

Abstract
Cytogenetic analysis provides important information on the genetic mechanisms of cancer. The Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (Mitelman DB) is the largest catalog of acquired chromosome aberrations, presently comprising >70,000 cases across multiple cancer types. Although this resource has enabled the identification of chromosome abnormalities leading to specific cancers and cancer mechanisms, a large-scale, systematic analysis of these aberrations and their downstream implications has been difficult due to the lack of a standard, automated mapping from aberrations to genomic coordinates. We previously introduced CytoConverter as a tool that automates such conversions. CytoConverter has now been updated with improved interpretation of karyotypes and has been integrated with the Mitelman DB, providing a comprehensive mapping of the 70,000+ cases to genomic coordinates, as well as visualization of the frequencies of chromosomal gains and losses. Importantly, all CytoConverter-generated genomic coordinates are publicly available in Google BigQuery, a cloud-based data warehouse, facilitating data exploration and integration with other datasets hosted by the Institute for Systems Biology Cancer Gateway in the Cloud (ISB-CGC) Resource. We demonstrate the use of BigQuery for integrative analysis of Mitelman DB with other cancer datasets, including a comparison of the frequency of imbalances identified in Mitelman DB cases with those found in The Cancer Genome Atlas (TCGA) copy number datasets. This solution provides opportunities to leverage the power of cloud computing for low-cost, scalable, and integrated analysis of chromosome aberrations and gene fusions in cancer.
Keywords: Cytogenetics, Cloud-based analysis, Mitelman database, Genomics
1. INTRODUCTION
1.1. Cytogenetics
Chromosome aberrations are a characteristic feature of neoplasia, and cytogenetic analyses of tumor cells have been instrumental for our understanding that cancer is a genetic disorder at the cellular level. Acquired chromosome abnormalities, numerical and structural, leading to escape from normal regulation of growth, apoptosis, and differentiation, have been reported in more than 70,000 cases of neoplasia comprising all major cancer entities. A steadily increasing number of characteristic aberrations that are associated with distinctive tumor types have been identified1; some are even pathognomonic, thus serving as important theoretical and clinical biomarkers. The growing understanding of the clinical significance of the genetic constitution of tumor cells has gradually led to an increasing emphasis on such features in the classification of neoplasms. As a consequence, cytogenetic data and/or their molecular equivalents have been incorporated as important, sometimes necessary, parameters in the WHO classifications of hematologic disorders2, central nervous system (CNS) tumors3, and bone and soft tissue tumors4.
1.2. Balanced and unbalanced chromosome aberrations
Chromosome aberration may be dichotomized into those (translocations, inversions, insertions) that are seemingly balanced, i.e., the aberrations are not associated with any net gain or loss of genetic material, and those that are unbalanced (e.g., ploidy shifts, numerical changes, deletions, duplications, and non-reciprocal translocations); often, balanced and unbalanced rearrangements co-exist in tumor cells. Of the balanced structural chromosomal aberrations that have been characterized at the genomic level, practically all have been shown to give rise to either deregulation, usually over-expression, of a cancer-causing gene in one of the breakpoints, or the creation of an abnormal cancer-initiating hybrid gene through fusion of parts of two genes, one in each breakpoint5. More than 1,000 oncogenic gene fusions in malignant hematologic disorders and solid tumors have by now been identified as a consequence of cytogenetically identified chromosomal abnormalities.
The introduction of deep sequencing or massively parallel sequencing (MPS) has revolutionized the possibilities to detect gene fusions without any pre-existing knowledge of the genetic constitution and has led to an enormous increase in the numbers of identified gene fusions in different cancer types6–8. The results have dramatically changed the gene fusion landscape; a plethora – more than 30,000 – new gene fusions, the great majority involving previously unsuspected genes, has been identified through MPS. Recent evidence, albeit indirect, indicates that a substantial subset of MPS-detected fusions may be stochastic events5,9, and a major challenge will be to verify by functional studies which of the alleged gene fusions are primary, pathogenetically important, and which are non-consequential “noise” abnormalities.
The great majority of all chromosomal changes in cancer are, however, unbalanced and lead to gain or loss of genetic material; more than 95% of all malignant epithelial tumors, the predominant cancer type in humans in terms of morbidity and mortality, display aneuploidy resulting in genomic imbalances. Gained/amplified segments have been shown to harbor oncogenes, and deleted segments to contain tumor suppressor genes1. Sophisticated new model systems have produced important insights into how aneuploidy develops and how imbalances may affect cells, but how they contribute to tumorigenesis is largely still unknown10,11,12. It is consequently important to map and validate all imbalances that characterize cancer cells.
1.3. Database content
The Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (Mitelman DB) is a continuously updated catalog that relates cytogenetic changes and their genomic consequences, in particular gene fusions, to tumor characteristics, based either on individual cases or associations. The database presently (January 19, 2023) contains information on >75,000 karyotypes and >33,000 unique gene fusions affecting >14,000 genes. It is important to note that the cytogenetic and sequencing studies complement each other in that the former focus on the relatively uncommon hematologic malignancies (leukemias and lymphomas) and bone and soft tissue tumors, the latter on the most common malignant epithelial tumors. It is striking that whereas 85% of the cases in the cytogenetic database are hematologic disorders or mesenchymal tumors, more than 80% of the cases with gene fusions are malignant epithelial neoplasms.
The karyotypes of individual cells as revealed by cytogenetic methods provide information not always captured by sequencing which gives an aggregated picture of the entire tumor cell population. Intratumor heterogeneity, clonal evolution, ploidy levels, and mechanisms of origin (for example, loss of genetic material may originate by many different chromosome abnormalities such as deletions, dicentric chromosomes, and unbalanced translocations) are examples of parameters more easily identified by cytogenetics. However, so far, no reliable computational tool to analyze the cytogenetic nomenclature has been available. An attempt to map systematically all imbalances in 3,185 solid tumors reported up to 1995 was made by Mertens et al.13 Today, with >70,000 cases, it would be an arduous task to try to perform such an analysis without the help of a computer program that is capable of converting cytogenetic nomenclature into genomic coordinates.
1.4. Computational analysis of karyotypes
Several methods to parse cytogenetic nomenclature14–16 have been recently developed. ISCN-SNAKE14 has been used to compare a select number of malignant neoplasms in the Mitelman DB with sequence data from The Cancer Genome Atlas (TCGA) project and was found to have a very good correspondence as regards chromosome gains, amplifications, and heterozygous deletions, indicating that cytogenetically identified aberrations, in spite of their lower resolution level, are in fact on par with genomic analyses. CytoGPS16 parses ISCN17 karyotypes into a machine-readable format and converts it into a binary Loss-Gain-Fusion (LGF) model which allows researchers to process automatically thousands of karyotypes. The recently developed CytoConverter18, which can precisely specify a chromosomal location according to its distance from the end of the chromosome, offers such a possibility. This user-friendly web-based tool allows users to input any number of human karyotypes and obtain the genomic coordinates of all gains and losses implied by each of the karyotypes. CytoConverter is the only existing program that focuses on conversion to genomic coordinates.
In this work we developed a cloud-based resource, the Mitelman DB web application, that combines Cytoconverter and the Mitelman DB. Cytoconverter’s interconversion functionality further increases accessibility to the comprehensive data in the cloud. Moreover, we demonstrate the effectiveness of CytoConverter as a solution to the lack of standard mapping from aberrations to genomic coordinates, a major issue obstructing large-scale genomic analyses. This was achieved through integration with the Mitelman DB, and by providing examples that compare and combine Mitelman DB data with common datasets available through the ISB-CGC.
2. MATERIAL AND METHODS
2.1. Mitelman database
The present work was based on the July 27, 2022 release of the database containing information on 73,930 karyotypes with 53,901 different abnormalities and 33,457 unique gene fusions affecting 14,061 different genes. Figure 1 shows the increase of data contained in the database since data collection was initiated. Table 1 shows the numbers of cytogenetically abnormal cases and identified fusion genes among various tumor entities. It should be kept in mind that tumor karyotypes often contain more than one abnormality; in particular, malignant solid tumors may be extremely complex with as many as 50 subclones and more than 100 distinct aberrations within the same tumor. The total number of abnormalities thus exceeds 400,000 and is hence almost six times higher than the number of cases. Table 2 shows the distribution of the malignant neoplasms according to organ involvement. The Mitelman DB, including genomic coordinates translated by CytoConverter, is stored in Google’s BigQuery data warehouse. BigQuery provides users with the ability to access, combine, and analyze large datasets using SQL.
Figure 1.
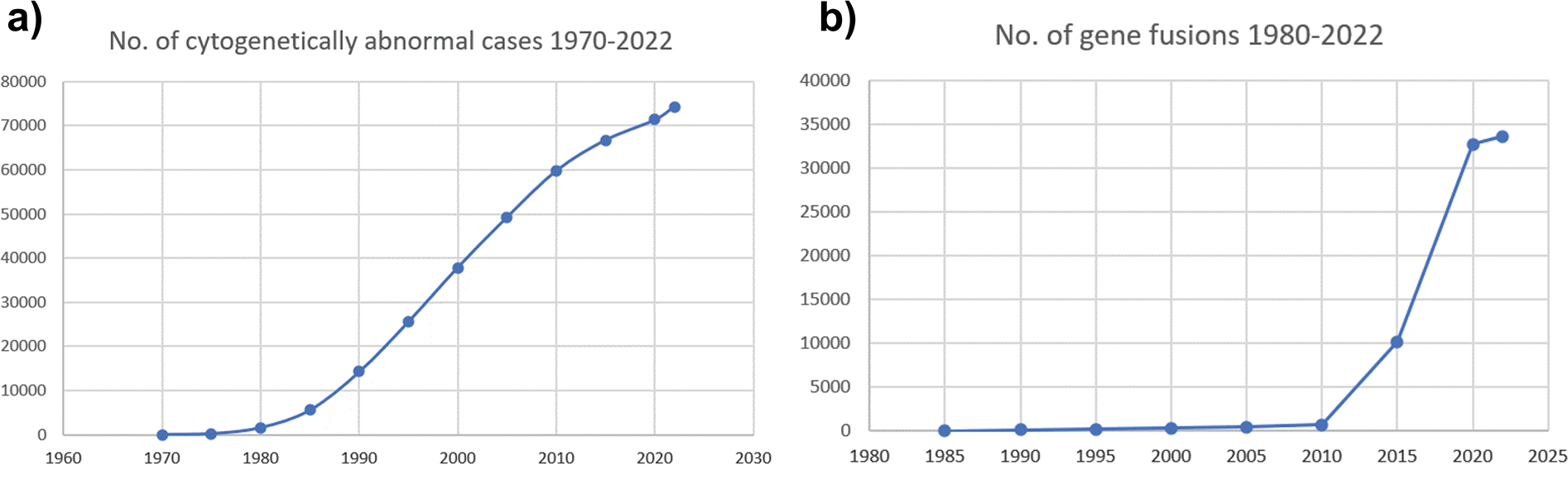
a) Number of neoplasms with clonal chromosome aberrations and b) number of gene fusions in the Mitelman DB since data collection was initiated.
Table 1.
Number of cytogenetically abnormal cases and fusion genes in the Mitelman database.
| Tumor type | No. of cytogenetically abnormal cases | No. of fusion genes* |
|---|---|---|
| Hematologic disorders | ||
| Undifferentiated and biphenotypic leukemia | 605 | 35 |
| Acute myeloid leukemia | 20,205 | 620 |
| Myelodysplastic syndromes | 5,497 | 71 |
| Chronic myeloproliferative disorders | 5,957 | 87 |
| Acute lymphoblastic leukemia | 11,873 | 760 |
| Plasma cell neoplasms | 2,331 | 543 |
| Mature B-cell neoplasms | 9,444 | 420 |
| Mature T- and NK-cell neoplasms | 1,412 | 44 |
| Hodgkin lymphoma | 250 | 13 |
| Miscellaneous hematopoietic/lymphoid neoplasms | 214 | 57 |
| Hematologic disorders - Total | 57,788 | 2,382 |
| Solid tumors | ||
| Benign solid tumors | ||
| Epithelial neoplasms | 1,156 | 79 |
| Mesenchymal neoplasms | 2,838 | 160 |
| Melanocytic neoplasms | 24 | 3 |
| Benign solid tumors - Total | 4,018 | 237 |
| Malignant solid tumors | ||
| Epithelial neoplasms | 6,066 | 24,918 |
| Germ cell neoplasms | 509 | 117 |
| Neuroglial neoplasms | 942 | 1,845 |
| Embryonal nervous system neoplasms | 638 | 180 |
| Melanocytic neoplasms | 352 | 1,682 |
| Mesenchymal neoplasms | 3,117 | 951 |
| Malignant solid tumors - Total | 11,217 | 28,230 |
The total numbers of fusion genes do not add up because each gene fusion is only counted once but may be found in distinct tumor entities.
Table 2.
Distribution of cytogenetically investigated abnormal cases and fusion genes in malignant tumors by organ involvement arranged in decreasing order of frequency according to global cancer incidence rates20.
| Tumor type | No. of cytogenetically abnormal cases | No. of fusion genes |
|---|---|---|
| Breast | 886 | 6,547 |
| Lung | 470 | 4,334 |
| Prostate | 208 | 2,288 |
| Skin | 269 | 1,721 |
| Colon | 404 | 750 |
| Stomach | 189 | 1,270 |
| Liver | 144 | 1,146 |
| Uterus | 323 | 2,272 |
| Esophagus | 44 | 610 |
| Thyroid | 143 | 288 |
| Bladder | 190 | 1,551 |
| Lymphoma | 12,012 | 517 |
| Pancreas | 180 | 331 |
| Leukemia | 46,468 | 1,912 |
| Kidney | 1,769 | 740 |
| Oral cavity | 249 | 403 |
| Ovary | 499 | 2,247 |
| Brain | 946 | 1,845 |
| Salivary glands | 135 | 60 |
| Soft tissue | 2,063 | 253 |
| Bone | 1,054 | 75 |
2.2. Generation of genomic coordinates from Mitelman DB data.
To generate genomic coordinates from the Mitelman DB, we used an updated version of CytoConverter with improved parsing ability. In the new version, ploidy processing does not require the chromosome count in the first field to be exact. Instead, the ploidy is determined via estimation of unaccounted chromosomes and with the bounds specified by ISCN17. The abbreviation of translocations is now permitted as long as the given translocation has been defined previously and no other unique translocation with different breakpoints bears the exact same chromosomes that were translocated. Improved parsing for clones is now implemented; CytoConverter is now able to transform previously used aberrations into consecutive clones.
In order to calculate the genomic coordinates of the gain or loss in each karyotype, all the karyotypes in the Mitelman DB were extracted. Before being converted, each karyotype was validated that it was in the proper format by a syntax checker tool, for details see User Guide in the Mitelman DB. Invalid aberrations within the karyotype were not included in the genomic imbalances analysis. If the karyotype could not be parsed at all, the entire karyotype was excluded. Among the 122,535 cytogenetically abnormal clones in the database, a total 99,970 (81.6%) were acceptable for analysis. The validated karyotype was then run against the CytoConverter tool using an R terminal for the conversion. The generated results were formatted and loaded into the BigQuery dataset that are part of the Mitelman DB web application. This dataset, “mitelman-db:prod” is publicly accessible in the Google Cloud Platform.
2.3. Dataset integration and analysis
We implemented two notebooks that use the BigQuery tables containing Mitelman DB and CytoConverter results in combination with other datasets available in Institute for Systems Biology Cancer Gateway in the Cloud (ISB-CGC). These notebooks are implemented in Google’s Jupyter Notebook application, Colaboratory (Colab) which uses Python to explore and analyze BigQuery tables. Colab can be used for free with limited computational resources, which is sufficient to run the analysis implemented in the notebooks.
Specifically, the notebooks compare frequencies of imbalances obtained from the Mitelman DB and those computed with TCGA. Pearson correlation was used to compare data from Mitelman DB and from TCGA chromosomal aberrations. The Pearson correlation coefficient computation was implemented as SQL User Defined Functions allowing to perform the entire statistical analysis in the cloud.
3. RESULTS
3.1. The Mitelman DB web application
The web application of the Mitelman DB leverages Google’s cloud resources to enable exploration of the Mitelman DB and the converted karyotype results generated from CytoConverter. Using the CytoConverter results, the application provides the option of viewing the genomic coordinate information for either individual karyotypes or for multiple karyotypes in a search result.
For individual karyotypes, the corresponding chromosome and its start and end position are given with the type of imbalance (gain or loss). The information is shown in both tabular and ideogram formats. A screenshot of the Karyotype Info page is shown in Figure 2 as an example. The chromosome abnormalities are shown in a table that includes the clone number, genomic coordinates of the chromosome (start and end position), and the imbalance type (gain or loss). The overall gain or loss in chromosomes can also be visualized in an ideogram (Figure 3).
Figure 2.
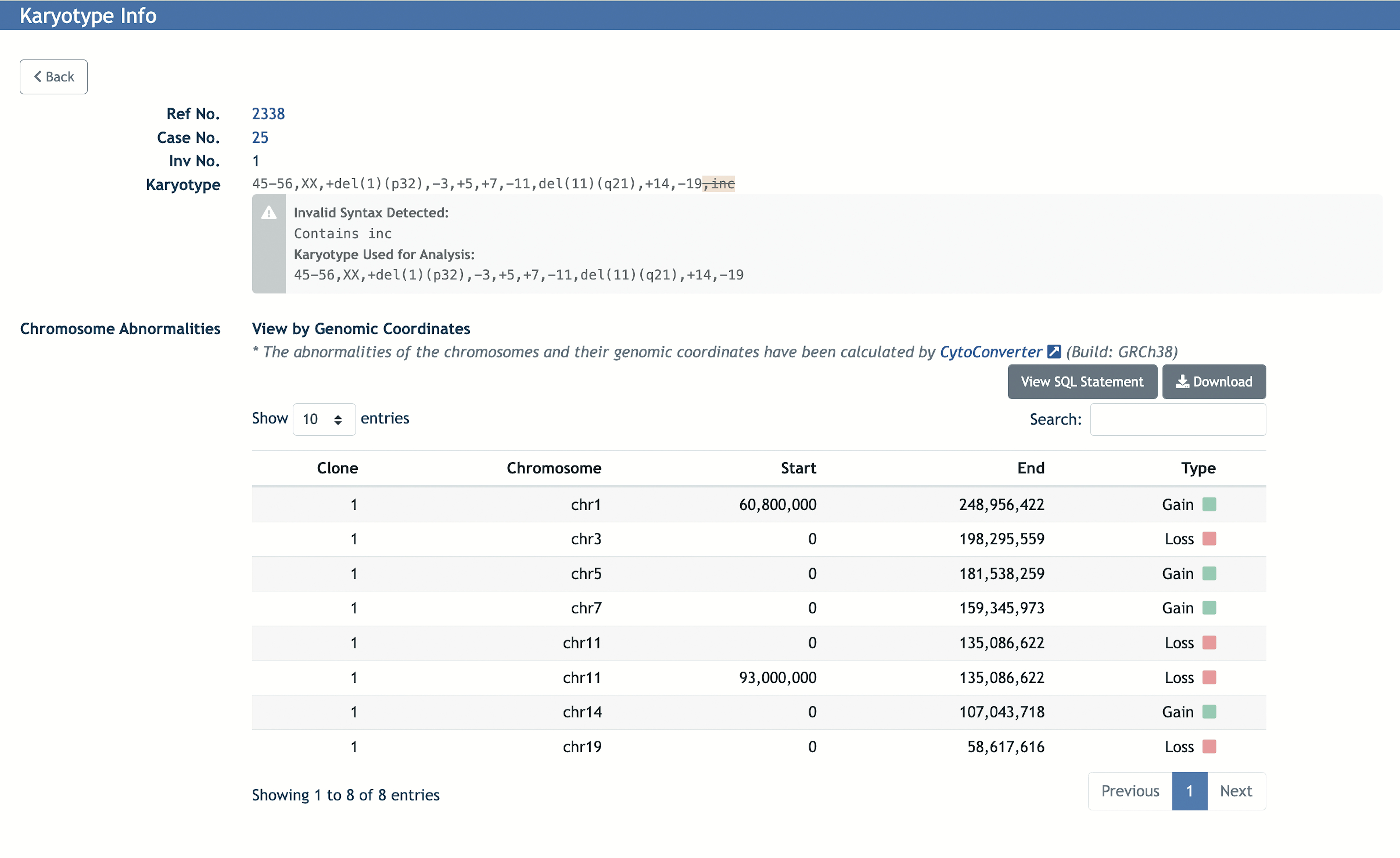
An example of a Karyotype Info page from the Mitelman DB web application, displaying the karyotype sample information and its chromosome abnormalities generated by CytoConverter.
Figure 3.
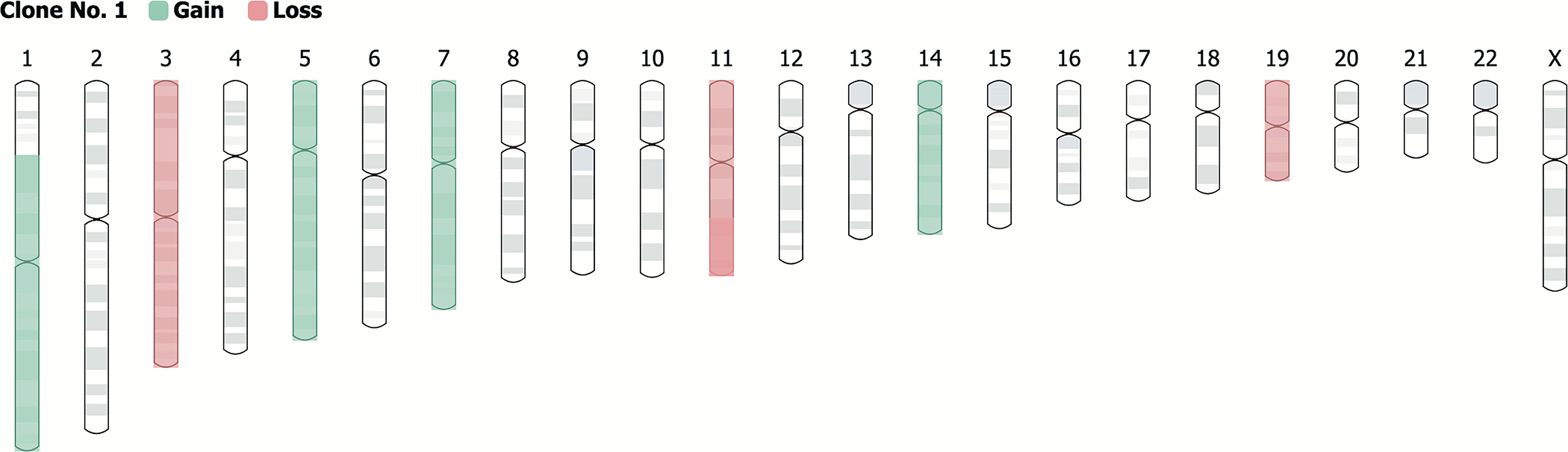
The overall gains and losses in chromosomes can also be visualized in an ideogram for each case.
For analysis of multiple samples, net imbalances across the selected group are available in chart, ideogram, or tabular format. Information includes the chromosome affected, start and end positions, and whether the segment has been lost or gained. Figure 4 shows an example of a result screen in the Mitelman DB, after running an overall chromosomal imbalance analysis from a Cases Cytogenetic search result.
Figure 4.
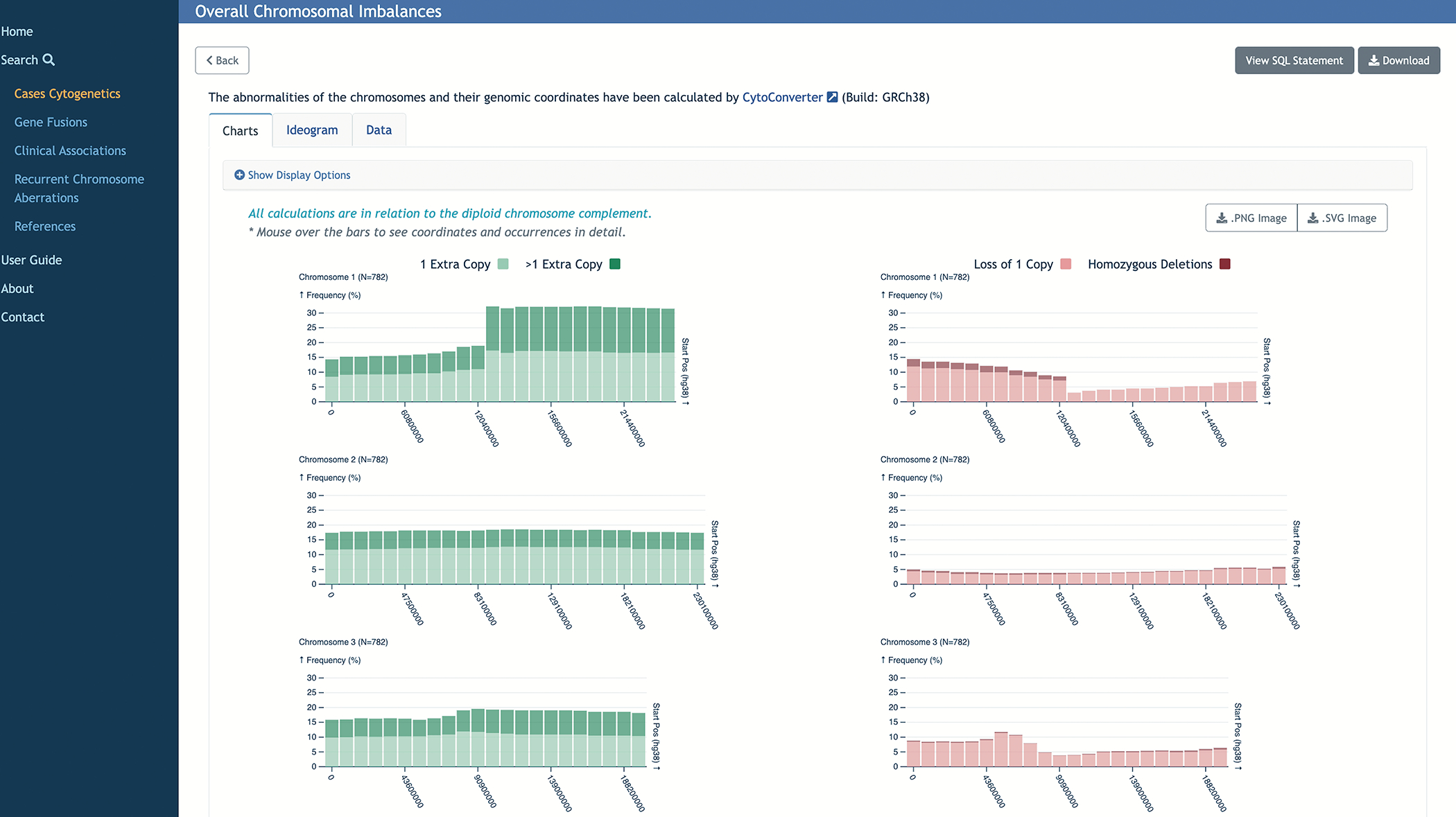
For multiple karyotypes in the Cases Cytogenetics search results, net imbalances across the selected group are available in chart, ideogram or tabular format.
3.2. Cloud-based analysis of the Mitelman DB
In addition to the Mitelman DB web application, users can leverage Google’s cloud computation resources to customize their analyses. The Mitelman DB, including genomic coordinates translated by CytoConverter, is stored in Google’s BigQuery data warehouse which provides users with the ability to access, combine, and analyze large datasets using SQL, Python, and R. We implemented two Python notebooks that demonstrate usage of the Mitelman DB in combination with other datasets available in the ISB-CGC set of BigQuery tables. The notebooks are hosted in ISB-CGC GitHub repository and that can be easily run in Google’s Jupyter Notebook application, Colaboratory (Colab), which allows users to write and execute Python in their browsers. These examples provide a framework to researchers for performing data mining of chromosome aberrations using common bioinformatics tools. They can easily be adapted to add user’s own data, as well as be supplemented and/or combined with other analyses.
The notebooks compare the frequencies of copy number changes in the Mitelman DB with those calculated from TCGA data for three well-known deletions: breast cancer (Chromosome 1), kidney adenocarcinoma (Chromosome 3), and acute myeloid leukemia (Chromosome 5). In agreement with Denomy et al.14 we found similar patterns of gains and losses in the two data sets (Figure 5). Moreover, Table S1 (Supplementary material) shows the Pearson correlation of frequencies computed from Mitelman DB and TCGA-BRCA for each chromosome. According to the analysis most of the significant correlations of Table S1 are also significant in the results of Denomy et al.14 with few exceptions, likely due to different resolution levels.
Figure 5.
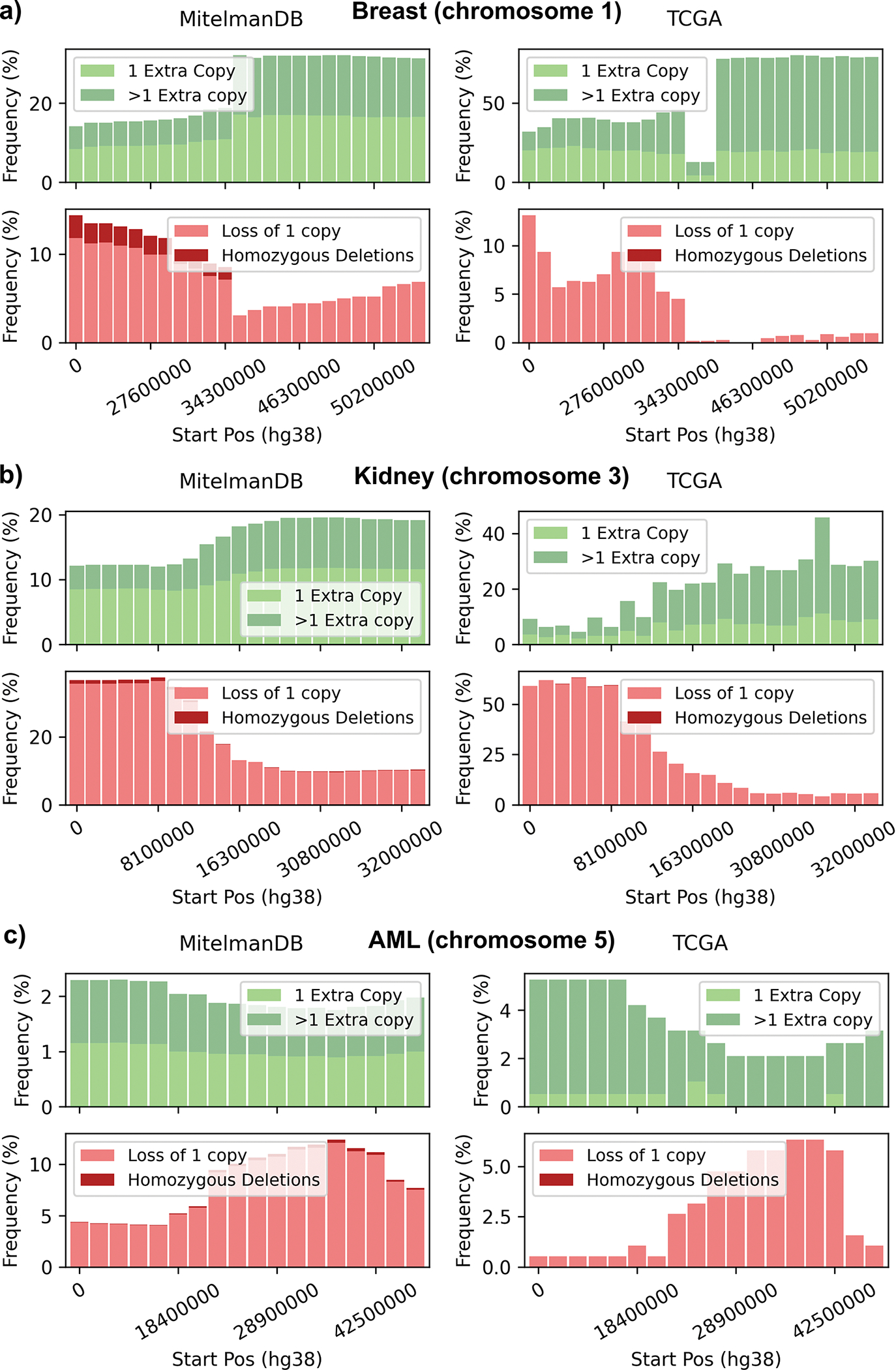
Frequencies of gains, deletions, amplifications, and homozygous deletions in a) chromosome 1 in breast cancer, b) chromosome 3 in kidney adenocarcinoma, and c) chromosome 5 in acute myeloid leukemia (AML), calculated from CytoConverter-generated Mitelman DB data (left panels) and TCGA CNV data (right panels).
4. DISCUSSION
The Mitelman DB provides a rich source of cancer genomic information that is complementary to recent genomic sequencing datasets of cancer. In the present work, the Mitelman DB has been integrated with CytoConverter, a recently developed web-based tool that generates genomic coordinates from the karyotypes. The integration is implemented in a user-friendly web application in which users can explore the Mitelman DB and view the genomic coordinate information for either individual or multiple karyotypes. For individual karyotypes, the corresponding chromosome, its start and end positions, and the type of imbalance (gain or loss) is given by the web application. For multiple karyotypes in the search results, net imbalances across the selected group are displayed in chart, ideogram, or tabular format; information includes the chromosomes affected, start and end positions, and whether the segment has been lost or gained.
We anticipate that the cloud-based resource will be of considerable value as it offers several benefits to the scientific community. The proposed resource provides a reliable, updated computational tool that links karyotypes to genomic coordinates, allowing a systematic analysis of the Mitelman DB and integration with other large datasets of cancer, to uncover previously unrecognized patterns in cancer genetics. Moreover, all CytoConverter-generated genomic coordinates are publicly available in Google BigQuery, a cloud-based data warehouse, facilitating data exploration and integration with other datasets hosted by the ISB-CGC Cloud Resource19, such as TCGA, PanCancer Atlas, the Human Tumor Atlas Network (HTAN), and the Clinical Proteomic Tumor Analysis Consortium (CPTAC). This feature provides opportunities to leverage the power of cloud computing for low-cost, scalable, and integrated analysis of chromosome aberrations and gene fusions in cancer.
BigQuery also allows users to access, combine, and analyze large datasets using SQL, Python, and R. We implemented cloud-based examples that demonstrate how the Mitelman DB can be combined with other datasets in the ISB-CGC set of BigQuery tables. Specifically, we compared the frequencies of imbalances in three cancer types in the Mitelman DB with those available in TCGA, finding an overall good correlation regarding gains, amplifications, and heterozygous deletions.
Supplementary Material
6. ACKNOWLEDGMENTS
Elaine Lee, Boris Aguilar, John Phan, Ronald Taylor, Kawther Abdilleh, David Pot, and William Longabaugh were funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN261201400008C and ID/IQ Agreement No. 17X146 under Contract No. HHSN261201500003I. Thomas LaFramboise was funded by grants from the U.S. National Institutes of Health (R01CA217992, R01LM013067, and R21CA249138).
Felix Mitelman, Bertil Johansson, and Fredrik Mertens were funded by the Swedish Cancer Society and the Swedish Childhood Cancer Foundation. We thank Jeppe Thaning for excellent computational support of the Mitelman DB.
Footnotes
CONFLICTS OF INTEREST
Dr. Taylor contributed to this article in his personal capacity. The views expressed are his own and do not necessarily represent the views of the National Institutes of Health or the United States Government.
SOFTWARE AND DATA AVAILABILITY
The Mitelman web application can be accessed from https://mitelmandatabase.isb-cgc.org
The Mitelman dataset and all CytoConverter-generated genomic coordinates are hosted on the ISB-CGC17 (https://isb-cgc.org/) in existing Google BigQuery tables which are publicly available (dataset ID: mitelman-db.prod). Example use cases, implemented in Jupyter notebooks, describing how to access and use these tables are available on the ISB-CGC Github repository of notebooks (https://github.com/isb-cgc/Community-Notebooks).
- Frequency of gains and losses in the Mitelman DB and TCGA: https://github.com/isb-cgc/Community-Notebooks/blob/master/MitelmanDB/Exploring_and_comparing_MitelmanDB_CytoConverter_and_TCGA_datasets.ipynb
- Pearson correlations comparing the Mitelman DB and TCGA: https://github.com/isb-cgc/Community-Notebooks/blob/master/MitelmanDB/Correlations_MitelmanDB_and_TCGA_datasets.ipynb
REFERENCES
- 1.Heim S, Mitelman F (eds). Cancer Cytogenetics: Chromosomal and Molecular Genetic Aberrations of Tumor Cells. Wiley Blackwell, New York; 2015. [Google Scholar]
- 2.WHO Classification of Tumours Editorial Board. Haematolymphoid tumours [Internet; beta version ahead of print]. Lyon (France): International Agency for Research on Cancer; 2022. [cited December 29, 2022]. (WHO classification of tumours series, 5th ed.; vol. 11). Available from: https://tumourclassification.iarc.who.int/chapters/63. [Google Scholar]
- 3.Louis DN, Reifenberger PA, von Deimling A, et al. (eds.) WHO Classification of Tumours of the Central Nervous System. IARC Press, Lyon;2016. [Google Scholar]
- 4.WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th Ed. IARC Press, Lyon; 2020. [Google Scholar]
- 5.Mertens F, Johansson B, Fioretos T, Mitelman F. The emerging complexity of gene fusions in cancer. Nat Rev Cancer. 2015;15(6):371–381. [DOI] [PubMed] [Google Scholar]
- 6.Gao Q, Liang WW, Foltz SM, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 2018;23(1):227–238.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu X, Wang Q, Tang M, et al. TumorFusions: an integrative resource for cancer-associated transcript fusions. Nucleic Acids Res. 2018;46(D1):D1144–D1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.PCAWG Transcriptome Core Group, Calabrese C, Davidson NR, et al. Genomic basis for RNA alterations in cancer. Nature. 2020;578(7793):129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johansson B, Mertens F, Schyman T, Björk J, Mandahl N, Mitelman F. Most gene fusions in cancer are stochastic events. Genes Chromosomes Cancer. 2019;58(9):607–611. [DOI] [PubMed] [Google Scholar]
- 10.Chunduri NK, Storchová Z. The diverse consequences of aneuploidy. Nat Cell Biol. 2019;21(1):54–62. [DOI] [PubMed] [Google Scholar]
- 11.Ben-David U, Amon A. Context is everything: aneuploidy in cancer. Nat Rev Genet. 2020;21(1):44–62. [DOI] [PubMed] [Google Scholar]
- 12.Steele CD, Abbasi A, Ashigul Islam SM, et al. Signatures of copy number alterations in human cancer. Nature. 2022;606(7916):984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mertens F, Johansson B, Höglund M, Mitelman F. Chromosomal imbalance maps of malignant solid tumors: a cytogenetic survey of 3185 neoplasms. Cancer Res. 1997;57(13):2765–2780. [PubMed] [Google Scholar]
- 14.Denomy C, Germain S, Haave B, et al. Banding Together: A systematic comparison of The Cancer Genome Atlas and the Mitelman Databases. Cancer Res. 2019;79(20):5181–5190. [DOI] [PubMed] [Google Scholar]
- 15.Abrams ZB, Tally DG, Abruzzo LV, Coombes KR. RCytoGPS: An R Package for reading and visualizing cytogenetics data. doi: 10.1101/2021.03.16.389791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abrams ZB, Zhang L, Abruzzo LV, et al. CytoGPS: a web-enabled karyotype analysis tool for cytogenetics. Bioinformatics. 2019;35(24):5365–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGowan-Jordan Jean, Hastings Ros J., and Moore Sarah, eds. ISCN 2020: An International System for Human Cytogenomic Nomenclature (2020). Reprint Of: Cytogenetic and Genome Research 2020, Vol. 160, No. 7–8. Karger, S, 2020. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, LaFramboise T. CytoConverter: a web-based tool to convert karyotypes to genomic coordinates. BMC Bioinformatics. 2019;20(1):467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reynolds SM, Miller M, Lee P, et al. The ISB Cancer Genomics Cloud: A flexible cloud-based platform for cancer genomics research. Cancer Research. 2017;77(21):e7–e10. doi: 10.1158/0008-5472.can-17-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.