


Abstract
Structural integrity of the hepatitus C virus (HCV) 5′ UTR region that includes the internal ribosome entry site (IRES) element is known to be essential for efficient protein synthesis. The functional explanation for this observation has been provided by the recent evidence that binding of several cellular factors to the HCV IRES is dependent on the conservation of its secondary structure. In order to better define the relationship between IRES activity, protein binding and RNA folding of the HCV IRES, we have focused our attention on its major stem–loop region (domain III) and the binding of several cellular factors: two subunits of eukaryotic initiation factor eIF3 and ribosomal protein S9. Our results show that binding of eIF3 p170 and p116/p110 subunits is dependent on the ability of the domain III apical stem–loop region to fold in the correct secondary structure whilst secondary structure of hairpin IIId is important for the binding of S9 ribosomal protein. In addition, we show that binding of S9 ribosomal protein also depends on the disposition of domain III on the HCV 5′ UTR, indicating the presence of necessary interdomain interactions required for the binding of this protein (thus providing the first direct evidence that tertiary folding of the HCV RNA does affect protein binding).
INTRODUCTION
Translation initiation in the hepatitis C virus (HCV) (1) is under the control of an internal ribosome entry site (IRES) starting at nt 20–45 of the 341 nt long 5′ untranslated region (5′ UTR) and extending ~30 nt in the core protein sequence (2–9; for a recent review see 10). The 5′ UTR and the initial core region fold upon themselves to form several characteristic stem–loop domains (I, II, III and IV), held together by a helical structure and a pseudoknot which are highly conserved in all virus isolates (11,12). Mapping of the IRES boundaries has shown that the sequence and structural integrity of domains II, III and IV is important for driving efficient protein translation (5,6,8,12–18) with domain I not participating directly in the formation of the IRES itself but possibly playing a role in inhibiting its activity (8). In addition, non-structured regions of the HCV 5′ UTR (19) and long-range RNA–RNA interactions (20) can also be important to preserve translation efficiency. To date, the better characterized cellular factors which bind to the IRES and may affect its activity are: p52 (La antigen) (21), PTB (although its exact role is controversial and may be implicated in regulation of HCV translation by acting outside the IRES region) (22–24), an unidentified 25 kDa protein (14), S9 ribosomal protein (25,26), hnNRP L (27), poly(C)-binding protein (28) and eukaryotic translation initiation factor eIF3 (25,26,29). These studies provide both a better understanding of the mechanism of HCV IRES functioning and additional insight concerning the general field of RNA–protein interactions. In fact, it has become increasingly clear that in order to achieve correct RNA–protein recognition two factors often have to be conserved: nucleotide sequence integrity and correct RNA secondary structure. Indeed, with regard to the secondary structure, protein binding may also be needed to promote its correct folding since it has recently been reported that binding of PTB to the EMCV 5′ UTR is essential for IRES activity only when there is a necessity to stabilize the native secondary structure (30).
Previously, we have described that the binding of two subunits (p170 and p116/p110) of eIF3 to HCV RNA is important for translation initiation (29). Using artificial mutants we identified the binding region as the apical stem–loop region of domain III and showed that direct disruption of its secondary structure leads to a loss in IRES activity (29). This result was in accordance with a chemical and enzymatic footprinting study performed on a binary eIF3–IRES complex which reported the binding of four eIF3 subunits, p170, p116, p66 and p47, and identified the binding site by footprinting analysis as consisting of the large central helix of domain III and hairpins IIIa, IIIb and IIIc (26). It was therefore interesting to further investigate the relationship between IRES activity and correct folding of this domain, evidently a complex issue since we had already described (29) that inversion of stem–loop IIIb sequence resulted in a mutant (5′ S/L) which had unexpectedly maintained IRES activity. This mutant had retained the capacity to bind p170, although not p116/p110, and the fact that one binding site was conserved offered a likely explanation for the lack of significant variations in the level of IRES activity (29).
In this study we have engineered several mutants which carry selected portions of domain III and each mutant was comprehensively analyzed for a variety of characteristics: ability to drive IRES translation in vivo and in vitro, correct folding of the apical stem–loop and ability to bind the eIF3 subunits previously identified (29). Our results show that only one mutant comprising domain III sequence from nt 145 to nt 248 was capable of binding p170 and p116/p110 and, in keeping with this result, the RNase digestion pattern of its apical stem–loop showed that the RNA secondary structure was identical to that of wild-type domain III. The fact that this mutant was not capable of driving IRES translation prompted us to look for additional cellular factors binding to the IIId element and to investigate the importance of this stem–loop for IRES activity. In particular, we focused our attention on the interaction of our mutants with S9 ribosomal protein, whose binding has already been seen to be affected by deletion of hairpin IIa and IIIc (25). This second part of the study shows that domain IIId structure is indeed implicated in S9 ribosomal protein binding and that its binding can also be affected by the relative disposition and orientation of the major HCV stem–loop domains.
MATERIALS AND METHODS
Plasmid construction of HCV 5′ UTR mutants
The strategy used to swap the positions of different domains of HCV 5′ UTR using a template sequence has already been described in detail elsewhere (29). The domain II region missing from 5′ MscStu (nt 23–102) was amplified and inserted back in the MscI site of this plasmid (obtaining plasmid 5′ domII) using two sense and antisense primers (5′-ggg-cgacactccacc-3′ and 3′ oligo: 5′-aggctgcacgacact-3′). In the StuI site we then inserted the amplified sequences of domain III using the following 5′ and 3′ primers: 5′-accccccctcccgggggtctgcgg-3′, 5′-cagtctcgcgggggca-3′ for mutant 5′ domIII (145–248), 5′-accccccctcccgggaattgccagga-3′, 5′-caatctccaggcata-3′ for mutant 5′ domIII (172–227) and 5′-accccccctcccggggaccgggtc-3′, 5′-cgagcgggttgatccaa-3′ for mutant 5′ domIII (184–213) (see Fig. 1A for a schematic diagram of each mutant). All these primers contained 5′ leader sequences aimed at restoring the nucleotides deleted during the formation of the MscI and StuI site. All these different mutants were inserted in the pSV GH bicistronic expression system (31) for transfection experiments in COS-1 cells as already described (29) and in the XbaI/HindIII restriction enzyme sites of pBluescript II KS+ (Stratagene). An in vitro transcription and translation assay (Novagen) was performed according to manufacturer’s instructions using 2 µg of each Bluescript plasmid. Mutant 5′ S/L, 5′ MscStu, and the wild-type 5′ UTR sequence (5′ wt) have already been described in detail elsewhere (29). Mutants of subdomain IIId were performed using the following 5′ and 3′ sets of primers: 5′-tagtgttgggtgtgatagccgcttgtggtac-3′, 5′-tcacacccaacactactcggctagcagtctc-3′ for mutant IIId disrupted (5′ domIIId disr.), 5′-gcgcttgggtgtgatagccgcttgtggtac-3′, 5′-atcacacccaagc-gctcttccgtagcagtctc-3′ for mutant IIId repaired (5′ domIIId rep.) and 5′-gcgctgggttgtgatgagccgcttgtggtac-3′, 5′-atcacaacccag-cgctttccgtagcagtctc-3′ for mutant IIId S/L (5′ domIIId S/L). These primers were used to amplify and insert back the mutated domIIId in the wild-type HCV 5′ UTR as described previously for the 5′ S/L mutant (29).
Figure 1.

(A) Schematic representation of the domain III mutants used in this study. The arrows indicate how each mutant was obtained from the original 5′ wt and template sequence (5′ MscStu). The three novel mutants contain different regions of domain III: 145–248, 172–227 and 184–213. (B) The IRES activity of each mutant as assayed in transfection in COS-1 cells. The arrows indicate the position of the processed (23 kDa) and unprocessed (25 kDa) HCV core protein. The HCV core protein was recognized using MAb B12.F8 (35) and visualized by ECL staining. (C) In vitro transcription and translation assay of all three mutants including 5′ wt and mutant 5′ S/L. The arrow indicates the unprocessed 35S-labeled HCV core protein (visualized by autoradiography).
Transcription of the pBluescript II KS+ plasmids
All plasmids described in this study were linearized by digestion with HindIII and transcription was performed with T7 RNA polymerase (Stratagene) in the presence of [α-32P]UTP and purified on a Nick column (Pharmacia) according to the manufacturers’ instructions. The labeled RNAs were then precipitated and resuspended in RNase-free water. The specific activities of labeled RNA preparations were ~4 × 106 c.p.m./µg of RNA. Ribosomal salt wash extract from COS-1 and the binding conditions for the UV-crosslinking assay using [α-32P]UTP labeled RNA probes (1 × 106 c.p.m. per incubation) have already been described in detail in a previous work (29). The samples were loaded on an 8 and 12% (as stated in the figure legends) SDS–PAGE gel which was subsequently dried and exposed to Kodak X-Omat AR films for 12–24 h. Films were then scanned on a Macintosh G3 workstation using Adobe Photoshop and printed using a Phaser 400 printer.
Enzymatic analysis of RNA secondary structure
In order to investigate the secondary structure of the domain III mutants we used single- and double-strand specific RNases. RNA was transcribed using T7 RNA polymerase (Stratagene) from the Bls KS+ domIII (145–248), domIII (172–227), domIII (184–213), 5′ wt and 5′ S/L mutants with HindIII. Reaction mixes (100 µl final vol) contained 1 µg of RNA and 0.02 U of RNase V1 (Pharmacia Biotech), or 0.5 U of RNase T1 (Sigma) in buffer A (10 mM Tris pH 7.5, 10 mM MgCl2, 50 mM KCl), or 20 U of S1 nuclease (Pharmacia Biotech) in buffer B (buffer A plus 1 mM ZnSO4). The RNA was digested at 30°C for 15 min in a water bath and a control aliquot of RNA without the addition of RNases was processed simultaneously with the digested samples. Reactions were stopped by extraction with phenol/chloroform and ethanol precipitated in 0.3 M sodium acetate (pH 5.2). The pellet was resuspended in 3 µl of water and RNase cleavage sites were identified by primer extension with a domain IV specific end-labeled oligonucleotide primer (5′-ggtgcacggtctacgaga-3′). In a total volume of 5 µl, 10 ng of 32P-labelled primer was hybridized to the resuspended RNA in RT buffer (50 mM Tris, pH 8.3, 3 mM MgCl2, 75 mM KCl). The solution was heated at 65°C for 5 min and allowed to cool for 5 min at room temperature. To each reaction we then added 15 µl of a solution in RT buffer containing 0.2 U of M-MLV reverse transcriptase (Gibco BRL), 2 µl of dNTP mix 5 mM and 2 µl of 0.1 M DTT. The mixture was held at 42°C for 30 min and then 2 µg of RNase A were added and the mixture incubated for a further 30 min at 37°C. It was then phenol/chloroform extracted and ethanol precipitated in 0.3 M sodium acetate (pH 5.2). The samples (enzymatically digested RNA, control reaction and a sequencing reaction using the same primer used for the RT reaction) were loaded on a 6% polyacrylamide denaturing gel (8 M urea, 1× TBE) which was subsequently run for 1–3 h at 1800 V, dried, and exposed to Kodak X-Omat AR films for 12–24 h. Films were then scanned on a Macintosh G3 workstation using Adobe Photoshop and printed using a Phaser 400 printer.
Assembly and sucrose density gradients of binary IRES–40S complexes
The purification of 40S ribosomal subunits and the assembly of binary IRES–40S ribosomal complexes were performed essentially as described by Pestova et al. (32). Briefly, 40S ribosomal subunits were prepared from HeLa extracts by precipitation for 4 h at 4°C and 30 000 r.p.m. in a Beckman 60Ti rotor and resuspended in buffer A (20 mM Tris–HCl pH 7.6, 2 mM DTT and 6 mM MgCl2) with 0.25 M sucrose and 150 mM KCl to a concentration of 40 A260 U/ml. This suspension was incubated with 1 mM puromycin (Sigma) for 10 min at 0°C and then for 10 min at 37°C before addition of KCl to 0.5 M final concentration. The 40S and 60S ribosomal subunits were then separated by centrifugation of 2 ml aliquots of this suspension through a 10–30% sucrose gradient in buffer A with 0.5 M KCl for 17 h at 4°C and 22 000 r.p.m. using a Beckman SW28 rotor. The 40S subunits were precipitated from the pooled gradient fractions by centrifugation for 18 h at 4°C and 50 000 r.p.m. in a 60Ti rotor. Pellets were resuspended in buffer B (20 mM Tris–HCl pH 7.6, 0.2 mM EDTA, 10 mM KCl, 1 mM DTT, 1 mM MgCl2 and 0.25 M sucrose) to a final concentration of 60 A260 U/ml. Ribosomal complexes were assembled by incubating 1 µg of labeled RNAs for 10 min at 30°C in a 100 µl reaction volume that contained buffer E (2 mM DTT, 100 mM potassium acetate, 20 mM Tris pH 7.6) with 2.5 mM magnesium acetate, 100 U of RNasin (Promega), 1 mM ATP and 6 pmol of 40S subunits. The complexes were resolved by centrifugation through a 10–30% sucrose gradient in buffer E with 6 mM magnesium acetate for 16 h at 4°C and 24 000 r.p.m. using a Beckman SW41 rotor. The radioactivity of gradient fractions was determined by Cerenkov counting.
RESULTS
We have previously shown that an isolated domain III sequence RNA (nt 134–290) binds two eIF3 subunits (29) even when transcribed apart from the rest of the entire HCV 5′ UTR sequence. This result is in agreement with a different study showing that a binary complex between the entire eIF3 complex and the full HCV IRES sequence protects from footprinting analysis only stem–loops IIIa, IIIb and IIIc of this domain (26). Taken together, these data were indicative that the apical portion of domain III is the only HCV 5′ UTR region involved with eIF3 binding to the IRES. Therefore, in order to better define these observations we have prepared three additional mutants which carry selected portions of this domain: nt 145–248, 172–227 and 184–213 (Fig. 1A). First of all, considering that all these sequences were inserted back in the exact position of the original wild-type domain III (nt 134–290) it was interesting to measure if any of these mutants had retained some IRES activity. Figure 1B shows that in an in vitro transcription and translation assay and in a transfection assay in COS-1 cells using a bicistronic system (Fig. 1C) all three mutants failed to stimulate IRES activity as estimated by production of a reporter HCV core protein. It should be noted that transfection efficiencies in Figure 1C, as measured by growth hormone production from the first cistron, were all similar, indicating that all RNAs were equally stable. As positive controls we used both the wild-type sequence (5′ wt) and the previously described mutant (5′ S/L) in which the apical region sequence (IIIb) had been inverted without occurring in a loss of IRES activity (29). It should be noted that in the in vitro assay only the unprocessed form of the core protein can be detected and that in both assays the molecular weight of this protein is slightly higher than the HCV native core protein (25 as opposed to 23 kDa) due to the addition in our constructs of a C-terminal tag sequence (33).
A necessary control to understand the inability of these mutants to drive IRES translation is represented by the analysis of their secondary structure (and comparing it with the wild-type structure). Although the secondary structure of HCV 5′ UTR wild-type domain III (shown in Fig. 2) has already been extensively validated based on computer predictions and confirmed by mutational analysis/RNase probing (11,12), we decided to repeat this analysis in order to obtain a reference pattern to compare it with that of the mutants. As shown in Figure 2 (right panel) the cleavage data obtained in the domain III region (nt 134–290) are in very good agreement with the already existing model (Fig. 2, left panel). In particular, strong T1 and S1 cleavages (indicating single-stranded regions) can be detected in correspondence to the apical loop (IIIb) and the lateral IIIa and IIId loops, with the exception of the proposed loop IIIc. Interestingly, a prominent S1 cleavage can also be detected in the lateral bulge which was described to be protected in footprinting experiments using the entire eIF3 complex (26). Furthermore, all the V1 cleavages (indicating double-stranded regions) detected in this analysis are entirely consistent with the proposed stem structures.
Figure 2.

Enzymatic determination of the RNA secondary structure of HCV 5′ UTR domain III. In vitro transcribed RNA was enzymatically digested with S1 nuclease, T1 and V1 RNases and reversely transcribed. The RT products were then separated on a 6% polyacrylamide sequencing gel and a sequencing reaction performed with the same primer used for the RT reaction was run in parallel to precisely determine the cleavage sites. Squares, circles and triangles indicate S1 nuclease, RNase T1 and V1 cleavage sites, respectively. Black, shaded and white symbols indicate high, medium and low cleavage intensities. The vertical bars indicate the proposed loop regions of IIIa, IIIb, IIIc and IIId. No enzyme was added to the reaction mixture in lane N. The observed cleavage sites (panel on the right) are reported on the proposed schematic diagram of domain III (panel on the left). Reported on this panel are also the portions of domain III inserted on the template sequence to obtain the three novel mutants.
The same procedure was then used to map the secondary structure of each mutant and the results of this analysis are shown in Figure 3. Only mutant 5′ domIII (145–248) shows an apical cleavage pattern very similar to that of the wild-type RNA. Indeed, not only are the single-strand cleavages in the loop are conserved but also the V1 specific cleavages in the stem immediately underneath. On the other hand, the apical loop region in mutants 5′ domIII (172–227) and 5′ domIII (184–213) shows very few, if any, single-strand specific cleavages, showing that a conformational change had occurred. Interestingly, the structure of the 5′ S/L mutant shows that in the IIIb mutated region the two G residues in the loop IIIb (present in inverted positions with respect to 5′ wt) were recognized by RNase T1 and that even the mutated stem region presented V1 cleavages, confirming the maintenance of a stem–loop structure similar (but not identical) to 5′ wt. Therefore, although the 5′ S/L mutant presents a very extensive primary sequence mutation its conformation resembles more the wild-type pattern than the two 5′ domIII (172–227) and 5′ domIII (184–213) mutants.
Figure 3.

RNA secondary structure of the apical portion of each domain III mutants with respect to the wild-type and 5′ S/L sequence. In vitro transcribed RNAs were enzymatically analyzed as described in the legend to Figure 2. Only the apical stem–loop region of domain III is shown and the putative IIIb loop portion is indicated by the vertical bar. The structure of mutants 5′ domIII (172–227) and 5′ domIII (184–213) are crossed to indicate that the stem–loop configuration is not consistent with the RNase analysis. In addition this figure shows the RNA secondary structure of domain III of the 5′ S/L mutant (its modified loop IIIb sequence is indicated by an asterisk).
It was then interesting to determine how the variation in secondary structure of these mutants was reflected in their ability to bind the previously described p170 and p116/p110 subunits. Figure 4A shows the UV-crosslinking assay of each 32P-labeled RNA with a ribosomal salt wash extract from COS-1 cells loaded on an 8% SDS–PAGE gel to better visualize high molecular weight proteins. This analysis shows that, as expected, mutant 5′ domIII (145–248) was capable of binding both p170 and p116/p110 (although with lower efficiency than 5′ wt) whilst no binding could be detected for the other two mutants 5′ domIII (172–227) and 5′ domIII (184–213). The fact that mutant 5′ domain III (145–248) showed a structure and binding pattern identical to that of the wild-type sequence but was completely incapable of IRES activity made it an ideal target to identify essential cellular factors that bind to (or whose binding is affected by) the lower missing portion of domain III (principally stem–loop IIId) and which could account for this loss. This possibility was enhanced by the fact that, in addition to eIF3, ribosomal protein S9 had also been detected as binding to domain III (25,26). In particular, binding of this protein was reported to be reduced by deletions of hairpins IIa and IIIc but not by deletion of hairpin IIIb (25). Figure 4B shows a competition analysis performed on the 5′ wt labeled RNA using a ribosomal salt wash extract with 5′ wt and 5′ domIII (145–248) cold RNA as competitor (in 2, 5 and 10 molar excess) and loaded on a 12% SDS–PAGE gel to visualize low molecular weight proteins. In this analysis, a protein of 25 kDa apparent molecular weight, consistent with the reported S9 ribosomal protein molecular weight observed in previous studies (25), was found to bind the 5′ wt sequence and could be competed away by cold 5′ wt RNA but not by cold 5′ domIII (145–248) RNA.
Figure 4.

UV-crosslinking analysis used to identify proteins binding to the wild-type HCV RNA and the domain III mutants. (A) UV-crosslinking assay (loaded on an 8% SDS–PAGE gel) using a COS-1 ribosomal salt wash extract with all the mutant UTRs carrying different portions of domain III. The arrow indicates the position of p170 and p116/p110. To better visualize the binding of these subunits we used 5 µg of cold 5′ MscStu template RNA as competitor (which had already been reported not to bind p170 and p116/p110). (B) Competition analysis (loaded on a 12% SDS–PAGE gel) of the proteins bound by labeled 5′ wt with cold 5′ wt and 5′ domIII (145–248) RNA. The arrow indicates the 25 kDa protein that binds labeled 5′ wt and can be competed by cold 5′ wt but which cannot be competed by cold 5′ domIII (145–248) RNA.
In order to better investigate the binding requirements for this protein we then prepared a set of mutants (Fig. 5A) which involved inversion of stem–loop IIId sequence (5′ IIId S/L), disruption of the principal stem (5′ IIId disr.) and its compensatory mutation (5′ IIId rep.). Analogously to the apical stem–loop mutants these mutants were analyzed for IRES activity both in vivo (Fig. 5A) and in vitro (Fig. 5B). The results show that none of these mutants displayed IRES activity, including the one bearing the intended compensatory mutation.
Figure 5.

(A) Schematic representation of the stem–loop IIId mutants 5′ domIIId (disr.), 5′ domIIId (rep.) and 5′ domIIId S/L. (B) Transfection in COS-1 cells of these mutant 5′ UTRs and the arrows indicate the position of the processed (23 kDa) and unprocessed (25 kDa) HCV core protein. The HCV core protein was recognized using MAb B12.F8 (35) and visualized by ECL staining. (C) In vitro transcription and translation assay of all mutants. The arrow indicates the unprocessed 35S-labelled HCV core protein (visualized by autoradiography).
Analysis of the secondary structure of each mutant is shown in Figure 6 and in keeping with the loss in IRES activity both the conformation of the disrupted mutant and that of the putatively repaired mutant fail to mimic that of the wild-type sequence or the 5′ S/L mutant. The fact that even the supposedly repaired mutant fails to fold again correctly highlights the fact that it is very difficult to predict beforehand the effect of engineered mutations. Figure 6 also shows the secondary structure of the IIId stem–loop of mutant 5′ S/L and that it is identical to that of 5′ wt (although this was an expected result considering that the engineered mutation was located in the apical IIIb stem–loop).
Figure 6.

RNA secondary structure of the stem–loop IIId mutants with respect to the wild-type sequence 5′ wt and the 5′ S/L mutant. In vitro transcribed RNAs were enzymatically analyzed as described in the legend to Figure 2. Only the lower region of domain III is shown and the putative IIId loop is indicated by a vertical bar. The structure of mutants 5′ domIIId (disr.), 5′ domIIId (rep.) and 5′ domIIId S/L are crossed to indicate that the original stem–loop configuration is not consistent with the RNase analysis.
The results of the structural analysis were reflected in the UV-crosslinking assay, as shown in Figure 7: only 5′ wt and 5′ S/L bind the 25 kDa protein, whilst none of the IIId mutants is capable of doing so. Binding of S9 to the 5′ S/L (whose mutation involved only hairpin IIIb) is in keeping with published data that report a reduction of p25 UV crosslinking following deletion of hairpins IIa and IIIc but not by deletion of hairpin IIIb (25). Our results thus provide a likely reason for the loss in IRES activity experienced by the correctly folded 5′ domIII (145–248) mutant and also report for the first time that stem–loop IIId affects the binding of S9 ribosomal protein to HCV 5′ UTR.
Figure 7.
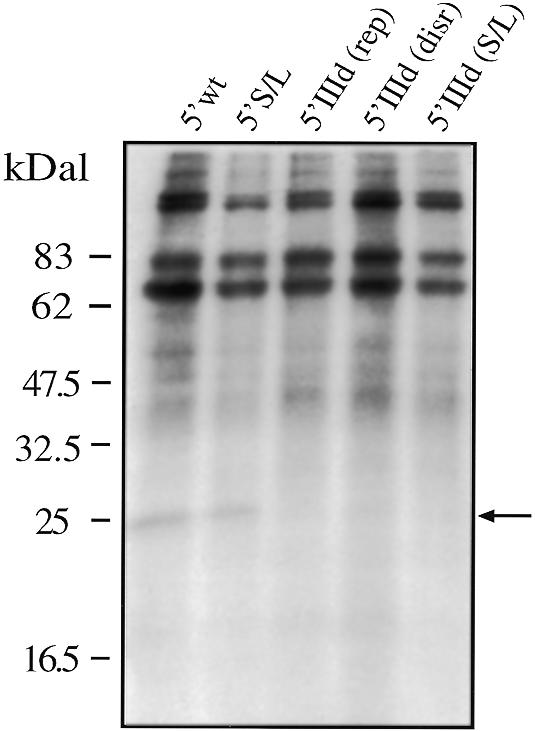
UV-crosslinking assay (loaded on a 12% SDS–PAGE gel) using a COS-1 ribosomal salt wash extract with all the mutant UTRs carrying different mutations in stem–loop IIId. Binding of the 25 kDa protein (indicated by an arrow) could be detected only for the wild-type sequence (5′ wt) and for mutant 5′ S/L. No binding could be observed with the 5′ domIIId (disr.), 5′ domIIId (rep.) and 5′ domIIId S/L mutant. To better visualize this binding we used 5 µg of cold 5′ MscStu RNA as competitor.
The fact that distant stem–loops (IIa, IIIc and in the present work IIId) have been found to affect S9 ribosomal protein binding suggests that, unlike p170 and p116/p110, the binding requirements for this protein might not be present on a single domain. A simple experiment to test this hypothesis was to see if S9 ribosomal protein binding can still be detected on a HCV 5′ UTR in which the sequences of hairpins IIa, IIIc and IIId are present (and unmutated) but in different orientations with respect to the wild-type. Therefore, we used the mutants already described in a previous paper in which the position of domain II and domain III on the HCV 5′ UTR had been inverted (Fig. 8A) and which did not possess any IRES activity. The results of the UV-crosslinking analysis performed on these mutants (Fig. 8B) shows that only the wild-type sequence (5′ wt) can bind S9 whilst mutants 5′ MscStu domII, 5′ MscStu domIII and 5′ MscStu domIII/domII do not bind this protein. It is important to note that both 5′ MscStu domIII and 5′ MscStu domIII/domII have already been reported (29) to bind both p170 and p116/p110, and that a secondary structure analysis of their domain IIId structure showed that this hairpin presented a cleavage pattern identical to that of the wild-type structure (data not shown).
Figure 8.

(A) Mutated UTRs used in the part of the study concerning the effect of swapping the positions of domain II and III on the binding of protein S9. How the different mutants were obtained from the original 5′ wt sequence is described in detail elsewhere (29). Major structural domains are indicated by Roman numerals I–IV. (B) UV-crosslinking assay (loaded on a 12% gel SDS–PAGE) using a COS-1 ribosomal salt wash extract with all the mutant UTRs carrying different combinations of domain II or III in swapped positions. The arrow indicates the 25 kDa protein observed to bind only to 5′ wt. No binding could be observed with 5′ MscStu domII, 5′ MscStu domIII and 5′ MscStu domIII/domII.
Finally, the failure to detect UV crosslinking of the S9 protein in our previous mutants could be due to two reasons. The first and simpler explanation is that the 40S ribosomal subunit does not actually bind to the mutant RNAs. Alternatively, the mutant RNAs might be able to bind to the 40S ribosomal subunit but in a different orientation compared to the wild-type RNA. In order to differentiate between these two possibilities we performed sucrose density gradient analysis of the wild-type RNA and of representative mutants in the presence of purified 40S subunits. Previous studies have already shown that in order to obtain binding of the 40S ribosomal subunit to the HCV IRES no additional initiation factors are required (25). The results shown in Figure 9 demonstrate that a ribosomal complex can only be detected with the wild-type HCV IRES, whilst mutant 5′ domIII (145–248) and mutant 5′ IIId rep. (see Figs 1 and 5, respectively) do not bind to the 40S ribosomal subunit.
Figure 9.

Binary IRES–40S ribosomal complex formation on HCV wild-type IRES and selected mutants. Assays were performed on a 10–30% sucrose density gradient with labeled RNAs and purified 40S ribosomal subunits. Assays were done using 5′ wt (A), 5′ domIII (145–248) (B) and 5′ IIId (rep) (C) mutant RNAs. Sedimentation was from right to left. The position of the binary complexes is indicated by an arrow.
DISCUSSION
Functional and mutational studies performed on the HCV 5′ UTR region coupled with data obtained from UV crosslinking and structural analyses have allowed us to gain substantial insight concerning HCV IRES function, as recently reviewed in (10). In particular, several cellular factors have been found to specifically bind its structure and, in this respect, the HCV IRES has been recently shown to share a similarity with the mechanism of translation initiation in prokaryotes (25). In this study we have further characterized the binding to the HCV 5′ UTR of two subunits of eukaryotic initiation factor eIF3 and of the ribosomal protein S9 with regards to conservation of RNA secondary structure. In addition, we have investigated the possible influence of RNA tertiary folding on their binding, providing direct evidence that it plays a role in the binding of S9 ribosomal protein to the HCV IRES.
The effect of secondary structure on the binding of the eIF3 subunits has already been preliminarily analyzed in a previous study in which we showed that binding could still occur in an isolated domain III sequence and that it could be impaired by mutations which were predicted to disrupt the RNA folding of the apical stem–loop (29). In this study we report that binding of the two previously described eIF3 subunits (p170 and p116/p110) can be achieved even when significant lower portions of domain III are missing and that the key element in this recognition is provided by whether or not the apical stem–loop RNA sequence is able to fold in the correct secondary structure. In fact, RNase mapping of the 5′ UTR secondary structure shows that nt 145–248 can fold autonomously in the correct RNA conformation and can then bind both subunits: an observation that definitively identifies this region as possessing the minimal sequence and structural features necessary for recognition of these subunits. This result is in agreement with the footprinting data previously published concerning an eIF3–HCV IRES binary complex which did not detect any protection in the HCV IRES outside the apical domain III region (26). It is also of particular interest the observation that mutants which contain sizeable portions of the apical stem–loop sequences previously identified as important for this interaction (nt 172–227 and 184–213) were not able to bind either p170 or p116/p110. This observation was not totally expected if we consider the fact that the previously described 5′ S/L mutant (in which the sequence of the apical stem–loop was inverted) had retained the ability to bind p170 but had lost the ability to bind p116/p110 (29). Therefore, if the recognition of p170 and p116/p110 had been predominantly dependent on the primary sequence some binding could still have taken place in the two 5′ domIII (172–227) and 5′ domIII (184–213) mutants. The fact that no binding occurred finds an explanation in the secondary structure of these mutants which, as opposed to the one of mutant 5′ S/L, revealed that in their case the structure of the apical stem–loop had not been preserved. On the other hand, the structure of the 5′ S/L mutant had conserved an unexpected similarity (considering the extent of the primary nucleotide changes) to the wild-type conformation. Taken together, these results show that conservation of secondary structure is a fundamental necessity to obtain protein binding and that it possesses just as great an importance as the primary nucleotide sequence. In particular, our results show that conserving one without the other (irrespectively) is not sufficient to obtain correct binding.
The fact that the correctly folded 5′ domIII (145–248) mutant was not capable of IRES translation (although its binding and structural profile are most similar to those of 5′ wt, even more so than those observed for the 5′ S/L mutant) was also indicative that the missing lower sequence is probably involved in the binding of some other essential cellular factor. This hypothesis was strengthened by the previous identification of ribosomal protein S9 (25) binding to HCV domain III. In accordance with this study we have identified a 25 kDa protein, whose molecular weight is consistent with the molecular weight of the S9 ribosomal protein, that binds to the 5′ wt sequence but which cannot be competed away by the 5′ domIII (145–248) mutant.
An increasing body of evidence shows that the majority of hairpins located on the HCV 5′ UTR are involved in the formation of the RNA–protein interactions that allow the IRES translation to take place. Up to now, hairpins IIIb (29), IIIc (15,25), and recently IIIe (18), have been demonstrated to be important for efficient IRES activity. Therefore, we investigated whether the integrity of hairpin IIId was also important for IRES activity and whether its disruption affected S9 ribosomal protein binding. Our results show that mutations of hairpin IIId which cause both sequence and structural changes lead to undetectable levels of IRES activity in conjunction with the loss of S9 protein binding. The analysis of IRES–40S binary ribosomal complex showed that mutant 5′ domIII (145–248) and mutant 5′ IIId rep. do not bind to the 40S ribosomal subunit, thus providing a functional explanation for the lack of S9 binding. Notably, it is also indicative that mutant 5′ S/L, which was observed to be capable of IRES translation, showed a IIId cleavage pattern similar to that of the wild-type sequence and can bind S9, providing an additional explanation of why this mutation did not affect the translational activity.
The fact that binding of the S9 ribosomal protein was already reported to be affected by deletion of hairpins IIa, IIIc (25) and IIId (this paper) suggested that the binding requirements for S9 are also extended to the tertiary folding of the RNA structure. In this respect, using small angle X-ray crystallography Kieft et al. have shown that the HCV IRES possesses indeed a higher order structure of spatially organized recognition domains in the absence of cellular factors (34). Therefore, the S9 interaction with the tertiary configuration was further analyzed by studying the binding pattern in mutants already used in a previous work where the position of wild-type domain II and domain III had been inverted on a 5′ UTR template without introducing mutations in their primary sequence (29). Our analysis shows that the simple change of orientation of these domains with conservation of sequence and RNA structure can abrogate binding of S9. Most indicatively, we have already reported that the disposition of domain III on the 5′ UTR had no effect on p170 and p116/p110 binding (29), highlighting a clear difference in the binding requirements between the eIF3 and S9 cellular factors. In conclusion, the emerging picture of the HCV IRES architecture is that of a highly organized 3D structure where the binding of proteins required for translation initiation needs not only an intact domain structure but also correct spatial configuration of the domains, a clear indication of the high degree of specialization evolved by the HCV IRES to bind essential cellular factors.
REFERENCES
- 1.Choo Q.L., Kuo,G., Weiner,A.J., Overby,L.R., Bradley,D.W. and Houghton,M. (1989) Science, 244, 359–362. [DOI] [PubMed] [Google Scholar]
- 2.Kamoshita N., Tsukiyama-Kohara,K., Kohara,M. and Nomoto,A. (1997) Virology, 233, 9–18. [DOI] [PubMed] [Google Scholar]
- 3.Tsukiyama-Kohara K., Iizuka,N., Kohara,M. and Nomoto,A. (1992) J. Virol., 66, 1476–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C., Sarnow,P. and Siddiqui,A. (1993) J. Virol., 67, 3338–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang C., Sarnow,P. and Siddiqui,A. (1994) J. Virol., 68, 7301–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang C., Le,S.Y., Ali,N. and Siddiqui,A. (1995) RNA, 1, 526–537. [PMC free article] [PubMed] [Google Scholar]
- 7.Fukushi S., Katayama,K., Kurihara,C., Ishiyama,N., Hoshino,F.B., Ando,T. and Oya,A. (1994) Biochem. Biophys. Res. Commun., 199, 425–432. [DOI] [PubMed] [Google Scholar]
- 8.Rijnbrand R., Bredenbeek,P., van der Straaten,T., Whetter,L., Inchauspe,G., Lemon,S. and Spaan,W. (1995) FEBS Lett., 365, 115–119. [DOI] [PubMed] [Google Scholar]
- 9.Reynolds J.E., Kaminski,A., Kettinen,H.J., Grace,K., Clarke,B.E., Carroll,A.R., Rowlands,D.J. and Jackson,R.J. (1995) EMBO J., 14, 6010–6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hellen C.U.T. and Pestova,T.V. (1999) J. Viral Hepatitis, 6, 79–87. [DOI] [PubMed] [Google Scholar]
- 11.Brown E.A., Zhang,H., Ping,L.H. and Lemon,S.M. (1992) Nucleic Acids Res., 20, 5041–5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Honda M., Brown,E.A. and Lemon,S.M. (1996) RNA, 2, 955–968. [PMC free article] [PubMed] [Google Scholar]
- 13.Buratti E., Gerotto,M., Pontisso,P., Alberti,A., Tisminetzky,S.G. and Baralle,F.E. (1997) FEBS Lett., 411, 275–280. [DOI] [PubMed] [Google Scholar]
- 14.Fukushi S., Kurihara,C., Ishiyama,N., Hoshino,F.B., Oya,A. and Katayama,K. (1997) J. Virol., 71, 1662–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang S., Collier,A.J. and Elliott,R.M. (1999) J. Virol., 73, 2359–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Honda M., Beard,M.R., Ping,L.H. and Lemon,S.M. (1999) J. Virol., 73, 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang L.H., Hsieh,C.L., Yen,A., Chung,Y.L. and Chen,D.S. (1998) Biochem. Biophys. Res. Commun., 252, 455–460. [DOI] [PubMed] [Google Scholar]
- 18.Psaridi L., Georgopoulou,U., Varaklioti,A. and Mavromara,P. (1999) FEBS Lett., 453, 49–53. [DOI] [PubMed] [Google Scholar]
- 19.Varaklioti A., Georgopoulou,U., Kakkanas,A., Psaridi,L., Serwe,M., Caselmann,W.H. and Mavromara,P. (1998) Biochem. Biophys. Res. Commun., 253, 678–685. [DOI] [PubMed] [Google Scholar]
- 20.Honda M., Rijnbrand,R., Abell,G., Kim,D. and Lemon,S.M. (1999) J. Virol., 73, 4941–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ali N. and Siddiqui,A. (1997) Proc. Natl Acad. Sci. USA, 94, 2249–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaminski A., Hunt,S.L., Patton,J.G. and Jackson,R.J. (1995) RNA, 1, 924–938. [PMC free article] [PubMed] [Google Scholar]
- 23.Ali N. and Siddiqui,A. (1995) J. Virol., 69, 6367–6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ito T. and Lai,M.M. (1999) Virology, 254, 288–296. [DOI] [PubMed] [Google Scholar]
- 25.Pestova T.V., Shatsky,I.N., Fletcher,S.P., Jackson,R.J. and Hellen,C.U. (1998) Genes Dev., 12, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sizova D.V., Kolupaeva,V.G., Pestova,T.V., Shatsky,I.N. and Hellen,C.U. (1998) J. Virol., 72, 4775–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hahm B., Kim,Y.K., Kim,J.H., Kim,T.Y. and Jang,S.K. (1998) J. Virol., 72, 8782–8788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spangberg K. and Schwartz,S. (1999) J. Gen. Virol., 80, 1371–1376. [DOI] [PubMed] [Google Scholar]
- 29.Buratti E., Tisminetzky,S., Zotti,M. and Baralle,F.E. (1998) Nucleic Acids Res., 26, 3179–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaminski A. and Jackson,R.J. (1998) RNA, 4, 626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicholson R., Pelletier,J., Le,S.Y. and Sonenberg,N. (1991) J. Virol., 65, 5886–5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pestova T.V., Hellen,C.U. and Shatsky,I.N. (1996) Mol. Cell. Biol., 16, 6859–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buratti E., Baralle,F.E. and Tisminetzky,S.G. (1998) Cell. Mol. Biol. (Noisy-le-grand), 44, 505–512. [PubMed] [Google Scholar]
- 34.Kieft J.S., Zhou,K., Jubin,R., Murray,M.G., Lau,J.Y. and Doudna,J.A. (1999) J. Mol. Biol., 292, 513–529. [DOI] [PubMed] [Google Scholar]
- 35.Cerino A., Boender,P., La Monica,N., Rosa,C., Habets,W. and Mondelli,M.U. (1993) J. Immunol., 151, 7005–7015. [PubMed] [Google Scholar]