T; Ryu I, Carbonylation Reactions of Alkyl Iodides through the Interplay of Carbon Radicals and Pd Catalysts. Acc. Chem. Res 2014, 47, 1563–1574. [DOI] [PubMed] [Google Scholar]
- 29.Torres GM; Liu Y; Arndtsen BA, A dual light-driven palladium catalyst: Breaking the barriers in carbonylation reactions. Science 2020, 368, 318–323. [DOI] [PubMed] [Google Scholar]
- 30.Roslin S; Odell LR, Palladium and visible-light mediated carbonylative Suzuki–Miyaura coupling of unactivated alkyl halides and aryl boronic acids. Chem. Comm 2017, 53, 6895–6898. [DOI] [PubMed] [Google Scholar]
- 31.Sardana M; Bergman J; Ericsson C; Kingston LP; Schou M; Dugave C; Audisio D; Elmore CS, Visible-Light-Enabled Aminocarbonylation of Unactivated Alkyl Iodides with Stoichiometric Carbon Monoxide for Application on Late-Stage Carbon Isotope Labeling. J. Org. Chem 2019, 84, 16076–16085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sargent BT; Alexanian EJ, Palladium-Catalyzed Alkoxycarbonylation of Unactivated Secondary Alkyl Bromides at Low Pressure. J. Am. Chem. Soc 2016, 138, 7520–7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mondal R; Okhrimenko AN; Shah BK; Neckers DC, Photodecarbonylation of α-Diketones: A Mechanistic Study of Reactions Leading to Acenes. J. Phys. Chem. B 2008, 112, 11–15. [DOI] [PubMed] [Google Scholar]
- 34.Peng P; Wang C; Shi Z; Johns VK; Ma L; Oyer J; Copik A; Igarashi R; Liao Y, Visible-light activatable organic CO-releasing molecules (PhotoCORMs) that simultaneously generate fluorophores. Org. Biomol. Chem 2013, 11, 6671–6674. [DOI] [PubMed] [Google Scholar]
- 35.Escorihuela J; Das A; Looijen WJE; van Delft FL; Aquino AJA; Lischka H; Zuilhof H, Kinetics of the Strain-Promoted Oxidation-Controlled Cycloalkyne-1,2-quinone Cycloaddition: Experimental and Theoretical Studies. J. Org. Chem 2018, 83, 244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji X; Wang B, Strategies toward Organic Carbon Monoxide Prodrugs. Acc. Chem. Res 2018, 51, 1377–1385. [DOI] [PubMed] [Google Scholar]
- 37.Ji X; Zhou C; Ji K; Aghoghovbia RE; Pan Z; Chittavong V; Ke B; Wang B, Click and Release: A Chemical Strategy toward Developing Gasotransmitter Prodrugs by Using an Intramolecular Diels–Alder Reaction. Angew. Chem. Int. Ed. Engl 2016, 55, 15846–15851. [DOI] [PubMed] [Google Scholar]
- 38.Poziomek EJ; Kronenberg ME; Havinga E, Chemical consequences of the hydration of 1,2-diketones. Recl. Trav. Chim. Pays-Bas 1966, 85, 791–792. [Google Scholar]
- 39.Wright MA; Wright JA, PhotoCORMs: CO release moves into the visible. Dalton Trans. 2016, 45, 6801–6811. [DOI] [PubMed] [Google Scholar]
- 40.Elgattar A; Washington KS; Talebzadeh S; Alwagdani A; Khalil T; Alghazwat O; Alshammri S; Pal H; Bashur C; Liao Y, Poly(butyl cyanoacrylate) nanoparticle containing an organic photoCORM. Photochem. Photobiol. Sci 2019, 18, 2666–2672. [DOI] [PubMed] [Google Scholar]
- 41.Friis SD; Lindhardt AT; Skrydstrup T, The Development and Application of Two-Chamber Reactors and Carbon Monoxide Precursors for Safe Carbonylation Reactions. Acc. Chem. Res 2016, 49, 594–605. [DOI] [PubMed] [Google Scholar]
- 42.Li H; Yang M; Qi Y; Xue J, Ligand-Free Pd-Catalyzed Carbonylative Cross-Coupling Reactions under Atmospheric Pressure of Carbon Monoxide: Synthesis of Aryl Ketones and Heteroaromatic Ketones. Eur. J. Org. Chem 2011, 2011, 2662–2667. [Google Scholar]
- 43.Iizuka M; Kondo Y, Remarkable ligand effect on the palladium-catalyzed double carbonylation of aryl iodides. Chem. Comm 2006, 1739–1741. [DOI] [PubMed] [Google Scholar]
- 44.Martinelli JR; Watson DA; Freckmann DMM; Barder TE; Buchwald SL, Palladium-Catalyzed Carbonylation Reactions of Aryl Bromides at Atmospheric Pressure: A General System Based on Xantphos. J. Org. Chem 2008, 73, 7102–7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neumann KT; Laursen SR; Lindhardt AT; Bang-Andersen B; Skrydstrup T, Palladium-Catalyzed Carbonylative Sonogashira Coupling of Aryl Bromides Using Near Stoichiometric Carbon Monoxide. Org. Lett 2014, 16, 2216–2219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.