

Abstract
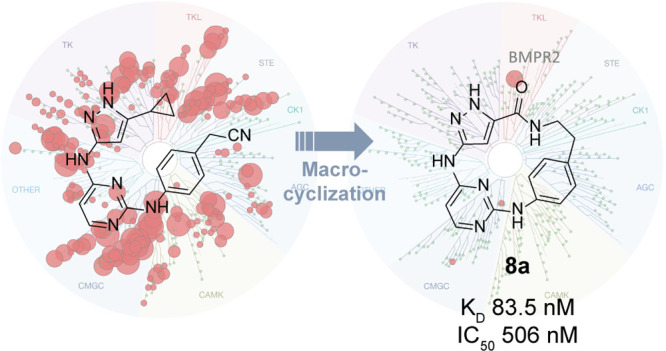
Bone morphogenetic protein (BMP) signaling is mediated by transmembrane protein kinases that form heterotetramers consisting of type-I and type-II receptors. Upon BMP binding, the constitutively active type-II receptors activate specific type-I receptors by transphosphorylation, resulting in the phosphorylation of SMAD effector proteins. Drug discovery in the receptor tyrosine kinase-like (TKL) family has largely focused on type-I receptors, with few inhibitors that have been published targeting type-II receptors. BMPR2 is involved in several diseases, most notably pulmonary arterial hypertension, but also contributes to Alzheimer’s disease and cancer. Here, we report that macrocyclization of the promiscuous inhibitor 1, based on a 3-amino-1H-pyrazole hinge binding moiety, led to a selective and potent BMPR2 inhibitor 8a.
Keywords: kinase assay, BMPR2, BMP signaling, kinase inhibitor, macrocycle, macrocyclic inhibitor
The human kinome encodes more than 500 kinases, which have been subdivided into diverse groups and families based on the sequence homology of their kinase domains.1 Protein kinases catalyze the reversible phosphorylation of specific substrates and are key regulators of cell proliferation, differentiation, metabolism, and apoptosis.2,3 Dysregulation of these intracellular signaling pathways is responsible for various diseases, in particular cancer.4 Inhibition of this impaired kinase function by targeting the ATP-binding pocket of the kinase of interest using kinase inhibitors can be used to treat the disease of interest. The major difficulty for conventional type-I and type-II kinase inhibitors is to achieve appropriate selectivity. Macrocyclization of a linear pharmacophore through a linker offers an opportunity to increase the selectivity of the acyclic analogue and provides a relatively new strategy for the development of selective kinase inhibitors.5 Thus, in recent years, macrocyclization has gained increasing scientific interest in drug discovery and medicinal chemistry. Macrocycles are limited in conformational flexibility, which increases selectivity by providing a locked three-dimensional structure that fits into a kinase of interest and not into other kinase active sites, and this reduces the entropic costs upon binding and provides a higher potency.6 In addition, biological and physiochemical properties can be tuned by cyclization compared to the acyclic counterpart.7,8 Furthermore, compared to optimized conventional inhibitors, macrocyclic inhibitors could have lower molecular weight and may have more favorable membrane permeability. Similarly, improved blood–brain permeability was observed for the macrocycle lorlatinib compared with the acyclic analogue crizotinib.9 The increasing number of drug candidates and the numerous publications in recent years have demonstrated that macrocyclization is an efficient strategy to improve the potency, target selectivity, as well as biochemical properties and offers a great opportunity for the discovery of new drugs.5,10−12
The bone morphogenetic protein receptor type-II (BMPR2) is a serine/threonine receptor kinase and belongs to the tyrosine kinase-like (TKL) group. BMPR2, together with diverse type-I receptors, binds bone morphogenetic proteins (BMPs), which are members of the TGF-β superfamily. Binding of these ligands results in the formation of heterotetrameric complexes consisting of two type-I and two type-II receptor homodimers. The type-II dimer activates the type-I complex by phosphorylation at its so-called GS-box.13 The activated type-I homodimer subsequently phosphorylates R-SMAD proteins (SMADs 1, 5, 8) and activates them, so that they can interact with Co-SMAD (SMAD4). This SMAD heterotrimer complex translocates into the nucleus and regulates the transcription of BMP target genes (Id1–Id4).14,15 In addition to this canonical signaling pathway, BMPR2 is also involved in noncanonical signaling pathways. For example, BMPR2 is involved in activating the ERK, MAPK, LIMK, NOTCH, and Wnt signaling pathways.16 BMPR2 expression can be found in many different tissues.17 It is important for vascular homeostasis, maintenance of pulmonary artery endothelial cell barrier function, and endothelial inflammatory response.18−20 Impaired BMP signaling has been associated with various diseases such as pulmonary arterial hypertension,21 vascular pathogenesis,22 cancer,23 and Alzheimer’s disease.24,25
To date, several inhibitors have been described for the type-I receptors, including chemical probes for ALK4/526 and ALK1/2.27 Few inhibitors have been described for the type-II receptor kinases such as BMPR2. Database searches identified only a small number of inhibitors targeting BMPR2 in the nanomolar range. Most of them are promiscuous kinase inhibitors such as JNJ-28312141, sunitinib, and nintedanib, with KD values of 310, 570, and 56 nM, respectively (Figure 1A).28 Dorsomorphin, an AMPK inhibitor, also targets BMPR2 with a potent IC50 value of 74 nM; however, it also potently affects the type-I receptor kinases ALK2, ALK3, and ALK6 (IC50 = 68, 95, and 235 nM, respectively).23 In 2013, the structure–activity relationship of pyrazolo[1,5-a]pyrimidine-based inhibitors derived from the dorsomorphin scaffold was published. These compounds affect BMPR2 in the low nanomolar range, but they also showed potent inhibition of ALK1, ALK2, ALK3, TGFBR2, and VEGFR2.29 More recently, Modukuri et al. have identified selective benzimidazole-based potent inhibitors (CDD-1115, CDD-1653) of BMPR2 by using a DNA-encoded chemistry technology (Figure 1A).30
Figure 1.

A. Chemical structures of JNJ-28312141, nintedanib, dorsomorphin, sunitinib, CDD-1115, and CDD-1653, with the corresponding KD and IC50 values reported for BMPR2.23,28,30 B. Selectivity profile of 1 at 1 μM. The chemical structure of 1 is shown beside the dendrogram.31 C. Waterfall plot of the kinome-wide screening data measured for 1 at 1 μM. D. Compound 1 cocrystallized with VRK1 (PDB: 3OP5).31,32 The hinge region is colored light blue, the P-loop red, the altered DFG motif green, and the αC-helix yellow, and compound 1 is illustrated in orange. E. Synthetic strategy used for the development of macrocyclic inhibitors based on 1.
Compound 1 is a promiscuous kinase inhibitor developed by Statsuk et al. A kinome-wide scan against 468 recombinant human protein kinases emphasized the promiscuous behavior of 1, which potently inhibits 262 kinases (Figure 1B, C). In this report, type-I and type-II BMP-receptor kinases were potently targeted. Among others, 1 showed strong binding to ALK5 (KD = 324 nM). The crystal structure in complex with VRK1 revealed an interaction of the 3-aminopyrazole moiety with D132 and F134 of the ATP binding pocket, whereas the pyrimidine faced the hydrophobic pocket (Figure 1D).31 In this study, we report the development of macrocyclic kinase inhibitors based on the structure of the promiscuous inhibitor 1 to target the understudied type-II receptor kinase BMPR2. To accomplish this, functional groups for the macrocyclization were introduced and different linker moieties were varied (Figure 1E).
The macrocycles 8a–e were synthesized as shown in Scheme 1. Starting material 2 reacted with 2,4-dichloropyrimidine (3) in a nucleophilic substitution to obtain compound 4 with a yield of 16%. Various linkers were attached by a second nucleophilic substitution as previously described.35 The yields were in a moderate range from 31% to 63%. Cleavage of the Boc-group followed by a saponification with lithium hydroxide led to the precursors 7a–e. The macrocyclization in the last step was done via an amide coupling, using hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) to obtain macrocycles 8a–e.
Scheme 1. Synthesis of the Macrocycles 8a–e.
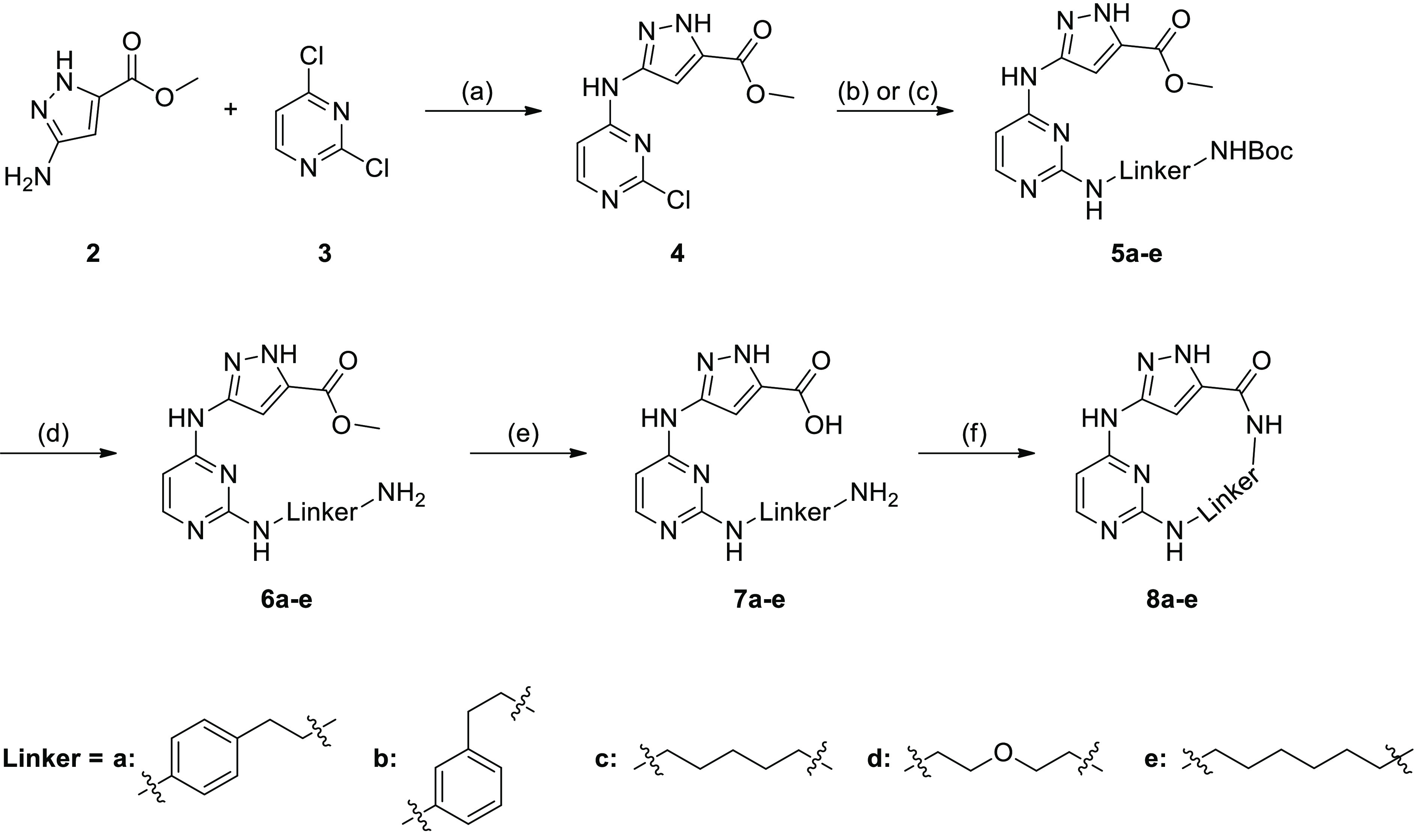
Reagents and conditions: (a) Et3N, isopropanol, 72 h, 50 °C; (b) Et3N, ethanol, microwave, 5 h, 120 °C; (c) HCl, ethanol, reflux, 18 h; (d) TFA, DCM, 0 °C to rt, oN; (e) LiOH·H2O, THF, H2O, 18 h, 50 °C; (f) HATU, DIPEA, DMF, 18 h, rt to 70 °C.
The synthesized macrocycles were measured in a differential scanning fluorimetry (DSF) assay to investigate their selectivity profile.33 A positive ΔTm compared to the ligand-free protein indicates stabilization of the protein by binding of the compound. For this purpose, an internal panel of 90 kinases was used with staurosporine (9) as a positive control. 1 was resynthesized, according to the synthesis published by Statsuk et al.,31 and its selectivity was assessed using a DSF assay. Macrocycle 8a showed an interesting profile by binding only three kinases with ΔTm shifts >5 °C. While its BMP2K binding was negligible compared to that of 9, GSK3B and BMPR2 were stabilized by 8a with 8.4 and 5.8 °C, respectively, and were further evaluated. 8b with an exchanged attachment point on the aromatic linker and 8c harboring an aliphatic C5 linker appeared to be less selective than compound 8a, with 17 and 10 stabilized kinases with ΔTm ≥ 5 °C, respectively. Interestingly, even a small change in the linker, from the aliphatic C5 linker in 8c, replacing a carbon with an oxygen to give compound 8d, resulted in an inactive compound. By extending the linker by one carbon atom, 8e regained selectivity compared to 8c. DSF data revealed stabilization of BMP2K, GSK3B, and STK3 by 8e with ΔTm shifts >5 °C, although the ΔTm shifts were lower compared to those with 9 (Table S1).
Compound 8a was selected as the most promising candidate for further characterization due to its good stabilization and selectivity, and together with lead structure 1, the binding affinities were determined by ITC (Figure 2). The ITC data revealed potent binding of compounds 1 and 8a to BMPR2, with KD values of 186 nM and 83.5 nM, respectively. However, thermodynamic differences between the binding of acyclic lead structure 1 and the macrocycle 8a could be identified. The binding of 1 was essentially enthalpy-driven (ΔH), balanced by unfavorable binding entropy changes (TΔS). In contrast, the binding of 8a was changed by an advantageous entropic contribution (TΔS) resulting in an overall lower binding constant. The thermodynamic profile suggests that the conformational constraints together with beneficial hydrophobic interactions of the macrocyclic inhibitor resulted in a favorable binding entropy change.
Figure 2.

Isothermal titration calorimetry (ITC) data for binding of 1 (panel A) and 8a (panel B) to the kinase domain of BMPR2 revealed nanomolar binding affinity with KD values of 186 nM and 83.5 nM, respectively.
After confirming the binding of macrocycle 8a to BMPR2 by two orthogonal binding assays, we were more interested in a functional assay, using an ADP-Glo (Promega, Madison, WI, USA) assay to determine the enzymatic inhibition (IC50) values for BMPR2 (Figure 3A). The IC50 values of 1, 8a, 8b, and 8c were in good agreement with the rank order of the measured DSF assay data. Compound 1, which showed the strongest stabilization in the DSF (9.2 °C), also has the lowest IC50 value (36.2 nM) in the ADP-Glo assay. The IC50 values for 8a–c ranged from 461 nM to 630 nM (Figure 3A). To determine the potential off-target activity of compounds 1, 8a, and 8c, we screened these compounds in a NanoBRET (Promega, Madison, WI, USA) assay on GSK3A/B (Figure 3A). The promiscuous inhibitor 1 revealed high cellular potency for both off-target kinases, with an IC50 value of 4.0 nM. The DSF assay also showed stabilization of GSK3B, with a ΔTm of 8.4 °C for compound 8a, but this was much less pronounced compared to 14.0 °C for compound 1. The NanoBRET assay revealed only weak cellular binding for GSK3A/B, with EC50 values of 10.9 μM and 33.6 μM. Activity on GSK3B was additionally measured in digitonin-lysed cells to rule out potential cell penetration limitations of the macrocycles. The EC50 value on GSK3B in the lysed mode was 2.3 μM for 8a, while 8c did not show activity against GSK3A/B, with EC50 values >45 μM for both kinases in intact cells. 8c also showed only weak potency (EC50 = 3.5 μM for GSK3B) in the lysed mode (Figure 3A).
Figure 3.

A. Enzyme kinetic IC50 values of 1, 8a, 8b, and 8c for BMPR2. The values were determined using an ADP-Glo assay. IC50 values for GSK3A and GSK3B were determined using NanoBRET. B. Selectivity data of 8a against an in-house DSF panel of 90 kinases. C. Table summarizing the top hits of the DSF selectivity screen of 8a as absolute values and with respect to the reference staurosporine. *Lestaurtinib was used as a reference compound. D. Selectivity data of 8a against a panel of 468 kinases using the KINOMEscan (Eurofins/DiscoverX at 1 μM). E. Waterfall plot illustrating the selectivity of 8a. F. Top hits of the KINOMEscan of 8a (for full list see Table S2).
After our in-house DSF panel revealed a promising selectivity profile (Figure 3B) and acceptable potency for BMPR2, 8a was selected for further selectivity profiling, using the ScanMAX KINOMEscan assay platform (Eurofins Scientific) (Figure 3C–E). Gratifyingly, 8a exhibited exclusive selectivity for BMPR2 in this comprehensive panel, with a selectivity score (S35) of 0.01 (screened at 1 μM). The data confirmed the activity of 8a at BMPR2, with just a few additional off-targets, e.g., oncogenic FLT mutant, and weak activity for GSK3A, JNK1, as well as RIOK2 (Figure 3C). In addition to the already determined weak activity of macrocycle 8a on GSK3A/B, we also examined the activity on the two other potential off-targets using the thermal shift assay and NanoBRET assay. Gratifyingly, only a low stabilization of JNK1 and RIOK2 in the thermal shift assay as well as only weak activity in the NanoBRET assay (Table S3) was shown, which confirmed the excellent selectivity profile of 8a.
To understand the potential binding mode, a docking study was performed with macrocycle 8a on BMPR2. The docking result revealed an ATP mimetic binding mode of 8a. The pyrazole moiety formed two hydrogen bonds with the backbone of Y282. A further hydrogen bond was observed between the amine linking the pyrimidine moiety and the aromatic ring with the backbone oxygen of the residue S350. Additional hydrophobic interactions have been noticed with I209, V217, and L340. The choice of the sterically demanding linker in compound 8a presumably forces the three aromatic ring systems out of planarity, leading to a strong ring strain of the macrocycle and reducing the flexibility to adopt different conformations (Figure 4). Unfortunately, attempts to cocrystallize BMPR2 with 1 or 8a failed, preventing us from determining experimental structures of 1 and 8a.
Figure 4.
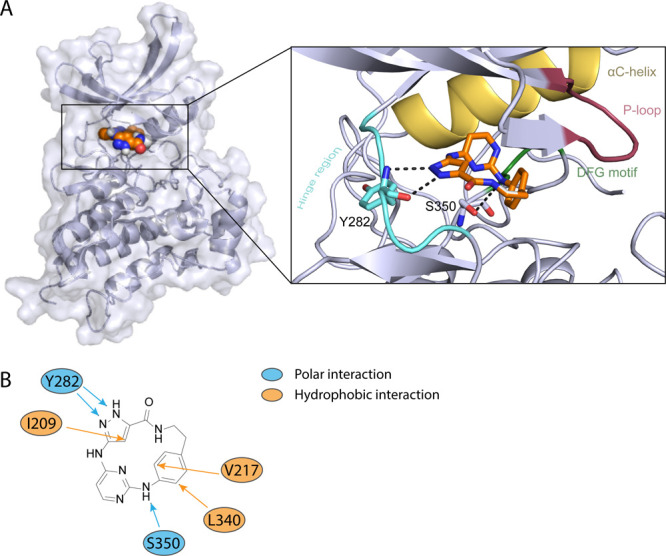
A. Binding mode of 8a with the kinase domain of BMPR2 determined by in silico docking. Macrocycle 8a was docked into the active conformation of BMPR2 (PDB: 6UNP). Hinge region is highlighted in light blue, P-loop red, DFG motif green, and αC-helix yellow, and the compound 8a is illustrated in orange. Docking poses were viewed by PyMOL, and protein–ligand interactions were analyzed using the PLIP.34 B. Interactions between the inhibitor and the amino acid residues of the protein kinase. Blue indicates a polar interaction and orange a hydrophobic interaction between the inhibitor 8a and BMPR2.
In summary, we have developed a novel series of macrocyclic kinase inhibitors possessing a 3-amino-1H-pyrazole scaffold, derived from the highly promiscuous kinase inhibitor 1. Rigidization using a macrocyclic approach of the 3-amino-1H-pyrazole scaffold with different linkers allowed us to strongly manipulate the selectivity profile of the promiscuous kinase inhibitor 1. With the aromatic linker in compound 8a, we developed a potent (IC50 = 506 nM) and selective BMPR2 inhibitor. Using ITC, we were able to show that the binding was enthalpically and entropically driven, whereas compound 1 exhibited mainly enthalpically driven binding to BMPR2. A docking study supports the result in which an ATP mimetic interaction with the hinge was observed. The docking model suggested that we succeeded in rotating the two heterocycles out of the planarity expected in their open form. This conformation was enforced by a short and rigid linker present in compound 8a. In this study, we demonstrated that macrocyclization is a powerful tool to enhance the selectivity profile of unselective acyclic compounds. Compound 8a represents a promising starting point that requires further characterization to better assess its potential in the cellular context, as the data so far are all in vitro data for BMPR2. The recently published compounds CDD-1115 and CDD-1653 are also very potent and selective in vitro compounds; however, there was a large discrepancy between the in vitro IC50 values and the in cellulo activity. Here, macrocycle 8a represents an additional chemotype that would be worth evaluating in the same context, to see if the discrepancy between the in vitro and in cellulo data is related to the compounds or is a target-related effect. Nonetheless, at this point we have identified a potent and selective BMPR2 inhibitor that represents an encouraging in vitro tool compound.
Acknowledgments
The authors are grateful for support by the Structural Genomics Consortium (SGC), a registered charity (no. 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN grant 875510], Janssen, Merck KGaA (aka EMD in Canada and U.S.), Pfizer, and Takeda. B.-T.B. also received support from the collaborative research center CRC 1399 “Mechanisms of drug sensitivity and resistance in small cell lung cancer”. A.K. would like to acknowledge funding from the Frankfurt Cancer Institute (FCI), an institute supported by LOEWE.
Glossary
Abbreviations
- BMP
bone morphogenetic protein
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- DSF
differential scanning fluorimetry
- ERK
extracellular-signal regulated kinase
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- ITC
isothermal titration calorimetry
- MAPK
mitogen-activated protein kinase
- oN
overnight
- rt
room temperature
- SMAD
small mothers against decapentaplegic
- TEA
triethylamine
- TFA
trifluoroacetic acid
- TGF
transforming growth factor
- TKL
tyrosine kinase-like
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00127.
Details of preparation, characterization, and evaluation experiments of compounds 4–8 (PDF)
Author Contributions
J.A.A., S.K., and T.H. designed the project; J.A.A. synthesized the compounds; G.W. performed the docking and ITC measurements; B.-T.B. performed ADP-Glo measurements; L.M.B. performed NanoBRET measurements; A.D.K. performed ITC measurements; A.K. provided the proteins for the DSF assay; S.K. supervised the research. The manuscript was written by J.A.A., S.K., and T.H. with contributions from all co-authors.
The authors declare the following competing financial interest(s): L.M.B. is a cofounder and B.-T.B. is a cofounder and the CEO of the Contract Research Organization CELLinib GmbH, Frankfurt, Germany.
Supplementary Material
References
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S. Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Gross S.; Rahal R.; Stransky N.; Lengauer C.; Hoeflich K. P. Targeting Cancer with Kinase Inhibitors Find the Latest Version: Targeting Cancer with Kinase Inhibitors. J. Clin. Invest. 2015, 125 (5), 1780–1789. 10.1172/JCI76094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks S. K. Genomic Analysis of the Eukaryotic Protein Kinase Superfamily: A Perspective. Genome Biol. 2003, 4, 111–118. 10.1186/gb-2003-4-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicenas J.; Zalyte E.; Bairoch A.; Gaudet P. Kinases and Cancer. Cancers 2018, 10 (3), 63. 10.3390/cancers10030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrhein J. A.; Knapp S.; Hanke T. Synthetic Opportunities and Challenges for Macrocyclic Kinase Inhibitors. J. Med. Chem. 2021, 64 (12), 7991–8009. 10.1021/acs.jmedchem.1c00217. [DOI] [PubMed] [Google Scholar]
- Fang Z.; Song Y.; Zhan P.; Zhang Q.; Liu X. Conformational Restriction: An Effective Tactic in ‘Follow-on’-Based Drug Discovery. Future Med. Chem. 2014, 6 (8), 885–901. 10.4155/fmc.14.50. [DOI] [PubMed] [Google Scholar]
- Mallinson J.; Collins I. Macrocycles in New Drug Discovery. Future Med. Chem. 2012, 4 (11), 1409–1438. 10.4155/fmc.12.93. [DOI] [PubMed] [Google Scholar]
- Driggers E. M.; Hale S. P.; Lee J.; Terrett N. K. The Exploration of Macrocycles for Drug Discovery — an Underexploited Structural Class. Nat. Rev. Drug Discovery 2008, 7, 608–624. 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- Johnson T. W.; Richardson P. F.; Bailey S.; Brooun A.; Burke B. J.; Collins M. R.; Cui J. J.; Deal J. G.; Deng Y.-L.; Dinh D.; Engstrom L. D.; He M.; Hoffman J.; Hoffman R. L.; Huang Q.; Kania R. S.; Kath J. C.; Lam H.; Lam J. L.; Le P. T.; Lingardo L.; Liu W.; McTigue M.; Palmer C. L.; Sach N. W.; Smeal T.; Smith G. L.; Stewart A. E.; Timofeevski S.; Zhu H.; Zhu J.; Zou H. Y.; Edwards M. P. Discovery of (10R)-7-Amino-12-Fluoro-2,10,16-Trimethyl-15-Oxo-10,15,16,17-Tetrahydro-2H-8,4-(Metheno)Pyrazolo[4,3-h][2,5,11]-Benzoxadiazacyclotetradecine-3-Carbonitrile (PF-06463922), a Macrocyclic Inhibitor of Anaplastic Lymphoma Kinase (ALK) and c-Ros O. J. Med. Chem. 2014, 57 (11), 4720–4744. 10.1021/jm500261q. [DOI] [PubMed] [Google Scholar]
- Engelhardt H.; Böse D.; Petronczki M.; Scharn D.; Bader G.; Baum A.; Bergner A.; Chong E.; Döbel S.; Egger G.; Engelhardt C.; Ettmayer P.; Fuchs J. E.; Gerstberger T.; Gonnella N.; Grimm A.; Grondal E.; Haddad N.; Hopfgartner B.; Kousek R.; Krawiec M.; Kriz M.; Lamarre L.; Leung J.; Mayer M.; Patel N. D.; Simov B. P.; Reeves J. T.; Schnitzer R.; Schrenk A.; Sharps B.; Solca F.; Stadtmüller H.; Tan Z.; Wunberg T.; Zoephel A.; McConnell D. B. Start Selective and Rigidify: The Discovery Path toward a Next Generation of EGFR Tyrosine Kinase Inhibitors. J. Med. Chem. 2019, 62 (22), 10272–10293. 10.1021/acs.jmedchem.9b01169. [DOI] [PubMed] [Google Scholar]
- Ma J.; Sanchez-Duffhues G.; Caradec J.; Benderitter P.; Hoflack J.; ten Dijke P. Development of Small Macrocyclic Kinase Inhibitors. Future Med. Chem. 2022, 14 (6), 389–391. 10.4155/fmc-2021-0342. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Fang R.; Rao Q. An Insight into the Medicinal Chemistry Perspective of Macrocyclic Derivatives with Antitumor Activity: A Systematic Review. Molecules 2022, 27 (9), 2837. 10.3390/molecules27092837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel J.; Sebald W.; Groppe J. C.; Mueller T. D. Intricacies of BMP Receptor Assembly. Cytokine Growth Factor Rev. 2009, 20 (5–6), 367–377. 10.1016/j.cytogfr.2009.10.022. [DOI] [PubMed] [Google Scholar]
- Fessel J. P.; Loyd J. E.; Austin E. D. The Genetics of Pulmonary Arterial Hypertension in the Post-BMPR2 Era. Pulm. Circ. 2011, 1 (3), 305–319. 10.4103/2045-8932.87293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikuad A.; Thangaratnarajah C.; von Delft F.; Bullock A. N. Structural Consequences of BMPR2 Kinase Domain Mutations Causing Pulmonary Arterial Hypertension. Sci. Rep. 2019, 9 (1), 18351. 10.1038/s41598-019-54830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andruska A.; Spiekerkoetter E. Consequences of BMPR2 Deficiency in the Pulmonary Vasculature and beyond: Contributions to Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19 (9), 2499. 10.3390/ijms19092499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig B. L.; Imamura T.; Okadome T.; Cox G. N.; Yamashita H.; Ten Dijke P.; Heldin C. H.; Miyazono K. Cloning and Characterization of a Human Type II Receptor for Bone Morphogenetic Proteins. Proc. Natl. Acad. Sci. U. S. A. 1995, 92 (17), 7632–7636. 10.1073/pnas.92.17.7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B.; Xu G.; Yu Y.; Lin J. The Role of TGF-β or BMPR2 Signaling Pathway-Related MiRNA in Pulmonary Arterial Hypertension and Systemic Sclerosis. Arthritis Res. Ther. 2021, 23 (1), 288. 10.1186/s13075-021-02678-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C. W.; Song H.; Kumar S.; Nam D.; Kwon H. S.; Chang K. H.; Son D. J.; Kang D. W.; Brodie S. A.; Weiss D.; Vega J. D.; Alberts-Grill N.; Griendling K.; Taylor W. R.; Jo H. Anti-Inflammatory and Antiatherogenic Role of Bmp Receptor Ii in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33 (6), 1350–1359. 10.1161/ATVBAHA.112.300287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton V. J.; Ciuclan L. I.; Holmes A. M.; Rodman D. M.; Walker C.; Budd D. C. Bone Morphogenetic Protein Receptor II Regulates Pulmonary Artery Endothelial Cell Barrier Function. Blood 2011, 117 (1), 333–341. 10.1182/blood-2010-05-285973. [DOI] [PubMed] [Google Scholar]
- Pousada G.; Lupo V.; Cástro-Sánchez S.; Álvarez-Satta M.; Sánchez-Monteagudo A.; Baloira A.; Espinós C.; Valverde Di. Molecular and Functional Characterization of the BMPR2 Gene in Pulmonary Arterial Hypertension. Sci. Rep. 2017, 7 (1), 1923. 10.1038/s41598-017-02074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derwall M.; Malhotra R.; Lai C. S.; Beppu Y.; Aikawa E.; Seehra J. S.; Zapol W. M.; Bloch K. D.; Yu P. B. Inhibition of Bone Morphogenetic Protein Signaling Reduces Vascular Calcification and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32 (3), 613–622. 10.1161/ATVBAHA.111.242594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiramongkolchai P.; Owens P.; Hong C. C. Emerging Roles of the Bone Morphogenetic Protein Pathway in Cancer: Potential Therapeutic Target for Kinase Inhibition. Biochem. Soc. Trans. 2016, 44 (4), 1117–1134. 10.1042/BST20160069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews L.; Adame A.; Patrick C.; DeLaney A.; Pham E.; Rockenstein E.; Hansen L.; Masliah E. Increased BMP6 Levels in the Brains of Alzheimer’s Disease Patients and APP Transgenic Mice Are Accompanied by Impaired Neurogenesis. J. Neurosci. 2010, 30 (37), 12252–12262. 10.1523/JNEUROSCI.1305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef H.; Morgenthaler A.; Schlesinger C.; Bugaj L.; Conboy I. M.; Schaffer D. V. Age-Associated Increase in BMP Signaling Inhibits Hippocampal Neurogenesis. Stem Cells 2015, 33 (5), 1577–1588. 10.1002/stem.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke T.; Wong J. F.; Berger B. T.; Abdi I.; Berger L. M.; Tesch R.; Tredup C.; Bullock A. N.; Müller S.; Knapp S. A Highly Selective Chemical Probe for Activin Receptor-like Kinases ALK4 and ALK5. ACS Chem. Biol. 2020, 15 (4), 862–870. 10.1021/acschembio.0c00076. [DOI] [PubMed] [Google Scholar]
- SGC, Chemical Probes, https://www.thesgc.org/chemical-probes.
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29 (11), 1046–1051. 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Engers D. W.; Frist A. Y.; Lindsley C. W.; Hong C. H.; Hopkins C. R. Synthesis and Structure-Activity Relationships of a Novel and Selective Bone Morphogenetic Protein Receptor (BMP) Inhibitor Derived from the Pyrazolo[1.5-a]Pyrimidine Scaffold of Dorsomorphin: The Discovery of ML347 as an ALK2 versus ALK3 Selective MLPCN. Bioorg. Med. Chem. Lett. 2013, 23 (11), 3248–3252. 10.1016/j.bmcl.2013.03.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modukuri R. K.; Monsivais D.; Li F.; Palaniappan M.; Bohren K. M.; Tan Z.; Ku A. F.; Wang Y.; Madasu C.; Li J. Y.; Tang S.; Miklossy G.; Palmer S. S.; Young D. W.; Matzuk M. M. Discovery of Highly Potent and BMPR2-Selective Kinase Inhibitors Using DNA-Encoded Chemical Library Screening. J. Med. Chem. 2023, 66, 2143–2160. 10.1021/acs.jmedchem.2c01886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statsuk A. V.; Maly D. J.; Seeliger M. A.; Fabian M. A.; Biggs W. H.; Lockhart D. J.; Zarrinkar P. P.; Kuriyan J.; Shokat K. M. Tuning a Three-Component Reaction for Trapping Kinase Substrate Complexes. J. Am. Chem. Soc. 2008, 130 (51), 17568–17574. 10.1021/ja807066f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couñago R. M.; Allerston C. K.; Savitsky P.; Azevedo H.; Godoi P. H.; Wells C. I.; Mascarello A.; de Souza Gama F. H.; Massirer K. B.; Zuercher W. J.; Guimarães C. R. W.; Gileadi O. Structural Characterization of Human Vaccinia-Related Kinases (VRK) Bound to Small-Molecule Inhibitors Identifies Different P-Loop Conformations. Sci. Rep. 2017, 7 (1), 7501. 10.1038/s41598-017-07755-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov O.; Niesen F. H.; Knapp S. Kinase Inhibitor Selectivity Profiling Using Differential Scanning Fluorimetry. Methods Mol. Biol. 2012, 795, 109–118. 10.1007/978-1-61779-337-0_7. [DOI] [PubMed] [Google Scholar]
- Adasme M. F.; Linnemann K. L.; Bolz S. N.; Kaiser F.; Salentin S.; Haupt V. J.; Schroeder M. PLIP 2021: Expanding the Scope of the Protein-Ligand Interaction Profiler to DNA and RNA. Nucleic Acids Res. 2021, 49 (W1), W530–W534. 10.1093/nar/gkab294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrhein J. A.; Berger L. M.; Tjaden A.; Krämer A.; Elson L.; Tolvanen T.; Martinez-Molina D.; Kaiser A.; Schubert-Zsilavecz M.; Müller S.; Knapp S.; Hanke T. Discovery of 3-Amino-1H-pyrazole-Based Kinase Inhibitors to Illuminate the Understudied PCTAIRE Family. Int. J. Mol. Sci. 2022, 23 (23), 14834. 10.3390/ijms232314834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.