


Abstract
Activation of Adriamycin by formaldehyde leads to the formation of drug–DNA adducts in vitro and these adducts stabilise the DNA to such a degree that they function as virtual interstrand cross-links. The formation of these virtual interstrand cross-links by Adriamycin was investigated in MCF-7 cells using a gene-specific interstrand cross-linking assay. Cross-linking was measured in both the nuclear-encoded DHFR gene and in mitochondrial DNA (mtDNA). Cross-link formation increased linearly with Adriamycin concentration following a 4 h exposure to the drug. The rate of formation of Adriamycin cross-links in each of the genomes was similar, reaching maximal levels of 0.55 and 0.4 cross-links/10 kb in the DHFR gene and mtDNA respectively, following exposure to 20 µM Adriamycin for 8 h. The interstrand cross-link was short lived in both DNA compartments, with a half-life of 4.5 and 3.3 h in the DHFR gene and mtDNA respectively. The kinetics of total Adriamycin adduct formation, detected using [14C]Adriamycin, was similar to that of cross-link formation. Maximal adduct levels (30 lesions/10 kb) were observed following incubation at 20 µM drug for 8 h. The formation of such high levels of adducts and cross-links could therefore be expected to contribute to the mechanism of action of Adriamycin.
INTRODUCTION
Despite numerous new approaches, the treatment of cancer continues to rely mainly on surgery, radiotherapy and chemotherapy (1). Of the fifty or so anticancer agents in current clinical use (2), one of the most widely used is Adriamycin (1–3), also known as doxorubicin. It exhibits a good response to a range of tumors (breast, stomach, acute leukaemia, lymphomas, multiple myelomas, sarcomas and bone tumors), and is used as both a single agent as well as in combination chemotherapy (1). Because of the many side effects of this drug (e.g. nausea, vomiting, diarrhoea, myelosuppression and a dose-limiting cardiotoxicity), there have been numerous attempts to obtain improved derivatives with enhanced activity or reduced side effects. Despite intense efforts over several decades, and the synthesis or isolation of over 2000 derivatives, the search for a significantly improved derivative has failed (3), due largely to a lack of understanding of the critical molecular events involved in the mechanism of action of this drug (3–5).
Since Adriamycin exhibits a wide range of cellular effects, it is likely that no single mechanism of action will account for all of the observed clinical and cellular responses (3,6). There is a large body of evidence to show that the dominant cellular target is DNA (5–7), resulting in two major types of DNA damage: DNA adducts (6–8) and protein-associated single- and double-strand DNA breaks (7,9). Although there is evidence to suggest that topoisomerase II is a primary target, leading to impairment of this enzyme and hence to protein-associated DNA breaks, this phenomenon is diminished at clinical levels of drug treatment (10) where the intracellular concentration of Adriamycin is typically of the order of 5 µM (and is sustained at these relatively high levels for many days, with a half-life of 4–5 days) (11). Furthermore, there is little evidence of the involvement of topoisomerase II in some tumors (12). While it is likely that impairment of topoisomerase II contributes to the mechanism of action of Adriamycin to some degree, it is equally likely that other mechanisms (such as the formation of adducts) contribute, particularly at the drug dosage levels routinely employed for chemotherapy. Recently the cytotoxicity of a range of derivatives of Adriamycin was found to be proportional to the extent of formation of DNA interstrand cross-links (ICLs) in HeLa cells (13). This result therefore raises the possibility that a major mechanism of action involves the formation of cross-links with DNA.
In vitro transcription assays have previously been used to clarify the nature of the interaction of Adriamycin with DNA, and the formation of drug-induced DNA adducts was observed almost exclusively at GpC sequences (14). A variety of experimental approaches have subsequently shown that the adducts at these sites contribute to an Adriamycin-induced interstrand cross-link at GpC sequences, and involve the exocyclic amino group of guanine (15,16). The rate of formation of adducts in vitro was the same as the rate of formation of ICLs (5,16,17) suggesting that the adducts are in fact ICLs. It has recently been confirmed that the adducts and ICLs are one and the same lesion (18,19). Electrospray mass spectral studies of oligonucleotides containing multiple GpC drug binding sites have revealed that the cross-links are mediated by formaldehyde which forms slowly under the reaction conditions employed for cross-link formation (18,19). Formaldehyde reacts with the amino group of Adriamycin to form a Schiff base which then reacts with the N2 of guanine to form a monoadduct (18,19). The structure has now been characterised by 2D NMR which has revealed that the drug intercalates adjacent to the GC site, and that the single adduct stabilises the duplex by the equivalent of 40 intercalated drug molecules or, alternatively, by an additional 12 hydrogen bonds (20). The structure of the lesion is essentially identical to the X-ray structure of the adduct discovered by accident some years ago (21). Because the monoadduct has the functionality of an interstrand cross-link, the term virtual cross-link has been used to describe this lesion (18,19). The Adriamycin cross-links are both heat and alkali labile (22), consistent with the known lability of the formaldehyde-mediated aminal link (N–C–N), and the absence of a second covalent link to complete the cross-link.
Although DNA adducts and cross-links induced by Adriamycin at GpC sequences have now been well characterised in vitro, and have been detected indirectly in bulk DNA in cells (13), the genomic targets and molecular characteristics of these lesions are unknown. We present here the use of a gene-specific interstrand cross-linking assay which shows that Adriamycin-induced virtual interstrand cross-links form at a similar rate and to a similar extent in both nuclear and mitochondrial DNA (mtDNA), and have a similar half-life in both compartments. Furthermore, total cellular adducts also form at a similar rate.
MATERIALS AND METHODS
Materials
Radionucleotides, [α-32P]dCTP and [α-32P]UTP (3000 Ci/mmol) and [14-14C]Adriamycin hydrochloride (53.0 mCi/mmol) were purchased from Amersham. A QIAamp blood kit was obtained from Qiagen. Restriction enzymes and a random primed labelling kit were supplied by Boehringer Mannheim.
Cells
MCF-7 cells were grown in DMEM medium (Trace Scientific) and 10% fetal bovine serum (Gibco BRL), supplemented with 0.1 mg/ml streptomycin and 100 U/ml penicillin.
DNA probes
The pBH31R1.8 plasmid probe for the DHFR gene, which detects a 22 kb fragment including the 5′-end of the gene (23), was provided by Dr V. A. Bohr (National Institute on Aging, NIH, Baltimore, MD). The 1.8 kb EcoRI fragment containing exons I and II of the DHFR gene was isolated from pBH31R1.8 and labelled using a random primed labelling kit (Boehringer Mannheim) and [α-32P]dCTP. The mitochondrial probe was a gift from Dr C. A. Filburn (National Institute on Aging). The strand-specific mitochondrial probe was prepared by generating run-off transcripts from the T7 promoter in the presence of [α-32P]UTP.
Drug treatment
Cells were seeded in 10 cm dishes at a density of 2.5 × 106 cells/dish ∼16 h prior to the experiment to ensure exponential growth of the cells at the time of drug treatment. Cells were incubated with increasing concentrations of Adriamycin in complete medium for 4 h. Cells were subsequently washed twice in PBS and removed from the plate using trypsin. The cells were pelleted and washed a further two times in PBS. Total genomic DNA was isolated using a QIAamp Blood kit (Qiagen, Germany) with slight modifications to the protocol. The cell lysis step was conducted at 50°C for 30 min to minimise the loss of the heat labile drug cross-links.
Detection of ICLs
Total DNA (2.5 µg) was digested either with HindIII (to release the 22 kb DHFR fragment) or with BamHI (to linearise the mitochondrial genome) for 2 h at 37°C. The DNA was extracted once with phenol, once with chloroform and ethanol precipitated. The pellet was resuspended in 10 µl TE and denatured by the addition of 20 µl loading buffer containing 90% formamide (final formamide concentration of 60%) and 0.1% bromophenol blue and incubation at 60°C for 5 min. The samples were quenched on ice and immediately loaded onto a 0.5% agarose gel and electrophoresed overnight at 30 V in TAE buffer.
The DNA was transferred to nylon (Hybond N+, Amersham) and fixed to the membrane by UV cross-linking (Stratalinker, Stratagene). The membranes were hybridised overnight with 32P-labelled probes under standard conditions (24). Quantitation was performed using a PhosphorImager and ImageQuant software (Molecular Dynamics, Sunnyvale, CA). The ICL frequency was calculated from the zero class of the Poisson distribution as described previously (25).
Adriamycin adduct formation
MCF-7 cells prepared as described above were incubated with 20 µM Adriamycin (10 µM [14C]Adriamycin and 10 µM unlabelled Adriamycin) for times up to 8 h. At various time intervals the cells were washed, harvested and the total genomic DNA isolated as described above. The DNA was subsequently extracted twice with phenol and once with chloroform. In order to allow selective precipitation of DNA from contaminating RNA, ammonium acetate was added to a final concentration of 2.5 M followed by the addition of two volumes of ethanol. The DNA was recovered by centrifugation, the pellet washed, dried and resuspended in 100 µl of TE buffer. The DNA concentration was calculated using a Cary 118 spectrophotometer. Total genomic DNA (50 µl) was added to 1 ml of OptiPhase Hisafe3 scintillation cocktail and the incorporation of 14C-labelled drug into the DNA was determined by scintillation counting on a Wallac 1410 Liquid Scintillation Counter.
RESULTS
Techniques generally employed for the detection of DNA ICLs in cells involve the use of harsh procedures including high temperature or alkali (25,26). As the stability of the Adriamycin cross-link in vitro is severely compromised under such conditions (22), we initially sought to develop a mild total genomic DNA isolation procedure to facilitate the stable extraction of Adriamycin-induced DNA ICLs from cells in culture. After testing several commercially available genomic DNA extraction kits, it was found, using 14C-labelled Adriamycin, that the adducts were most stable to extraction using the QIAamp Blood kit (Qiagen) (data not shown). Modification of the QIAamp protocol such that the cell lysis step was performed at 50°C, further increased the recovery of Adriamycin cross-links.
Dependence of cross-link formation on Adriamycin concentration
Once conditions for the extraction of cross-linked DNA had been optimised, the concentration dependence of interstrand cross-linking was then investigated. The MCF-7 breast adenocarcinoma cell line was treated with increasing concentrations of Adriamycin for 4 h. The cells were then harvested and total genomic DNA isolated. The DNA was then analysed for Adriamycin-induced DNA ICLs in the nuclear encoded DHFR gene and in mtDNA using a gene-specific cross-linking assay (25). In order to maximise the detection of cross-links, conditions employed for DNA denaturation were modified to 60°C for 5 min in 60% formamide, based on the known instability of Adriamycin cross-links in vitro (22).
The results of the cross-linking assay are shown in Figure 1. As seen in both the DHFR gene (Fig. 1A) and mtDNA (Fig. 1B) blots, DNA from cells incubated in the absence of Adriamycin was completely denatured under the conditions employed and migrated at single-strand DNA molecular weight. DNA from cells treated with Adriamycin showed a concentration-dependent increase in DNA migrating as double-strand DNA, consistent with the formation of Adriamycin-induced ICLs. A low level of cross-linking was apparent in both the DHFR gene fragment and mtDNA at 7.5 µM Adriamycin, while at 60 µM Adriamycin most of the DNA fragments migrated as cross-linked DNA.
Figure 1.
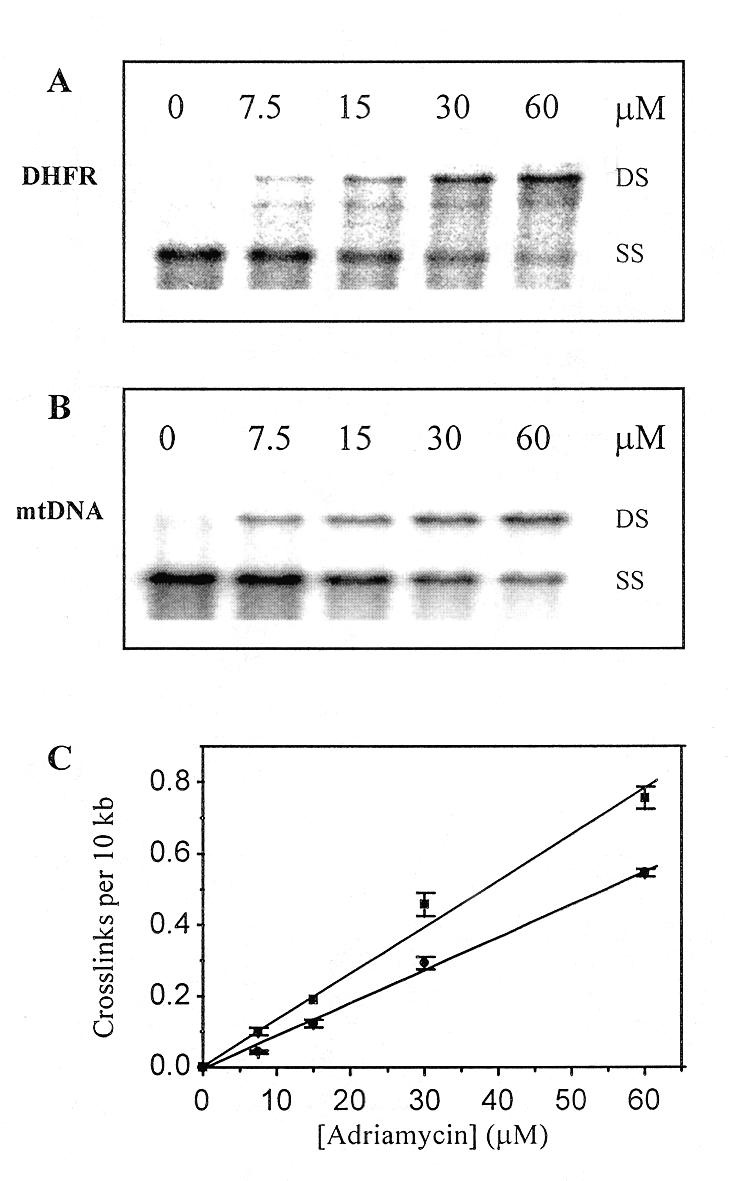
Adriamycin concentration-dependent formation of ICLs. MCF-7 cells were treated with 0–60 µM Adriamycin for 4 h. Total genomic DNA was isolated, restricted and then denatured in formamide and resolved on a 0.5% agarose gel. (A) Representative Southern blot of HindIII-digested genomic DNA fragments probed for the 22 kb 5′-DHFR gene fragment. (B) Representative blot of BamHI-digested DNA fragments probed for mtDNA where DS is double strand DNA and SS is single strand DNA. (C) PhosphorImager analysis was used to quantitate the formation of Adriamycin-induced cross-links in the DHFR gene (square) and mtDNA (circle). Data were derived from two separate blots from each of two biological experiments and the cross-link values are presented as the means ± SE.
The blots were quantitated and the frequency of cross-links formed in both the DHFR gene fragment and mtDNA was determined using the Poisson equation (25). The results were normalised to cross-links per 10 kb and are shown in Figure 1C. Cross-linking increased linearly as a function of Adriamycin concentration in both the DHFR gene and in mtDNA. Maximal cross-link levels of 0.76 cross-links/10 kb were observed in the DHFR gene at 60 µM Adriamycin. In contrast, a 30% lower level of cross-links was detected in mtDNA (0.55 cross-links/10 kb) at the same drug concentration.
Time dependence of cross-link formation
Because of the known slow rate of formation of Adriamycin crosslinks in vitro, it was important to establish if a similar phenomenon existed in cells. The dependence of Adriamycin cross-linking on incubation time was therefore investigated. MCF-7 cells were incubated with 20 µM Adriamycin for up to 8 h before total genomic DNA was isolated and analysed for the formation of Adriamycin-induced DNA ICLs. As shown in Figure 2, cross-links increased as a function of time in both the DHFR gene (Fig. 2A) and mtDNA (Fig. 2B). Quantitation of cross-linking in both fragments (Fig. 2C) clearly demonstrates that the kinetics of cross-link formation in both the nuclear gene and mtDNA is similar, suggesting that uptake of the drug into either the nucleus or mitochondria is not a limiting factor to cross-link formation. Cross-links increased rapidly up to ~5 h, after which levels began to plateau. At 8 h, where maximal cross-linking was observed, 30% more cross-links were present in the DHFR fragment than in mtDNA, consistent with the result obtained in Figure 1.
Figure 2.
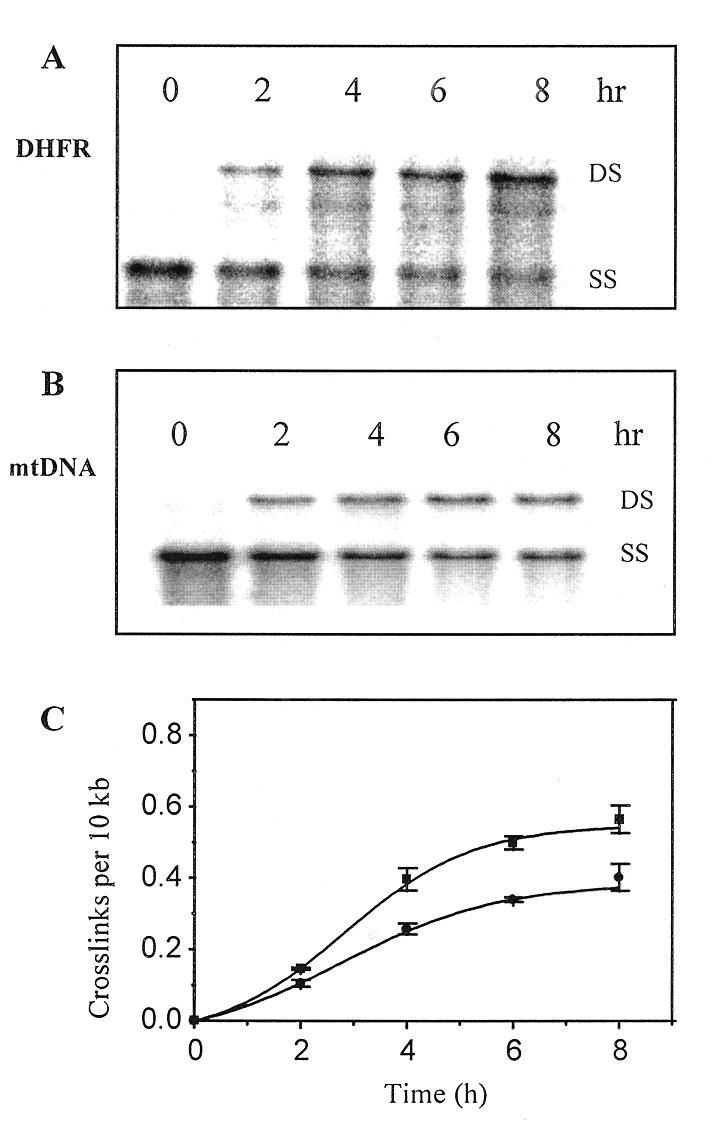
Reaction time dependent formation of Adriamycin induced ICLs. MCF-7 cells were treated with 20 µM Adriamycin for 0–8 h as shown, then total genomic DNA was isolated, restricted, denatured and resolved on a 0.5% agarose gel. The DNA was transferred to nylon and probed with either the DHFR gene probe [HindIII-restricted DNA (A)] or the mtDNA probe [BamHI-restricted DNA (B)]. (C) PhosphorImager analysis was used to quantitate the time-dependent formation of Adriamycin-induced cross-links in the DHFR gene (square) and mtDNA (circle). Data were derived from two separate blots from each of two biological experiments and are presented as the means ± SE.
Stability of cross-links
As Adriamycin adducts are unstable in vitro, we sought to investigate the stability of these lesions in cells. The cells were therefore treated with Adriamycin to induce an initial cross-link frequency before the drug was removed, and then incubated in drug-free medium for up to 8 h. The DNA was then extracted and analysed for remaining cross-links. As the contribution of DNA replication to the total DNA present becomes significant after about half the cell doubling time, times beyond 8 h were not analysed. The results of the cross-linking assay (Fig. 3) clearly show that cross-links are short lived in cells and decrease significantly as a function of time in both the DHFR fragment (Fig. 3A) and mtDNA (Fig. 3B), consistent with the known loss of Adriamycin-induced cross-links from bulk DNA in HeLa cells (27). The time-dependent loss of the Adriamycin cross-links was quantitated and is shown as a first order plot in Figure 3C. The kinetics of cross-link decay in both the DHFR gene and mtDNA was similar, and consistent with a first order kinetic process with a half-life of 4.7 and 3.3 h in each fragment respectively. These values are also similar to that detected for the decay of Adriamycin-induced ICLs in vitro, where the half-life of interstrand cross-links was shown to be 4.7 h (22).
Figure 3.
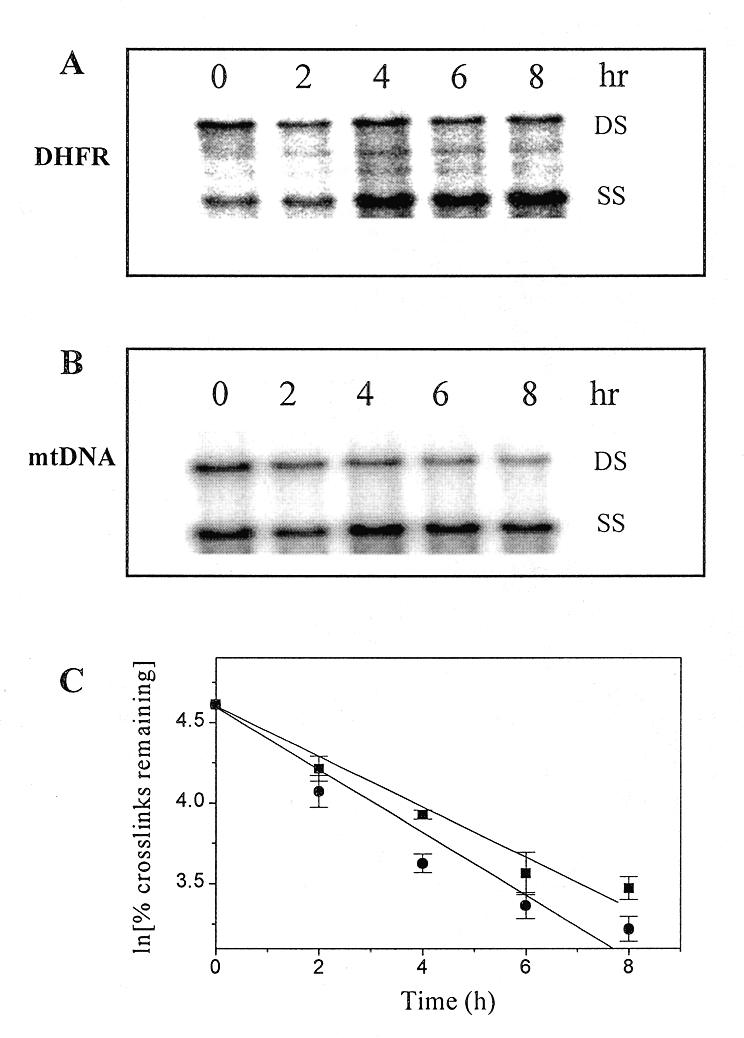
Stability of Adriamycin-induced ICLs. MCF-7 cells were treated with 70 µM Adriamycin for 90 min. The drug was then removed and the cells were either harvested immediately (lane 0) or incubated in drug-free medium for times up to 8 h as shown. Purified, restricted and denatured total genomic DNA was resolved on 0.5% agarose and then transferred to nylon. (A) Representative blot of HindIII-digested genomic fragments probed with the DHFR gene probe. (B) Representative blot of BamHI-digested genomic fragments probed for mtDNA. (C) PhosphorImager analysis was used to quantitate the time-dependent decrease in Adriamycin cross-links in the DHFR gene (square) and mtDNA (circle). Data were derived from two separate blots each from at least two biological experiments and are presented as the means ± SE.
Formation of 14C-labelled Adriamycin adducts
14-14C-labelled Adriamycin was used to investigate the formation of total drug adducts in cells. DNA isolated from cells incubated in the presence of [14C]Adriamycin for up to 8 h was analysed for the incorporation of radiolabelled drug. To eliminate the possibility of carry-over of residual intercalated drug from the genomic DNA extraction procedure, the isolated DNA was extracted twice with phenol and once with chloroform before scintillation analysis. The Adriamycin adduct frequency was calculated per 10 kb and is expressed as a function of time (Fig. 4A). As observed with the formation of cross-links, drug adduct levels increased rapidly up to ~5 h before beginning to plateau. The absolute level of Adriamycin adducts formed was very high, reaching up to 30 adducts per 10 kb, some 50-fold higher than ICL levels. Since there was an apparent discrepancy between the levels of adducts formed as compared to cross-links, this may reflect differences in the assay conditions required for the detection of adducts and cross-links. In contrast to the direct measurement of 14C incorporation used for the detection of adducts, the detection of cross-links required additional DNA restriction digestion and DNA denaturation in formamide, each of which is likely to contribute to significant loss of the lesion (22). To test the contribution of these additional processes to adduct decay, DNA was isolated from MCF-7 cells treated with 10 µM [14C]Adriamycin and 10 µM unlabelled Adriamycin. The DNA was then subjected to the same assay conditions used for detection of cross-links. This treatment resulted in an ∼7-fold loss of adducts, showing that the additional processes involved in detection of cross-links are indeed detrimental.
Figure 4.

Time-dependent formation and stability of Adriamycin adducts. (A) Cells were incubated with 20 µM Adriamycin (10 µM Adriamycin; 10 µM [14-14C]Adriamycin) for up to 8 h. Total genomic DNA was isolated and purified by extraction with phenol and chloroform. Drug-induced DNA adducts were quantitated by scintillation counting. (B) MCF-7 cells were treated with 70 µM Adriamycin (60 µM Adriamycin; 10 µM [14-14C]Adriamycin) for 90 min. The drug was then removed and the cells were either harvested immediately or incubated in drug-free medium for times up to 8 h as shown. Total adducts were quantitated by scintillation analysis and plotted as a first order logarithmic decay.
Stability of [14C]Adriamycin adducts
In order to confirm that total Adriamycin adducts behave similarly to Adriamycin cross-links (consistent with the view that adducts and cross-links represent the same lesions), MCF-7 cells were treated in an identical manner as described above (stability of cross-links) except that [14-14C]Adriamycin was used at a concentration of 10 µM and unlabelled drug was used at a final concentration of 60 µM. DNA was isolated from Adriamycin-treated cells for up to 8 h post-incubation and then analysed for the incorporation of radiolabelled drug (Fig. 4B). This experiment confirmed that total adducts are short-lived, displaying a first order kinetic decay with a half-life of ∼8.1 h. This value is similar to the 7.4 h half-life of loss of [14C]Adriamycin adducts in vitro (22), consistent with the loss of a single population of adducts over the 8 h time frame of this study.
DISCUSSION
While the formation of DNA cross-links by Adriamycin has been well characterised in vitro, the current study provides the first direct evidence for the formation of Adriamycin cross-links in subcellular DNA compartments. In vitro, Adriamycin cross-links have been observed to form almost exclusively at GpC sequences and are heat and alkali labile (15,16,22). Recently the structure of the Adriamycin lesion was shown to be a virtual cross-link in which an intercalated Adriamycin molecule is covalently bound through a labile aminal link to the exocyclic amine of guanine in a 5′-GpC sequence to yield a monoadduct (18,19). The monoadduct is further strongly stabilised by additional hydrogen bonding between the intercalated drug and guanine on the opposite DNA strand, to yield a functional (virtual) cross-link (20). The unusual structure of the Adriamycin cross-link readily explains the observed lability of the lesion and why such cross-links have been difficult to detect in cells. Conventional genomic DNA extraction and cross-link detection protocols have routinely utilised heat or alkali, conditions which are known to result in labilisation of the Adriamycin cross-link (18,22). Therefore, on the basis of the known chemistry and the observed in vitro stability of the cross-link, we have developed methods for the direct and quantitative analysis of cross-link formation in specific genomic DNA fragments from cells treated with Adriamycin.
Adriamycin cross-linking in nuclear DNA and mtDNA
The results of the cross-linking assay clearly demonstrate the formation of Adriamycin cross-links in the transcriptionally active DHFR gene and in mtDNA. Both the nuclear encoded gene and mtDNA demonstrated a similar susceptibility to cross-linking, evident by the similar profiles for drug concentration dependence, formation kinetics and stability, clearly indicating that both environments are favourable for cross-link formation.
The 30% lower level of crosslinks detected in mtDNA as compared with the nuclear gene may be due to the increased accumulation of drug into the nucleus as compared to mitochondria (28,29). While uptake of Adriamycin into mitochondria has been less well studied than uptake into the nucleus, the drug has been shown to accumulate to significant levels in mitochondria in vivo, primarily due to its affinity for cardiolipin (30,31). Indeed, impairment of mitochondrial activity through interactions at the level of the mitochondrial membranes has been implicated in the cardiotoxicity of Adriamycin (6).
Alternatively, as in vitro studies have clearly revealed the role of formaldehyde in Adriamycin cross-link formation (18,19), the availability of formaldehyde (or other cellular aldehydes which may also yield a virtual cross-link) may differ between these two cellular compartments, thus altering their relative susceptibility to the formation of these lesions. There are several reasons to support the notion that formaldehyde may act as a key intermediate in the formation of Adriamycin-induced ICLs in tumor cells: (i) several studies have shown elevated formaldehyde levels in some tumors as compared to normal cells (19); (ii) the formaldehyde–Adriamycin complex (doxoform) is more cytotoxic than Adriamycin (32); and (iii) formaldehyde can be generated by oxidation of a variety of cellular sources, one of which is the drug itself (19), thus potentially yielding a local supply of formaldehyde in close proximity to the DNA-localised drug. However, at this stage it should be concluded that while Adriamycin ICLs in tumors may involve formaldehyde, this has not yet been confirmed, and it is possible that other aldehydes or other mechanisms may be involved.
Stability of ICLs and [14C]Adriamycin adducts
Adriamycin cross-links were lost at similar rates from both nuclear DNA and mtDNA upon removal of the drug from the medium (Fig. 3); however, 14C adducts were lost at a slower rate from total genomic DNA (Fig. 4B). Some difference may be expected between the half-lives as measured by these two assays since the measurement of 14C adducts reflects loss from the total genomic DNA pool, whereas the cross-links were assessed in only a single gene (DHFR) or small coding region (mtDNA).
The cellular removal of ICLs requires the concerted activity of the nucleotide excision and recombinational repair pathways. While the decay in cross-links could potentially be attributed to the activity of DNA repair pathways, mitochondria are known to be defective in the repair of DNA interstrand cross-links (33,34). It is therefore possible that this result reflects the known instability of the lesion (22) rather than processing of the damage by the repair proteins. It is interesting to note that anthramycin, which also binds the exocyclic amino group of guanine via an aminal link to form a similar type of adduct to the Adriamycin cross-link (35), shows a similar lability in nucleotide excision repair deficient cells as observed in vitro, and this loss of adducts has also been suggested to reflect the inherent lability of the lesion (36).
The DNA adduct induced by anthramycin results in only minimal distortion of the DNA helix and is poorly recognised and, hence, poorly removed by the repair proteins. As the Adriamycin cross-link also does not significantly distort the DNA helix (20,21), then by analogy with anthramycin (36), the cross-link may also be poorly recognised by the repair surveillance proteins, thus resulting in minimal repair of the ICL. The results of the current study which show a similar stability of the cross-link in nuclear and mitochondrial DNA, despite the differences in ICL repair capacity of these compartments, together with the information that the half-life of crosslinks in vitro is similar (22), indeed suggests that no significant repair of the Adriamycin cross-link occurred in the repair proficient nucleus over the time course studied.
Are ICLs involved in the biological activity of Adriamycin?
The Adriamycin concentration used for much of this work was relatively high to ensure that quantitation of the number of DNA fragments per DNA fragment was statistically meaningful (25). The instability of the lesion results in significant losses during the preparation of drug-treated genomic DNA for cross-linking analysis (2 h incubation at 37°C and denaturation at 60°C for 5 min in 60% formamide). The actual cross-link levels are therefore expected to be substantially higher than those detected by the cross-linking assay. Despite the detection of high levels of Adriamycin ICLs, the number of adducts detected was even 50-fold higher (Figs 2C and 4) and this difference can be attributed partially to experimental differences in the two assays employed to measure ICLs and adducts. While adduct analysis involved the direct analysis of 14C-labelled Adriamycin into bulk genomic DNA, the ICL assay required multiple incubations which have resulted in an ∼7-fold loss of cross-links. The additional unexplained loss of cross-links is ∼7-fold and this may be explained by one or more of the following factors. It is possible that re-annealing of the large fragments (22 and 16.5 kb for DHFR and mitochondrial respectively) used for Adriamycin cross-link detection may require the presence of more than one cross-link per DNA fragment. Furthermore, the DHFR and mitochondrial fragments probed represent a minor proportion of actively transcribed DNA and an even lower proportion of total genomic DNA. This is potentially significant since lesion frequencies may vary markedly in different genomic regions depending on accessibility to the local chromatin structure.
Since previous in vitro studies have shown that Adriamycin adducts and cross-links are essentially the same lesion (18,22), the 30 adducts/10 kb detected (Fig. 4A) indicates a maximum cross-linking level of 30 cross-links/10 kb after exposure to Adriamycin for 8 h, equivalent to 107 cross-links/genome. Under these cell culture conditions (where the concentration of Adriamycin in the media is initially 20 µM), the drug accumulates in cells and the intracellular drug concentration reaches levels of 300–600 µM (37,38). Assuming that the number of cross-links is proportional to the drug level, at clinical intracellular concentrations (5 µM) (11) the estimated number of cross-links per genome is then 105. As this represents a level 100-fold higher than required for cytotoxicity by other cross-linking agents (39), further investigation of the cellular consequences of such lesions is clearly warranted.
Conclusions
In summary, we have shown that when cells in culture are exposed to micromolar levels of Adriamycin, this results in the formation of an extensive number of ICLs. The significance of interstrand cross-linking has been documented by a strong correlation between DNA cross-linking capacity and the cytotoxicity for a series of anthracycline derivatives (13). In contrast to this apparent mechanism of action, stabilisation of topoisomerase II-associated DNA strand breaks has also been well documented following exposure of cells to low levels of Adriamycin (9). However, the absence of a clear correlation between DNA strand breaks and cytotoxicity suggests that other factors are likely to be involved. Since both of these lesions (ICLs and protein-associated DNA strand breaks) occur at clinical drug levels, it is likely that both contribute to the mechanism of action of Adriamycin to some degree. Irrespective of the relative significance of the two lesions, it is clear from the present results that Adriamycin forms ICLs in both genomic compartments of the cell and the cellular consequences of such damage warrants further detailed study.
Acknowledgments
ACKNOWLEDGEMENTS
This work was carried out with support from the Australian Research Committee. We thank Farmitalia Carlo Erba (Milan) for the supply of Adriamycin, and Dr V. A. Bohr (National Institute on Aging, NIH, Baltimore, MD) for supplying the plasmid pBH31R1.8
REFERENCES
- 1.DeVita V.T., Hellman,S. and Rosenberg,S.A. (1993) Cancer Principles and Practice in Oncology, 4th Edn, Vols 1 and 2. J.B. Lippincott, Philadelphia, PA.
- 2.Schacter L.P., Anderson,S., Canetta,R.M., Kelley,S., Nicaise,C., Onetto,N., Rozencweig,M., Smaldone,L. and Winograd,B. (1992) Semin. Oncol., 19, 613–621. [PubMed] [Google Scholar]
- 3.Weiss R.B. (1992) Semin. Oncol., 19, 670–686. [PubMed] [Google Scholar]
- 4.Cummings J., Willmott,N., Hoey,B.M., Marley,E.S. and Smyth,J.F. (1992) Biochem. Pharmacol., 44, 2165–2174. [DOI] [PubMed] [Google Scholar]
- 5.Phillips D.R. and Cullinane,C. (1999) In Creighton, T.E. (ed.), Encyclopedia of Molecular Biology. John Wiley and Sons, New York, NY, pp. 68–72.
- 6.Myers C.E., Mimnaugh,E.G., Yeh,G.C. and Sinha,B.K. (1988) In Lown,J.W. (ed.), Anthracyclines and Anthracenedione-based Anticancer Agents. Elsevier, Amsterdam, The Netherlands, pp. 527–570.
- 7.Myers C.E. (1992) Cancer Chemother. Biol. Resp. Modif., 13, 45–52. [PubMed] [Google Scholar]
- 8.Phillips D.R., Cullinane,C., Trist,H. and White,R.J. (1990) In Pullman,B. and Jortner,J. (eds), Molecular Basis of Specificity in Nucleic Acid-Drug Interactions. Kluwer Academic, Dordrecht, The Netherlands, pp. 137–155.
- 9.Holm C., Covey,D., Kerrigan,K.W. and Pommier,Y. (1991) In Pomesil,M. and Kohn,K. (eds), DNA Topoisomerases in Cancer. Oxford University Press, New York, pp. 161–171.
- 10.Fornari F.A., Jarvis,W.D., Orr,M.S., Randolph,S., Grant,S. and Gerwitz,D.A. (1996) Biochem. Pharmacol., 51, 931–940. [DOI] [PubMed] [Google Scholar]
- 11.Speth P.A.J., Linssen,P.C.M., Boezeman,J.B.M., Wessels,J.M.C. and Haanen,C. (1986) J. Chromatogr., 377, 415–422. [DOI] [PubMed] [Google Scholar]
- 12.Pratt W.B., Ruddon,R.W., Ensminger,W.D. and Maybaum,J. (1994) The Anticancer Drugs, 2nd Edn. Oxford University Press, New York, pp. 160–164.
- 13.Skladanowski A. and Konopa,J. (1994) Biochem. Pharmacol., 47, 2279–2287. [DOI] [PubMed] [Google Scholar]
- 14.Phillips D.R., White,R.J. and Cullinane,C. (1989) FEBS Lett., 246, 233–240. [DOI] [PubMed] [Google Scholar]
- 15.Cullinane C. and Phillips,D.R. (1990) Biochemistry, 29, 5638–5646. [DOI] [PubMed] [Google Scholar]
- 16.Cullinane C., van Rosmalen,A. and Phillips,D.R. (1994) Biochemistry, 33, 4632–4638. [DOI] [PubMed] [Google Scholar]
- 17.Cutts S. and Phillips,D.R. (1995) Nucleic Acids Res., 23, 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taatjes D.J., Gaudiano,G., Resing,K. and Koch,T.H. (1996) J. Med. Chem., 39, 4135–4138. [DOI] [PubMed] [Google Scholar]
- 19.Taatjes D.J., Gaudiano,G., Resing,K. and Koch,T.H. (1997) J. Med. Chem., 40, 1276–1286. [DOI] [PubMed] [Google Scholar]
- 20.Zeman S.M., Phillips,D.R. and Crothers,D.M. (1998) Proc. Natl Acad. Sci. USA, 95, 11561–11565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang A.H.-J., Gao,Y.-G., Liaw,Y.-C. and Li,Y. (1991) Biochemistry, 30, 3812–3815. [DOI] [PubMed] [Google Scholar]
- 22.van Rosmalen A., Cullinane,C., Cutts,S.M. and Phillips,D.R. (1995) Nucleic Acids Res., 23, 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhen W., Evans,M.K., Haggerty,C.M. and Bohr,V.A. (1993) Carcinogenesis, 14, 919–924. [DOI] [PubMed] [Google Scholar]
- 24.Sambrook J., Frisch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, NY.
- 25.Vos J.M. (1988) In Friedberg,E.C. and Hanawalt,P.C. (eds), DNA Repair: A Laboratory Manual of Research Procedures. Marcel Dekker, New York, Vol. 3, pp. 367–398.
- 26.Kohn K., Hartley,J.A. and Mattes,W.B. (1981) In Freidberg,E.C. and Hanawalt,P.C. (eds), DNA Repair: A Laboratory Manual of Research Procedures. Marcel Dekker, New York, Vol. 1 Part B, pp. 370–401.
- 27.Skladanowski A. and Konopa,J. (1994) Biochem. Pharmacol., 47, 2269–2278. [DOI] [PubMed] [Google Scholar]
- 28.Kawai H., Minamiya,Y., Kitamura,M., Matsuzaki,I., Hashimoto,M., Suzuki,H. and Abo,S. (1997) Cancer, 79, 214–219. [DOI] [PubMed] [Google Scholar]
- 29.Meschini S., Molinari,A., Calcabrini,A., Citro,G. and Arancia,G. (1994) J. Microsc., 176, 204–210. [DOI] [PubMed] [Google Scholar]
- 30.Nicolay K., Timmers,R.J.M., Spoelstra,E., van der Neut,R., Fok,J.J., Huigen,Y.M., Verkleij,A.J. and de Kruijff,B. (1984) Biochim. Biophys. Acta, 778, 359–371. [DOI] [PubMed] [Google Scholar]
- 31.Nicolay K., Fok,J.J., Voorhout,W., Post,J.A. and de Kruijff,B. (1986) Biochim. Biophys. Acta, 887, 35–41. [DOI] [PubMed] [Google Scholar]
- 32.Fenick D.J., Taatjes,D.J. and Koch,T.H. (1997) J. Med. Chem., 40, 2452–2461. [DOI] [PubMed] [Google Scholar]
- 33.LeDoux S.P., Wilson,G.L., Beecham,E.J., Stevnsner,T., Wassermann,K. and Bohr,V.A. (1992) Carcinogenesis, 13, 1967–1973. [DOI] [PubMed] [Google Scholar]
- 34.Cullinane C. and Bohr,V.A. (1998) Cancer Res., 58, 1400–1404. [PubMed] [Google Scholar]
- 35.Petrusek R.L., Anderson,G.L., Garner,T.F., Fannin,Q.L., Kaplan,D.J., Zimmer,S.G. and Hurley,L.H. (1981) Biochemistry, 20, 1111–1119. [DOI] [PubMed] [Google Scholar]
- 36.Petrusek R.L., Uhlenhopp,E.L., Duteau,N. and Hurley,L.H. (1982) J. Biol. Chem., 257, 6207–6216. [PubMed] [Google Scholar]
- 37.Johnson B.A., Cheang,M.S. and Goldenberg,G.J. (1986) Cancer Res., 46, 218–233. [PubMed] [Google Scholar]
- 38.Capranico G., Soranzo,C. and Zunino,F. (1986) Cancer Res., 46, 5499–5503. [PubMed] [Google Scholar]
- 39.Lawley P.D. and Phillips,D.H. (1996) Mutat. Res., 355, 13–40. [DOI] [PubMed] [Google Scholar]