

Abstract
Oncogenic mutations in KRAS drive common metabolic programmes that facilitate tumour survival, growth and immune evasion in colorectal carcinoma, non-small-cell lung cancer and pancreatic ductal adenocarcinoma. However, the impacts of mutant KRAS signalling on malignant cell programmes and tumour properties are also dictated by tumour suppressor losses and physiological features specific to the cell and tissue of origin. Here we review convergent and disparate metabolic networks regulated by oncogenic mutant KRAS in colon, lung and pancreas tumours, with an emphasis on co-occurring mutations and the role of the tumour microenvironment. Furthermore, we explore how these networks can be exploited for therapeutic gain.
The RAS family of proteins is ubiquitously expressed in mammals and consists of KRAS, NRAS and HRAS isoforms1. RAS proteins drive signalling through growth and survival pathways, such as the phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) cascades2. Point mutations in genes encoding RAS proteins are common across numerous cancer types3,4. Among these, KRAS is most frequently mutated in colorectal carcinoma (CRC; 40%), non-small-cell lung cancer (NSCLC; 35%) and pancreatic ductal adenocarcinoma (PDA; more than 95%)5, and its associated oncogenic activity is implicated in all the hallmarks of cancer6,7. KRAS mutations render the protein constitutively active, thereby driving signalling through progrowth and anti-apoptotic pathways in the absence of stimulation. Thus, in principle, KRAS has considerable potential as an oncology drug target. Indeed, substantial progress has been made in recent years in developing potent, KRAS-G12C isoform-specific inhibitors2,6,8,9. Tempering this optimism, a significant percentage of oncogenic mutant KRAS (KRAS*) tumours are not driven by G12C mutations, and the complexity of RAS signalling still presents a significant therapeutic obstacle.
Despite KRAS mutations being common among colon, lung and pancreas tumours, nuanced aspects of KRAS signalling and tissue physiology differentially influence how KRAS* impacts cancer biology. Expression levels of KRAS*, as well as the specific mutations of KRAS, influence its activity and also differ between the tissues of origin10–12. For example, NSCLC more frequently harbours the tobacco smoking-associated KRAS-G12C mutation, while the KRAS-G12D mutation is predominant in PDA5. In contrast to PDA and NSCLC, where KRAS* is the disease-initiating mutation, KRAS mutations are secondary hits in CRC progression13–15. The expression levels and activity of KRAS* can also differ on the basis of mutation type10.
Adding further complexity, tissue-specific, co-occurring mutations cooperate with KRAS* to dictate downstream effector signalling16, and the unique physiological function, architecture and cellular composition of the tissue of origin impact crosstalk between KRAS*-driven cancer cells and surrounding parenchymal, stromal and immune cell types17–19. Thus, despite sharing the expression of KRAS*, CRC, NSCLC and PDA develop distinct phenotypes with unique tumour microenvironments (TMEs) that regulate tumour properties and response to therapy.
In the past decade, a wealth of studies have delineated how KRAS* signalling can reprogramme cancer cell metabolism to support tumour growth. In addition, tumours arising in different tissues have unique metabolic programmes and associated dependencies. Because KRAS* promotes a number of targetable common and/or tissue-specific pathways across cancer types, an alternative approach to directly targeting KRAS* is to block KRAS* metabolic effector pathways (REFS2,20). Furthermore, the exposure of tissue-specific supporting cell types to the dysregulated metabolism of KRAS*-driven cancer cells can affect their own metabolism and function. In this Review, we therefore examine how KRAS* orchestrates the metabolism of CRC, NSCLC and PDA. We discuss the intrinsic metabolism of KRAS*-driven cancer cells and metabolic interactions in the broader TME. Finally, we describe novel metabolic vulnerabilities garnered from these studies and the associated therapeutic inroads.
Cancer cell metabolism
Common metabolic pathways between cancers
KRAS* drives bioenergetic, biosynthetic and redox programmes, many of which in a manner independent of tumour type21,22. These programmes build biomass and support an overall energetic state within the cancer cell favourable to deregulated proliferation (FIG. 1).
Fig. 1 |. KRAS* rewires cancer cell metabolism.
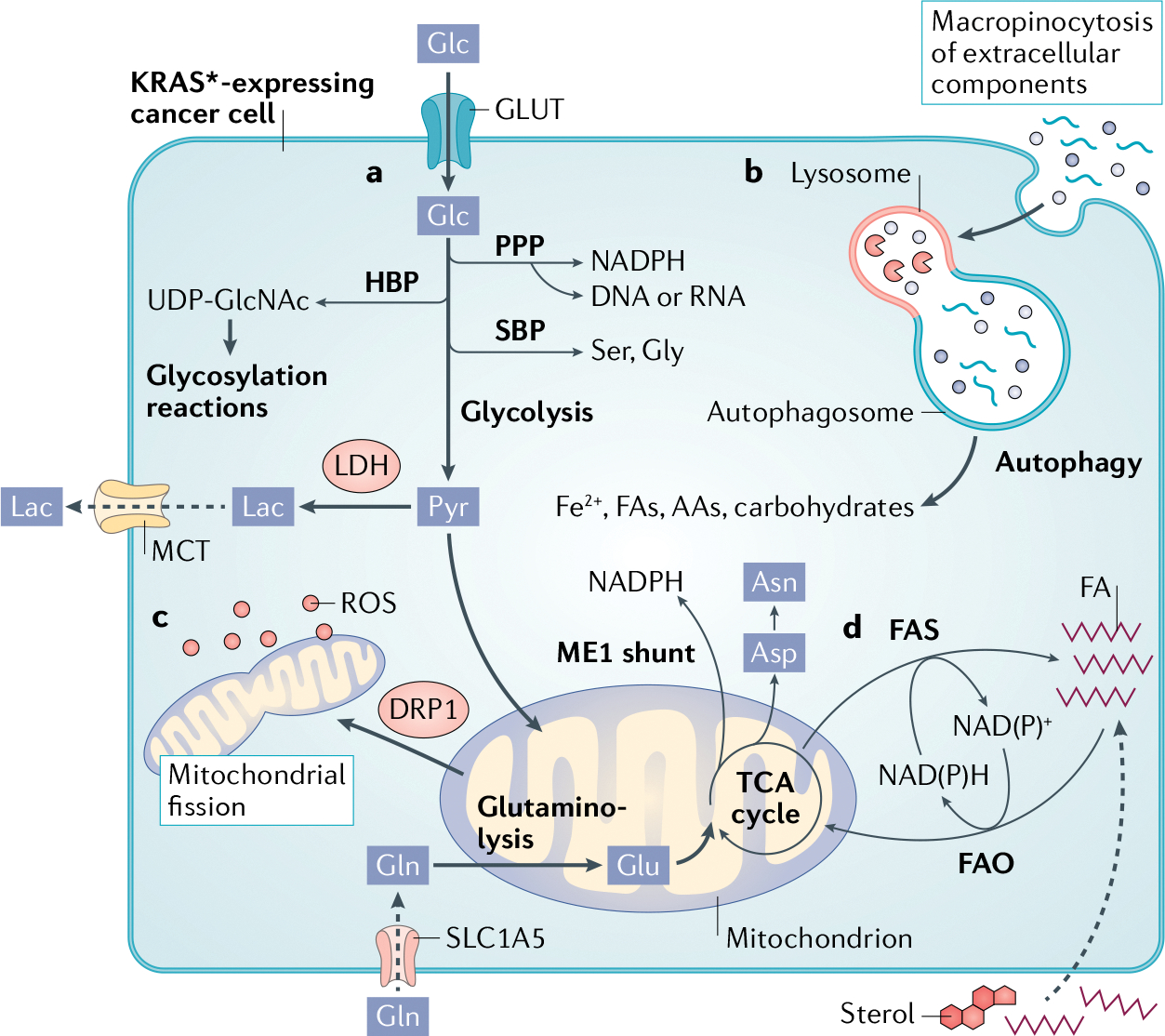
a | Glucose (Glc) transport via glucose transporters (GLUTs) and flux through glycolysis is upregulated to provide intermediates for branching biosynthetic pathways. The hexosamine biosynthetic pathway (HBP) produces uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) for glycosylation. The two arms of the pentose phosphate pathway (PPP) generate nicotinamide adenine dinucleotide phosphate (NADPH) as well as ribose bases for nucleotide synthesis. Flux through the serine biosynthesis pathway (SBP) generates the amino acids serine (Ser) and glycine (Gly). To maintain redox balance, increased expression of lactate dehydrogenase (LDH) converts pyruvate (Pyr) to lactate (Lac), which is transported out of the cell via monocarboxylate transporters (MCTs) to sustain glycolysis. Pyruvate also contributes carbon to the tricarboxylic acid (TCA) cycle in mitochondria. Glutamine (Gln) is imported by SLC1A5 and converted to glutamate (Glu) by glutaminase 1 (GLS1) and fuels the TCA cycle. Mutant KRAS (KRAS*) diverts TCA cycle intermediates through malic enzyme 1 (ME1) for NADPH production, as well as synthesis of amino acids such as aspartate (Asp) and asparagine (Asn). b | KRAS* cells are dependent on nutrient-scavenging pathways, such as macropinocytosis and autophagy, whereby free biosynthetic precursors are released for utilization by cancer cells. c | KRAS* regulates mitochondrial dynamics through DRP1 to fine-tune mitochondrial fission and maintain optimal reactive oxygen species (ROS) levels for signalling, while avoiding toxicity. d | Depending on the intracellular redox state, KRAS* cells use fatty acid synthesis (FAS) to regenerate NAD(P)+ and synthesize lipids or fatty acid oxidation (FAO), whereby lipids are used for energy and NAD(P)H production. AA, amino acid; FA, fatty acid; Fe2+, iron.
Central carbon metabolism.
KRAS* signalling confers a competitive advantage to cancer cells through increased glucose uptake and elevated flux through glycolysis, concurrently fuelling numerous branching biosynthetic pathways23–29. Cells harbouring KRAS* highly express glucose transporters (GLUTs), leading to efficient glucose uptake30,31 (FIG. 1). The flux of glucose through glycolysis is elevated due to KRAS*-regulated expression and/or activity of key glycolytic enzymes32,33. Several glycolytic intermediates are shunted into biosynthetic pathways essential for nucleotide production, amino acid synthesis or glycosylation reactions23,34, the activity of which is coordinated by KRAS* signalling35–38 (FIG. 1). To prevent an unfavourable cellular oxidation state and glycolytic stalling, KRAS* also promotes the rapid reduction of glycolysis-derived pyruvate to lactate23,39,40 (FIG. 1). Lactate transporters are expressed in a KRAS*-dependent manner and efficiently export lactate from the cell40–42 (FIG. 1). As a whole, this glycolytic phenotype driven by KRAS* supports malignant progression and correlates with poorer prognoses in patients with KRAS*-driven cancers40,43.
Mitochondria.
Mitochondria are hubs for biosynthesis44–46, regulate signalling and gene expression, and are implicated in tumour initiation and progression47–49. KRAS* modulates the total mitochondrial content and function through induction of mitophagy, and disrupted mitophagy delays tumour progression50. Mitochondrial metabolism and flux of electrons through the electron transport chain (ETC) generate reactive oxygen species (ROS)51. Mitochondrial ROS are required for KRAS*-driven tumour growth in mouse models52. However, damaged mitochondria are inefficient, and aberrant or excessive mitochondrial metabolism can lead to toxic ROS levels. As noted, KRAS* signalling leads to removal of defective mitochondria through mitophagy and prevents excessive levels of ROS50. Additionally, KRAS* signalling is vital for fine-tuning mitochondrial dynamics, maintaining an optimal balance of mitochondrial fission or mitochondrial fusion53–55 (FIG. 1).
Amino acids and protein synthesis.
In addition to their classical role in protein biosynthesis, amino acids can also be a valuable fuel. For example, glutamine is a vital source of carbon and nitrogen for tumours56. KRAS* also promotes alternative glutamine catabolism, leading to production of nicotinamide adenine dinucleotide phosphate (NADPH)26,57 (FIG. 1). Cultured colon, lung and pancreatic cancer cells expressing KRAS* have been shown to be ‘addicted’ to glutaminolysis57–59 (FIG. 1) for growth and survival. While glutamine is the most abundant amino acid in serum56, in vitro studies are often conducted in media with supraphysiological concentrations of glutamine and lacking stromal or immune compartments. These factors have contributed to an important debate on the degree of this dependency on glutaminolysis in vivo (which substantially differs from that determined in vitro), as well as its tissue specificity56,58,60,61. Nevertheless, the expression of mitochondrial glutamate transporters was elevated in KRAS* tumour tissue compared with matched non-transformed tissue, and blockade of mitochondrial glutamate transport in cells with KRAS* decreased the abundances of tricarboxylic acid (TCA) cycle intermediates, disrupted redox homeostasis and impaired proliferation both in vitro and in tumour xenografts62.
Glutamine anaplerosis is also important for the production of amino acids such as aspartate and asparagine from TCA cycle intermediates, which are critical for nucleotide and protein synthesis (FIG. 1)63–65 (non-peer reviewed data in REF.64). Aside from glutamine catabolism, recent work in cell lines and mouse models demonstrated that glutamine synthesis is crucial to PDA, as glutamine serves as a vital source of nitrogen needed to produce nucleotides and other amino acids66. Clearly, the role of glutamine metabolism in KRAS* tumours is complex and requires further study.
Nutrient scavenging.
Dysregulated growth and lack of proper vasculature leads to oxygen and nutrient deprivation in tumours. Therefore, cancer cells often rely on scavenging and recycling pathways to fuel biosynthesis and combat cellular stressors67. RAS pathway activation induces macropinocytosis68,69, which provides an array of fuel sources that can be degraded in the lysosome, endowing cancer cells with the building blocks to support biomass generation in nutrient-deprived conditions70 (FIG. 1). Recent studies have illustrated that the regulation of macropinocytosis by KRAS requires a complex interplay with other signalling cascades67,70–75.
Autophagy regulated in cancer cells expressing KRAS* and is a crucial scavenging pathway under acute nutrient deprivation, by which intracellular nutrients can be diverted to meet metabolic demands required for survival76–83 (FIG. 1). Therefore, during culture in buffered saline lacking essential nutrients, limited mitochondrial substrate availability can lead to dangerous levels of ROS and depleted nucleotide pools. In this setting, autophagic flux provided KRAS*-driven cancer cells with glutamine and glutamate to fuel the TCA cycle and support nucleotide production84. Interestingly, blockade of downstream KRAS* signalling through ERK or RAF promoted autophagic activity, suggesting a potential mechanism by which cancer cells might sustain metabolism even when KRAS* signalling is targeted directly85,86. In a broader context, autophagy within the host has also been shown to maintain circulating glucose and arginine levels, and ablation of the autophagic machinery systemically led to regression of KRAS* tumours in mice, although tumours with other oncogenic drivers were similarly affected87,88.
Lipid metabolism.
Proliferating cells require fatty acid synthesis (FAS) to generate lipids for processes such as membrane synthesis. Lipids derived from intracellular stores or through extracellular uptake can also be utilized as a fuel source through fatty acid oxidation (FAO) to generate ATP89. FAS requires reducing potential, while FAO generates reducing potential. Therefore, the redox state of a cancer cell can determine whether FAS or FAO is predominant (FIG. 1). In studies with human and murine models of NSCLC, tumours showed increased expression of FAS-related enzymes and were sensitive to FAS inhibition compared with either the normal lung or non-KRAS* lung adenocarcinoma90–93. Conversely, in another study, KRAS* promoted fatty acid uptake and FAO in mouse models of NSCLC, and deletion of a key FAO enzyme impaired tumour growth94. Additionally, in PDA, KRAS* induces scavenging of environmental lipids or utilization of previously stored lipid droplets, both in cell culture and in mouse KRAS*-expressing allografts, under conditions that are unfavourable for FAS such as hypoxia or during invasion95–97. These findings highlight that lipid metabolism is regulated not by KRAS* alone but also by other contextual factors, such as the energetic state of the cell, the tissue of origin or the experimental model.
Dependence on co-occurring mutations
Co-occurring mutations in oncogenic drivers or tumour suppressors cooperate with KRAS* to shape the phenotype of a cancer cell, and these depend on the tissue of origin (Box 1).
Box 1 |. Tissue-specific metabolism: oncogenic KRAS and the cell of origin.
The physiological properties and functions of an organ influence tissue metabolism. These include oxygen tension, microbiota presence and composition, vascularity and immunosurveillance. The cell within a tissue that experiences the initial oncogenic hit and drives tumorigenesis is known as the cell of origin250. The intrinsic metabolic programmes in the cell of origin, or those available through dedifferentiation events, can be hijacked by oncogenic mutant KRAS (KRAS*) to support transformation. Collectively, the metabolic parameters in the tissues and the cell of origin interact with KRAS* to impose distinct features, some of which enact unique metabolic vulnerabilities.
Colorectal cancer
LGR5+ intestinal stem cells residing in the crypt are a major cell of origin in colon cancer. In these cells, loss of the tumour suppressor adenomatous polyposis coli (APC) and overactive β-catenin–Wnt pathway activity drive adenoma formation251. These cells rely on fatty acid oxidation for self-renewal and regeneration, which is similarly utilized by transformed stem cells following loss of APC to enhance tumorigenicity106,107. As KRAS mutations follow loss of APC, an existing question remains as to how KRAS* changes fatty acid metabolism to support the progression of adenomas to carcinomas14.
Non-small-cell lung cancer
Bronchiolar, alveolar type II and bronchoalveolar stem cells have been postulated as the cell of origin in lung cancer252–255. Susceptibility to transformation is complex and influenced by cell type, physical location, co-occurring mutations and immune signalling256–258. A common requirement for transformation is the ability to adapt to oxidative stress, as KRAS* induces high levels of reactive oxygen species and the lung is a naturally oxidative environment. Indeed, loss-of-function Kelch-like ECH-associated protein 1 (KEAP1) mutations and gain-of-function nuclear factor erythroid 2-related factor 2 (NRF2) mutations lead to overactive NRF2 signalling in lung cancer relative to other tumour types. In the KRAS* lung, these additional mutations confer a survival advantage on transformed cells to buffer reactive oxygen species due to an oxygenated environment.
Pancreatic ductal adenocarcinoma
In the normal pancreas, acinar cells avidly consume amino acids to synthesize digestive enzymes, whereas ductal cells are primarily responsible for transport of peptides and hormones. Acinar cells in the pancreas transdifferentiate into ductal cells in response to injury or KRAS mutation and are the presumed cell of origin in pancreatic ductal adenocarcinoma, although ongoing investigations have also implicated ductal cells174,175,259–261. In the transformed state, KRAS* pancreatic ductal adenocarcinoma cells possess metabolic programmes inherent to both acinar and ductal cells. This could explain why acinar-derived tumours demonstrate a ductal-like programme of decreased uptake and metabolism of branched-chain amino acids178.
APC and KRAS* in CRC metabolism.
Loss-of-function (LOF) mutations in the gene encoding the tumour suppressor adenomatous polyposis coli (APC) initially drive tumorigenesis in ~80% of CRCs14. APC is a negative regulator of the transcription factor β-catenin. APC loss in colon epithelial cells results in the activation and nuclear translocation of β-catenin, which then promotes aberrant WNT signalling and early adenoma formation. Subsequent oncogenic activation of KRAS* occurs in 40% of colon adenomas and enables maximal activation of β-catenin and WNT signalling and progression to carcinoma98. One metabolic mechanism that supports this finding is KRAS*-induced production of succinate from glutamine in CRC cells in vitro. This in turn inhibits demethylases, resulting in hypermethylation and increased expression of WNT pathway components, along with elevated β-catenin activity99 (FIG. 2a).
Fig. 2 |. KRAS* synergizes with co-occurring mutations to direct metabolism.
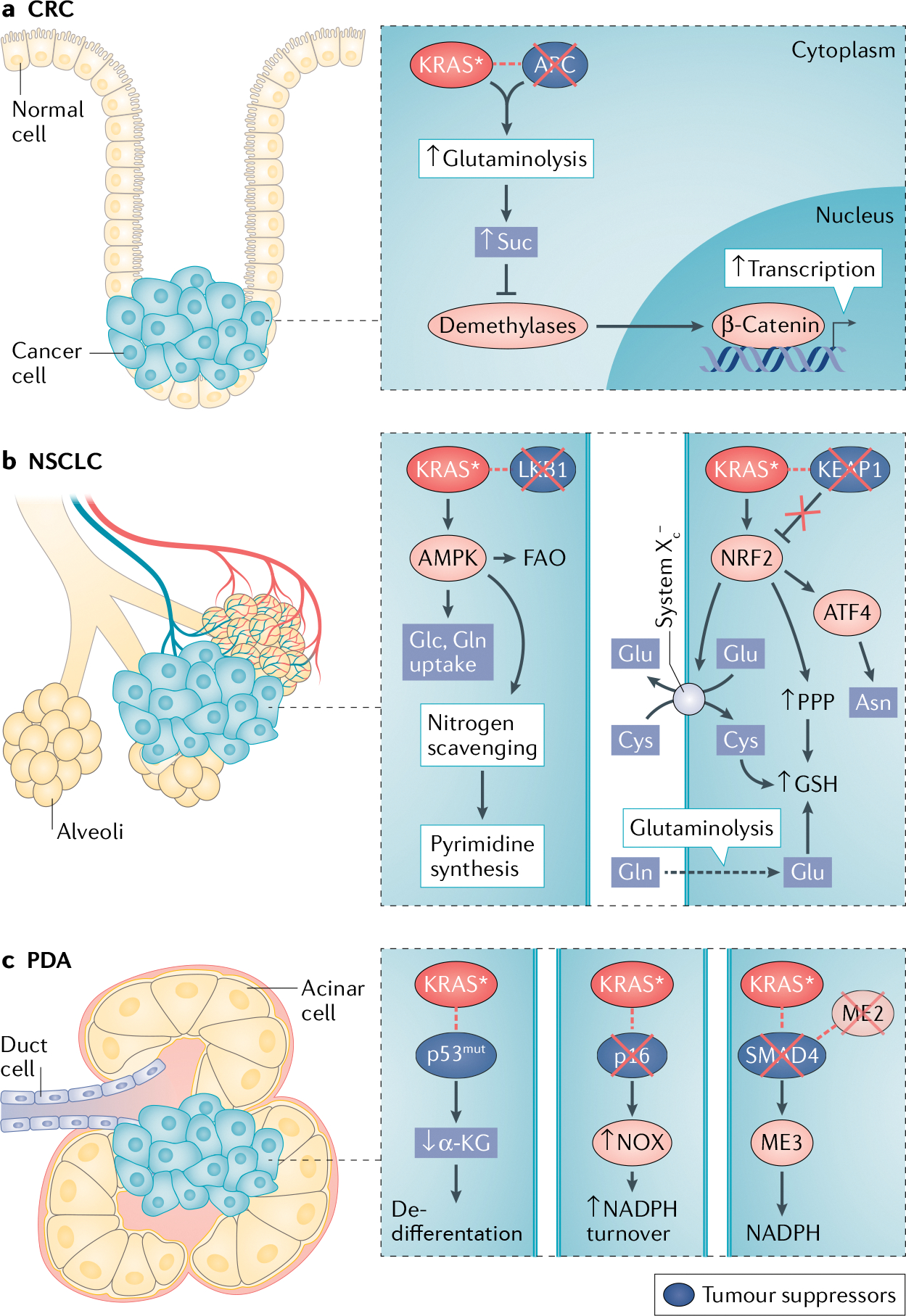
a | In colorectal carcinoma (CRC), mutant KRAS (KRAS)*-mediated glutamine (Gln) catabolism increases levels of the tricarboxylic acid cycle intermediate succinate (Suc), which inhibits the activity of α-ketoglutarate (αKG)-dependent demethylases. Hypermethylation of Wnt pathway target genes promotes their expression and cooperates with β-catenin transcriptional activity for maximal activation downstream of loss of adenomatous polyposis coli (APC). b | In non-small-cell lung cancer (NSCLC), aberrant activity of adenosine 5′-monophosphate-acivated kinase (AMPK), due to loss of liver kinase B1 (LKB1), increases fatty acid oxidation (FAO) and uptake of glucose (Glc) and glutamine (left). Scavenging of ammonia from the urea cycle supplies nitrogen needed for pyrimidine synthesis. Loss of Kelch-like ECH-associated protein 1 (KEAP1) activates the nuclear factor erythroid 2-related factor 2 (NRF2) antioxidant transcriptional programme. Increased glutamate (Glu) produced from glutamine through glutaminolysis is exchanged for extracellular cystine (cysteine dimer; Cys–Cys) through system XC−, which is also upregulated by NRF2. Cysteine is incorporated into glutathione (GSH) for protection from reactive oxygen species. NRF2 also stimulates production of GSH through the pentose phosphate pathway (PPP) and regulates asparagine (Asn) production through activating transcription 4 (ATF) activity (right). c | In pancreatic ductal adenocarcinoma (PDA), mutations of p53 (p53mut) lead to reduced αKG levels and impaired demethylase activity, affecting epigenetic programmes and cell state (left). Loss of the tumour suppressor p16 leads to increased NADH oxidase (NOX) expression and activity, which is important for nicotinamide adenine dinucleotide phosphate (NADPH) turnover and redox balance (middle). Loss of the SMAD4 genetic locus coincides with loss of malic enzyme 2 (ME2), rendering cells dependent on compensatory ME3 activity for NADPH production (right).
MYC is a target of β-catenin and cooperates with KRAS* during malignant transformation in CRC100. Indeed, MYC is required for activation of the majority of WNT target genes following loss of APC101. An in-depth metabolomics analysis of CRC cells demonstrated a role for MYC mediated metabolic reprogramming in early adenoma formation in tumours from patients102. Furthermore, APC loss induced the expression of metabolic target genes of MYC103. These data show that loss of APC requires MYC for induction of oncogenic programmes, yet how KRAS* and MYC cooperate in CRC following loss of APC warrants deeper investigation. As an aside, MYC activity has also been shown to be essential in KRAS*-driven NSCLC and PDA, and there is considerable overlap in the metabolic programmes induced by KRAS* and MYC, emphasizing the complex interplay between these two oncogenic drivers23,100,104,105.
Fatty acid metabolism in APC-null progenitor cells also plays a role in tumorigenesis. A high-fat diet induces FAO in intestinal stem and progenitor cells in mice, and FAO increases stem cell function and renewal in mice and in ex vivo organoid cultures106,107. These more numerous progenitor cells are susceptible to oncogenic transformation, and subsequent loss of APC combined with elevated FAO resulted in increased tumour formation and progression106. However, cooperation between loss of APC and KRAS* in fatty acid metabolism and stem cell fate remains to be elucidated.
Colorectal tumours at the primary site display heterogeneity in KRAS status, as not all transformed cells harbour KRAS mutations. Therefore, it is reasonable to assume that remarkable metabolic heterogeneity exists within a colorectal tumour. Because KRAS* is sufficient to drive invasive and metastatic disease progression108, it is tempting to speculate that targeting KRAS*-driven metabolism could preferentially block invasion and cause regression of metastatic tumours. In mouse models, aggressive tumours arise from cells harbouring both KRAS* and LOF APC mutations compared with tumours driven by either mutation alone, suggesting a highly complex genotype-dependent interaction109–111. An area that has not been well studied in CRC is cooperative network regulation by LOF APC mutations and KRAS* that promote metabolic pathways critical for tumour progression. Additional work into intratumoural metabolic differences based on the driving oncogenic events could uncover a differential dependence on metabolic pathways for primary tumour maintenance or metastasis of CRC.
LKB1 and KEAP1 loss synergize with KRAS* in lung tumour metabolism.
STK11 encodes the tumour suppressor liver kinase B1 (LKB1) and is lost in approximately 20% of NSCLCs15,16. LKB1 enacts its tumour suppressive programme primarily through phosphorylation and activation of adenosine 5′-monophosphate-activated protein kinase (AMPK) and related AMPK family members112–115. In this manner, the LKB1–AMPK axis senses energetic stress and acts as a metabolic brake under unfavourable nutrient conditions113,116,117. In line with this, loss of LKB1 in human NSCLC cell lines induced an aberrant metabolic programme supportive of malignant progression, with increased consumption and metabolism of glucose and glutamine118 (FIG. 2b). Dysregulated AMPK activity is also required for tumour growth in vivo as loss of AMPK in a KRAS*, LKB1-deficient autochthonous mouse model of NSCLC reduced tumour burden119.
During energetic stress, AMPK promotes FAO112, a pathway also utilized by normal lung epithelial cells during starvation120. Suggesting a role for AMPK activity, KRAS*, LKB1-deficient lung tumours relied on FAO following acute energy crisis caused by deletion of autophagic components, a finding that was not observed in NSCLC with p53 mutations121. AMPK activity following loss of LKB1 also induces scavenging of nitrogen from urea cycle ammonia for incorporation into pyrimidines and is required for the growth of KRAS*-driven, LKB1-deficient human NSCLC cell lines and xenografts122 (FIG. 2b). As a side note, some PDAs have also been shown to harbour LOF mutations in STK11, and preclinical studies in mouse models of PDA demonstrated that LKB1 loss cooperates with KRAS* to drive tumour progression123–125. Further work remains to determine whether the metabolic programmes found in KRAS*-driven, LKB1-deficient NSCLC are relevant for PDA.
Approximately 20% of patients with KRAS*-driven NSCLC have tumours that harbour LOF mutations in the gene encoding Kelch-like ECH-associated protein 1 (KEAP1), the negative regulator of nuclear factor erythroid 2-related factor 2 (NRF2)15,16,126. KEAP1 loss promotes NSCLC progression127, and conversely, loss of NRF2 blocks tumour growth in autochthonous mouse models of both NSCLC and PDA128. It has been shown that NRF2 induces an antioxidant transcriptional programme in NSCLC cell lines and in human tumours in part through modulation of activating transcription factor 4 (ATF4)129. Similarly, KRAS* promotes expression of asparagine synthase (ASNS) and asparagine production through ATF4 (REF.65) (FIG. 2b), demonstrating the cooperation between KRAS* and NRF2 following KEAP1 deletion. In the normal lung, production of NAPDH through the pentose phosphate pathway (PPP) is necessary for protection against free radicals, detoxification reactions and lipid synthesis130,131. NRF2 activity due to loss of KEAP1 also enhances PPP flux and production of NADPH and glutathione (GSH) in human NSCLC cell lines132 Remarkably, despite the essentiality of the PPP for KRAS*-driven tumours, a recent study demonstrated that loss of a key PPP enzyme did not abrogate growth of CRC or NSCLC tumour models, suggesting KRAS* can adapt to loss of PPP activity via compensatory metabolic pathways133.
Indeed, KEAP1-mutant cells can utilize glutaminolysis as an alternative to the PPP to produce GSH and to replenish intracellular glutamate pools127,134. In particular, NRF2 can induce transcription of SLC7A11, which encodes for xCT, the rate limiting subunit of the glutamate––cystine exchanger system XC−. While this NRF2-driven glutamate–cystine exchange promotes cystine import and GSH production, it also depletes intracellular glutamate, which is also needed for the TCA cycle. Therefore, cancer cells with overactive NRF2 rely on glutaminolysis through glutaminase 1 (GLS1) to maintain sufficient glutamate pools to both import cystine and fuel the TCA cycle135 (FIG. 2b). Conversely, wild-type KEAP1 NSCLC cells were insensitive to GLS1 inhibition in vivo due to compensatory incorporation of glucose into the TCA cycle, as opposed to relying on glutamine for anapleurosis58. Collectively, these data illustrate how co-occurring mutations can drive metabolic heterogeneity in lung tumours and highlight the importance of characterizing metabolic dependencies in genetic subtypes of lung cancer136.
Loss of TP53, INK4A or SMAD4 cooperates with KRAS* to direct PDA metabolism.
LOF mutations in the TP53 gene encoding the tumour suppressor p53 in the context of KRAS* are observed in about 50% of PDAs137,138. As mentioned previously, KRAS*-driven cells rely on nutrient scavenging and recycling pathways6,67,68,81. Surprisingly, an initial study in KRAS*-driven PDA lacking p53 showed inhibition of autophagy with hydroxychloroquine actually increased tumour growth in comparison with KRAS*-driven tumours with p53 (REF.139). A follow-up study using a mouse model of PDA with Trp53 loss of heterozygosity, one that more closely resembles the genetic evolution as well as the tissue and immune architecture of human disease, showed that autophagy is critical for PDA metabolism independently of p53 status140. Furthermore, an earlier study established that autophagy is required oxidative phosphorylation, ROS modulation and tumour growth in PDA, regardless of p53 status141.
p53 has also been shown to play a role in the epigenetic regulation of cell state in PDA through modulation of the TCA cycle intermediate α-ketoglutarate (αKG) in orthotopic allografts, autochthonous mouse models and human PDA. Decreased levels of αKG resulting from loss of p53 impair the activity of certain chromatin-modifying enzymes, ultimately inducing a dedifferentiated cell state with enhanced cellular fitness. Conversely, re-expression of wild-type p53 leads to elevated levels of αKG and promotes a differentiated, less aggressive state142. This is an important mechanism by which p53 suppresses tumour progression in PDA (FIG. 2c).
The tumour suppressor p16 (INK4A) is inactivated in more than 90% of KRAS* PDAs143. Loss of p16 in the context of KRAS* cell lines in vitro resulted in upregulation of levels of NADPH oxidase 4 (NOX4) and increased glycolytic flux compared with cells expressing p16 (REF.144). Thus, NOX4 activity, which sustains glycolysis by oxidizing NADPH, and resulting from loss of p16, supports the broader metabolic programmes induced by KRAS* (FIG. 2c).
The genomic locus for the tumour suppressor SMAD4 is deleted in more than 50% of KRAS* PDAs143. This tumour suppressor loss is a non-specific process that often results in the concurrent deletion of neighbouring chromosomal genes. In the case of SMAD4, the gene encoding mitochondrial malic enzyme 2 (ME2) is in close genomic proximity and is co-deleted in ~25% of cases. ME2 converts malate to pyruvate and is required for NADPH production145. KRAS* PDA cells can compensate for ME2 loss through ME3 activity, and this genomic context creates a vulnerability for inhibition of ME3 in ME2-null cells145 (FIG. 2c). This is an important example in which a co-occurring genetic event indirectly influences KRAS* metabolism and provides a novel therapeutic target.
It is important to note that these highlighted co-occurring mutations are not exclusive to the tumour types in which they are discussed here. For instance, TP53 is also commonly mutated in colon and lung tumours, and the metabolic perturbations induced by mutant p53 have been reviewed extensively elsewhere14,146,147. Nor are the highlighted mutations the only co-occurring mutations found in these tumours. Each tumour is a complex amalgam of unique and sometimes rare mutations all working in tandem with KRAS* to drive aberrant metabolism. We have aimed to use these studies in colon, lung and pancreas tumours to demonstrate the importance of co-occurring mutations in shaping tumour metabolism. Given the genetic heterogeneity seen in patients, much work remains to both model this heterogeneity and identify metabolic pathways that can be targeted in certain genetic subtypes of these diseases.
Tissue-dependent features
Even though we can rapidly identify the driving mutations in a patient’s tumour using advanced genomics and high-throughput sequencing, we must also consider the effects of the cell of origin and tissue constraints on the biology of the tumour. While the metabolic features discussed in the following sections are not exclusive to the indicated tumour type, there are instances in which KRAS*-transformed cells co-opt the metabolic machinery in the tissues of origin. Here we discuss how this is influenced by tissue structure and how this impacts nutrient and oxygen access.
Hypoxia, inflammation and iron in the colon.
The oxygen tension in the colonic lumen can be as much as ten times lower than that in oxygenated blood or tissue (that is, ~10 mmHg)148 (FIG. 3a). This hypoxic lumen supports growth of facultative anaerobic microorganisms, and consequently symbiotic metabolic interactions with colonocytes149 (BOX 2), which in certain contexts, such as dysbiosis or wound healing, can drive inflammatory signalling in the colon150.
Fig. 3 |. Tissue specific metabolism hijacked by KRAS*.
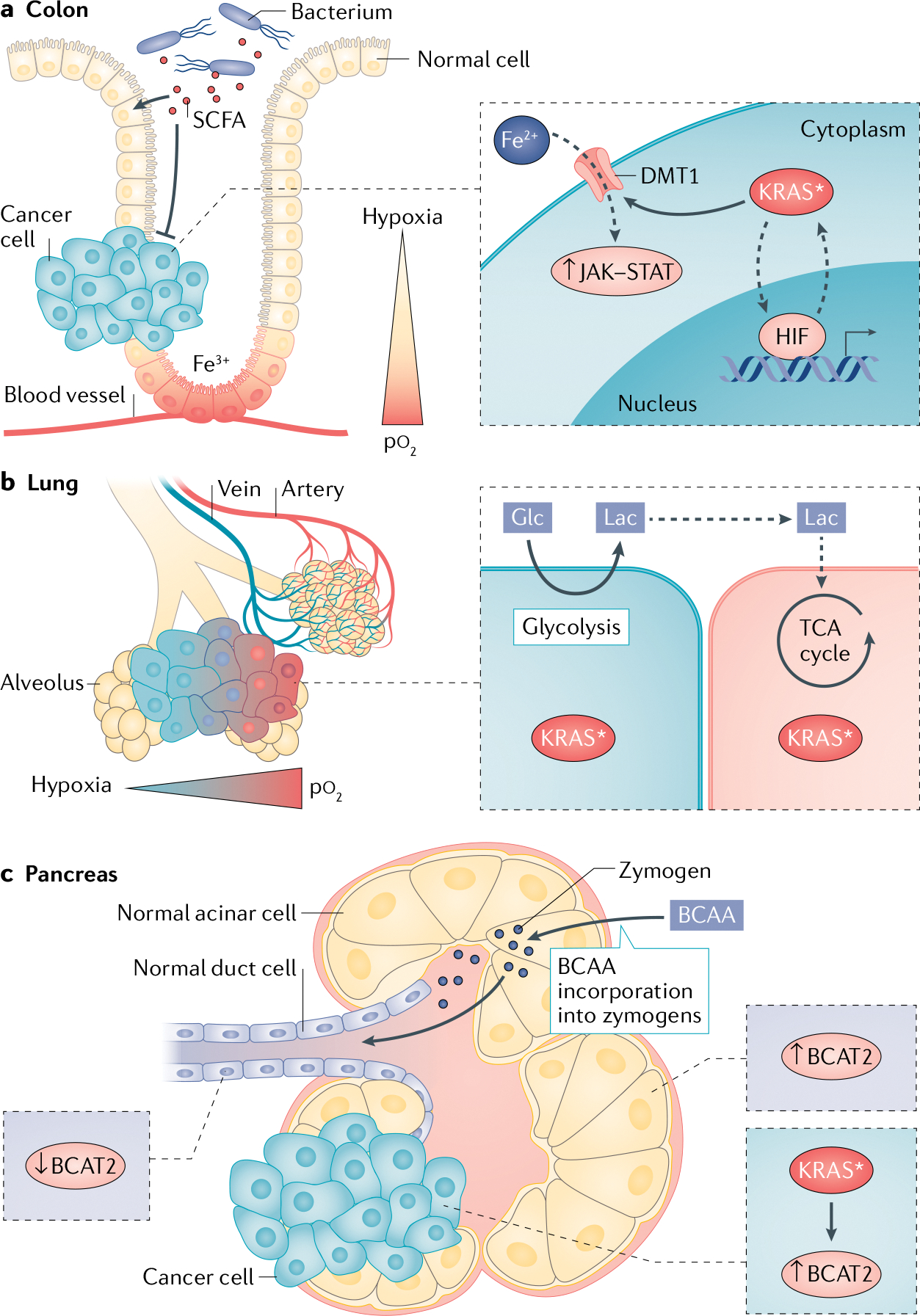
a | Hypoxic gradients in the colon stabilize hypoxia-inducible factors (HIFs) in normal colonocytes, which can then cooperate with mutant KRAS (KRAS*) following transformation. The hypoxic lumen also promotes colonization by short-chain fatty acid (SCFA)-producing bacteria. SCFAs are used by heathy colon cells for fuel and are prohibitive for development of colon tumours. Normal colonocytes are essential for iron (Fe3+) absorption, and iron (Fe2+) uptake through divalent metal transporter 1 (DMT1) is utilized by KRAS* colon cancer cells to drive protumorigenic JAK–STAT signalling. b | Normal lung epithelial cells demonstrate high glucose (Glc) uptake and export of lactate (Lac). In non-small-cell lung cancer, KRAS* cancer cells located far from a blood vessel in hypoxia utilize glycolysis and produce lactate, which can be taken up by tumour cells nearer to the blood vessel and utilized for oxidative respiration. c | Zymogen-producing acinar cells are branched-chain amino acid (BCAA) avid to obtain building blocks for peptides and digestive enzymes and display elevated branched-chain amino acid transaminase 2 (BCAT2) expression. BCAT2 levels are lower in normal ductal cells. During transformation by KRAS*, acinar cells dedifferentiate into ductal cells and progress to pancreatic ductal adenocarcinoma. KRAS* stabilizes and increases BCAT2 expression in cancer cells, demonstrating how the duct-like pancreatic ductal adenocarcinoma cells retain acinar cell metabolic programmes. pO2, oxygen tension; TCA, tricarboxylic acid.
Box 2 |. Metabolic interactions between oncogenic KRAS and the microbiota.
Symbiotic interactions between commensal microorganisms and host cells maintain tissue homeostasis. For instance, colonocytes in the normal colon rely on oxidative phosphorylation and deplete the lumen of oxygen, fostering the growth of obligate anaerobes that process dietary fibre into by-products for epithelial cells262. Some microbial products, such as the short-chain fatty acid (SCFA) butyrate, inhibit proliferation and renewal of stem cells located at the base of intestinal crypts, so colonocytes in the upper crypt consume and oxidize SCFAs for fuel as well as to protect the intestinal stem cells263.
Exposure to the SCFA butyrate produced by obligate anaerobes also caused cell cycle arrest in colon cancer cells in vitro264. Colon cells transformed by mutant KRAS (KRAS*) upregulate glycolysis and lactate release and decrease oxygen consumption. Elevated lactate levels in the lumen and increased oxygenation skew the microbiota towards facultative anaerobes that do not produce SCFAs from fibre262. In this manner, KRAS* signalling might lead to avoidance of the growth-inhibitory effects of butyrate.
Transformation by KRAS* disrupts tissue architecture in the colon, lung and pancreas and rewires metabolism. This induces dysbiosis to promote tumour progression and suppress the innate and adaptive immune systems265–268. Increased KRAS*-driven ERK and phosphatidylinositol 3-kinase (PI3K) signalling can lead to enriched commensal bacteria in lung tumours269. In the diseased lung, changes to the metabolome positively correlated with the bacterial load in the lower airways, indicating the microbiota could also shape the metabolic environment encountered by cancer cells270.
Future work will investigate how KRAS*-dysregulated metabolism shapes the tumour microbiota. For example, an understudied area concerns how the microbiota affects the composition of metabolites that regulate immune responses to cancer. Additionally, systemic changes to the broader host microbiota resulting from KRAS*-driven tumours could reciprocally affect the nutrients available to tumours.
Both hypoxia and chronic inflammation can contribute to the development of CRC. Hypoxia-inducible factors (HIFs) modulate the cellular response to low oxygen tension and serve to integrate oxygen sensing, metabolism and inflammation150–153. They are thus essential for proper functioning of cells in the colon152,154–156. Some evidence indicates an overlap between KRAS*-regulated and HIF-regulated genes in CRC157,158. Hypoxia increases expression of KRAS* in CRC cell lines159, and KRAS* reciprocally stabilizes HIF1α via several effector pathways, indicating a potential positive-feedback loop between HIFs and KRAS*160–162 (FIG. 3a), thereby promoting CRC cell survival in hypoxia. It also renders CRC cells dependent on HIF activity, as loss of HIF1α or HIF2α in CRC xenografts and autochthonous models has been shown to impair tumour cell proliferation163.
A crucial function of the colon is to absorb nutrients from the diet. Iron is required for red blood cell development and systemic delivery of oxygen and is a cofactor in numerous metabolic reactions. Iron import is regulated primarily by HIF2α in the colon164,165. In CRC, HIF2α-mediated iron import through divalent metal transporter 1 (DMT1) promoted JAK–STAT signalling and tumour growth in vivo166,167 (FIG. 3a). Currently the role of KRAS* in iron metabolism is unclear, although haem iron intake is correlated with an increased risk of colon tumours harbouring KRAS* (REFS157,158). Indeed, The Cancer Genome Atlas data revealed that in CRC, KRAS* potentiates the expression of iron importers166 (FIG. 3a). Therefore, KRAS* may initiate a feedforward cycle via regulation of HIF2α to sustain high iron levels for CRC growth. Further work on iron metabolism in CRC is warranted, especially given the recent interest in ferroptosis168–170 and the discovery that ferroptosis-inducing compounds are selectively lethal in cells engineered to express oncogenic HRAS-G12V171.
Oxygen gradients and alternative fuel sources in the lung.
The lung is essential for oxygen transfer and transport throughout the body120,130,131. Despite its high oxygen tension and proximity to oxygen-laden blood vessels, oxygen gradients still develop throughout the lung148 (FIG. 3b). Interestingly, work with tissue slices and perfused lungs decades ago discovered the normal lung is glucose avid and releases lactate130,131. This finding was confirmed recently by isotope tracing and metabolomics analysis172 in normal lungs of pigs showing that glucose-derived lactate was released in large quantities from the lung. Lactate was a major source of carbon for the TCA cycle within lung cells (in contrast to other organs such as the pancreas and colon)172. It is tempting to speculate that cells further from blood vessels convert glucose to lactate, which can be used by cells closer to oxygenated blood vessels as an alternative fuel source120 (FIG. 3b). The release of lactate from one cell population and its use as a carbon source in other cells seems to occur in NSCLC as well. Isotopic tracing experiments with labelled glucose in human patients demonstrated that NSCLC tumours are metabolically heterogeneous in their glucose utilization136. More perfused areas of the tumours used alternative fuel sources such as lactate, while poorly perfused regions relied on glycolysis and glucose incorporation into the TCA cycle136,173 (FIG. 3b).
Branched-chain amino acid metabolism in the pancreas.
Recent work has shown that PDA can arise from either acinar or ductal cells depending on mutational profiles and stromal cues. This has fuelled a critical debate regarding the putative cell of origin in PDA, although a common theory ascribes the origin of PDA to the dedifferentiation programme known as acinar-ductal metaplasia174,175. In normal physiology, acinar cells of the exocrine pancreas produce copious peptides and enzymes to aid digestion, making the pancreas highly amino acid avid to obtain the building blocks for these zymogens172,176 (FIG. 3c). Compared with ductal cells, acinar cells display elevated expression of the mitochondrial protein branched-chain amino acid transaminase 2 (BCAT2) and rapidly incorporate branched-chain amino acids (BCAAs) into protein176,177 (FIG. 3c). Despite low levels of BCAT2 in normal ductal cells, BCAT2 expression increased as KRAS*-transformed cells progressed from an acinar to a ductal fate177. Indeed, overexpression of KRAS in normal pancreatic ductal cells stabilized BCAT2 expression, and inhibition of BCAT2 hindered the formation of early pancreatic lesions in a genetic mouse model of tumorigenesis driven by KrasG12D (REF.177). These results suggest a model by which normal acinar cells express BCAT2 to regulate incorporation of BCAAs into proteins to perform their cellular functions, while KRAS*-harbouring ductal cells hijack BCAT2 catabolic activity to meet biosynthetic and energetic demands (FIG. 3c).
However, unlike in this model of PDA driven solely by KRAS*, tumours with both KRAS* and loss of p53 displayed low BCAA catabolism, and the growth of subcutaneous allografts in mice was not affected by dual loss of BCAT1 and BCAT2 (REF.178). Furthermore, a recent study found that pancreatic cancer-associated fibroblasts (CAFs) in the tumour stroma synthesized and released BCAAs, and that BCAT activity in human PDA cells was required for the use of BCAAs for oxidation and protein synthesis179. These seemingly contradictory findings regarding BCAA metabolism showcase the complex interplay between KRAS*, co-occurring mutations and environmental context.
Comparative studies of metabolic pathways.
In some studies, KRAS*-driven metabolic pathways have been directly contrasted between cancer types in different tissues. For instance, while glutamate oxaloacetate transaminase 1 (GOT1) is essential in PDA for protection from ROS, and GOT1 inhibition sensitized PDA cell lines to radiation in vitro and in murine xenografts, CRC cells were not affected by inhibition of GOT1 (REF.180). Interestingly, the aforementioned study demonstrating BCAA metabolism is not an essential pathway in PDA concurrently showed that NSCLC tumours also driven by KRAS* and LOF p53 had elevated flux through the BCATs and relied on BCAAs for tumour growth178, emphasizing the tissue dependence of the metabolic phenotype in cancer cells driven by the same genotype. Finally, the tissue of origin dictates whether a cancer cell depends on the Preiss–Handler de novo NAD+ synthesis or NAD salvage pathway181. Tumours arising in the pancreas have greater amplifications in Preiss–Handler pathway enzymes compared with those of the lung or colon, suggesting PDA depends more on NAD synthesis than salvage in comparison with NSCLC or CRC181. These studies collectively demonstrate that contextual cues such as the TME, cell of origin and model system must be considered in addition to oncogenic drivers when tumour metabolism is being studied.
Metabolism in the TME
KRAS*-driven programmes in cancer cells can shape the cellular composition and thus the vascularity and architecture of the TME. This, in turn, impacts the availability of nutrients and oxygen, while establishing gradients of pH and metabolic by-products, collectively dictating the metabolic nature of the TME182. Furthermore, the depletion or accumulation of metabolites, resulting from aberrant cancer metabolism, has ramifications for the function of surrounding immune and stromal cells182–186. To manage the metabolic challenges imposed by the TME, cancer and non-cancer cells participate in cooperative metabolic interactions that support tumour growth, as well as competitive metabolic interactions that promote tumour survival by limiting nutrient availability to, and thus the activity of, the antitumour immune system182,187,188 (FIG. 4).
Fig. 4 |. Interactions in the KRAS* TME.
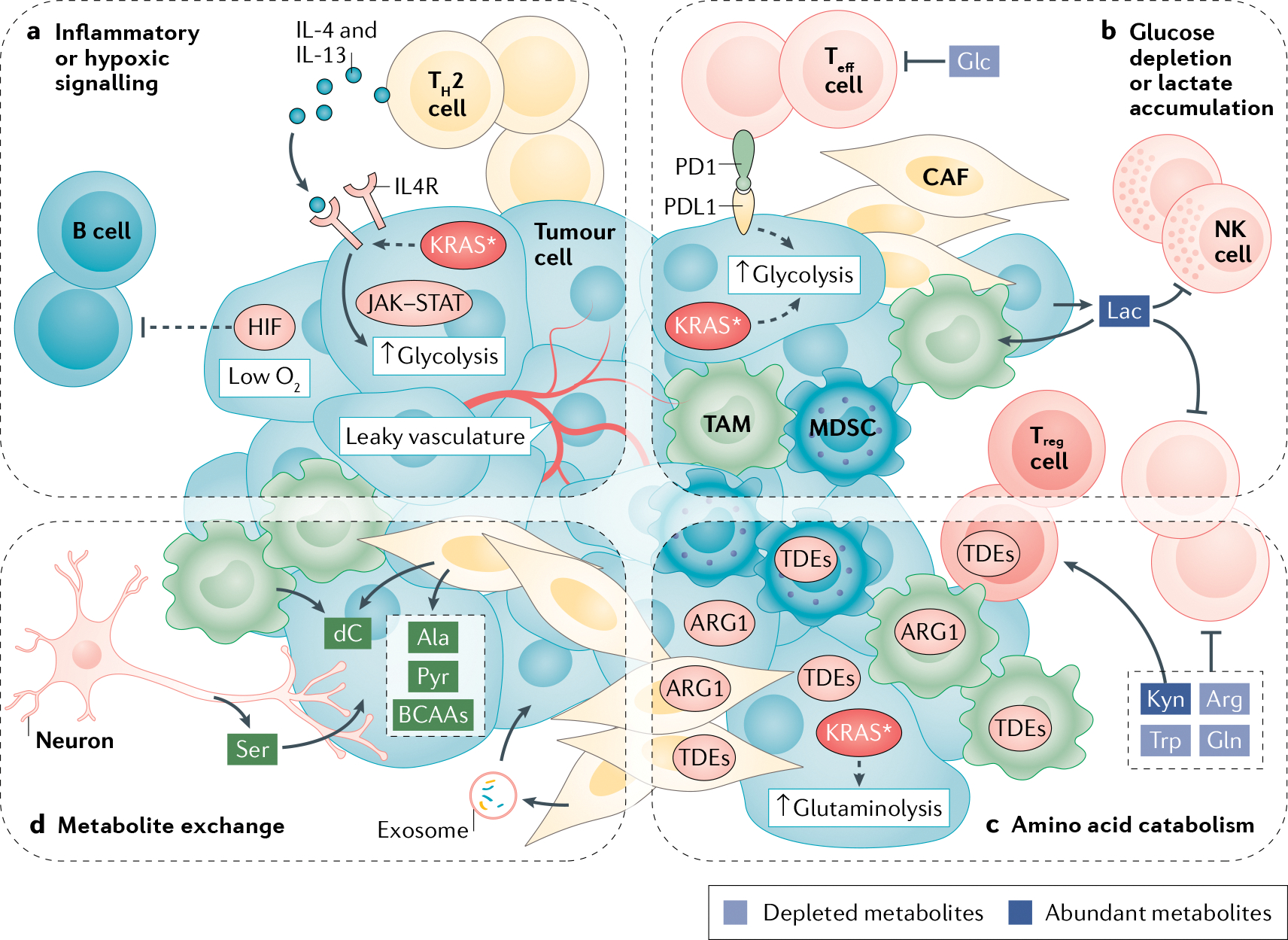
a | T helper 2 (TH2) cells in the tumour microenvironment (TME) release type I cytokines. Mutant KRAS (KRAS*) increases expression of their receptors on the surface of cancer cells. Downstream signalling through JAK–STAT activates MYC to increase expression of glycolytic enzymes. Hypoxia-inducible factor (HIF) activity excludes B cells from the TME. b | Glucose (Glc) is required by effector T cells (Teff cells) to mount an antitumour immune response, yet glucose is depleted due to elevated glycolysis in KRAS*-expressing cancer cells and cancer-associated fibroblasts (CAFs). Expression of programmed death ligand 1 (PDL1) on cancer cells and binding with its PD1 receptor on Teff cells also stimulates glycolysis in cancer cells. Increased levels of lactate (Lac) released into the extracellular space by cancer cells and taken up by other cells further inhibits glycolysis in Teff cells and natural killer (NK) cells, while polarizing macrophages to a tumour-associated macrophage (TAM) state. c | Elevated glutaminolysis and glutamine (Gln) uptake in KRAS* cells depletes glutamine, which is required by Teff cells. Expression of tryptophan-degrading enzymes (TDEs) on regulatory T cells (Treg cells), CAFs, TAMs and cancer cells degrades tryptophan (Trp) to its derivative kynurenine (Kyn), which promotes Treg cell activity. KRAS* cancer cells are auxotrophic for arginine (Arg) and express arginase 1 (ARG1) to break down arginine in the TME. ARG1 is also expressed in CAFs and TAMs. d | Induction of autophagy in CAFs increases release of alanine (Ala) for uptake and use by cancer cells. CAFs also release pyruvate (Pyr), branched-chain amino acids (BCAAs) and metabolite-filled exosomes to support dysregulated KRAS* metabolism in cancer cells. Deoxycytidine (dC) released from CAFs and TAMs is taken up by cancer cells and blunts chemotherapy efficacy. Innervating neurons release serine (Ser) to support protein translation in cancer cells under serine deprivation. MDSC, myeloid-derived suppressor cell.
Hypoxia and inflammation
Solid tumours are marked by a prolonged, dysregulated inflammatory response189. KRAS* drives inflammatory signalling early in tumour progression and cooperates with exogenous factors such as tobacco or pollutants in the lung, the microbiota in the colon and secretory enzymes in the pancreas to generate an inflammatory environment that potentiates oncogenic signalling190–193. Similarly, KRAS* drives aberrant proliferation and signalling, which contributes to tumour hypoxia182.
KRAS* signalling, tumour hypoxia and the activation of HIFs remodel the immune microenvironment in the tumour. For example, hypoxia in a poorly perfused, late-stage KRAS*-driven tumour drives expression of HIF1α, which led to B cell exclusion from the tumour in a mouse model of PDA194,195 (FIG. 4a). This could be speculated as a mechanism by which KRAS* leads to recruitment of immune cells to support tumour initiation under conditions of sufficient oxygen and low HIF expression, and exclusion of metabolically competitive immune cells via HIF activity late in tumour progression when oxygen and nutrients are scarce.
Proinflammatory signalling in the TME enhances dysregulated metabolic programmes in PDA. For instance, upon recruitment to the PDA tumour stroma, CD4+ T cells differentiate into the T helper 2 (TH2) subtype, which creates an immunosuppressive environment by promoting the anti-inflammatory phenotype of tumour-associated macrophages (TAMs)196. Of note, TH2 cell-derived IL-4 and IL-13 signalling supports the metabolism of PDA cells. In a mouse model of PDA, KRAS* upregulated surface expression of IL-4Rα197, rendering cancer cells more sensitive to TH2 cell-derived cytokines. This in turn elevated MYC activity via JAK–STAT signalling and increased glycolytic gene expression and flux in cancer cells, thereby contributing to tumour growth197 (FIG. 4a). Future work with KRAS*-driven mouse models of NSCLC and CRC could determine whether these phenotypes are unique to PDA or dependent on KRAS* across tumour types.
Glucose competition
Proliferative cell types in the TME, such as effector immune cells, are sensitive to the glucose-depleted state common in, but not exclusive to, KRAS*-driven tumours184,185,188. Low glucose levels in the TME resulting from elevated uptake and utilization by cancer cells prevent expansion of and cytokine release by T cells (FIG. 4b). Also, expression of glycolytic enzymes in samples from patients with NSCLC was inversely correlated with T cell infiltration198. Mechanistically, interactions between programmed death ligand 1 (PDL1) on murine CRC cancer cells and PD1 on T cells can stimulate glycolysis in cancer cells187 (FIG. 4b). This suggests that immune checkpoint blockade could dampen glycolysis in cancer cells, increasing glucose availability for effector T cells.
CAFs undergo metabolic reprogramming upon activation, marked by increased glycolysis and dependence on glucose183 (FIG. 4b). In support of this, CAFs from human CRC tissue had increased expression of glycolytic enzymes and decreased TCA cycle activity compared with normal fibroblasts199. Co-injection of these CAFs with CRC cell lines in mouse subcutaneous xenografts promoted tumour growth199. Isotope tracing studies similarly demonstrated that pancreatic CAFs are highly glycolytic41. Thus, while depriving antitumour immune cells of glucose clearly conferred a survival advantage on cancer cells as reported in several studies, limiting access of CAFs to glucose could impair tumour-promoting interactions between cancer cells and CAFs. Therefore, further work remains to determine how CAFs and cancer cells engage in cooperative glucose metabolism in a TME with limited glucose.
Lactate accumulation
Rapid glucose metabolism through glycolysis in KRAS*-driven cancer cells leads to an accumulation of lactate in the TME (FIG. 4b). Lactate is transported by the family of monocarboxylate transporters (MCTs)200,201. Interestingly, in tissue from primary PDAs, expression of the lactate export protein MCT4 correlated with expression of immunosuppressive markers in the TME202. In support of an immunosuppressive role of extracellular lactate in tumours, a recent study discovered that macrophages exposed to lactate-high medium conditioned by KRAS*-driven CRC cells, or supplemented with exogenous lactate, demonstrated increased expression of genes associated with the TAM anti-inflammatory state in contrast with macrophages cultured in normal growth media203 (FIG. 4b). Moreover, the accumulation of intracellular lactate in T cells blocked glycolytic flux, rendering T cells unable to perform cytotoxic functions against cancer cells204 (FIG. 4b). Furthermore, lactate in the TME of colon liver metastases induced apoptosis in infiltrating natural killer cells due to increased intracellular pH and production of mitochondrial ROS205 (FIG. 4b). Collectively, these data demonstrate how lactate, a metabolic by-product of KRAS*-driven metabolism in cancer cells, creates an immunosuppressive TME.
Regulation of amino acid levels
Effector T cells require specific amino acids to mount an antitumour response185. The metabolic pathways in KRAS*-expressing cancer cells lead to depletion of certain amino acids from the TME (FIG. 4c). Specifically, increased glutamine uptake and metabolism mediated by KRAS* depletes glutamine from the TME57,206,207 (FIG. 4c). This process deprives T cells of a fuel source vital for their activation, because effector T cells also rely on glutamine to proliferate and produce cytokines during an antitumour response207.
Tryptophan is another important amino acid for T cell responses, and its depletion limits effector T cell infiltration and expansion208–212. Although direct links between KRAS* driving tryptophan depletion remain to be uncovered, tryptophan is depleted from the TME in colon, lung and pancreas tumours due to the activity of tryptophan-degrading enzymes expressed by cancer cells, CAFs, myeloid-derived suppressor cells, and regulatory T cells185,207,212,213 (FIG. 4c).
There is considerable competition for the amino acid arginine in the KRAS*-shaped TME182 (FIG. 4c). Arginine catabolism or conversion serves as a source of nitrogen through the urea cycle, and generates proline, polyamines and/or nitric oxide. Arginine thus contributes to the biosynthetic capacity of tumour cells and/or the activity of proinflammatory or anti-inflammatory immune cells185,214. Lung and pancreatic cancer cells are unable to synthesize arginine as they do not express argininosuccinate synthetase (ASS) and therefore depend on exogenous arginine214. Colon cancer cells, however, express ASS and can generate arginine, providing them with a survival advantage in an arginine-depleted TME214. In terms of immune cells in the TME, proinflammatory M1-like macrophages have been shown to generate nitric oxide from arginine via inducible nitric oxide synthase (iNOS) to perform an antitumour cytotoxic response, whereas TAMs express arginase 1 (ARG1) to break down arginine into polyamines, which supports the anti-inflammatory response185. In addition, CAFs, and myeloid-derived suppressor cells in the TME also express ARG1195,215 (FIG. 4c). As effector T cells require arginine for full activation and cytotoxic responses207, these processes, combined with increased uptake of arginine by arginine-auxotrophic cancer cells, suppress the inflammatory, antitumour immune response182,191,203,212.
Cooperative interactions between cancer cells and the tumour stroma compensate for limited amino acid availability (FIG. 4d). As mentioned earlier, during nutrient deprivation, autophagy can be deployed to recycle critical metabolites. Indeed, CAFs in both NSCLC and PDA displayed elevated autophagy compared with normal fibroblasts216,217. This can be to the advantage of cancer cells, because, for example, elevated autophagy in pancreatic CAFs increased the availability of alanine to PDA cells, which was released by CAFs and taken up by PDA cells to fuel the TCA cycle217 (FIG. 4d). Pancreatic CAFs have also been shown to package numerous metabolites, including amino acids, into exosomes for delivery to cancer cells218 (FIG. 4d). Moreover, recent work demonstrated that pancreatic CAFs display elevated synthesis and release of BCAAs to be taken up and metabolized by PDA cells179, as highlighted earlier and in FIG. 4d.
Lastly, an emerging area of research concerns the role of the nervous system in regulating tumour metabolism. Pancreatic tumours are highly innervated, and the presence of neurons correlates with more aggressive disease and poorer outcomes219. A recent study revealed that under conditions of serine deprivation in mice with PDA, such as a serine-free diet, neurons were recruited to the tumour and synthesized and released serine for uptake by PDA cells (FIG. 4d). This allowed translation of proteins in cancer cells to proceed uninterrupted220. While this study was specific to PDA, future work in this emerging area could reveal whether this mechanism is also active in NSCLC or CRC.
Dysregulated lipid metabolism
The composition of lipids in the TME is affected by fatty acid metabolism of KRAS*-driven cancer cells, which access intracellular lipid stores or take up extracellular lipids91,97. This leads to the accumulation or depletion of lipid species in the TME, which can affect the metabolism of surrounding cells. For instance, long-chain fatty acids are abundant in the pancreatic TME221. In a mouse model of PDA, cytotoxic CD8+ T cells infiltrating PDAs imported long-chain fatty acids. However, these T cells had low expression of the enzyme critical for breakdown of long-chain fatty acids, and therefore fatty acids accumulated in T cells, leading to metabolic exhaustion and impaired cytotoxic activity221. T cells possess remarkable metabolic plasticity, and it is possible that signalling induced by KRAS* in cancer cells indirectly prevents utilization of alternative fuel sources in T cells, although this remains to be proven mechanistically207. New lipidomics technologies for studying lipid metabolism will enable their further characterization in other non-cancer cell types in the TME.
Targeting metabolism in KRAS* tumours
KRAS*-driven tumours have a remarkable ability to develop resistance to inhibition of KRAS* even after ablation of the oncogenic driver222–225. For instance, PI3K-mediated reactivation of the MAPK pathway, as well as focal adhesion kinase signalling, has been shown to drive resistance to KRAS* genetic silencing in vitro and in vivo226,227. (Of note, there is evidence suggesting in vitro culture conditions underestimate the essentiality of KRAS* for in vivo tumour growth228.) Furthermore, a KRAS-G12C-specific chemical inhibitor effectively inhibited in vitro and in vivo tumour growth, emphasizing caution is required when one is drawing inferences from genetic silencing of KRAS* (REF.228). Given the current lack of drugs targeting other KRAS mutants, an important strategy to consider is to target downstream metabolic effector pathways alone, in combinations with other effectors, or even in combination with inhibition of KRAS* to block resistance mechanisms inherent to cancer cells or driven by the TME.
Drug-induced vulnerabilities
Cancer cells can utilize metabolic mechanisms to compensate for loss of KRAS*. Therefore, genetic ablation of KRAS* in a mouse model of PDA led to tumour regression, but a subset of cancer cells persisted and reconstituted the tumour. These residual cells relied on oxidative phosphorylation (as opposed to glycolysis), inhibition of which in combination with oncogene ablation prevented tumour recurrence222.
As mentioned earlier, scavenging mechanisms are upregulated by KRAS* and are potential targets in combination with KRAS* inhibition. For example, knockdown of KRAS* or ERK inhibition in PDA cells in vitro led to an increase in autophagy, potentially to manage the bioenergetic defects imposed by MAPK pathway inhibition85,86,222. This notion was confirmed by blocking autophagy with chloroquine in combination with ERK inhibitors, which slowed PDA growth in mice and increased apoptosis in PDA cells in vitro85. Combining autophagy and ERK inhibition in PDA is now being examined in clinical trials (NCT04214418 and NCT04386057). Furthermore, targeting the regulatory machinery or signalling pathways associated with macropinocytosis inhibits tumour progression in NSCLC and PDA70,229, suggesting this strategy could undermine the ability of KRAS* cancer cells to survive under metabolic stress.
Like the cell intrinsic pathways, the TME can also support tumour growth after loss of KRAS* signalling. For example, pancreatic TAMs have been shown to release TGFβ, which drove tumour progression via SMAD4 in mice after doxycycline-inducible KRAS* had been extinguished223. Furthermore, short hairpin RNA-mediated knockdown of IL-4Rα or JAK chemical inhibition in KRAS* PDA cells abrogated the TH2 cell-derived cytokine cascade discussed earlier, and reduced tumour progression in mice197. While these findings were shown in PDA, similar mechanisms might occur in lung and colon tumours.
Vulnerabilities from co-occurring mutations
The inhibition of mechanisms induced by co-occurring mutations that cooperate with KRAS* is another avenue to consider when metabolism is being targeted in KRAS*-driven tumours. LKB1 loss mediated activation of AMPK in cell lines and rendered tumours sensitive to phenformin in a mouse model of KRAS*-driven NSCLC. This ETC-inhibiting drug selectively induced apoptosis and slowed growth in LKB1-deficient cells both in vitro and in vivo in comparison with NSCLC cells with loss of p53 (REF.230). Also, inhibiting the pyrimidine synthesis enzyme CPS1 in LKB1-deficient, KRAS*-driven human NSCLC cell lines (as compared with cell lines with either LKB1 loss of KRAS* alone) led to cellular DNA damage, cell cycle stalling and impaired tumour growth in murine xenografts122. Inhibition of another pyrimidine synthesis enzyme, deoxythymidylate kinase (DTYMK), was synthetic lethal in human and mouse LKB1-deficient lung cancer cells in vitro compared with wild-type LKB1 NSCLC cell lines231. Similarly, LKB1-deficient, KRAS*-driven mouse NSCLC cells cultured ex vivo were sensitized to inhibition of the serine biosynthesis pathway or DNA methylation, while cells with either KRAS* alone or co-occurring loss of p53 remained unaffected125. Additionally, inhibition of the hexosamine biosynthetic pathway enzyme glutamine–fructose 6-phosphate transaminase (isomerizing) 2 (GFPT2) with azaserine was a unique vulnerability for KRAS–LKB1-co-mutated NSCLC cell lines, xenografts and genetic tumour models compared with NSCLC with mutant p53 (REF.34). Finally, loss of the autophagic gene Atg7 was synthetic lethal in a KRAS*, LKB1-deficient NSCLC mouse model compared with KRAS*, p53-deficient tumours, suggesting that LKB1-deficient NSCLC tumours may be selectively sensitive to inhibition of autophagy121.
In NSCLC, KEAP1 mutations rendered mouse NSCLC orthotopic allografts and patient-derived xenografts dependent on glutaminolysis and sensitive to chemical inhibition of GLS1 with CB-839, while tumours with active KEAP1 were unaffected58,127. CB-839 is currently being tested in a clinical trial (NCT03872427). In addition, the overactive NRF2 antioxidant programme resulting from KEAP1 deficiency depleted intracellular glutamate levels in mouse NSCLC cells. As glutamate is needed to synthesize non-essential amino acids (NEAAs), including serine and glycine, glutamate depletion rendered these cells reliant on exogenous NEAAs. Thus, dietary intervention with a serine and glycine-free diet to deprive tumours of these NEAAs slowed mouse KEAP1-mutant allograft growth, yet this had no effect on wild-type KEAP1 tumours. Additionally, GLS1 inhibition with CB-839 further reduced intracellular glutamate levels and synergized with the NEAA-free diet in these models232. This is one example of how a specific dietary intervention informed by tumour genotype can improve metabolic therapies downstream of KRAS*.
Cooperative interactions in the stroma
The KRAS*-driven remodelling of the TME observed in PDA dictates much of the biology of these tumours138. Therefore, PDA is an optimal model in which to study metabolic interactions with the TME, and a wealth of literature exists on reprogramming the pancreatic tumour stroma. The activation and expansion of CAFs from quiescent fibroblasts drives the desmoplastic response in pancreatic tumours233, and CAFs can contribute to therapy resistance mechanisms in PDA. One of these mechanisms involves expression of the vitamin D receptor (VDR) in CAFs. Treatment of PDA-bearing mice with the VDR agonist calcipotriol caused CAFs to revert to an inactivated cell state, induced stromal remodelling and improved delivery of the chemotherapeutic gemcitabine233. Another mechanism of treatment resistance involves the release of pyruvate by CAFs (FIG. 4d). CAF-derived pyruvate decreased the efficacy of ETC complex I inhibitors in cancer cells, and blocking pyruvate import increased sensitivity to these therapeutics in vitro (non-peer reviewed data in REF.41). As mentioned previously, interactions with cancer cells induce CAFs in NSCLC and PDA to increase autophagy and supply cancer cells with critical metabolites216–218. Treatment of mice bearing KRAS*-driven tumours with autophagy inhibitors could block not only cancer cell metabolism but could also limit the availability of nutrients derived from the tumour stroma by reprogramming CAFs to a less activated state234.
CAFs have also been shown to release deoxycytidine (dC)235. From a study on TAMs in KRAS*-driven PDA, we know that dC can be released by TAMs and contribute to gemcitabine resistance236. Gemcitabine is structurally similar to dC, and TAM-derived dC was shown to compete with gemcitabine for uptake and metabolism by PDA cells, thereby lessening this chemotherapeutic’s efficacy. Thus, ablation of TAMs from tumours or targeting dC production in TAMs increased the response of PDA tumours to gemcitabine236. As dC secretion from CAFs might contribute to chemoresistance, these findings indicate that multiple cell types may cooperate to provide resistance to PDA cells to chemotherapy (FIG. 4d).
Lastly, as mentioned earlier, neurons recruited to the pancreatic TME release serine to support PDA metabolism. This interaction was impaired by treatment with a neuron-specific TRK inhibitor, to block innervation, in combination with a serine-free diet, and together these blocked tumour growth in PDA mouse models220.
Myriad cell types exist in KRAS* tumours. We have only recently begun to disentangle cooperative metabolic crosstalk between cancer cells and the TME, and future work has the potential to identify additional opportunities to block these supportive interactions.
Harnessing the immune system
The advent of immunotherapy has improved treatments for some cancers and demonstrated that the immune system can be reprogrammed to recognize and attack cancer cells237. However, colon, lung and pancreas tumours with KRAS* are among the least responsive cancers to immune checkpoint-based therapy238,239. This is in part due to non-metabolic factors such as the type of oncogenic driver, the overall tumour mutational burden or the degree of microsatellite instability (MSI), as, for example, MSI-high CRCs typically respond better to immunotherapy than MSI-low tumours238. The metabolic changes in the TME induced by KRAS* also suppress the antitumour immune system. Targeting these metabolic networks to reprogramme immune cells and/or prevent the depletion of metabolites required for an immune response has considerable potential to increase the efficacy of immunotherapy in these cancers.
The glutamine antagonist 6-diazo-5-oxo-l-norleucine (DON) broadly inhibits glutaminolysis by blocking the activities of glutamine deamidases. Because KRAS* induced dependence on glutamine in colon cancer cells, mice bearing syngeneic transplants of murine colon tumour cells were treated with DON to analyse its effect on tumour growth240. Surprisingly, DON treatment resulted not only in impaired mouse CRC allograft growth but also in an increased number of tumour-infiltrating T cells in comparison with untreated tumour-bearing mice240. While DON impaired glutamine metabolism in both cancer cells and T cells, T cells displayed remarkable metabolic plasticity and compensated for the lack of glutamine-derived carbon in the TCA cycle by incorporating glucose-derived or acetate-derived carbons instead240–242. This endowed T cells with a robust proliferative and energetic response and conferred a long-lasting memory phenotype240,242. Immune checkpoint inhibition with anti-PD1 synergized with DON treatment to inhibit tumour progression240. KRAS*-driven NSCLCs that harbour co-occurring mutations in KEAP1 and/or LKB1 are often resistant to immune checkpoint-based therapy243–246. Thus, a prodrug derivative of DON is now in clinical trials for treatment of multiple cancers, and a phase II trial will test its efficacy in KEAP1-mutant and LKB1-mutant NSCLC (NCT04471415).
Similarly, a potential synergy between GLS1 inhibition and immune checkpoint blockade is currently being tested in a phase II clinical trial in KEAP1-mutant tumours (NCT04265534). Future studies will need to determine how LKB1 and KEAP1 mutations might promote immune evasion in NSCLC and identify novel or existing metabolic therapies that can enhance antitumour immune responses in combination with immune checkpoint blockade.
Immunologically ‘cold’ PDAs are also resistant to immune checkpoint blockade247. A recent study demonstrated that activity of autophagy in PDA in vivo leads to the selective lysosomal degradation of major histocompatibility complex class I (MHC-I) in cancer cells248. MHC-I is required for antigen presentation on cancer cells and recognition by the immune system. Thus, MHC-I downregulation allowed PDA cells to evade the antitumour immune system. Additionally, autophagy was implicated in preventing T cell killing through modulation of TNF signalling249. Indeed, in mouse models of PDA, autophagy inhibition with chloroquine synergized with both anti-PD1 and anti-CTLA4 to inhibit tumour progression248.
In summary, targeting the metabolism of KRAS* tumours can provide nutrients to the immune system to increase the number and heighten the activity of the antitumour effector cells, priming tumours for immune checkpoint blockade therapy.
Conclusion
A better understanding of effector pathways downstream of KRAS*, such as dysregulated metabolism, could lead to enhanced therapeutic interventions alone or in combination with direct inhibition of KRAS*. This will require future work to disentangle metabolic pathways mediated by specific KRAS mutations and synergy with co-occurring mutations, as well contextual cues determined by the tissue of origin.
We are only beginning to appreciate how KRAS* signalling directly and indirectly shapes the broader metabolism of the TME. Continuing to identify metabolic networks that support KRAS*-driven metabolism, mediate resistance to therapy and suppress the immune system will identify opportunities to cut off nutrient sources, enhance chemotherapy and/or promote antitumour immune responses. Cutting-edge approaches such as in vivo isotope tracing, spatially resolved mass spectrometry and single-cell sequencing technologies will be critical in these endeavours. Moving forwards, exploiting metabolic networks in CRC, NSCLC and PDA according to both genetic and environment contexts is a promising therapeutic strategy for improving the treatment of KRAS*-driven tumours.
Acknowledgements
S.A.K. is supported by NIH award F31CA247457. T.P. is supported by NIH grants R37CA222504 and R01CA227649 and American Cancer Society Research Scholar Grant RSG-17-200-01-TBE. Y.M.S. is supported by NIH grants R01CA245546 and R01CA148828. C.A.L. is supported by NIH grants R37CA237421, R01CA248160 and R01CA244931.
Glossary
- Glycolysis
The enzymatic oxidation of glucose to pyruvate, which produces energy and carbon for bioenergetic and biosynthetic processes.
- Oxidation state
In cellular metabolism, the availability of electron acceptors as determined by the breakdown or production of metabolites.
- Mitophagy
Selective targeting and degradation of mitochondria, often those that are defective or excessive.
- Electron transport chain (ETC)
A series of electron-accepting enzymes embedded in the inner mitochondrial membrane responsible for producing the proton-motive force needed for energy production.
- Reactive oxygen species (ROS)
Small, oxygen-containing molecules that are highly reactive due to the electron-accepting nature of oxygen.
- Mitochondrial fission or mitochondrial fusion
A process by which mitochondria are combined (fusion) or divided into smaller fragments (fission).
- Glutaminolysis
The enzymatic breakdown of the amino acid glutamine.
- Tricarboxylic acid (TCA) cycle
A series of mitochondrial enzymatic reactions that oxidize metabolic intermediates to produce reducing equivalents that drive the electron transport chain and intermediates for the synthesis of lipids and amino acids.
- Anaplerosis
Replenishment of tricarboxylic acid cycle intermediates.
- Macropinocytosis
Regulated, non-specific engulfment of the extracellular space by the cellular plasma membrane to obtain nutrients.
- Autophagy
Degradation of intracellular metabolites and organelles to eliminate damaged cellular components and/or provide basic metabolic building blocks.
- Urea cycle
A series of chemical reactions involving nitrogen-containing metabolites essential for recycling nitrogen or excreting toxic ammonia waste as urea.
- Glutathione (GSH).
Cellular antioxidant composed of glutamate, cysteine and glycine important for quenching damaging reactive oxygen species levels.
- Cystine
The water-soluble dimer of the amino acid cysteine.
- System XC−
Amino acid antiporter system that exchanges glutamate for cystine.
- Oxidative phosphorylation
Oxygen-dependent process by which the proton-motive force between the inner mitochondrial membrane space and matrix is used to generate ATP.
- Hypoxia-inducible factors (HIFs).
Transcription factors, stabilized under conditions of low oxygen levels, targeting the promoters of genes containing hypoxia response elements.
- Ferroptosis
An oxidative, iron-dependent form of programmed cell death.
- Isotope tracing
Application of metabolites in which stable isotopes are incorporated to follow the metabolism or fate of a given nutrient (for example via mass spectroscopy-based metabolomics).
- Acinar–ductal metaplasia
The morphological and transcriptional programmes by which acinar cells dedifferentiate into ductal cells in response to injury or oncogenic stress.
- Pyrimidine synthesis
The metabolic pathway producing the nucleotides uridine, cytidine and thymidine.
- Microsatellite instability (MSI).
An inherent propensity for genomic mutations caused by malfunctioning DNA repair machinery.
- Deamidases
Enzymes that catalyse the removal of amido groups.
Footnotes
Competing interests
T.P. has received honoraria and consulting fees from Calithera Biosciences and research support from Dracen Pharmaceuticals and Agios Pharmaceuticals. C.A.L. has received consulting fees from Astellas Pharmaceuticals and is an inventor on patents pertaining to KRAS-regulated metabolic pathways, redox control pathways in pancreatic cancer and targeting the GOT1 pathway as a therapeutic approach. Y.M.S. and S.A.K. have no competing interests to declare.
Peer review information
Nature Reviews Cancer thanks C. Der, P. Dey, J.Y. Guo and Ö. Yilmaz for their contribution to the peer review of this work.
References
- 1.Mo SP, Coulson JM & Prior IA RAS variant signalling. Biochem. Soc. Trans. 46, 1325–1332 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore AR, Rosenberg SC, McCormick F & Malek S RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov. 19, 533–552 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prior IA, Lewis PD & Mattos C A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanchez-Vega F et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 173, 321–337.e310 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prior IA, Hood FE & Hartley JL The frequency of ras mutations in cancer. Cancer Res. 80, 2969–2974 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox AD, Fesik SW, Kimmelman AC, Luo J & Der CJ Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 13, 828–851 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Ledford H Cancer: the Ras renaissance. Nature 520, 278–280 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Cox AD, Der CJ & Philips MR Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin. Cancer Res. 21, 1819–1827 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li S, Balmain A & Counter CM A model for RAS mutation patterns in cancers: finding the sweet spot. Nat. Rev. Cancer 18, 767–777 (2018). [DOI] [PubMed] [Google Scholar]
- 11. Zafra MP et al. An in vivo Kras allelic series reveals distinct phenotypes of common oncogenic variants. Cancer Discov. 10, 1654–1671 (2020). This study generated mice harbouring a series of Kras mutations in the pancreas or lung and reported significant differences in transformative capabilities and responses to treatments.
- 12.Serebriiskii IG et al. Comprehensive characterization of RAS mutations in colon and rectal cancers in old and young patients. Nat. Commun. 10, 3722 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins MA et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Invest. 122, 639–653 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fearon ER & Vogelstein B A genetic model for colorectal tumorigenesis. Cell 61, 759–767 (1990). [DOI] [PubMed] [Google Scholar]
- 15.Herbst RS, Morgensztern D & Boshoff C The biology and management of non-small cell lung cancer. Nature 553, 446–454 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Skoulidis F & Heymach JV Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 19, 495–509 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crawford HC, Pasca di Magliano M & Banerjee S Signaling networks that control cellular plasticity in pancreatic tumorigenesis, progression, and metastasis. Gastroenterology 156, 2073–2084 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peddareddigari VG, Wang D & Dubois RN The tumor microenvironment in colorectal carcinogenesis. Cancer Microenviron. 3, 149–166 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lambrechts D et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 24, 1277–1289 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Zeitouni D, Pylayeva-Gupta Y, Der CJ & Bryant KL KRAS mutant pancreatic cancer: no lone path to an effective treatment. Cancers 8, 45 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bryant KL, Mancias JD, Kimmelman AC & Der CJ KRAS: feeding pancreatic cancer proliferation. Trends Biochem. Sci. 39, 91–100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kerr EM, Gaude E, Turrell FK, Frezza C & Martins CP Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 531, 110–113 (2016). This study demonstrates that a gain in copy number of mutant Kras late in lung tumour progression results in substantial metabolic rewiring.
- 23. Ying H et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670 (2012). This study illustrates how oncogenic Kras signalling regulates metabolism through MAPK signalling and MYC activity in pancreatic cancer.
- 24.Yun J et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325, 1555–1559 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pupo E, Avanzato D, Middonti E, Bussolino F & Lanzetti L KRAS-driven metabolic rewiring reveals novel actionable targets in cancer. Front. Oncol. 9, 848 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaglio D et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 7, 523 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kerr EM & Martins CP Metabolic rewiring in mutant Kras lung cancer. FEBS J. 285, 28–41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hutton JE et al. Oncogenic KRAS and BRAF drive metabolic reprogramming in colorectal cancer. Mol. Cell Proteom. 15, 2924–2938 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aguilera O et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 7, 47954–47965 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iwamoto M et al. Regulation of 18F-FDG accumulation in colorectal cancer cells with mutated KRAS. J. Nucl. Med. 55, 2038–2044 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Kawada K et al. Relationship between 18F-fluorodeoxyglucose accumulation and KRAS/BRAF mutations in colorectal cancer. Clin. Cancer Res. 18, 1696 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Amendola CR et al. KRAS4A directly regulates hexokinase 1. Nature 576, 482–486 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H et al. Inhibition of glycolytic enzyme hexokinase II (HK2) suppresses lung tumor growth. Cancer Cell Int. 16, 9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim J et al. The hexosamine biosynthesis pathway is a targetable liability in KRAS/LKB1 mutant lung cancer. Nat. Metab. 10.1038/s42255-020-00316-0 (2020). This study identifies loss of LKB1 in KRAS* lung cancer results in an increased dependency on the hexosamine biosynthesis pathway, providing a prime example for how co-occurring mutations cooperate with KRAS* to direct metabolism.
- 35.Santana-Codina N et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 9, 4945 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ali ES et al. ERK2 phosphorylates PFAS to mediate posttranslational control of de novo purine synthesis. Mol. Cell 78, 1178–1191.e1176 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mattaini KR, Sullivan MR & Vander Heiden MG The importance of serine metabolism in cancer. J. Cell Biol. 214, 249–257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maddocks ODK et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Xie H et al. Targeting lactate dehydrogenase–a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 19, 795–809 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCleland ML et al. Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin. Cancer Res. 19, 773–784 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Kerk SA et al. The pancreatic tumor microenvironment buffers redox imbalance imposed by disrupted mitochondrial metabolism. Preprint at bioRxiv 10.1101/2020.08.07.238766 (2020). [DOI] [Google Scholar]
- 42.Baek G et al. MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep. 9, 2233–2249 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Graziano F et al. Glycolysis gene expression analysis and selective metabolic advantage in the clinical progression of colorectal cancer. Pharmacogenomics J. 17, 258–264 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Martinez-Reyes I & Chandel NS Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weinberg SE & Chandel NS Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 11, 9–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schvartzman JM, Thompson CB & Finley LWS Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 217, 2247–2259 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vasan K, Werner M & Chandel NS Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 32, 341–352 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kong H & Chandel NS Regulation of redox balance in cancer and T cells. J. Biol. Chem. 293, 7499–7507 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chandel NS Evolution of mitochondria as signaling organelles. Cell Metab. 22, 204–206 (2015). [DOI] [PubMed] [Google Scholar]
- 50. Humpton TJ et al. Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discov. 9, 1268–1287 (2019). This study finds that in mouse models of PDA KRAS* regulates mitochondrial dynamics and function to optimize ROS signalling and maximize bioenergetic programmes.
- 51.Sullivan LB & Chandel NS Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2, 17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Weinberg F et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl Acad. Sci. USA 107, 8788–8793 (2010). This is among the first studies to illustrate how mutant Kras influences metabolism.
- 53.Chauhan SS et al. PIM kinases alter mitochondrial dynamics and chemosensitivity in lung cancer. Oncogene 39, 2597–2611 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kashatus JA et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 57, 537–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu M et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 10.1172/jci.insight.126915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hensley CT, Wasti AT & DeBerardinis RJ Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Invest. 123, 3678–3684 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Son J et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Davidson SM et al. Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528 (2016). This study extensively details the differential utilization of glucose and glutamine in lung cancer cells between in vitro and in vivo models.
- 59.Miyo M et al. Metabolic adaptation to nutritional stress in human colorectal cancer. Sci. Rep. 6, 38415 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuneva MO et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 15, 157–170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mayers JR & Vander Heiden MG Famine versus feast: understanding the metabolism of tumors in vivo. Trends Biochem. Sci. 40, 130–140 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wong CC et al. SLC25A22 promotes proliferation and survival of colorectal cancer cells With KRAS mutations and xenograft tumor progression in mice via intracellular synthesis of aspartate. Gastroenterology 151, 945–960 e946 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Toda K et al. Metabolic alterations caused by KRAS mutations in colorectal cancer contribute to cell adaptation to glutamine depletion by upregulation of asparagine synthetase. Neoplasia 18, 654–665 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Halbrook CJ et al. Clonal heterogeneity supports mitochondrial metabolism in pancreatic cancer. Preprint at bioRxiv 10.1101/2020.05.15.098368 (2020). [DOI] [Google Scholar]
- 65. Gwinn DM et al. Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L-asparaginase. Cancer Cell 33, 91–107.e106 (2018). This study shows that under nutrient deprivation, signalling downstream of KRAS* in KEAP1-deficient lung cancer increases asparagine production and that these tumours are sensitive to asparagine depletion via asparaginase.
- 66.Bott AJ et al. Glutamine anabolism plays a critical role in pancreatic cancer by coupling carbon and nitrogen metabolism. Cell Rep. 29, 1287–1298. e1286 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamphorst JJ et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 75, 544–553 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Commisso C et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 (2013). This early work engineers normal cell types to express KRAS* and identifies that KRAS* upregulates macropinocytosis to non-specifically consume extracellular nutrients.
- 69.Ramirez C, Hauser AD, Vucic EA & Bar-Sagi D Plasma membrane V-ATPase controls oncogenic RAS-induced macropinocytosis. Nature 576, 477–481 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hodakoski C et al. Rac-mediated macropinocytosis of extracellular protein promotes glucose independence in non-small cell lung cancer. Cancers 10.3390/cancers11010037 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.King B, Araki J, Palm W & Thompson CB Yap/Taz promote the scavenging of extracellular nutrients through macropinocytosis. Genes Dev. 34, 1345–1358 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Palm W, Araki J, King B, DeMatteo RG & Thompson CB Critical role for PI3-kinase in regulating the use of proteins as an amino acid source. Proc. Natl Acad. Sci. USA 114, E8628–E8636 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y & Commisso C Macropinocytosis in cancer: a complex signaling network. Trends Cancer 5, 332–334 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee SW et al. EGFR-Pak signaling selectively regulates glutamine deprivation-induced macropinocytosis. Dev. Cell 50, 381–392 e385 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hobbs GA et al. Atypical KRAS(G12R) mutant is impaired in PI3K signaling and macropinocytosis in pancreatic cancer. Cancer Discov. 10, 104–123 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kimmelman AC & White E Autophagy and tumor metabolism. Cell Metab. 25, 1037–1043 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perera RM et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Perera RM & Bardeesy N Pancreatic cancer metabolism: breaking it down to build it back up. Cancer Discov. 5, 1247–1261 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang A et al. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov. 8, 276–287 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang S et al. Autophagy inhibition dysregulates TBK1 signaling and promotes pancreatic inflammation. Cancer Immunol. Res. 4, 520–530 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Guo JY et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470 (2011). This work provides evidence that mutant RAS upregulates autophagy to provide nutrients and sustain metabolic programmes during acute nutrient stress.
- 82.Amaravadi RK, Kimmelman AC & Debnath J Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 9, 1167–1181 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.White E Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 27, 2065–2071 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guo JY et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 30, 1704–1717 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bryant KL et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 25, 628–640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.White E Blockade of RAF and autophagy is the one-two punch to take out Ras. Proc. Natl Acad. Sci. USA 116, 3965–3967 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Poillet-Perez L et al. Autophagy maintains tumour growth through circulating arginine. Nature 563, 569–573 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Karsli-Uzunbas G et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4, 914–927 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Currie E et al. Cellular fatty acid metabolism and cancer. Cell Metab. 18, 153–161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Singh A et al. De novo lipogenesis represents a therapeutic target in mutant Kras non-small cell lung cancer. FASEB J. 10.1096/fj.201800204 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gouw AM et al. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc. Natl Acad. Sci. USA 114, 4300–4305 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Svensson RU et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 22, 1108–1119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zaytseva YY et al. Increased expression of fatty acid synthase provides a survival advantage to colorectal cancer cells via upregulation of cellular respiration. Oncotarget 6, 18891–18904 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Padanad MS et al. Fatty acid oxidation mediated by acyl-CoA synthetase long chain 3 is required for mutant KRAS lung tumorigenesis. Cell Rep. 16, 1614–1628 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kamphorst JJ et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl Acad. Sci. USA 110, 8882–8887 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guillaumond F et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl Acad. Sci. USA 112, 2473–2478 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rozeveld CN, Johnson KM, Zhang L & Razidlo GL KRAS controls pancreatic cancer cell lipid metabolism and invasive potential through the lipase HSL. Cancer Res. 80, 4932 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Phelps RA et al. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell 137, 623–634 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wong CC et al. In colorectal cancer cells with mutant KRAS, SLC25A22-mediated glutaminolysis reduces DNA demethylation to increase WNT signaling, stemness, and drug resistance. Gastroenterology 10.1053/j.gastro.2020.08.016 (2020). [DOI] [PubMed] [Google Scholar]
- 100.Stine ZE, Walton ZE, Altman BJ, Hsieh AL & Dang CV MYC, Metabolism, and Cancer. Cancer Discov. 5, 1024 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sansom OJ et al. Myc deletion rescues Apc deficiency in the small intestine. Nature 446, 676–679 (2007). [DOI] [PubMed] [Google Scholar]
- 102.Satoh K et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc. Natl Acad. Sci. USA 10.1073/pnas.1710366114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Venkateswaran N et al. MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 33, 1236–1251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Soucek L et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 27, 504–513 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sodir NM MYC instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Discov. 10, 588–607 (2020). [DOI] [PubMed] [Google Scholar]
- 106.Beyaz S et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531, 53–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mihaylova MM et al. Fasting activates fatty acid oxidation to enhance intestinal stem cell function during homeostasis and aging. Cell Stem Cell 22, 769–778.e764 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Boutin AT et al. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes Dev. 31, 370–382 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Feng Y et al. Mutant kras promotes hyperplasia and alters differentiation in the colon epithelium but does not expand the presumptive stem cell pool. Gastroenterology 141, 1003–1013.e1010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hinoi T et al. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 67, 9721 (2007). [DOI] [PubMed] [Google Scholar]
- 111.Tang J et al. Trp53 null and R270H mutant alleles have comparable effects in regulating invasion, metastasis, and gene expression in mouse colon tumorigenesis. Lab. Invest. 99, 1454–1469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jeon SM, Chandel NS & Hay N AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shackelford DB & Shaw RJ The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9, 563–575 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Murray CW et al. An LKB1-SIK axis suppresses lung tumor growth and controls differentiation. Cancer Discov. 9, 1590–1605 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hollstein PE et al. The AMPK-related kinases SIK1 and SIK3 mediate key tumor-suppressive effects of LKB1 in NSCLC. Cancer Discov. 9, 1606–1627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shaw RJ et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl Acad. Sci. USA 101, 3329–3335 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shaw RJ et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Faubert B et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc. Natl Acad. Sci. USA 111, 2554–2559 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Eichner LJ et al. Genetic analysis reveals AMPK is required to support tumor growth in murine kras-dependent lung cancer models. Cell Metab. 29, 285–302 e287 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu G & Summer R Cellular metabolism in lung health and disease. Annu. Rev. Physiol. 81, 403–428 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bhatt V et al. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes Dev. 33, 150–165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kim J et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546, 168–172 (2017). This work shows that lung tumours with KRAS* and LKB1 deficiency utilize an unconventional pathway to recycle nitrogen for use in vital nucleotide production.
- 123.Su GH et al. Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am. J. Pathol. 154, 1835–1840 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Morton JP et al. LKB1 haploinsufficiency cooperates with Kras to promote pancreatic cancer through suppression of p21-dependent growth arrest. Gastroenterology 139, 586–597.e5976 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kottakis F et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539, 390–395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Campbell JD et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 48, 607–616 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Romero R et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 23, 1362–1368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.DeNicola GM et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.DeNicola GM et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 47, 1475–1481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tierney DF Lung metabolism and biochemistry. Annu. Rev. Physiol. 36, 209–231 (1974). [DOI] [PubMed] [Google Scholar]
- 131.Fisher AB Intermediary metabolism of the lung. Env. Health Perspect. 55, 149–158 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mitsuishi Y et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79 (2012). [DOI] [PubMed] [Google Scholar]
- 133.Ghergurovich JM et al. Glucose-6-phosphate dehydrogenase is not essential for K-ras–driven tumor growth or metastasis. Cancer Res. 80, 3820 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Galan-Cobo A et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 79, 3251–3267 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sayin VI et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife 10.7554/eLife.28083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hensley CT et al. Metabolic heterogeneity in human lung tumors. Cell 164, 681–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bailey P et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016). [DOI] [PubMed] [Google Scholar]
- 138.Ryan DP, Hong TS & Bardeesy N Pancreatic adenocarcinoma. N. Engl. J. Med. 371, 1039–1049 (2014). [DOI] [PubMed] [Google Scholar]
- 139.Rosenfeldt MT et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 504, 296–300 (2013). [DOI] [PubMed] [Google Scholar]
- 140.Yang A et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 4, 905–913 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yang S et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Morris JPT et al. alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573, 595–599 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Aguirre AJ et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 17, 3112–3126 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ju HQ et al. Mutant Kras- and p16-regulated NOX4 activation overcomes metabolic checkpoints in development of pancreatic ductal adenocarcinoma. Nat. Commun. 8, 14437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Dey P et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 542, 119–123 (2017). This study identifies that in PDA ME2 is near the SMAD4 locus and is lost concurrently with SMAD4 deletion, conferring a unique dependency for PDA on ME3 activity.
- 146.Collisson EA et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Berkers CR, Maddocks ODK, Cheung EC, Mor I & Vousden KH Metabolic regulation by p53 family members. Cell metabolism 18, 617–633 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ortiz-Prado E, Dunn JF, Vasconez J, Castillo D & Viscor G Partial pressure of oxygen in the human body: a general review. Am. J. Blood Res. 9, 1–14 (2019). [PMC free article] [PubMed] [Google Scholar]
- 149.Albenberg L et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 147, 1055–1063 e1058 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shay JE & Celeste Simon M Hypoxia-inducible factors: crosstalk between inflammation and metabolism. Semin. Cell Dev. Biol. 23, 389–394 (2012). [DOI] [PubMed] [Google Scholar]
- 151.McGettrick AF & O’Neill LAJ The role of HIF in immunity and inflammation. Cell Metab. 32, 524–536 (2020). [DOI] [PubMed] [Google Scholar]
- 152.Schofield CJ & Ratcliffe PJ Signalling hypoxia by HIF hydroxylases. Biochem. Biophys. Res. Commun. 338, 617–626 (2005). [DOI] [PubMed] [Google Scholar]
- 153.Semenza GL HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest. 123, 3664–3671 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Furuta GT et al. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J. Exp. Med. 193, 1027–1034 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Xie L et al. Hypoxia-inducible factor/MAZ-dependent induction of caveolin-1 regulates colon permeability through suppression of occludin, leading to hypoxia-induced inflammation. Mol. Cell Biol. 34, 3013–3023 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Manresa MC & Taylor CT Hypoxia inducible factor (HIF) hydroxylases as regulators of intestinal epithelial barrier function. Cell Mol. Gastroenterol. Hepatol. 3, 303–315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gilsing AM et al. Dietary heme iron and the risk of colorectal cancer with specific mutations in KRAS and APC. Carcinogenesis 34, 2757–2766 (2013). [DOI] [PubMed] [Google Scholar]
- 158.Chun SY et al. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1alpha and HIF-2alpha target genes. Mol. Cancer 9, 29 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zeng M, Kikuchi H, Pino MS & Chung DC Hypoxia activates the K-ras proto-oncogene to stimulate angiogenesis and inhibit apoptosis in colon cancer cells. PLoS ONE 5, e10966 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Richard DE, Berra E, Gothie E, Roux D & Pouyssegur J p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J. Biol. Chem. 274, 32631–32637 (1999). [DOI] [PubMed] [Google Scholar]
- 161.Mylonis I et al. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J. Biol. Chem. 281, 33095–33106 (2006). [DOI] [PubMed] [Google Scholar]
- 162.Kikuchi H, Pino MS, Zeng M, Shirasawa S & Chung DC Oncogenic KRAS and BRAF differentially regulate hypoxia-inducible factor-1alpha and −2alpha in colon cancer. Cancer Res. 69, 8499–8506 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dang DT et al. Hypoxia-inducible factor-1alpha promotes nonhypoxia-mediated proliferation in colon cancer cells and xenografts. Cancer Res. 66, 1684–1936 (2006). [DOI] [PubMed] [Google Scholar]
- 164.Taylor M et al. Hypoxia-inducible factor-2alpha mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology 140, 2044–2055 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Schwartz AJ et al. Hepatic hepcidin/intestinal HIF-2alpha axis maintains iron absorption during iron deficiency and overload. J. Clin. Invest. 129, 336–348 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Xue X et al. Iron uptake via DMT1 integrates cell cycle with JAK-STAT3 signaling to promote colorectal tumorigenesis. Cell Metab. 24, 447–461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Xue X et al. Hypoxia-inducible factor-2alpha activation promotes colorectal cancer progression by dysregulating iron homeostasis. Cancer Res. 72, 2285–2293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Stockwell BR & Jiang X The chemistry and biology of ferroptosis. Cell Chem. Biol. 27, 365–375 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Stockwell BR, Jiang X & Gu W Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. 30, 478–490 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Stockwell BR et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yang WS & Stockwell BR Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hui S et al. Quantitative fluxomics of circulating metabolites. Cell Metab. 32, 676–688 e674 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hui S et al. Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bailey JM et al. p53 mutations cooperate with oncogenic Kras to promote adenocarcinoma from pancreatic ductal cells. Oncogene 35, 4282–4288 (2016). [DOI] [PubMed] [Google Scholar]
- 175.Seino T et al. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell 22, 454–467. e456 (2018). [DOI] [PubMed] [Google Scholar]
- 176.Jang C et al. Metabolite exchange between mammalian organs quantified in pigs. Cell Metab. 30, 594–606 e593 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Li JT et al. BCAT2-mediated BCAA catabolism is critical for development of pancreatic ductal adenocarcinoma. Nat. Cell Biol. 22, 167–174 (2020). [DOI] [PubMed] [Google Scholar]
- 178. Mayers JR et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 353, 1161–1165 (2016). This work posits that lung and not pancreas tumours depend on BCAA metabolism, which illustrates that the environmental context and co-occurring mutations are also pivotal in dictating tumour metabolism.
- 179.Zhu Z et al. Tumour-reprogrammed stromal BCAT1 fuels branched-chain ketoacid dependency in stromal-rich PDAC tumours. Nat. Metab. 2, 775–792 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Nelson BS et al. Tissue of origin dictates GOT1 dependence and confers synthetic lethality to radiotherapy. Cancer Metab. 8, 1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chowdhry S et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 569, 570–575 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lyssiotis CA & Kimmelman AC Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 27, 863–875 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Schworer S, Vardhana SA & Thompson CB Cancer metabolism drives a stromal regenerative response. Cell Metab. 29, 576–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Olenchock BA, Rathmell JC & Vander Heiden MG Biochemical underpinnings of immune cell metabolic phenotypes. Immunity 46, 703–713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Leone RD & Powell JD Metabolism of immune cells in cancer. Nat. Rev. Cancer 20, 516–531 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Buck MD, Sowell RT, Kaech SM & Pearce EL Metabolic instruction of immunity. Cell 169, 570–586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Chang CH et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Andrejeva G & Rathmell JC Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metab. 26, 49–70 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.MacCarthy-Morrogh L & Martin P The hallmarks of cancer are also the hallmarks of wound healing. Sci. Signal. 13, eaay8690 (2020). [DOI] [PubMed] [Google Scholar]
- 190.Tape CJ et al. Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell 165, 910–920 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.di Magliano MP & Logsdon CD Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 144, 1220–1229 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Georges LM, Verset L, Zlobec I, Demetter P & De Wever O Impact of the microenvironment on tumour budding in colorectal cancer. Adv. Exp. Med. Biol. 1110, 101–111 (2018). [DOI] [PubMed] [Google Scholar]
- 193.Abe Y & Tanaka N The hedgehog signaling networks in lung cancer: the mechanisms and roles in tumor progression and implications for cancer therapy. Biomed. Res. Int. 2016, 7969286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lee KE et al. Hif1a deletion reveals pro-neoplastic function of B cells in pancreatic neoplasia. Cancer Discov. 6, 256–269 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Pylayeva-Gupta Y et al. IL35-producing B cells promote the development of pancreatic neoplasia. Cancer Discov. 6, 247–255 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.DeNardo DG et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Dey P et al. Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer cells utilizes cytokines from the tumor microenvironment. Cancer Discov. 10, 608–625 (2020). This work shows how cytokine signalling mediated by KRAS* from PDA cells to the immune compartment results in a compensatory mechanism by which immune signalling promotes glycolysis in the cancer cells.
- 198.Cascone T et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 27, 977–987 e974 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zhang D et al. Metabolic reprogramming of cancer-associated fibroblasts by IDH3alpha downregulation. Cell Rep. 10, 1335–1348 (2015). [DOI] [PubMed] [Google Scholar]
- 200.Halestrap AP The monocarboxylate transporter family–structure and functional characterization. IUBMB Life 64, 1–9 (2012). [DOI] [PubMed] [Google Scholar]
- 201.Halestrap AP & Wilson MC The monocarboxylate transporter family–role and regulation. IUBMB Life 64, 109–119 (2012). [DOI] [PubMed] [Google Scholar]
- 202.Hutcheson J et al. Immunologic and metabolic features of pancreatic ductal adenocarcinoma define prognostic subtypes of disease. Clin. Cancer Res. 22, 3606–3617 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Colegio OR et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Fischer K et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109, 3812–3819 (2007). [DOI] [PubMed] [Google Scholar]
- 205.Harmon C et al. Lactate-mediated acidification of tumor microenvironment induces apoptosis of liver-resident NK cells in colorectal liver metastasis. Cancer Immunol. Res. 7, 335–346 (2019). [DOI] [PubMed] [Google Scholar]
- 206.Zhao Y et al. Colorectal cancers utilize glutamine as an anaplerotic substrate of the TCA cycle in vivo. Sci. Rep. 9, 19180 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sugiura A & Rathmell JC Metabolic barriers to T cell function in tumors. J. Immunol. 200, 400–407 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Smith C et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. 2, 722–735 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hsu YL et al. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget 7, 27584–27598 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Brandacher G et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin. Cancer Res. 12, 1144–1151 (2006). [DOI] [PubMed] [Google Scholar]
- 211.Witkiewicz AK et al. Genotyping and expression analysis of IDO2 in human pancreatic cancer: a novel, active target. J. Am. Coll. Surg. 208, 781–789 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Holmgaard RB et al. Tumor-expressed IDO recruits and activates MDSCs in a treg-dependent manner. Cell Rep. 13, 412–424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Sadik A et al. IL4I1 is a metabolic immune checkpoint that activates the AHR and promotes tumor progression. Cell 182, 1252–1270.e1234 (2020). [DOI] [PubMed] [Google Scholar]
- 214.Szefel J, Danielak A & Kruszewski WJ Metabolic pathways of L-arginine and therapeutic consequences in tumors. Adv. Med. Sci. 64, 104–110 (2019). [DOI] [PubMed] [Google Scholar]
- 215.Zhang Y et al. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discov. 10, 422–439 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Chaudhri VK et al. Metabolic alterations in lung cancer-associated fibroblasts correlated with increased glycolytic metabolism of the tumor. Mol. Cancer Res. 11, 579–592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sousa CM et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zhao H et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 5, e10250 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Zahalka AH & Frenette PS Nerves in cancer. Nat. Rev. Cancer 20, 143–157 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Banh RS et al. Neurons release serine to support mRNA translation in pancreatic cancer. Cell 10.1016/j.cell.2020.10.016(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Manzo T et al. Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J. Exp. Med. 10.1084/jem.20191920 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Viale A et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632 (2014). This study discovers that certain PDA cells are resistant to extinguished KRAS* signalling through increased mitochondrial metabolism, demonstrating potential metabolic resistance mechanisms to targeting of KRAS*.
- 223.Hou P et al. Tumor microenvironment remodeling enables bypass of oncogenic KRAS dependency in pancreatic cancer. Cancer Discov. 10, 1058–1077 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kapoor A et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 158, 185–197 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Tanaka N et al. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. 10.1158/2159-8290.CD-21-0365 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Muzumdar MD et al. Survival of pancreatic cancer cells lacking KRAS function. Nat. Commun. 8, 1090 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Chen P-Y et al. Adaptive and reversible resistance to Kras inhibition in pancreatic cancer cells. Cancer Res. 78, 985 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Janes MR et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589.e517 (2018). [DOI] [PubMed] [Google Scholar]
- 229.Yao W et al. Syndecan 1 is a critical mediator of macropinocytosis in pancreatic cancer. Nature 568, 410–414 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Shackelford DB et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 23, 143–158 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Liu Y et al. Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer Discov. 3, 870 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.LeBoeuf SE et al. Activation of oxidative stress response in cancer generates a druggable dependency on exogenous non-essential amino acids. Cell Metab. 31, 339–350.e334 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Sherman MH et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159, 80–93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Yan Y et al. The effects and the mechanisms of autophagy on the cancer-associated fibroblasts in cancer. J. Exp. Clin. Cancer Res. 38, 171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Dalin S et al. Deoxycytidine release from pancreatic stellate cells promotes gemcitabine resistance. Cancer Res. 79, 5723–5733 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Halbrook CJ et al. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 29, 1390–1399 e1396 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ribas A & Wolchok JD Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Cristescu R et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 362, eaar3593 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Brahmer JR et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Leone RD et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 10.1126/science.aav2588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ma EH et al. Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8+ T cells. Immunity 51, 856–870 e855 (2019). [DOI] [PubMed] [Google Scholar]
- 242.Balmer ML et al. Memory CD8+ T cells balance pro- and anti-inflammatory activity by reprogramming cellular acetate handling at sites of infection. Cell Metab. 32, 457–467.e455 (2020). [DOI] [PubMed] [Google Scholar]
- 243.Hellmann MD et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 33, 843–852.e844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Skoulidis F et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 8, 822–835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Rizvi H et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J. Clin. Oncol. 36, 633–641 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Arbour KC et al. Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clin. Cancer Res. 24, 334 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Halbrook CJ, Pasca di Magliano M & Lyssiotis CA Tumor cross-talk networks promote growth and support immune evasion in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 315, G27–G35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Yamamoto K et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581, 100–105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Zhu XG et al. Functional genomics in vivo reveal metabolic dependencies of pancreatic cancer cells. Cell Metab. 10.1016/j.cmet.2020.10.017 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Visvader JE Cells of origin in cancer. Nature 469, 314–322 (2011). [DOI] [PubMed] [Google Scholar]
- 251.Barker N et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608–611 (2009). [DOI] [PubMed] [Google Scholar]
- 252.Kim CF et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121, 823–835 (2005). [DOI] [PubMed] [Google Scholar]
- 253.Xu X et al. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc. Natl Acad. Sci. USA 109, 4910–4915 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Mainardi S et al. Identification of cancer initiating cells in K-Ras driven lung adenocarcinoma. Proc. Natl Acad. Sci. USA 111, 255–260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Sutherland KD et al. Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc. Natl Acad. Sci. USA 111, 4952–4957 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Guerra C et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell 4, 111–120 (2003). [DOI] [PubMed] [Google Scholar]
- 257.Best SA et al. Synergy between the KEAP1/NRF2 and PI3K pathways drives non-small-cell lung cancer with an altered immune microenvironment. Cell Metab. 27, 935–943.e934 (2018). [DOI] [PubMed] [Google Scholar]
- 258.Best SA et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat. Commun. 10, 4190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Gidekel Friedlander SY et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 16, 379–389 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.De La O J-P et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl Acad. Sci. USA 10.1073/pnas.0810111105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Habbe N et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc. Natl Acad. Sci. USA 10.1073/pnas.0810097105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Litvak Y, Byndloss MX & Baumler AJ Colonocyte metabolism shapes the gut microbiota. Science 10.1126/science.aat9076 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Kaiko GE et al. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell 165, 1708–1720 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.McNabney SM & Henagan TM Short chain fatty acids in the colon and peripheral tissues: a focus on butyrate, colon cancer, obesity and insulin resistance. Nutrients 10.3390/nu9121348 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Leinwand J & Miller G Regulation and modulation of antitumor immunity in pancreatic cancer. Nat. Immunol. 21, 1152–1159 (2020). [DOI] [PubMed] [Google Scholar]
- 266.Pushalkar S et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 8, 403–416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Jin C et al. Commensal microbiota promote lung cancer development via γδ T cells. Cell 176, 998–1013.e1016 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Smith PM et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Tsay JJ et al. Airway microbiota is associated with upregulation of the PI3K pathway in lung cancer. Am. J. Respir. Crit. Care Med. 198, 1188–1198 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Segal LN et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat. Microbiol. 1, 16031 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]