


Abstract
The AP-2 family of transcriptional regulator proteins has three members, α, β and γ. AP-2α and γ are expressed in placenta and in the human trophoblast cell line JEG-3. AP-2 has been shown to regulate expression of the placental human chorionic gonadotropin (hCG) α- and β-subunit genes, however, previous work did not distinguish between the family members. Tryptic peptides of the AP-2 protein complexes purified from JEG-3 cells by oligo-affinity chromatography using the hCGα AP-2 site match the amino acid sequence of AP-2γ. The fact that AP-2γ is present at significant levels and binds the hCGα trophoblast-specific element suggests that AP-2γ is at least part of the binding complex in vivo and plays a role in regulating hCG expression. We show that mutation of each of four AP-2 binding sites within the hCGβ promoter decreases expression in transfection assays, demonstrating that all four sites are required for maximal expression in JEG-3 cells. Furthermore, we find differences in regulation of the family members: AP-2α mRNA levels increase in response to cAMP while AP-2γ mRNA levels do not. The demonstrated importance of the AP-2 sites in controlling hCGα and β expression and the likely involvement of more than one family member suggest that a balance in AP-2 proteins is involved in coordinate regulation of these genes. Moreover, many placenta-restricted genes are regulated by AP-2 proteins, thus members of this family may play an important overall role in placenta-specific expression.
INTRODUCTION
During human embryonic development, the reproductive hormone human chorionic gonadotropin (hCG) is a key messenger for embryo to maternal communication and plays an important role in the successful establishment and maintenance of pregnancy (1–3). hCG is a member of a family of heterodimeric glycoprotein hormones that includes luteinizing hormone and follicle-stimulating hormone, which are expressed in the pituitary gonadotropes, and thyroid-stimulating hormone, which is expressed in the pituitary thyrotropes. All members of this hormone family share a common α-subunit and have unique β-subunits. α-Subunit gene expression must be regulated in three disparate cell types in two different tissues: the trophoblasts of the placenta and the gonadotropes and thyrotropes of the anterior pituitary. In addition, its expression must be coordinated with that of a specific β-subunit.
Regulation of the hCG α-subunit gene in placental cells has been studied by transfection of human placental trophoblast cell lines derived from choriocarcinoma tumors that express hCG, such as JEG-3 and BeWo (4–6). A placenta-specific enhancer (7,8) contains three types of distinct cis-acting elements, a trophoblast-specific element (TSE, or upstream regulatory element, URE) that binds proteins of the activator protein-2 (AP-2) class, a GATA factor binding site that binds GATA-2 and GATA-3 (9), and two cAMP response elements that bind CREB. Full tissue-specific activity requires all three types of elements, each of which has little activity individually (7,10). The activity of the binding proteins at these sites is synergistic and the combination of these elements can confer tissue-specific expression to a heterologous promoter in an orientation- and distance-independent manner (7). In addition to tissue specificity (7,8,11), the CREs are required for full cAMP response of the human α-subunit gene (12). Two copies are present as an exact 18 bp direct repeat (7,13), each containing an 8 bp palindromic core sequence (TGACGTCA) which is identical to other characterized CREs (14). The GATA site also confers induction in response to cAMP and is the major tissue-specific element in the equine α-subunit gene, which has neither an AP-2 site nor a CRE (10). Here, we show that the AP-2 site also contributes to the cAMP responsiveness of hCGα gene expression.
Since the original description of the hCGα TSE/URE (7,8,15), similar sequences have been identified and implicated in control of other placenta-restricted genes, such as the placenta-specific exon I of the aromatase cytochrome P450 gene (16) and the murine adenosine deaminase gene (17). In addition, four adjacent TSEs are present in the 5′ regulatory region of the hCGβ5 gene (15). Further, AP-2 has been shown to play a major role in the activation of hCGβ expression (4,15). Since AP-2 regulates both the α- and β-subunits, it may be responsible for the coordinate expression of these genes in placenta (15).
Similarities between the DNA consensus sequence for the TSE and the consensus sequence recognized by the family of transcriptional AP-2 proteins exist, suggesting that their binding proteins are related. AP-2 family member proteins range in size from 48 to 52 kDa (18–22). They recognize the consensus sequence 5′-GCCCCAGGC-3′ present in the regulatory regions of a number of cellular and viral genes, including human metallothionein IIA (hMtIIa), human growth hormone, c-myc, the major histocompatibility complex H2Kb and the SV40 and bovine papilloma viral enhancers (20,21). AP-2 protein expression and/or their action on target genes have been shown to be regulated by retinoic acid, TPA, the calcium ionophore A23187 and cAMP (20,23,24). These proteins are tissue-specific factors and, to date, three family members have been described, AP-2α, AP-2β and AP-2γ.
In the adult mouse, AP-2α appears at low levels in the kidney, cerebellum, spleen and thymus. During mouse embryogenesis, AP-2α expression is limited both spatially and temporally, appearing principally in epithelial and neural crest cell lineages (25). Its expression pattern during murine development suggested a role for this factor in regulating genes involved in the development of the peripheral nervous system, face, limbs, skin and nephric tissue. Since the isolation and cloning of human AP-2α from HeLa cells, several isoforms have been described. One human isoform is an alternatively spliced mRNA that encodes a protein differing in the C-terminus (26). This protein, AP-2B, lacks the dimerization domain necessary for DNA binding. Meier et al. (27) described four differentially spliced isoforms of AP-2α present during murine embryogenesis. Furthermore, human and murine genomic and cDNA clones encoding a second AP-2-related transcription factor, AP-2β, have been isolated and appear to derive from a gene distinct from AP-2α. AP-2β shares high homology with AP-2α and both are expressed during embryogenesis. In general, patterns of AP-2α and AP-2β mRNA expression are observed to overlap. However, certain differences were also reported. AP-2β was predominant in the dorsal and anteriolateral primordium of the midbrain, while AP-2α was expressed in a distinct region of the anterior midbrain. The third family member AP-2γ is also derived from a distinct gene. In mice, all three AP-2 family genes have been reported to be expressed extra-embryonically in the trophoblastic tissue and to persist in the trophoblastic cells as AP-2 expression develops in the embryo (25). However, Shi et al. (28) showed that AP-2β levels were extremely low in both the placenta and the embryo. AP-2γ, in contrast, is abundantly expressed in placenta, suggesting a key role for this family member in placental gene regulation. AP-2 mRNA and protein are present in the human trophoblast cell line JEG-3. Johnson et al. (4) have recently reported that AP-2α is present in the hCGα TSE protein binding complex but their analysis actually did not distinguish between the three AP-2 family members. We provide evidence, including amino acid sequence identity, that AP-2γ is present in the AP-2 complex binding the TSEs. Additionally, we show that in contrast to regulation in HeLa cells, AP-2 family mRNA is induced by cAMP in placental cells, further supporting a role for AP-2 in hCG placenta-specific gene expression.
MATERIALS AND METHODS
TSE binding protein peptide analysis
The TSE binding protein complex was partially purified as described previously (15). Large scale purification from JEG-3 cells resulted in several micrograms of purified TSE binding complex that were then separated on a preparative SDS–PAGE gel and transferred to nitrocellulose for trypsin digestion (29). The amino acid sequences of three tryptic fragments were determined using an Applied Biosystems (ABI470) Protein Sequencer.
Nuclear extract preparation and gel mobility shift assays
Nuclear extracts were prepared according to the method of Dignam et al. (30), as described by Delegeane et al. (7), with the modification that precipitation was performed using 25–28% ammonium sulfate. The gel mobility shift assay was slightly modified from Sawadogo et al. (31). Binding reactions were performed in 20 µl containing 10 mM Tris–HCl pH 7.8, 50 mM KCl, 5 mM spermidine, 1 mM EDTA, 5 mM DTT, 0.5 mM PMSF, 1 mM benzamidine, 10% (v/v) glycerol, 0.25 µg/µl BSA and 0.1 µg/µl poly(dI-dC)·poly(dI-dC). Oligonucleotides were purified on acrylamide gels before use. The following double-stranded DNA oligonucleotides were used: 5′-gatcAAAAATGACCTAAGGGTTGAAACA-3′, corresponding to the human α-subunit TSE from –180 to –157 bp, and 5′-gatcCACACGGGCCGCGGGCGGTCAGTT-3′, which corresponds to the hMtIIa AP-2 binding site from –164 to –187 bp. The mutant TSE is identical to the TSE except for three base substitutions in the TSE binding recognition sequence, 5′-CC-TAAGGG-3′, so that this sequence reads 5′-CTGAGAG-3′ (15). All oligonucleotides were synthesized with 5′-GATC-3′ at their 5′-ends.
DNA probes were prepared by end-labeling the double-stranded oligonucleotides with T4 polynucleotide kinase and [γ-32P]dATP (6000 Ci/mmol). All binding reactions were carried out at room temperature and each reaction was preincubated for 5–10 min before the addition of probe (10 000 c.p.m., 1–2 fmol). To resolve the complexes, the reactions were applied to 5% native acrylamide gels (30:1 acrylamide:bis-acrylamide) in 0.25× TBE, 5% (v/v) glycerol.
RNA extraction and northern analysis
Total RNA was prepared from tumor cell lines using the acid guanidinium thiocyanate/phenol/chloroform method (32). RNAs were harvested at the times indicated in the figures. Northern blotting was performed as described by Maniatis et al. (33). Blots were hybridized with 32P-labeled AP-2α or AP-2γ cDNAs. The cDNA probes were generated as follows. For the AP-2α probe, ~1 µg of JEG-3 poly(A)+ RNA was converted to cDNA as described previously (34). JEG-3 cDNA (5 ng) was used in PCR amplifications with Taq polymerase, NEB buffer and 1 µM final concentration of each oligonucleotide primer. Primer 1, 5′-GAtCTAGAGGGGCATATCCGTTCAC-3′, is in the 5′-untranslated region, 30 bp upstream from the initiator codon. Primer 2, 5′GGtCtAGAGCCTCACTTTCTGTGC-3′, overlaps the 3′ stop codon. Lower case letters indicate nucleotide changes made to provide XbaI recognition sites. Following PCR, the expected 1354 bp fragment was gel isolated and cloned into the XbaI site of pICR20 (35). Nucleotide sequencing was performed by the Sanger–Messing dideoxy chain termination method (36) using [α-32P]dATP and modified T7 DNA polymerase (US Biochemical Corp., Cleveland, OH). The AP-2α probe used in Figure 7 represents the 529 bp fragment derived from an XbaI/SmaI digest.
Figure 7.
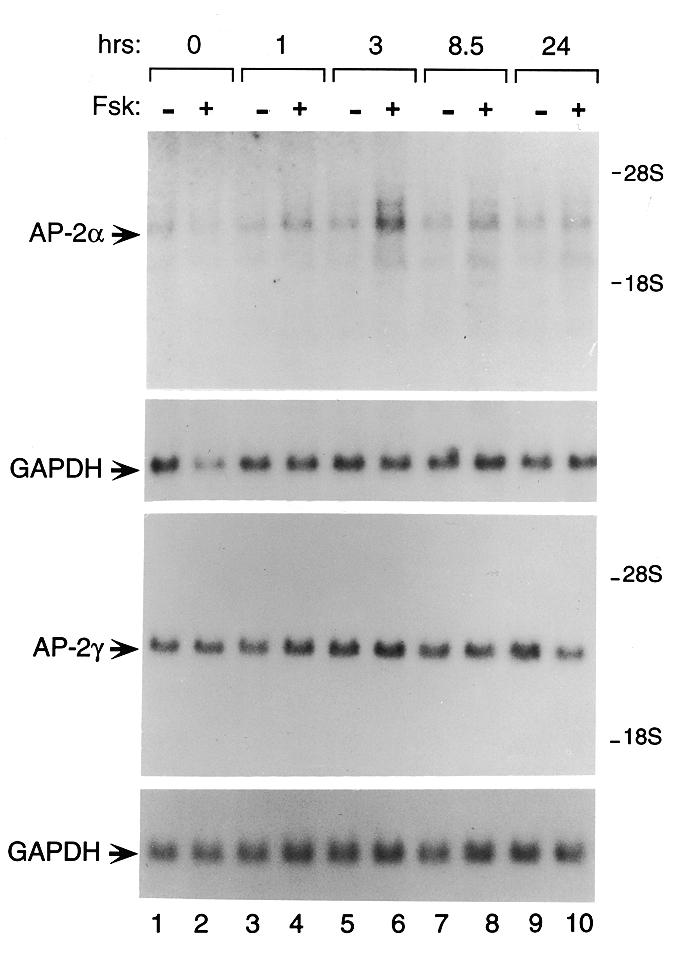
AP-2α mRNA expression is induced by forskolin in JEG-3 cells. JEG-3 cells were treated with either forskolin (10 µM) or ethanol vehicle for the times indicated in hours at the top of the figure. Ten micrograms of JEG-3 total RNA were electrophoresed on a formaldehyde gel, transferred onto nylon membrane and probed with 32P-labeled cDNA probes encoding the 5′-regions of either AP-2α or AP-2γ, as described in Materials and Methods. A radiolabeled probe encoding GAPDH was used as a loading control and for normalization.
A plasmid containing the full-length AP-2γ cDNA inserted into pBK-CMV (18) was kindly provided by Dr Ronald Weigel. The AP-2γ probe used in Figure 7 is the 410 bp fragment derived from a BamHI/BglI digest.
hCGβ plasmids
Construction of the hCGβ5 promoter truncations –361, –305, –279 and –78 have been described previously (15). Site-directed mutagenesis of the AP-2 binding sites was performed within the –361 hCGβ5 promoter directing transcription of the chloramphenicol acetytransferase (CAT) reporter gene. Three of the four mutants were constructed (mFP1, mFP4-1 and mFP4-2) using the Chameleon™ double-stranded, site-directed mutagenesis kit from Stratagene. The mFP2 mutant was obtained using PCR. Briefly, PCR primers incorporating the mutations were designed for both the sense and antisense strands of the promoter. These primers were used in separate PCR reactions with upstream and downstream vector primers to amplify sequences from the –361-CAT hCGβ wild-type expression vector. The PCR products were purified away from the primers and then annealed to one another. This annealed DNA, containing a certain proportion of heteroduplex strands, was amplified in a secondary PCR reaction with the upstream and downstream vector primers to generate a product whose sequence was identical to that of the vector except for the introduced internal mutation.
Oligonucleotides used in the mutagenesis are described below and shown in Figure 5A. All mutations were confirmed by the dideoxy chain termination sequencing method (36) in the presence of [α-32P]dATP (3000 Ci/mmol) and Sequenase (US Biochemical Corp.) under conditions described by the manufacturer.
Figure 5.



(A) Sequence assignment and quantity of the predominant amino acid identified at each position in three tryptic peptides of affinity-purified TSE binding protein. Quantity, phenylthiohydantoin amino acid (PTH-AA) detected (pmol); X, no unambiguous assignment could be made; (aa), tentative assignment (confidence <90%); *, quantification not possible. (B) Homology of the TSE binding complex peptides to AP-2 family member amino acid sequences. Comparison of tryptic peptides of partially purified TSE binding protein to homologous peptide sequences from mouse and human AP-2 protein family members is shown. Identical amino acids are underlined. Peptide 1 is identical to amino acids 360–369 of hAP-2γ, peptide 2 is identical to amino acids 342–347 of hAP-2γ and peptide 3 is identical to amino acids 122–138 of hAP-2γ. (C) Schematic diagram showing structural elements of AP-2 protein family members. The number of amino acids comprising AP-2α, AP-2β and AP-2γ, respectively, are shown on the right. Percentages below the diagram indicate degree of identity of each human family member to human AP-2α. The TSE binding protein peptides 1–3 are shown above the homologous regions of the AP-2 structure.
Oligonucleotides
Double-stranded oligonucleotides corresponding to the following regions in the hCGβ5 gene were used: mFP1 from –320 to –298, mFP2 from –302 to –274, mFP4-1 from –223 to –198 and mFP4-2 from –245 to –217. The mutants were identical to the wild-type sequence except that the residues at positions –310, –290, –231 and –215 were changed to T residues and those at –304, –284, –225 and –209 were changed to A residues.
Cell culture, transfection and CAT assays
JEG-3 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 U/ml), streptomycin (0.1 mg/ml) and 4.5 mg/ml glucose in an atmosphere with 5% CO2. Transfections were performed using calcium phosphate precipitates (37) containing 15 µg of CAT expression vector DNA and 1.5 µg of the internal control plasmid TKβ-galactosidase. JEG-3 cells were incubated with precipitates for 4–5 h and then rinsed and given fresh medium, with cells harvested a further 48 h later. Protein extracts were prepared by freeze–thawing as previously described (38). CAT assays (39) were performed as previously described and β-galactosidase assays were performed as described by Delegeane et al. (7). Units of CAT activity were normalized to the internal control β-galactosidase activity.
RESULTS
The TSE binding complex and AP-2 display identical DNA binding characteristics
Steger et al. (15) previously isolated a 56 kDa protein that binds the TSEs present in the enhancer regions of both the hCG α- and β-subunit genes. Figure 1 compares the TSE and AP-2 binding sites. The CCNNNGGG sequence, important for protein binding to the TSE, agrees well with the consensus sequence GCCNNNGGC that is recognized by AP-2 family members. Strong homology between the hCGα and hCGβ TSEs and AP-2 binding elements suggested that the TSE binding protein and AP-2 proteins may be related.
Figure 1.

The TSE is homologous to the binding sites recognized by AP-2. The core TSE sequence elements of the human CGα and CGβ5 subunit promoters are displayed aligned with the sequence elements that comprise AP-2 binding sites in the hMtIIa (20,22) and SV40 enhancer (22,45) regions as well as the general consensus sequence recognized by AP-2 (20,45). Stars indicate identity with the AP-2 consensus sequence.
The TSE binding protein was originally identified in the human placental cell line JEG-3 (7). AP-2α was identified and cloned from HeLa human epithelial cells (21). To investigate the relatedness of the TSE binding protein and the AP-2 family of proteins, we performed EMSA to compare the TSE binding activities of JEG-3 and HeLa cell nuclear extracts. As expected, a radiolabeled probe of the hCGα TSE produces a single shifted band when combined with JEG-3 nuclear extract (Fig. 2, lane 1). Interestingly, this band co-migrates with a complex formed between JEG-3 protein and a known AP-2 site from the hMtIIa gene (lane 5). In assays using nuclear extract from the AP-2-producing HeLa cell line, complexes formed with TSE and hMtIIa probes are indistinguishable (lanes 9 and 13). Specificity is also demonstrated by cross-competition with the appropriate unlabeled oligonucleotide. When added in 100-fold molar excess to the binding mixtures, unlabeled TSE or hMtIIa oligonucleotides compete with labeled probe to diminish the levels of radiolabeled complex (lanes 2, 3, 6, 7, 10, 11, 14 and 15). However, an unlabeled mutant TSE oligonucleotide, which is unable to bind TSE binding protein, fails to eliminate the complex in the EMSA (lanes 4, 8, 12 and 16).
Figure 2.
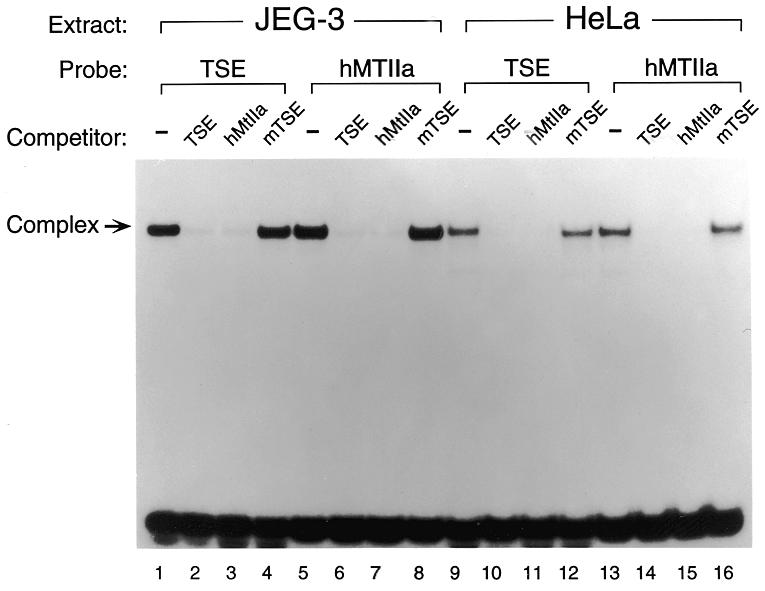
Comparison of the TSE binding activity in JEG-3 nuclear extract with AP-2 in HeLa nuclear extract. Electrophoretic mobility shift analysis of the TSE and the hMtIIa binding proteins in JEG-3 and HeLa cells. hMtIIa is a strong AP-2 binding site (20,45). Binding assays were performed with the indicated probes using ~1 µg of nuclear extract. As specific competitors, unlabeled TSE, hMtIIa and a TSE containing a mutation that abolishes binding (mTSE) were added at ~100-fold molar excess over the probe.
A polyclonal antibody raised against a C-terminal AP-2α peptide (polyclonal rabbit anti-human AP-2 antiserum from Santa Cruz Biotechnology, CA) has been shown to supershift complexes formed by TSE-containing oligonucleotides and proteins present in JEG-3 and HeLa cell extracts (4; data not shown). The antibody is predicted to cross-react with all three family members (AP-2α, β and γ) since this region is highly conserved. To further establish the identity of the TSE binding protein as an AP-2 family member, gel shift analyses were performed using affinity-purified protein rather than cell extract. Previously, we showed that affinity-purified TSE binding protein interacts strongly with the hCGα TSE and the TSE sites in hCGβ footprint region (FP) 2 and FP4, interacting less strongly with the site located in FP1 (15). Figure 3 shows that affinity-purified TSE binding protein forms a single complex with each of the following radiolabeled probes: the hCGα TSE, hMtIIa, the hCGβ5 TSE located in FP2 and the hCGβ5 TSE located in FP4. Each TSE complex is recognized and supershifted by addition of the anti-AP-2 antibody, while addition of rabbit IgG has no effect.
Figure 3.
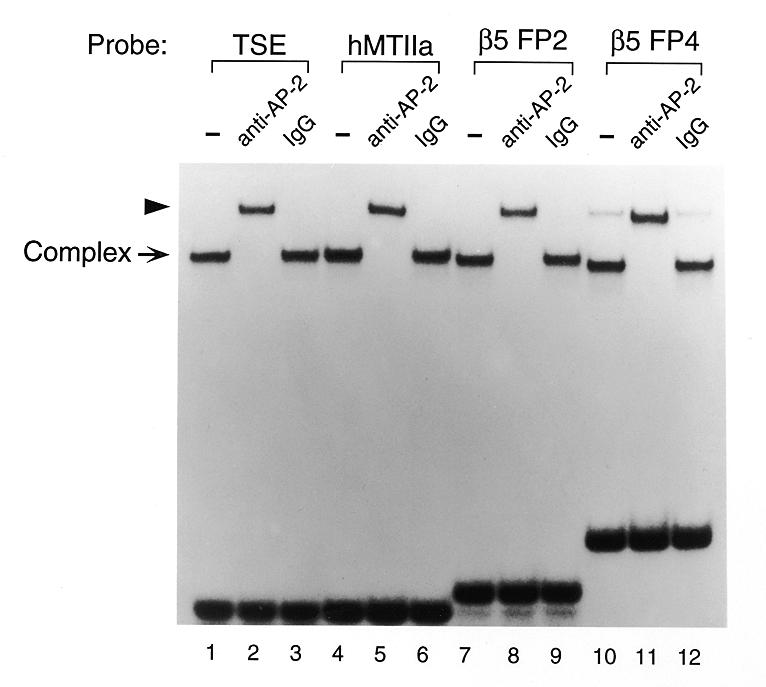
Affinity-purified TSE binding protein reacts with a polyclonal AP-2 antibody. EMSAs were performed using affinity-purified TSE binding protein. TSE binding protein was purified as described by Steger et al. (15). Either 1 µl of anti-AP-2 antibody (polyclonal rabbit anti-human AP-2 antibody from Santa Cruz Biotechnology, CA) or preimmune IgG was added to the reaction mixture. The arrowhead indicates a presumed supershift complex. TSEs from the hCGβ gene (β5) also form complexes that are supershifted by the anti-AP-2 antibody.
AP-2 sites play an important role in controlling hCGβ subunit gene expression
Previously, it has been shown that in the hCGβ subunit gene AP-2 proteins interact with a cluster of sites in a region important for expression in JEG-3 cells termed FP1, FP2 and FP4 (4,15). It was most likely that the multiple AP-2 binding sites at FP1, FP2 and FP4 are required in combination for activation. In fact, roles for FP2 and FP4 have been demonstrated (4). To test and rank the relative contribution of each of the four sites, we introduced mutations interrupting the four individual AP-2 binding sites in the 5′-flanking sequence of the hCGβ5 gene by site-directed mutagenesis. Separate mutations were made within FP4 since it has two elements that each bind AP-2 proteins. The mutations were introduced into the –361 hCGβ5 promoter on the CAT reporter and the plasmids were transfected into JEG-3 cells in parallel with the previously studied truncation plasmids to test the effects on hCGβ gene expression (Fig. 4A).
Figure 4.


Effects of AP-2 binding site mutations on hCGβ5 promoter activity. (A) Point mutations are shown in bold within each of the four AP-2 binding sites within the three footprints that bind AP-2 on the human CGβ5 subunit promoter. (B) Expression levels were determined by transfecting hCGβ5 promoter/CAT expression vectors containing mutations in each of the four individual AP-2 sites into JEG-3 cells. These were compared to the full-length wild-type promoter (–361), an empty vector control and a series of hCGβ5 promoter truncations previously studied (15). The presence of each of the wild-type TSE elements for each transfected plasmid is indicated by ovals. An X in that position indicates a mutation. Units of CAT activity were normalized to the internal control β-galactosidase activity. CAT/β-galactosidase activity values were normalized to the activity of the 361β-CAT wild-type plasmid. Values are the means ± SEM of at least three independent transfection experiments. Error bars represent SEM.
Mutation of each individual element decreased expression from the hCGβ5 promoter (Fig. 4B). The most effective mutation was mFP-2 but, notably, even the least effective mutation, mFP4-1, decreased expression by 60%. We also performed EMSAs demonstrating that these mutations eliminate binding by AP-2 proteins (data not shown).
Thus, all four of the hCGβ5 AP-2 sites are required for maximal placenta-specific expression in JEG-3 cells. In comparison to the 5′ truncations, deletion to –305, which deletes just one AP-2 site (FP1), decreases expression by 49%, while deletion to –279, which deletes two AP-2 sites, decreases expression by over 80%, similar to the internal mutation of the more important AP-2 sites (FP1, FP2 and FP4-2).
Tryptic peptides isolated from the TSE binding complex are homologous to AP-2γ
To determine the identity of the TSE binding complex, we previously purified the placental transcription factor that binds to the TSE to near homogeneity from JEG-3 cells (15). Three tryptic peptides from the partially purified material were sequenced. Figure 5A summarizes the pmol amounts of the predominant amino acid detected at each position. All sequence assignments were made with >90% confidence except for residues listed in parentheses, where the confidence level was <90%. The assignments listed were the major signal observed in each cycle, with background and impurities accounting for <10% of the major signal in all cases and <5% in most cases. Peptide 1 sequence is 10 amino acids in length, of which eight amino acids are exact matches to the entire family of AP-2 sequences (Fig. 5B), however, all 10 amino acids match human AP-2γ. Peptide 1 is in the span region of the helix–span–helix domain involved in AP-2 dimerization (Fig. 5C). A second peptide of six amino acids also shows 100% homology to human AP-2γ. This peptide, which is also located in the span domain, is less related to AP-2α and unrelated to AP-2β. In the third peptide sequence, all 17 amino acids match human AP-2γ, while fewer match with other members of the AP-2 family. Peptide 3 is found in the acidic activation domain, which is least conserved between family members. The 100% homology between all three TSE binding complex peptides and human AP-2γ leads to the conclusion that the TSE binding complex contains AP-2γ.
AP-2α and AP-2γ are expressed in JEG-3 placental trophoblast cells
Expression of AP-2 family mRNAs is restricted to a subset of tissues that include many neural, epidermal and urogenital tissues. Previously, in situ hybridization of mouse embryo sections showed that AP-2α mRNA is expressed extra-embryonically in trophoblastic tissue earlier than it is detected in the embryo proper and as expression becomes detectable in the embryo it also continues to persist in the trophoblastic cells (25). The presence of AP-2 proteins in JEG-3 nuclear extract and placental cells was demonstrated by western blot analysis and EMSA antibody supershift using the commercial antibody (4; data not shown). This AP-2 antibody supershifts the EMSA complexes on the hCGα and hCGβ subunit promoters and reacts with a band of 48–52 kDa from JEG-3 extract, the appropriate size for all three family members: AP-2α, AP-2β and AP-2γ. Therefore, it is not known if the band observed by western blot represents AP-2α, AP-2γ or both.
Northern analysis reveals at least five mRNAs in JEG-3 RNA that hybridize with a full-length AP-2α cDNA probe (data not shown). Because the RNA banding pattern in JEG-3 cells was not identical to that in HeLa (22) or PA-1 cells (26), it seemed possible that the TSE binding protein might be a placenta-specific isoform of AP-2α. To date, at least five additional isoforms of human AP-2α have been identified in HeLa and PA-1 cells (26,27). To test whether the AP-2α mRNAs present in JEG-3 cells might be distinct from the previously characterized AP-2α isolated from HeLa cells, we used PCR to clone the cDNA for AP-2α from JEG-3 mRNA. Oligonucleotides encoding the 5′- and 3′-termini of AP-2α were used for PCR (see Materials and Methods). However, after cloning and sequencing multiple AP-2α cDNAs from JEG-3 mRNA using PCR, the only functional open reading frame found was identical to that of AP-2α expressed in HeLa cells. While our primers would not have detected AP-2α variant 3, they were capable of detecting the other two AP-2α isoforms known to be expressed in HeLa cells (27).
The full-length AP-2α probe used previously hybridizes to all AP-2 mRNAs because of highly conserved 3′ sequences. To distinguish between AP-2 mRNAs, we used cDNA probes containing the 5′ divergent sequences of AP-2α and AP-2γ (Materials and Methods). This analysis shows that both AP-2α and AP-2γ mRNAs are expressed in JEG-3 cells (see Fig. 7). While the PCR and northern analysis suggest the possibility that AP-2α protein is present, to date only AP-2γ protein has been isolated from JEG-3 cells (Fig. 5). A unique probe targeting AP-2β was not used because AP-2β levels are extremely low in both the placenta and the embryo (28).
AP-2 contributes to cAMP-induced expression of the hCG α-subunit gene
Activation of a cAMP-regulated pathway increases hCGα gene expression as measured by transfection assay. The CRE and GATA elements have been shown to respond but the role of AP-2 has not been addressed. To investigate the role of the cis-acting elements and their interactions in cAMP-regulated expression of the α-subunit gene, we introduced base sub-stitutions into the placenta-specific enhancer that prevent binding to the AP-2 site, the GATA site and the CRE (4,9,15). The cAMP-induced expression of each site, along with different combinations of mutated sites, was assessed within the context of the intact promoter by performing transient expression assays (Fig. 6). As expected, the cAMP-induced response is mediated primarily through the CRE. In addition, as shown previously, the GATA site confers induction in response to cAMP (10). However, in the absence of both the CRE and GATA sites, stimulation of the cAMP pathway by forskolin induces AP-2-driven expression 6.5-fold, an effect that is lost upon mutation of the TSE. The ability of the hCGα AP-2 site (TSE) to mediate cAMP induction is consistent with the observation that AP-2 family members mediate cAMP and kinase C responsiveness in a number of genes (20,21). Accordingly, we tested whether forskolin affects AP-2 family mRNA levels in JEG-3 cells (Fig. 7, lanes 4, 6 and 8). Because the full-length probe reacts with all AP-2 mRNA species, probes specific for the unique sequences in the 5′-region were generated and used to distinguish between AP-2α and AP-2γ (as noted above). AP-2α mRNA is increased between 1 and 8.5 h following forskolin induction. At 3 h, AP-2α mRNA expression is 2- to 3-fold higher in cells treated with forskolin versus cells treated with ethanol vehicle. In contrast to AP-2α mRNA expression, which is induced in response to cAMP (Fig. 7, lanes 4, 6 and 8), AP-2γ levels remain constant regardless of forskolin induction (Fig. 7, lanes 4, 6 and 8).
Figure 6.

The TSE/AP-2 binding site contributes to cAMP-induced expression in JEG-3 cells. The transcriptional activity of wild-type and mutant α-subunit enhancer/promoter expression vectors was determined by transfection into the placental trophoblast cell line JEG-3. CAT expression plasmids contain the human α-subunit gene from –224 to +60 (with one of the two 18 bp CRE repeats deleted) with either wild-type or mutant CREs, TSEs and GATA elements. The presence of the wild-type TSE, GATA and CRE elements for each transfected plasmid is indicated by an oval, diamond and triangle, respectively. An X indicates a mutation. cAMP-regulated expression was analyzed by treating JEG-3 transfected cells with forskolin for the 12–14 h before harvest. Fold cAMP activation is the ratio of the induced transcriptional activity brought about by forskolin treatment divided by the basal activity for that individual expression vector. Units of CAT activity were normalized to the internal control β-galactosidase activity. CAT/β-galactosidase activity values were normalized to the basal activity of the wild-type α-subunit gene expression vector. All values are means of three independent transfection experiments, with error bars representing SEM.
DISCUSSION
The hCG α-subunit gene is coordinately regulated by the family of AP-2 proteins, the zinc finger transcriptional regulators GATA-2 and GATA-3 and the ubiquitous cAMP-sensitive activator CREB. In addition, AP-2 proteins are major regulators of the hCGβ gene and, as such, act to coordinate expression of both hCG subunit genes in placenta. AP-2 has now been found to regulate a number of placental genes, but the recent discovery that there is a family of AP-2 proteins, encoded by three independent genes (α, β and γ), makes it imperative that the roles of the individual proteins be determined. Previous studies point towards a possible role for AP-2α in hCGα and hCGβ regulation. However, we provide evidence, including amino acid sequence identity, that AP-2γ is present in the AP-2 protein complex binding the TSEs.
Several lines of evidence show that the protein that binds the hCG α- and β-subunit genes is a member of the AP-2 family (4,15). These data include similarities in size (52–56 kDa), DNA binding sites (CCNNNGG) and migration rate in EMSAs. In EMSAs, oligonucleotide probes containing the hCGα TSE or the hMtIIa AP-2 binding site are equally effective in competing for binding to TSEB or AP-2. Additionally, TSE–protein EMSA complexes are recognized by antibody specific for AP-2 family proteins. This antibody is directed against the C-terminus of AP-2α. Since it was created prior to the identification of AP-2γ and AP-2β, it was not designed to discriminate between the products of the three separate genes, and in fact is predicted to cross-react with all three. McPherson et al. (18) have demonstrated that the antibody is strongly cross-reactive with AP-2γ. Furthermore, the antibody will likely cross-react with AP-2β, due to the high homology of the three proteins in this C-terminal region.
hAP-2α 420SHTDNNAKSSDKEEKHRK437
hAP-2β 432SGEGPGSKTGDKEEKHRK449
hAP-2γ 433QSPADSNKTLEKMEKHRK450
AP-2α and AP-2β are even more closely related at the C-terminus than the α and γ proteins, thus the antibody does not distinguish between different AP-2 proteins in EMSA or western blot analysis. This is the same antibody used by Johnson et al. (4) to determine that AP-2α was the regulator of the hCG genes.
In mice, AP-2α and AP-2γ are expressed in both the embryo and the trophoblastic tissue during development. AP-2β is barely detectable in this tissue (28). AP-2 is also expressed in human trophoblasts. Primary cultures of human placental cytotrophoblasts show elevated levels of AP-2 mRNA upon differentiation into syncytiotrophoblasts in vitro; levels gradually increase over a 12 day period (4). Probes used in these experiments did not differentiate between family members. We have cloned the cDNA for AP-2α from JEG-3 cells and have partially purified AP-2γ from the same cell line, demonstrating expression of these genes in placental trophoblasts. In addition, we have shown by northern analysis, using probes that distinguish between AP-2α, AP-2β and AP-2γ, that AP-2α and AP-2γ are expressed in JEG-3 cells (AP-2β is known not to be present in placenta) (28).
AP-2 binding sites have been shown to convey induction by cAMP to their target genes. Previously, this effect could not be explained by quantitative changes in AP-2 expression. Luscher et al. (23) showed that expression of both AP-2 mRNA and protein in HeLa cells was not enhanced by agents that increase intracellular cAMP. Sequence analysis reveals that all three AP-2 family members contain a putative PKA phosphorylation site, suggesting the possibility of a post-translational mechanism involving phosphorylation. At least one example supports this mechanism. Park et al. (24) showed that AP-2 mediates cAMP responsiveness in the insulin induction of acetyl CoA carboxylase gene expression. In addition, they demonstrated that AP-2 is phosphorylated by PKA both in vitro and in vivo. Interestingly, phosphorylation does not affect binding activity of AP-2 to the DNA. These data correlate with previous observations that treatment of cells with forskolin, an agent that activates cAMP-dependent protein kinase, does not alter the binding of AP-2 to its regulatory element, suggesting that the change in transcriptional activity is mediated through a process other than simple binding to the AP-2 site.
Our studies demonstrate that, in contrast to previous examples, AP-2-mediated cAMP induction of placental genes is correlated with an increase in AP-2α mRNA expression. These data do not exclude the involvement of phosphorylation or other mechanisms in achieving cAMP-induced transactivation. If AP-2 is responsible for coordinate expression of placental genes, as implied by its role in hCG α- and β-subunit expression, then multiple mechanisms of activation may be required to achieve the level of complex control necessary. In fact, even regulation of the hCGβ gene alone is apparently highly complex, relying on four sites that contribute synergistically to activation of expression.
A widespread role in placenta-specific gene expression is evidenced by the demonstration that the AP-2 family transcription factors play important roles in a number of placental genes. For example, AP-2 regulates activity of the ovine P450 side chain cleavage (CYP11A1) promoter in trophoblast cell lines but not in adrenal (Y1) cells (16). It regulates the human 17β-hydoxysteroid dehydrogenase type 1 gene in JEG-3 cells in coordination with Sp1 and GATA factors (40). It regulates the germ cell alkaline phosphatase gene in JEG-3 cells, also in coordination with Sp1 (41). Another example involves transcription of the chorionic gonadotropin/luteinizing hormone receptor gene (42). Expression of this gene in both JEG-3 cells and placenta is dependent on an AP-2 site and an AP-2-like protein that form complexes with the proximal promoter. Additionally, Shi et al. (28) have shown that AP-2γ regulates murine adenosine deaminase gene expression during placental development. We and others (4,15; and studies herein) have shown that both the α- and β-subunit genes of hCG are also regulated by AP-2 family members for basal and cAMP-sensitive expression. Furthermore, the evidence shows the presence of both AP-2α and AP-2γ (28; and studies herein) in JEG-3 cells. While co-transfections with AP-2α expression vector will activate the hCG subunit genes (4), no direct evidence has demonstrated the presence of AP-2α in the TSE binding complex. Additionally, in mice lacking AP-2α, no placental defects are found (43,44). Our protein sequencing data firmly demonstrate the presence of AP-2γ in the complex binding the TSE, perhaps indicating a dominant or at least independent role for AP-2γ in placental regulation of the hCG genes. Thus, one or more of the AP-2 proteins may be important regulators for both hCG gene expression and for placenta-specific expression in general.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Janice S. Bailey and Kevin Y. Urayama for assistance with this work. This work was supported by grants from the March of Dimes and from the NIH (HD20377) to P.L.M., an American Heart Association fellowship to C.L. and an Association pour la Recherche sur le Cancer fellowship (ARC) to S.C.
REFERENCES
- 1.Simpson E.R. and MacDonald,P.C. (1981) Annu. Rev. Physiol., 43, 163–188. [DOI] [PubMed] [Google Scholar]
- 2.Flint A. (1983) Curr. Topics Exp. Endocrinol., 5, 75–95. [Google Scholar]
- 3.Talamantes F. and Ogren,L. (1988) In Knobil,E. and Neill,J.D. (eds), The Physiology of Reproduction. Raven Press, New York, NY, Vol. 2, pp. 2093–2144.
- 4.Johnson W., Albanese,C., Handwerger,S., Williams,T., Pestell,R.G. and Jameson,J.L. (1997) J. Biol. Chem., 272, 15405–15412. [DOI] [PubMed] [Google Scholar]
- 5.Kohler P.O. and Bridson,W.E. (1971) J. Clin. Endocrinol., 32, 683–687. [DOI] [PubMed] [Google Scholar]
- 6.Ringler G.E. and Strauss,J.F. (1990) Endocr. Rev., 11, 105–123. [DOI] [PubMed] [Google Scholar]
- 7.Delegeane A.M., Ferland,L.H. and Mellon,P.L. (1987) Mol. Cell. Biol., 7, 3994–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jameson J.L., Jaffe,R.C., Deutsch,P.J., Albanese,C. and Habener,J.F. (1988) J. Biol. Chem., 263, 9879–9886. [PubMed] [Google Scholar]
- 9.Steger D.J., Hecht,J.H. and Mellon,P.L. (1994) Mol. Cell. Biol., 14, 5592–5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steger D.J., Altschmied,J., Büscher,M. and Mellon,P.L. (1991) Mol. Endocrinol., 5, 243–255. [DOI] [PubMed] [Google Scholar]
- 11.Mellon P.L., Clegg,C.H., Correll,L.A. and McKnight,G.S. (1989) Proc. Natl Acad. Sci. USA, 86, 4887–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darnell R.B. and Boime,I. (1985) Mol. Cell. Biol., 5, 3157–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silver B.J., Bokar,J.A., Virgin,J.B., Vallen,E.A., Milsted,A. and Nilson,J.H. (1987) Proc. Natl Acad. Sci. USA, 84, 2198–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deutsch P.J., Hoeffler,J.P., Jameson,J.L., Lin,J.C. and Habener,J.F. (1988) J. Biol. Chem., 263, 18466–18472. [PubMed] [Google Scholar]
- 15.Steger D.J., Büscher,M., Hecht,J.H. and Mellon,P.L. (1993) Mol. Endocrinol., 7, 1579–1588. [DOI] [PubMed] [Google Scholar]
- 16.Yamada K., Harada,N., Honda,S.-I. and Takagi,Y. (1995) J. Biol. Chem., 270, 25064–25069. [DOI] [PubMed] [Google Scholar]
- 17.Shi D., Winston,J.H., Blackburn,M.R., Datta,S.K., Hanten,G. and Kellems,R.E. (1997) J. Biol. Chem., 272, 2334–2341. [PubMed] [Google Scholar]
- 18.McPherson L.A., Baichwal,V.R. and Weigel,R.J. (1997) Proc. Natl Acad. Sci. USA, 94, 4342–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bosher J.M., Totty,N.F., Hsuan,J.J., Williams,T. and Hurst,H.C. (1996) Oncogene, 13, 1701–1707. [PubMed] [Google Scholar]
- 20.Imagawa M., Chiu,R. and Karin,M. (1987) Cell, 51, 251. [DOI] [PubMed] [Google Scholar]
- 21.Mitchell P.J., Wang,C. and Tjian,R. (1987) Cell, 50, 847–861. [DOI] [PubMed] [Google Scholar]
- 22.Williams T., Admon,A., Lüscher,B. and Tjian,R. (1988) Genes Dev., 2, 1557–1569. [DOI] [PubMed] [Google Scholar]
- 23.Lüscher B., Mitchell,P.J., Williams,T. and Tjian,R. (1989) Genes Dev., 3, 1507–1517. [DOI] [PubMed] [Google Scholar]
- 24.Park K. and Kim,K.-H. (1993) J. Biol. Chem., 268, 17811–17819. [PubMed] [Google Scholar]
- 25.Mitchell P., Timmons,P., Hébert,J., Rigby,P. and Tjian,R. (1991) Genes Dev., 5, 105–119. [DOI] [PubMed] [Google Scholar]
- 26.Buettner R., Kannan,P., Imhof,A., Bauer,R., Yim,S.O., Glockshuber,R., Van Dyke,M.W. and Tainsky,M.A. (1993) Mol. Cell. Biol., 13, 4174–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meier P., Koedood,M., Philipp,J., Fontana,A. and Mitchell,P.J. (1995) Dev. Biol., 169, 1–14. [DOI] [PubMed] [Google Scholar]
- 28.Shi D. and Kellems,R.E. (1998) J. Biol. Chem., 273, 27331–27338. [DOI] [PubMed] [Google Scholar]
- 29.Fischer W.H., Karr,D., Jackson,B., Park,M. and Vale,W. (1991) Methods Neurosci., 6, 69–84. [Google Scholar]
- 30.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sawadogo M., Van Dyke,M.W., Gregor,P.D. and Roeder,R.G. (1988) J. Biol. Chem., 263, 11985–11993. [PubMed] [Google Scholar]
- 32.Chomczynski P. and Sacchi,N. (1987) Anal. Biochem., 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 33.Maniatis T., Fritsch,E.F. and Sambrook,J. (1982) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 34.Belsham D.D., Wetsel,W.C. and Mellon,P.L. (1996) EMBO J., 15, 538–547. [PMC free article] [PubMed] [Google Scholar]
- 35.Marsh J., Ertle,M. and Weykes,E. (1984) Gene, 32, 481–485. [DOI] [PubMed] [Google Scholar]
- 36.Sanger F., Nicklen,S. and Coulson,A.R. (1977) Proc. Natl Acad. Sci. USA, 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mellon P.L., Parker,V., Gluzman,Y. and Maniatis,T. (1981) Cell, 27, 279–288. [DOI] [PubMed] [Google Scholar]
- 38.Gorman C. (1985) In Glover,D.M. (ed.), DNA Cloning: A Practical Approach. IRL Press, Oxford, UK, Vol. II, pp. 143–190.
- 39.Seed B. and Sheen,J.Y. (1988) Gene, 67, 271–277. [DOI] [PubMed] [Google Scholar]
- 40.Piao Y.-S., Peltoketo,H., Vihko,P. and Vihko,R. (1997) Endocrinology, 138, 3417–3425. [DOI] [PubMed] [Google Scholar]
- 41.Wada N. and Chou,J.Y. (1993) J. Biol. Chem., 268, 14003–14010. [PubMed] [Google Scholar]
- 42.Hu Y.L., Lei,Z.M. and Rao,C.V. (1996) Endocrinology, 137, 3897–3905. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J., Hagopian-Donaldson,S., Serbedzija,G., Elsemore,J., Plehn-Dujowich,D., McMahon,A.P., Flavell,R.A. and Williams,T. (1996) Nature, 381, 238–241. [DOI] [PubMed] [Google Scholar]
- 44.Schorle H., Meier,P., Buchert,M., Jaenisch,R. and Mitchell,P.J. (1996) Nature, 381, 235–238. [DOI] [PubMed] [Google Scholar]
- 45.Williams T. and Tjian,R. (1991) Genes Dev., 5, 670–682. [DOI] [PubMed] [Google Scholar]