


Abstract
TAR, a 59 nt 5′-terminal hairpin in human immunodeficiency virus 1 (HIV-1) mRNA, binds viral Tat and several cellular proteins. We report that eukaryotic translation initiation factor 2 (eIF2) recognizes TAR. TAR and the AUG initiation codon domain, located well downstream from TAR, both contribute to the affinity of HIV-1 mRNA for eIF2. The affinity of TAR for eIF2 was insensitive to lower stem mutations that modify sequence and structure or to sequence changes throughout the remainder that leave the TAR secondary structure intact. Hence, eIF2 recognizes structure rather than sequence in TAR. The affinity for eIF2 was severely reduced by a 3 nt change that converts the single A bulge into a 7 nt internal loop. T1 footprinting showed that eIF2 protects nucleotides in the loop as well as in the strand opposite the A bulge. Thus, eIF2 recognizes the TAR loop and lower part of the sub-apical stem. Though not contiguous, these regions are brought into proximity in TAR by a bend in the helical structure induced by the UCU bulge; binding of eIF2 opens up the bulge context and apical stem. The ability to bind eIF2 suggests a function for TAR in HIV-1 mRNA translation. Indeed, the 3 nt change that reduces the affinity of TAR for eIF2 impairs the ability of reporter mRNA to compete in translation. Interaction of TAR with eIF2 thus allows HIV-1 mRNA to compete more effectively during protein synthesis.
INTRODUCTION
TAR (trans-activator response region) is a 59 nt hairpin loop structure (1) found at the 5′-end of all classes of mRNA encoded by the human immunodeficiency virus 1 (HIV-1), the etiological agent of acquired immune deficiency syndrome (AIDS) (2,3). This cis-acting sequence contains a binding site for the viral Tat protein that trans-activates HIV-1 gene expression during early transcriptional elongation of the viral RNA (reviewed in 4,5). A number of cellular proteins also interact directly with TAR, including the RNA-activated protein kinase, PKR, which phosphorylates the α-subunit of eukaryotic translation initation factor 2 (eIF2) (6), the PKR homolog TRBP (7,8), a sequence-specific, single-stranded DNA-binding protein Pur-α (9), RNA polymerase II and TRP-185 (10,11), a human chromosome 12-associated 83 kDa protein (12) and nuclear protein p140 (13). Currently, it is believed that the major role of TAR is to act as an RNA enhancer that controls HIV-1 transcription at initiation and elongation (reviewed in 14). A post-transcriptional role for binding of Tat to TAR has also been suggested (15–17). Indeed, TAR has the ability to activate PKR (18–20), thus causing translational down-regulation through phosphorylation and inactivation of eIF2. Other post-transcriptional functions, if any, remain to be elucidated.
During initiation of translation, eIF2 forms a ternary complex with Met-tRNAf and GTP that must bind to the 40S ribosomal subunit before binding of mRNA can occur (21). eIF2 also interacts directly with mRNA (22–33) through its β-chain (30–32). eIF2 protects specific sequences in mRNA that overlap with the ribosome binding site (26,33,34), consistent with the view that eIF2 guides the 40S ribosomal subunit to its binding site in mRNA. Indeed, eIF2 promotes selection of 5′-proximal translation initiation sites by ribosomes (35). Genetic evidence that eIF2 recognizes the initiation codon is provided by mutations in yeast eIF2, particularly in a zinc finger motif of the β-chain (36,37), that permit utilization of an altered initiation codon (36,38,39). The interaction between mRNA and eIF2 is relevant for translational control: the affinity of an mRNA for eIF2 correlates tightly with its ability to compete in translation while competition between different mRNAs is relieved by an excess of eIF2 (24,27).
Here we show that eIF2 recognizes and binds to the 5′-terminal TAR structure in HIV-1 mRNA. TAR and the AUG initiation codon context each contribute to the affinity of the viral mRNA for this initiation factor. eIF2 recognizes the TAR loop and the lower part of the sub-apical stem. Though not contiguous, these regions are brought into proximity in TAR by a bend in the helical structure induced by the UCU bulge; binding of eIF2 opens up the bulge context. The finding that TAR serves to bind an initiation factor suggests a direct function for this RNA structure in the translation of all classes of HIV-1 mRNA. Indeed, a mutation that reduces the affinity of TAR for eIF2 impairs the ability of reporter mRNA to compete in translation.
MATERIALS AND METHODS
HIV-1 RNA transcripts
pSVTAR59LUC was created by integration of an oligonucleotide consisting of HIV-1 TAR sequence 1–59 into the SmaI site of pBEH (40) and the XbaI–KpnI fragment from the firefly luciferase (LUC) gene into the respective sites of the same vector. A XhoI–KpnI fragment containing TAR and the luciferase gene from pSVTAR59LUC was inserted into pMT7T3 (41) digested with XhoI and KpnI, to generate pMT7T3TARLUC. Cleavage of pMT7T3TARLUC with NcoI, XbaI or both generated upon transcription a 77 nt T7 RNA transcript containing the 59 nt HIV-1 TAR sequence preceded by 13 and followed by 5 extraneous nucleotides (TAR), a 123 nt RNA transcript containing TAR and a 46 nt luciferase sequence including the AUG initiation codon (TAR–LUC) or the 46 nt luciferase RNA transcript (LUC), respectively. pHIVCG vectors, containing TAR and extending into HIV-1 gag, were used to generate unlabeled T7 transcripts as described (42): pHIVCG4 was digested with RsaI to yield HIV-1311, with HaeIII to yield HIV-1415 and with AvaII to yield HIV-11333 RNA; pHIVCG6 was digested with SacI to generate HIV-1311–612 RNA and pHIVCG7 was digested with HaeII to generate HIV-1415Δ RNA. pSP64/TARCAT, containing nucleotides 1–80 of the HIV-1 5′-UTR, and mutant plasmids pSP64/TAR3CAT, pSP64/TAR3RCAT and pSP64/TAR3R3CAT were linearized with HindIII and used to generate unlabeled SP6 RNA transcripts TAR, TAR 3, TAR 3R and TAR 3R3 as described (43).
pSVTARM5LUC, pSVTARM6LUC and pSVTARM7LUC were prepared by the procedure described for pSVTAR59LUC. pSVTARM5LUC was digested with XhoI and KpnI, yielding a TARM5LUC fragment that was cloned into pMT7T3 to generate the transcription vector pMT7T3TARM5LUC. The latter plasmid was linearized with NcoI and used as template for the 77 nt T7 RNA transcript TAR M5. A 2944 nt BglI–XhoI fragment, isolated from pSVTARM6LUC and pSVTARM7LUC, was ligated to a 1363 nt BglI–XhoI fragment generated from pMT7T3 to yield pMT7T3TARM6 and pMT7T3TARM7 that, linearized with NcoI, were used as template for 77 nt T7 RNA transcripts TAR M6 and TAR M7. Full-length TAR–LUC T7 transcripts, containing TAR or TAR M5 abutted to the open reading frame of luciferase mRNA and its 3′-UTR, were generated from pMT7T3TARLUC and pMT7T3TARM5LUC DNA, respectively, linearized with KpnI. Uniformly 32P-labeled T7 transcript containing the 5′-terminal 203 nt of IFN-γ mRNA was generated from NdeI-digested phIFN-γ-1 DNA, carrying a complete human IFN-γ cDNA sequence under the T7 promoter in pMBC-2T (41). Transcripts were analyzed on 1 or 2% agarose gels, using denatured MspI-digested pGEM3 DNA as marker.
Complex formation between eIF2 and TAR
Uniformly labeled T7 TAR RNA transcripts or 5′-terminal 203 nt IFN-γ mRNA transcript were synthesized using [α-32P]UTP (0.8 Ci/µmol) and 10 nM UTP for labeling. eIF2 was purified from a rabbit reticulocyte ribosomal salt wash by chromatography on DEAE–cellulose, phosphocellulose (28) and heparin–Sepharose (Pharmacia) and was ≥98% pure as judged by electrophoresis. Binding of mRNA by the eIF2 preparation was sensitive to inhibition by Met-tRNAf/GTP but not by uncharged tRNA/GTP. Complex formation between eIF2 and mRNA was assayed by electrophoretic mobility shift. Reaction mixtures of 20 µl contained [32P]mRNA (2.2 × 106 c.p.m./pmol), unlabeled competitor RNA as shown and eIF2 in binding buffer (50 mM KCl, 20 mM Tris–HCl, pH 7.8, 2 mM Mg acetate, 1 mM dithiothreitol). After incubation for 15 min at 30°C followed by incubation for 10 min on ice, samples were run for 5 h at 100 V and 4°C through 4% native polyacrylamide gels in 90 mM boric acid, 25 mM EDTA, 90 mM Tris base. Autoradiograms were scanned with a Umax Powerlook II scanner and band intensity was quantitated using NIH Image v.1.61 software.
Translation of mRNA
Translation mixtures containing BMV RNA (Promega), alone or together with TAR–LUC RNA transcript, 30 µl of micrococcal nuclease-treated, heme-supplemented rabbit reticulocyte lysate (Promega), amino acid mixture lacking methionine, 25 pmol of [35S]methionine (1.2 Ci/µmol) and 20 U of RNasin (Promega) were incubated for 60 min at 30°C. Aliquots of 5 µl were analyzed by 12.5% SDS–PAGE, soaking the gel in Amplify (Amersham) before autoradiography. Band intensity was quantitated using NIH Image v.1.61 software.
T1 footprinting of the TAR–eIF2 complex
5′-End-labeled 77 nt TAR T7 transcript was generated from unlabeled transcript (60 pmol) by dephosphorylation with calf alkaline phosphatase, incubation with 70 µCi of [γ-32P]ATP (3 Ci/µmol) and T4 polynucleotide kinase, 6% polyacrylamide/ 8 M urea gel electrophoresis and elution of labeled product. Labeled RNA (0.3 pmol) was incubated without or with eIF2 (0.12 pmol) in binding buffer for 15 min at 30°C followed by incubation for 10 min on ice and then digested for 30 min at 30°C with 0.01 U of RNase T1 (Worthington), phenol extracted and ethanol precipitated. The RNA was dissolved in loading buffer and analyzed on an 8% polyacrylamide sequencing gel.
Activation of PKR
TAR and TAR mutant M5 RNA transcripts were purified twice by gel electrophoresis, followed by chromatography on CF-11 cellulose (Whatman), washed with ethanol and eluted with water as described (44). Rabbit reticulocyte lysate ribosomal pellet was dissolved into 50 mM Tris–HCl, pH 7.4, and stored at –80°C (25). Activation of PKR was assayed in 20 µl reaction mixtures containing 20 mM Tris–HCl, pH 7.4, 90 mM KCl, 1 mM Mg acetate, 0.1 mM unlabeled ATP, 12 U of RNasin and 7 µCi of [γ-32P]ATP (3 Ci/µmol), ribosomal fraction and RNA transcript or double-stranded RNA (polyI:polyC) (Sigma). After incubation for 20 min at 30°C, the reaction was terminated by adding 20 µl of 2× SDS–PAGE loading buffer and heating for 5 min at 95°C; mixtures were analyzed by 10% SDS–PAGE.
RESULTS
Binding of HIV-1 TAR by eIF2
The mobility shift assay of Figure 1A shows complex formation between eIF2 and a 32P-labeled 77 nt RNA transcript, in which the 59 nt TAR stem–loop from the HIV-1 5′-UTR is preceded by 13 and followed by 5 extraneous nucleotides (Fig. 1C). Complex formation was competed out by unlabeled TAR transcript and was absent when 10-fold excess of BSA was substituted for eIF2 (Fig. 1B). Two complexes, C1 and C2, were consistently observed even at limiting eIF2 concentrations and persisted when all the RNA had been complexed, suggesting that they result from distinct secondary structures of the bound TAR RNA molecule.
Figure 1.
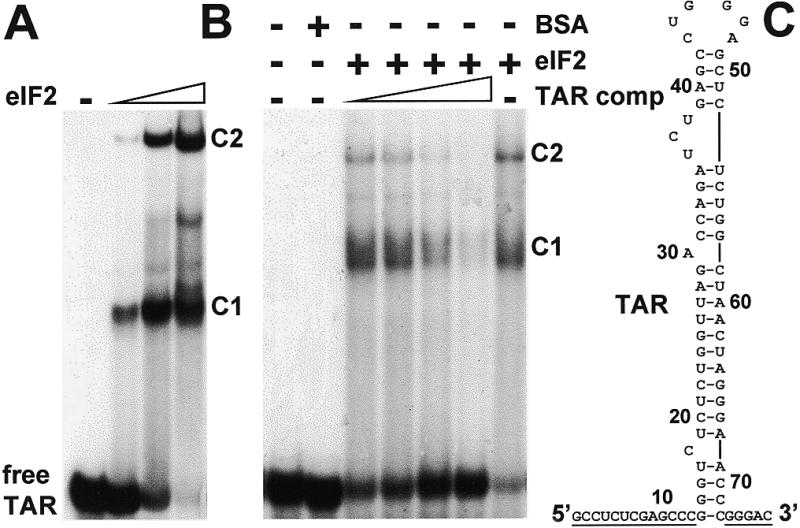
Complex formation between HIV-1 TAR and eIF2. (A) Uniformly 32P-labeled 77 nt T7 transcript (0.08 pmol, 1.25 × 105 c.p.m./pmol) containing TAR abutted by plasmid-derived nucleotides underlined in (C) was incubated without eIF2 or with 0.07, 0.2 or 0.3 pmol of eIF2. The reaction mixture was subjected to electrophoresis on a native gel to separate free TAR RNA from complexes C1 and C2. The autoradiogram is shown. (B) In a separate experiment, TAR RNA (0.1 pmol) and, where shown, 0.25 pmol of eIF2 were incubated in the absence or presence of unlabeled TAR RNA (0.5, 1, 2 or 4 pmol) (TAR competitor). Bovine serum albumin (BSA) served as protein control. Analysis was done as in (A).
TAR and the AUG initiation codon each contribute to the mRNA affinity for eIF2
In the gel shift analysis of Figure 2A, the affinity for eIF2 of different RNA transcripts, all originating at the 5′-end of TAR and extending to different points within HIV-1 gag mRNA (42), was studied by their ability to compete for binding to eIF2 with a model mRNA substrate, consisting of the 5′-terminal 203 nt of human IFN-γ mRNA that cover the 125 nt 5′-UTR and 78 nt of the open reading frame. We have shown that this mRNA fragment binds with high affinity to eIF2 (Ben-Asouli et al., unpublished results). The 5′-terminal HIV-1415 and HIV-11333 mRNA fragments, which contain both TAR and the AUG initiation codon at position 350, exhibited a significantly higher affinity for eIF2 than HIV-177 or HIV-1311, both of which contain TAR yet lack the AUG initiation codon, or than HIV-1311–612, which contains the AUG initiation codon context yet lacks TAR. Deletion of nucleotides 316–346 just preceding the AUG initiation codon in HIV-1415Δ, which are required for dimerization of the viral RNA during replication (42), had only a modest effect on affinity for eIF2 (Fig. 2B). These results were reproducible. The properties of the HIV-1 gag mRNA fragments, summarized in Figure 2C, show that TAR and the AUG initiation codon domain, which is well removed from TAR, each contribute to the binding affinity for eIF2. We conclude that the binding site for eIF2 in HIV-1 gag mRNA is composite.
Figure 2.
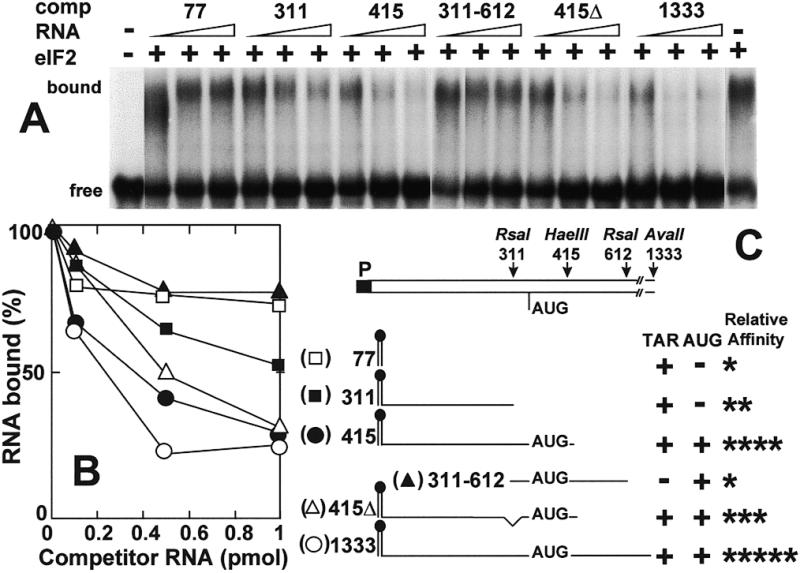
HIV-1 TAR and the AUG initiation codon each contribute to affinity for eIF2. (A) Uniformly 32P-labeled human IFN-γ mRNA 5′-terminal 203 nt transcript (0.08 pmol, 1.25 × 105 c.p.m./pmol) was incubated without eIF2 or with 0.3 pmol of eIF2, in the absence or presence of unlabeled HIV-1 T7 transcripts (0.1–1 pmol) (comp RNA) as shown schematically in (C). The autoradiogram shows free and bound RNA (A). Bound RNA is quantitated in (B). Relative affinities for eIF2 are summarized in (C).
This is borne out by study of the RNA constructs in Figure 3, consisting of 77 nt as in Figure 1C (TAR), the same sequence abutted to a 46 nt sequence containing the AUG initiation codon context of luciferase mRNA (TAR–LUC) or the latter sequence alone (LUC) (Fig. 3C). At low concentrations, TAR and LUC RNA each competed less effectively with labeled TAR transcript for eIF2 than did the fusion construct TAR–LUC RNA that carries both TAR and the AUG initiation codon domain (Fig. 3A, quantitated in B).
Figure 3.
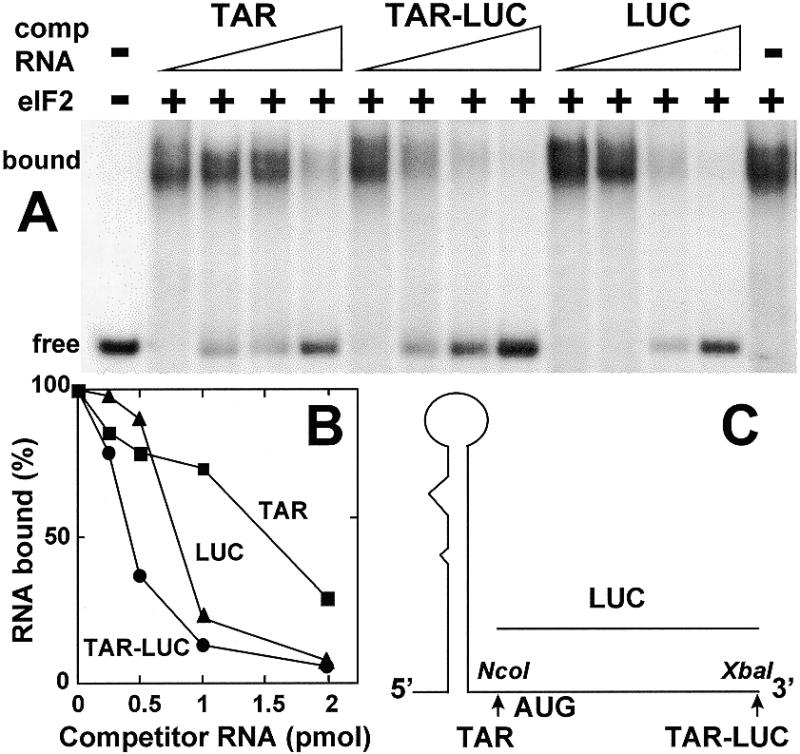
Role of TAR and the AUG initiation codon context in binding of eIF2. Uniformly 32P-labeled 77 nt TAR T7 transcript (0.08 pmol, 1.25 × 105 c.p.m./pmol) was incubated without eIF2 or with 0.12 pmol of eIF2, in the absence or presence of 0.25–2 pmol of unlabeled TAR, TAR–LUC or LUC T7 RNA transcript as shown schematically in (C). Electrophoresis on a native gel was used to separate free TAR RNA from complex. The autoradiogram shows free and bound RNA (A). Bound RNA is quantitated in (B).
Base pairing in the lower stem of TAR is not critical for binding of eIF2
In the experiment in Figure 4, we studied RNA in which the 5′-terminal 59 nt of TAR extend for another 21 nt into the HIV-1 5′-UTR (43). Mutations TAR 3 and TAR 3R create internal loops in the lower stem of HIV-1 TAR, while in the compensatory double mutant TAR 3R3 base pairing is restored (Fig. 4C) (43). The effect of these mutations on affinity for eIF2 was analyzed by mobility shift, through the ability of the unlabeled mutant RNAs to compete with labeled TAR (Fig. 4A and B). The mutations had little, if any, effect on the affinity of TAR for eIF2, since the ability to compete for eIF2 did not differ significantly between wild-type TAR and the mutant forms. Apparently, intact base pairing in the lower stem is not essential for binding of TAR to eIF2.
Figure 4.
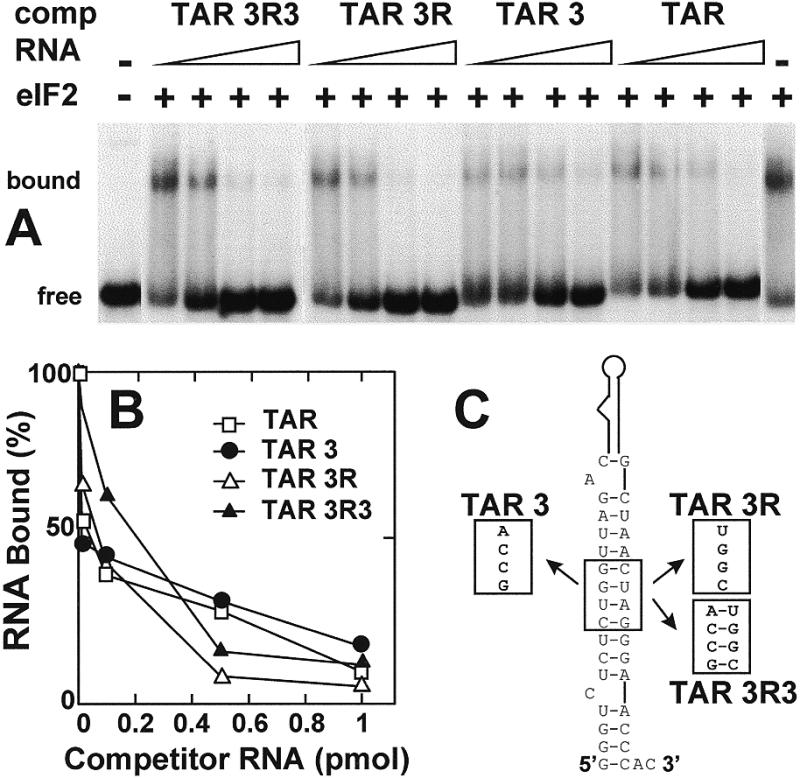
Effect of lower stem mutations in TAR on affinity for eIF2. Uniformly 32P-labeled human IFN-γ mRNA 5′-terminal 203 nt transcript (0.08 pmol, 1.25 × 105 c.p.m./pmol) was incubated without eIF2 or with 0.09 pmol of eIF2, in the absence or presence of 0.01–1 pmol of unlabeled 80 nt wild-type TAR SP6 transcript (TAR) or mutant forms as shown schematically in (C). The autoradiogram of a native gel shows free and bound RNA (A). Bound RNA is quantitated in (B).
A 3 nt change in TAR strongly reduces its affinity for eIF2
In the TAR mutation M5, 3 nt have been altered, extending the single nucleotide bulge at A30 in the wild-type TAR transcript into a 7 nt internal loop (Fig. 5D). This change had a pronounced negative effect on affinity of TAR for eIF2, whether judged by mobility shift competition analysis of TAR–eIF2 complex C1 or C2 (Fig. 5A–C). In contrast, mutant forms M6 and M7, in which the primary sequence was changed extensively but the secondary structure was maintained, competed for eIF2 with an affinity comparable to wild-type TAR. We conclude that eIF2 recognizes structure rather than sequence in TAR and that the M5 mutation affects a structural feature of TAR that is essential for its recognition by eIF2.
Figure 5.
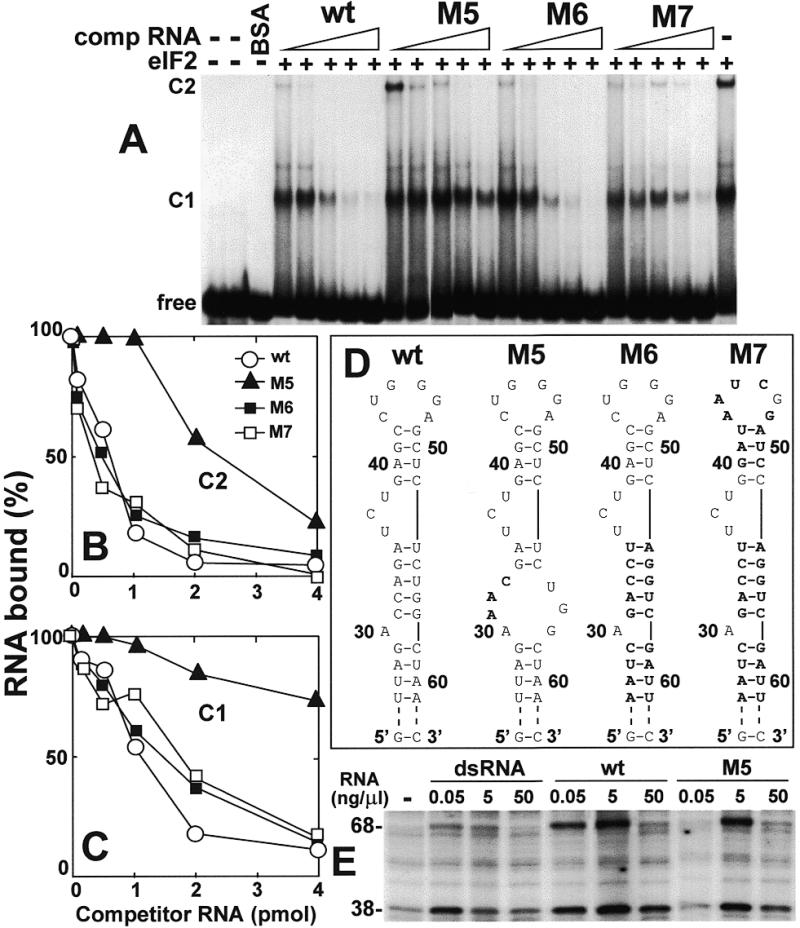
A 3 nt change in TAR strongly reduces its affinity for eIF2. Uniformly 32P-labeled 77 nt TAR T7 transcript (0.08 pmol, 1.25 × 105 c.p.m./pmol) was incubated without eIF2 or with 0.14 pmol of eIF2, in the absence or presence of 0.1–4 pmol of wild-type (wt) or mutant TAR T7 RNA transcript shown schematically in (D) (mutations are denoted in bold). Electrophoresis on a native gel was used to separate free TAR RNA from complexes C1 and C2 (A). C2 and C1 are quantitated in (B) and (C), respectively. In (E), activation of PKR by the indicated concentrations of dsRNA, wt or M5 TAR transcript was assayed by phosphorylation of the PKR (68 kDa) and eIF2α (38 kDa) bands. The autoradiogram is shown.
The M5 mutation reduces the ability of TAR to activate PKR
TAR RNA is able to activate the RNA-dependent eIF2 α-chain kinase, PKR, resulting in its autophosphorylation (68 kDa band) and in phosphorylation of the eIF2 α-chain (38 kDa band) in the ribosome fraction of rabbit reticulocyte lysate (Fig. 5E). As for double-stranded RNA, activation of PKR by TAR became less effective at high RNA concentrations. Although at 5 ng/µl wild-type TAR and M5 mutant RNA activated PKR to a similar extent, at a limiting concentration of 0.05 ng/µl M5 mutant RNA was less active than wild-type TAR in inducing phosphorylation of PKR.
The ability of TAR to activate PKR was impaired by the TAR 3 and TAR 3R mutations in the lower stem and restored in the compensatory mutant TAR 3R3 (19). Conversely, the affinity of TAR for eIF2 was severely reduced in TAR M5 RNA but not in TAR 3 or TAR 3R RNA (Figs 4 and 5). It follows that eIF2 and PKR recognize overlapping yet distinct regions in the TAR RNA structure.
The M5 mutation reduces the translation efficiency of TAR-containing mRNA
To examine whether the M5 mutation, which severely reduces the affinity of TAR for eIF2 (Fig. 5), also affects the translation efficiency of an mRNA carrying TAR, we studied the ability of mRNA trancripts carrying a 5′-terminal TAR or TAR M5 sequence abutted to full-length luciferase mRNA (cf. Fig. 3C) to compete in translation with bromegrass mosaic virus (BMV) RNA. To this end, BMV RNA (45) was translated in rabbit reticulocyte lysate in the presence of increasing amounts of luciferase mRNA carrying TAR or TAR M5 (Fig. 6). Although BMV RNA efficiently directed translation of the 20 kDa coat protein and a 35 kDa protein, wild-type TAR-containing mRNA was able to compete effectively with BMV RNA, as seen from a progressive decrease in the translation of both proteins in response to increasing concentrations of TAR–LUC mRNA. In contrast, TAR M5-containing luciferase mRNA was reproducibly a far weaker competitor (Fig. 6A and B). Whereas TAR–LUC mRNA inhibited BMV RNA translation by 3- to 4-fold, TAR M5–LUC mRNA did not reduce it significantly.
Figure 6.
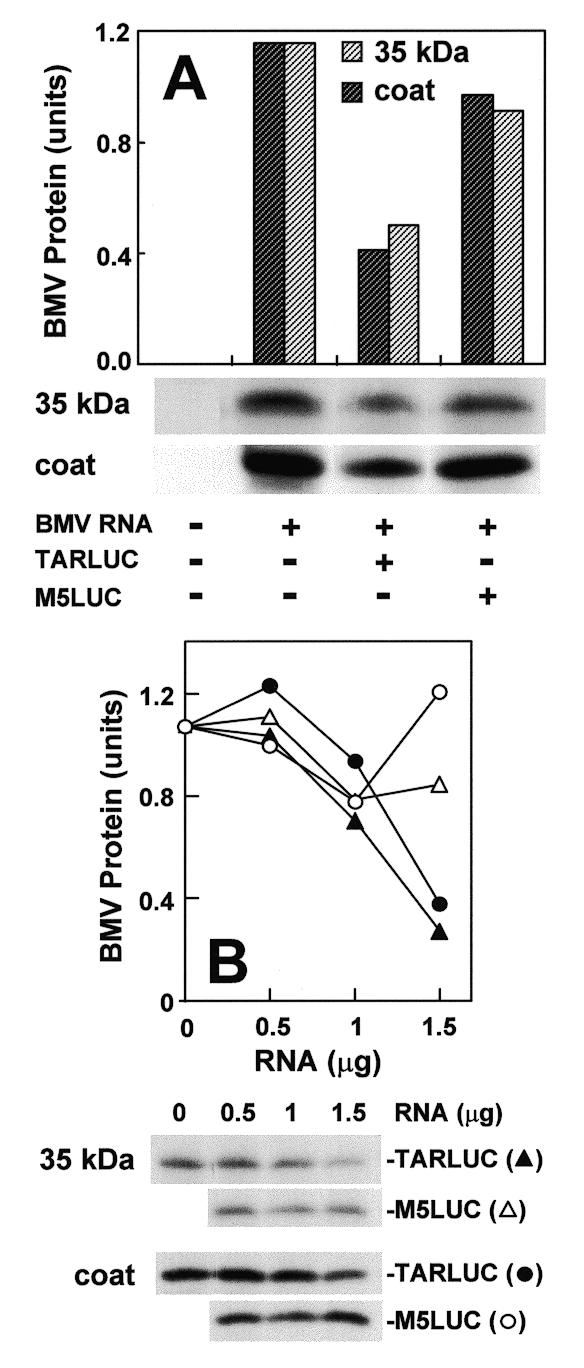
Translational competition by mRNA carrying 5′-terminal TAR or TAR M5 sequence. BMV RNA (1 µg) was translated in the reticulocyte lysate, in the absence or presence of TAR–LUC T7 transcript containing TAR (closed circle and triangle) or TAR M5 (open circle and triangle) abutted to the open reading frame of luciferase mRNA and its 3′-UTR: in (A) 1 µg; in (B) amounts indicated. After SDS–PAGE, the intensity of 35S-labeled 20 kDa coat (closed and open circle) and 35 kDa (closed and open triangle) BMV protein bands in the autoradiograms was quantitated in arbitrary units. To facilitate comparison, intensity of the 35 kDa band without competing mRNA was normalized to that of the coat protein band.
Interaction of the TAR structure with eIF2, which is impaired by the M5 mutation, is thus important for the translation efficiency of TAR-containing mRNA.
T1 footprint of eIF2 in TAR
To delineate the eIF2-binding site within TAR, T1 footprinting was performed on the native TAR–eIF2 complex (Fig. 7A and B). Binding of eIF2 protected both nucleotides in the loop (UGGG) from T1 attack and in the lower part of the second stem (G), opposite those altered by the M5 mutation, and enhanced the sensitivity to T1 of nucleotides in the apical stem and bulge region (Fig. 7C and D). Based on footprint analysis, eIF2 recognizes the TAR loop. Binding of eIF2 opens up the UCU bulge context.
Figure 7.
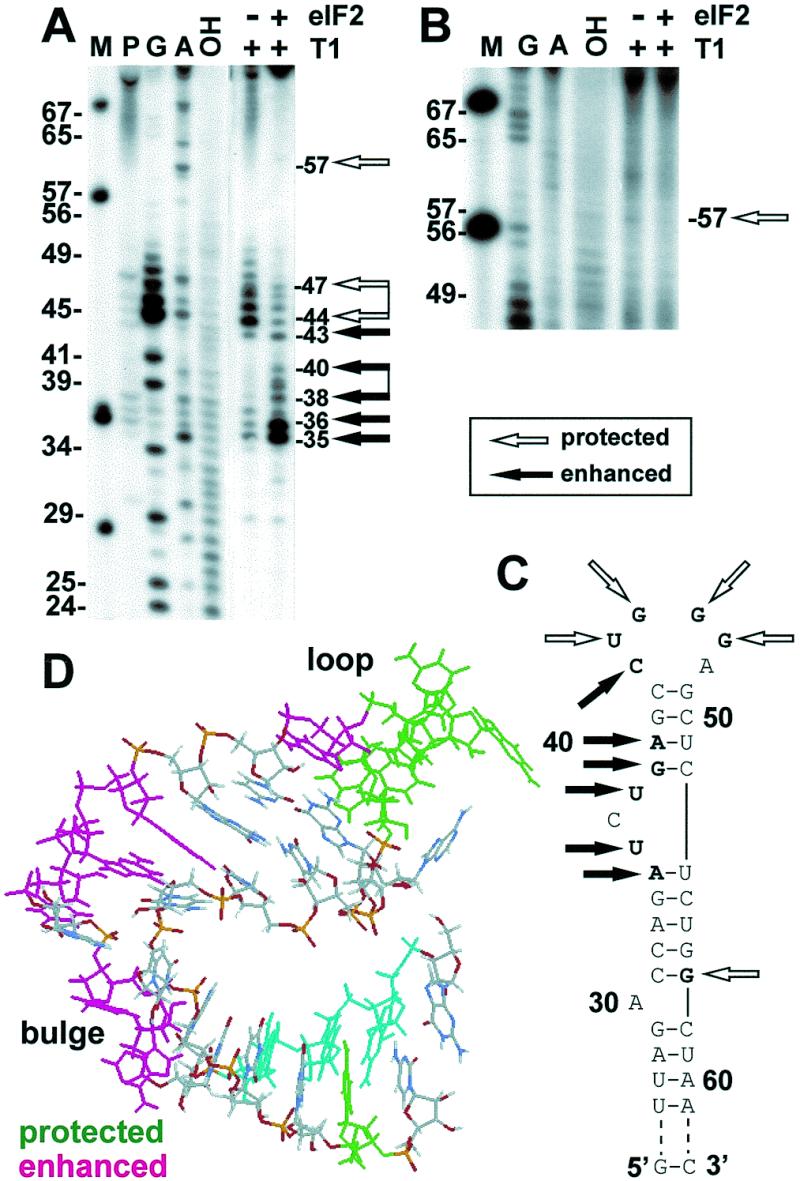
T1 footprint analysis of TAR–eIF2 complex. 5′-End-labeled 77 nt TAR T7 transcript (0.3 pmol, 3 × 104 c.p.m./pmol) was incubated without eIF2 or with 0.12 pmol of eIF2 and then digested with T1 nuclease (0.01 U). M, MspI-digested pGEM3 DNA marker; G, G ladder obtained by digesting the transcript, denatured at 50°C in 7 M urea, with T1 nuclease (1 U); A, a ladder obtained by digesting the denatured transcript with U2 nuclease (1.5 U); OH, alkaline digest. Autoradiograms of sequencing gels (A) and (B) represent separate experiments. (C) Nucleotides protected or sensitized by eIF2 to T1 attack are shown in bold. (D) Three-dimensional, heteronuclear NMR-derived structure of TAR corresponding to nucleotides 30–58 in (C) [pdb1anr.ent (57); in the model shown, the C31-G57 base pair was changed to G31-C57 and A30 to G30, causing it to pair with C58]. Positions protected or sensitized by eIF2 to T1 attack are marked. Nucleotides affected by the M5 mutation are colored cyan in (D).
Figure 7A also shows somewhat enhanced cleavage at positions 32–33 when eIF2 is present, although the increase is weak when compared to that of bands moving at positions 35–43. Likewise, binding of eIF2 may extend to residues beyond G57 (Fig. 7A and B) and thereby could affect the conformation of residues 32–33 on the opposite side of the stem. Clearly, such binding is weaker than that at the loop.
From Figure 7D it can be seen that the nucleotides in TAR that are recognized by eIF2, as judged by protection from nuclease attack (green) or by the M5 mutation (cyan), are brought into proximity by a bend in the helical structure induced by the UCU bulge. On the other hand, nucleotides sensitized to nuclease attack upon binding of eIF2 (magenta) face away from this region.
DISCUSSION
Our results show that eIF2 recognizes and binds to the 5′-terminal TAR structure in HIV-1 mRNA. TAR and the AUG initiation codon domain, which is located well downstream from TAR, each contribute to the affinity of the viral mRNA for this initiation factor. Thus, the binding site for eIF2 in HIV-1 mRNA is composite. The finding that TAR serves to bind an initiation factor suggests a direct function for this RNA structure in the translation of all classes of HIV-1 mRNA. Indeed, a mutation in TAR that reduces its affinity for eIF2 also impairs the ability of reporter mRNA to compete in translation. This shows that the interaction of TAR with eIF2 is important for the translation efficiency of HIV-1 mRNA.
The affinity of TAR for eIF2 is insensitive to mutations that modify sequence and base pairing in its lower stem or to extensive sequence changes throughout the remainder of TAR that do not affect secondary structure. Hence, eIF2 recognizes structure rather than sequence in TAR. In contrast, the affinity for eIF2 was severely reduced by a 3 nt change (the M5 mutation) that causes the single A bulge to be extended into a 7 nt internal loop, composed of 4 nt on one side and 3 nt on the other. This mutation affects a structural feature that is essential for recognition by eIF2. Binding of eIF2 protected nucleotides in the TAR loop as well as near the A bulge, in the strand opposite to that altered by the M5 mutation. Though not contiguous, these regions in TAR are brought into proximity by a bend in the helical structure induced by the UCU bulge (Fig. 7D). Binding of eIF2 induces changes in the conformation of the UCU bulge and its context as well as in the apical stem.
Both within HIV-1 gag mRNA, in which the initiating AUG is located 291 nt downstream from TAR, and within TAR–LUC RNA, in which TAR is fused directly to the AUG initiation codon context of luciferase, affinity for eIF2 is contributed by both TAR and the AUG initiation codon domain (Figs 2 and 3). In HIV-1 gag mRNA, chemical probing supports folding of this RNA such that the AUG initiation codon is brought into close proximity with the TAR domain (46). This analysis provides independent support for our conclusion that TAR and the AUG initiation codon domain create a composite binding site for eIF2 in HIV-1 mRNA. A role for the context of the AUG initiation codon is indicated by the finding that deletion of the dimerization domain just upstream of the AUG initiation codon, in HIV-1415Δ, resulted in a discernibly lower affinity for eIF2 (Fig. 2B).
HIV-1 mRNA causes activation of PKR through its TAR domain (18–20), resulting in inhibition of translation (18). This ability is sensitive (19) to mutations in the lower stem of TAR that disrupt base pairing, TAR 3 and 3R (see Fig. 4). In contrast, these mutations had little, if any, effect on binding of eIF2. Indeed, footprinting data in Figure 7 reinforce the conclusion that the region affected by these mutations is not involved in the interaction of TAR with eIF2. Apparently, intact base pairing in the lower stem is not essential for binding of TAR to eIF2. Conversely, affinity for eIF2 was severely affected by the M5 mutation yet ability to activate PKR was also reduced, supporting the interpretation that eIF2 and PKR recognize distinct, yet overlapping, regions in the TAR structure. Possibly, interaction of eIF2 and PKR with contiguous regions in TAR may facilitate phosphorylation of eIF2 by this kinase. In this context, it is of note that the Tat protein, which also binds to the upper part of TAR, can be phosphorylated by PKR and competes with eIF2 as a substrate for this kinase (47).
Mutations TAR 3 and TAR 3R create a novel, internal 8 nt loop, but this did not influence the affinity of TAR for eIF2. In contrast, the 7 nt internal loop generated by the M5 mutation had a major negative effect on ability of TAR to bind eIF2. This shows that it is not the internal loop in M5 as such but its specific location in TAR that interferes with the binding of eIF2. Recognition by eIF2 thus shows specificity. Only 3 nt were changed in M5 RNA; in contrast, far more extensive sequence changes introduced in the M6 and M7 RNAs (Fig. 5D), including all three positions affected in M5 and, for M7, nucleotides within the loop, did not reduce binding affinity for eIF2. Unlike for M5, however, the nucleotide substitutions in M6 and M7 did not perturb secondary structure. These results show that it is not the primary sequence of TAR per se that is recognized by eIF2, but its structure. The ability of eIF2 to protect the UGGG sequence in the TAR loop from T1 attack, together with the undiminished affinity for eIF2 of M7 TAR RNA in which three of these nucleotides as well as the protected G57 have been altered, again shows that for recognition of TAR, eIF2 is flexible in terms of primary nucleotide sequence.
eIF2 interacts directly with mRNA (22–33), where it protects specific sequences that overlap with the ribosome-binding site (26,33,34). The affinity of an mRNA for eIF2 correlates tightly with its ability to compete in translation while competition between different mRNAs is relieved by an excess of eIF2 (24,27). The mRNA-binding activity of eIF2 locates to the β-subunit (30–32). Involvement of the AUG initiation codon context for recognition of mRNA by eIF2 is supported by biochemical (35) and genetic studies (36,38,39) but the structural complexity and length of mRNA molecules have impeded a more direct analysis of the eIF2-binding domain in mRNA. The present results with TAR, whose structure has been resolved by nuclear magnetic resonance (48) and crystallography (49), offer the first detailed analysis of structural features in an mRNA that are important for recognition by eIF2 (Fig. 7D).
The translation efficiency of luciferase reporter mRNA carrying a 5′-terminal TAR structure was strongly impaired by the M5 mutation, which affects the affinity of TAR for eIF2. This was seen from a drastic reduction in the ability of the mRNA containing mutant TAR to compete with BMV RNA in translation (Fig. 6). Interaction of TAR with eIF2 thus allows HIV-1 mRNA to compete more effectively during protein synthesis.
The role of TAR in regulating the translation of HIV-1 mRNA is, however, more complex. Binding of eIF2 to the loop and bulge domains of TAR enhances the ability of the downstream mRNA to compete in translation. On the other hand, by activating PKR through its lower stem, TAR exerts a negative effect on translation (18,43); simultaneous interaction of TAR with PKR and eIF2 could even facilitate phosphorylation of eIF2 and thus promote its inactivation. As shown by the data in Figure 6, nonetheless, during translation in the reticulocyte lysate, the affinity of TAR for eIF2 contributes positively to translation efficiency.
Comparison of the modes of interaction of initiation factor eIF2 and of the transcription regulatory protein Tat with TAR is instructive. Tat binds at the UCU bulge (14) and forms an equimolar complex with TAR through electrostatic interaction of the basic Arg repeat of Tat with negatively charged phosphates surrounding the bulge; binding is further stabilized by hydrogen bonding in the major groove of the RNA (50,51). The bulge induces a bend in the RNA helix that distorts the local structure (Fig. 7D) and widens the major groove of the RNA to expose hydrogen bonding contacts that are important for binding of Tat (52–55). Binding of mRNA by eIF2 similarly involves basic domains; in this case, three runs of seven Lys residues, as well as a zinc finger motif (32,36). Like eIF2, Tat induces a conformational change in TAR that repositions the functional groups on the bases and the phosphate backbone that are critical for recognition. Tat causes the UCU residues in the bulge, which are loosely stacked into the helix in free RNA, to unstack (56), while a triple base interaction may be formed between the U36A40-U51 base pair (57). Upon binding of Tat, the major groove, which is open and accessible in free TAR, undergoes a transition to a more tightly packed structure that is folded around Arg side chains emanating from the Tat protein (58).
As seen above, binding of eIF2 to TAR also induces structural changes in the UCU bulge and its context as well as in the apical stem. However, major differences are seen between the binding of eIF2 and that of Tat. eIF2 recognizes nucleotides in the TAR loop, in addition to those in the stem region near the single A bulge affected by the M5 mutation, but does not bind directly to the UCU bulge. While Tat does not bind to the loop, interaction of Tat with cyclin T confers a requirement for sequences in the loop that are not recognized by Tat alone (59).
Acknowledgments
ACKNOWLEDGEMENTS
We thank J.-L. Darlix for HIV gag plasmids and N. Sonenberg for TAR 3, TAR 3R and TAR 3R3 plasmids and Christine Morelle for construction of pSVTAR59LUC and mutant forms M5–M7. This work was supported by grants from the German–Israeli Joint Programme in Biotechnology.
REFERENCES
- 1.Rosen C.A., Sodroski,J.G. and Haseltine,W.A. (1985) Cell, 41, 813–822. [DOI] [PubMed] [Google Scholar]
- 2.Barre-Sinoussi F., Chermann,J.C., Rey,F., Nugeyre,M.T., Chamaret,S., Gruest,J., Dauguet,C., Axler-Blin,C., Vezinet-Brun,F., Rouzioux,C., Rozenbaum,W. and Montagnier,L. (1983) Science, 220, 868–871. [DOI] [PubMed] [Google Scholar]
- 3.Gallo R.C., Salahuddin,S.Z., Popovic,M., Shearer,G.M., Kaplan,M., Haynes,B.F., Palker,T.J., Redfield,R., Oleske,J., Safai,B. et al. (1984) Science, 224, 500–503. [DOI] [PubMed] [Google Scholar]
- 4.Yankulov K. and Bentley,D. (1998) Curr. Biol., 8, R447–R449. [DOI] [PubMed] [Google Scholar]
- 5.Garber M.E. and Jones,K.A. (1999) Curr. Opin. Immunol., 11, 460–465. [DOI] [PubMed] [Google Scholar]
- 6.Carpick B.W., Graziano,V., Schneider,D., Maitra,R.K., Lee,X. and Williams,B.R.G. (1997) J. Biol. Chem., 272, 9510–9516. [DOI] [PubMed] [Google Scholar]
- 7.Gatignol A., Buckler-White,A., Berkhout,B. and Jeang,K.T. (1991) Science, 251, 597–600. [DOI] [PubMed] [Google Scholar]
- 8.Gatignol A., Buckler,C. and Jeang,K.T. (1993) Mol. Cell. Biol., 13, 2193–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chepenik L.G., Tretiakova,A.P., Krachmarov,C.P., Johnson,E.M. and Khalili,K. (1998) Gene, 210, 37–44. [DOI] [PubMed] [Google Scholar]
- 10.Wu-Baer F., Lane,W.S. and Gaynor,R.B. (1995) EMBO J., 14, 5995–6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu-Baer F., Lane,W.S. and Gaynor,R.B. (1996) J. Biol. Chem., 271, 4201–4208. [DOI] [PubMed] [Google Scholar]
- 12.Hart C.E., Saltrelli,M.J., Galphin,J.C. and Schochetman,G. (1995) J. Virol., 69, 6593–6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothblum C.J., Jackman,J., Mikovits,J., Shukla,R.R. and Kumar,A. (1995) J. Virol., 69, 5156–5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones K.A. and Peterlin,B.M. (1994) Annu. Rev. Biochem., 63, 717–724. [DOI] [PubMed] [Google Scholar]
- 15.Rosen C.A., Sodroski,J.G., Goh,W.C., Dayton,A.L., Lippke,J. and Haseltine,W.A. (1986) Nature, 319, 555–559. [DOI] [PubMed] [Google Scholar]
- 16.Cullen B.R. (1986) Cell, 46, 973–982. [DOI] [PubMed] [Google Scholar]
- 17.Rice A.P. and Mathews,M.B. (1988) Nature, 332, 551–553. [DOI] [PubMed] [Google Scholar]
- 18.Edery I., Petryshyn,R. and Sonenberg,N. (1989) Cell, 56, 303–312. [DOI] [PubMed] [Google Scholar]
- 19.Roy S., Agy,M., Hovanessian,A.G., Sonenberg,N. and Katze,M.G. (1991) J. Virol., 65, 632–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maitra R.K., McMillan,N.A., Desai,S., McSwiggen,J., Hovanessian,A.G., Sen,G., Williams,B.R. and Silverman,R.H. (1994) Virology, 204, 823–827. [DOI] [PubMed] [Google Scholar]
- 21.Hershey J.W.B. (1991) Annu. Rev. Biochem., 60, 717–755. [DOI] [PubMed] [Google Scholar]
- 22.Kaempfer R., Hollender,R., Abrams,W.R. and Israeli,R. (1978) Proc. Natl Acad. Sci. USA, 75, 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaempfer R., Rosen,H. and Israeli,R. (1978) Proc. Natl Acad. Sci. USA, 75, 650–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Segni G., Rosen,H. and Kaempfer,R. (1979) Biochemistry, 18, 2847–2854. [DOI] [PubMed] [Google Scholar]
- 25.Rosen H., Knoller,S. and Kaempfer,R. (1981) Biochemistry, 20, 3011–3020. [DOI] [PubMed] [Google Scholar]
- 26.Kaempfer R., Van-Emmelo,J. and Fiers,W. (1981) Proc. Natl Acad. Sci. USA, 78, 1542–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosen H., Di Segni,G. and Kaempfer,R. (1982) J. Biol. Chem., 257, 946–952. [PubMed] [Google Scholar]
- 28.Gonsky R., Lebendiker,M.A., Harary,R., Banai,Y. and Kaempfer,R. (1990) J. Biol. Chem., 265, 9083–9089. [PubMed] [Google Scholar]
- 29.Scheper G.C., Thomas,A.A.M. and Voorma,H.O. (1991) Biochim. Biophys. Acta, 1089, 220–226. [DOI] [PubMed] [Google Scholar]
- 30.Gonsky R., Itamar,D., Harary,R. and Kaempfer,R. (1992) Biochimie, 74, 427–434. [DOI] [PubMed] [Google Scholar]
- 31.Flynn A., Shatsky,I.N., Proud,C.G. and Kaminski,A. (1994) Biochim. Biophys. Acta, 1219, 293–301. [DOI] [PubMed] [Google Scholar]
- 32.Laurino J.P., Thompson,G.M., Pacheco,E. and Castilho,B.A. (1999) Mol. Cell. Biol., 19, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perez-Bercoff R. and Kaempfer,R. (1982) J. Virol., 41, 30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaempfer R. (1984) In Fraenkel-Conrat,H. and Wagner,R.R. (eds), Comprehensive Virology, Vol. 19. Regulation of Eukaryotic Translation. Plenum Press, New York, NY, pp. 99–175.
- 35.Dasso M.C., Milburn,S.C., Hershey,J.W.B. and Jackson,R.J. (1990) Eur. J. Biochem., 187, 361–371. [DOI] [PubMed] [Google Scholar]
- 36.Donahue T.F., Cigan,A.M., Pabich,E.K. and Valavicius,B.C. (1988) Cell, 54, 621–632. [DOI] [PubMed] [Google Scholar]
- 37.Pathak V.K., Nielsen,P.J., Trachsel,H. and Hershey,J.W. (1988) Cell, 54, 633–639. [DOI] [PubMed] [Google Scholar]
- 38.Cigan A.M., Pabich,E.K., Feng,L. and Donahue,T.F. (1989) Proc. Natl Acad. Sci. USA, 86, 2784–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dorris D.R., Erickson,F.L. and Hannig,E.M. (1995) EMBO J., 14, 2239–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Artelt P., Morelle,C., Ausmeier,M., Fitzek,M. and Hauser,H. (1988) Gene, 68, 213–219. [DOI] [PubMed] [Google Scholar]
- 41.Dirks W., Schaper,F. and Hauser,H. (1994) Gene, 149, 389–390. [DOI] [PubMed] [Google Scholar]
- 42.Darlix J.L., Gabus,C., Nugeyre,M.T., Clavel,F. and Barre-Sinoussi,F. (1990) J. Mol. Biol., 216, 689–699. [DOI] [PubMed] [Google Scholar]
- 43.Parkin N.T., Cohen,E.A., Darveau,A., Rosen,C., Haseltine,W. and Sonenberg,N. (1988) EMBO J., 7, 2831–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Circle D.A., Neel,O.D., Robertson,H.D., Clarke,P.A. and Mathews,M.B. (1997) RNA, 3, 438–448. [PMC free article] [PubMed] [Google Scholar]
- 45.Ahlquist P., Dasgupta,R. and Kaesberg,P. (1984) J. Mol. Biol., 172, 369–383. [DOI] [PubMed] [Google Scholar]
- 46.Baudin F., Marquet,R., Isel,C., Darlix,J.L., Ehresmann,B. and Ehresmann,C. (1993) J. Mol. Biol., 229, 382–389. [DOI] [PubMed] [Google Scholar]
- 47.Brand S.R., Kobayashi,R. and Mathews,M. (1997) J. Biol. Chem., 272, 8388–8397. [DOI] [PubMed] [Google Scholar]
- 48.Aboul-Ela F., Karn,J. and Varani,G. (1995) J. Mol. Biol., 253, 313–332. [DOI] [PubMed] [Google Scholar]
- 49.Ippolito J.A. and Steitz,T.A. (1998) Proc. Natl Acad. Sci. USA, 95, 9819–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frankel A.D. (1992) Protein Sci., 1, 1539–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gait M.J. and Karn,J. (1993) Trends Biochem. Sci., 18, 255–259. [DOI] [PubMed] [Google Scholar]
- 52.Weeks K.M., Ampe,C., Schultz,S.C., Steitz,T.A. and Crothers,D.M. (1990) Science, 249, 1281–1285. [DOI] [PubMed] [Google Scholar]
- 53.Weeks K.M. and Crothers,D.M. (1991) Cell, 66, 577–588. [DOI] [PubMed] [Google Scholar]
- 54.Colvin R.A. and Garcia-Blanco,M.A. (1992) J. Virol., 66, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delling U., Reid,L.S., Barnett,R.W., Ma,M.Y., Climie,S., Sumner-Smith,M. and Sonenberg,N. (1992) J. Virol., 66, 3018–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Puglisi J.D., Tan,R., Calnan,B.J., Frankel,A.D. and Williamson,J.R. (1992) Science, 257, 76–78. [DOI] [PubMed] [Google Scholar]
- 57.Puglisi J.D., Chen,L., Frankel,A.D. and Williamson,J.R. (1993) Proc. Natl Acad. Sci. USA, 90, 3680–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aboul-ela F., Karn,J. and Varani,G. (1996) Nucleic Acids Res., 24, 3974–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wei P., Garber,M.E., Fang,S.M., Fischer,W.H. and Jones,K.A. (1998) Cell, 92, 451–462. [DOI] [PubMed] [Google Scholar]