

Abstract
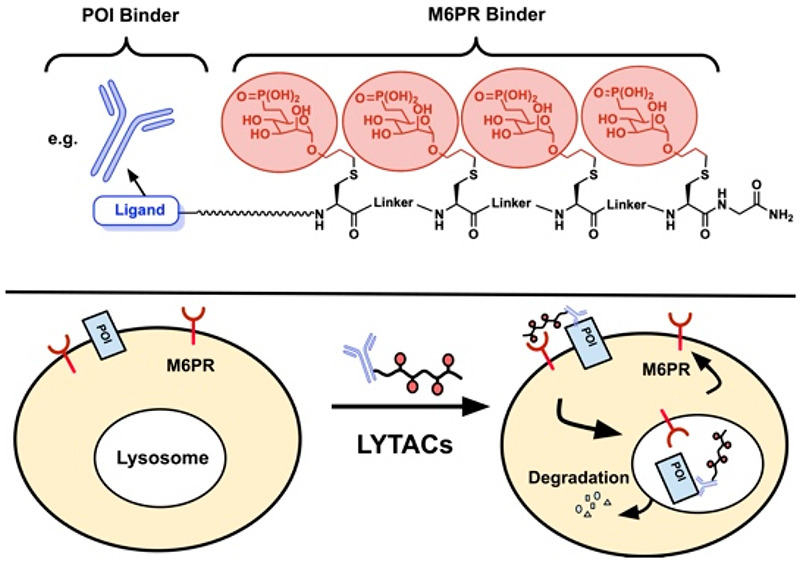
Lysosome targeting chimeras (LYTACs) are a new protein degradation strategy that has recently emerged. LYTACs utilize the native cell internalization process in the body to target and degrade therapeutically relevant extracellular proteins via the lysosomal pathways. The first lysosomal internalization receptor recently used for LYTACs is the mannose-6-phosphate receptor (M6PR). M6PR is expressed across most cell types, making it ideal for internalization and degradation of numerous extracellular proteins. Herein, we report the development of a series of structurally well-defined mannose-6-phosphonate (M6Pn)-peptide conjugates that are capable of linking to a variety of targeting ligands for proteins of interest and successfully internalizing and degrading those proteins through M6PR. This will greatly facilitate the development of M6Pn based LYTACs for therapeutic applications.
Keywords: Drug Discovery, Glycoconjugates, Lysosome, Mannose-6-Phosphate, Protein Degradation
The tagging of specific protein targets for degradation is a key area of chemistry and drug development that has become increasingly important as our knowledge of protein/ligand interactions improves. There are several routes available for targeted protein degradation, such as monofunctional small molecule inducers and heterobifunctional degraders.1 The latter, heterobifunctional degraders, are gaining more and more attention due to their ability to degrade a wide range of targets and their ability to degrade targets without requiring a binder that has functional activity with the protein of interest (POI). The primary utilization of this technique has so far been through PROTACs (Proteolysis Targeting Chimeras), wherein a ligand for E3 ligase is linked to a ligand for the POI. This molecule can then form a ternary complex with the POI and E3 ligase, which tags the POI for degradation via the ubiquitin-proteasome system. PROTACs have been widely used for the degradation of a large number of protein targets, such as the receptor-interacting protein kinase 2 (RIPK2),2 bromodomain-containing protein 4 (BRD4),3−5 and the estrogen and androgen receptors.6 However, PROTACs can only degrade cytoplasmic proteins or proteins with a binding domain inside the cell. About 40% of proteins in the proteome are outside the cells and many of them are associated with various diseases.7 It then becomes important to develop methods to efficaciously degrade those proteins that are beyond the reach of PROTACs and other degraders.
One such method has been found through lysosome targeting chimeras (LYTACs).8 LYTACs are heterobifunctional molecules that contain a ligand for the POI and a ligand for lysosome targeting receptor such as mannose-6-phosphate receptor (M6PR), which can complement existing strategies for intracellular protein degradation. LYTACs can potentially expand the range of targetable POIs to numerous extracellular proteins including secreted and cell membrane associated proteins. There are two main carbohydrate ligand/receptor interactions that have been used for LYTAC design: the asialoglycoprotein receptor (ASGPR) found in liver cells, which can be used to target and degrade circulating proteins and membrane proteins on the liver; and M6PR present throughout the body, allowing for the targeting and degradation of cell membrane associated proteins on different tissues. The binding of ASGPR to its ligand GalNAc has been well documented for drug delivery9 and was recently applied to the development of degraders by us10 and others11,12 with great success. M6PR and its ligand, mannose-6-phosphate (M6P), are not as well understood, which has led to many different approaches in designing general conjugates for drug delivery to utilize this interaction, with varying degrees of success.13−16 While it is known that multivalency is important in the M6P/M6PR binding relationship, the nature of that multivalent requirement is not specified, especially for LYTAC design. Further probing the M6P/M6PR interaction in a controlled manner is necessary to fully utilize it for targeted degradation.
Recently, M6P/M6PR was successfully applied in LYTACs by Bertozzi’s group, where a glycopolypeptide incorporating 20–90 mannose-6-phosphonates (M6Pn) was synthesized and then attached to an antibody for a POI.8 While this serves as an elegant example for the first application of M6P/M6PR in LYTAC development, the reported LYTACs employed large multivalences in M6Pn without well-defined structures, which are challenging for quality control in the drug development process. We herein report an improved M6PR-recruiting LYTAC design, which uses structurally well-defined M6PR ligands and simplifies the development and production process. Our new LYTAC platform contains a short peptide backbone decorated with several M6Pn units separated by various linkers for efficient uptake into the cell via the M6PR (Figure 1). The modular nature of the peptide backbone and the coupling chemistry used for conjugating the M6Pn to the peptide backbone allows for systematic investigation of the distance between M6Pn units, as well as the multivalency of M6Pn, giving further insights into the requirements of M6P/M6PR-mediated internalization and subsequent degradation. The synthetic route streamlines those previously explored for M6PR based lysosome targeting degraders and provides a more controlled and specific targeting of the M6PR that retains efficient uptake and degradation. During our studies, Wang’s group reported an elegant alternative for structurally well-defined M6PR ligands by employing a site-specific chemoenzymatic method to attach high affinity M6P glycan ligands to antibodies for targeted protein degradation.17
Figure 1.

Overview of M6Pn based LYTAC. The design is composed of several M6Pn units conjugated to a peptide backbone and separated by different linkers. This backbone can also be linked to a variety of ligands for target proteins.
In order to prepare multivalent ligands that are capable of both effectively targeting the M6PR and conjugating to the peptide backbone of the LYTAC, several considerations must be made. First, the natural 6-phosphoester of M6P has been shown to undergo hydrolysis in human serum,18 which would destroy the M6PR targeting ability of the conjugates in vivo, and potentially lead to unwanted binding to the mannose receptor on macrophages.19 This would require implementing some other connections to the phosphate, such as M6Pn with a stable carbon linker on the 6-position.20 Second, to avoid the tedious process of creating a protected M6Pn-conjugated amino acid building block that would be compatible with the Fmoc solid phase peptide synthesis used to create the peptide backbone, the M6Pn would need to be modified in some way to allow easy conjugation to the peptide backbone.
To address the first consideration, we elected to follow the previous LYTAC from Bertozzi’s group and install a phosphonate moiety in place of the native phosphate.8 The attachment of carbohydrates to the peptide scaffold is not an easy task due to the diverse functional groups in carbohydrates and peptides. This is the bottleneck in the recent syntheses of oligomeric M6P for drug delivery15 and M6Pn for LYTACs.8 After surveying a variety of options, we decided to address the second consideration using a thiol–ene reaction between a terminal alkene on the anomeric position of the M6Pn and a free thiol on the cysteine of the peptide backbone. The thiol–ene reaction can be carried out under mild conditions that are compatible with all functional groups we have. This reaction provides high yields and high reaction rates,22 with the added benefit of not needing any protection or manipulation of the peptide backbone side chains or the M6Pn. In addition, the resulting products can be used for cellular uptake studies without further manipulation, a strategy that has been used by us,23,24 and others,25 to evaluate linker effects for PROTACs.
The fully modified M6Pn 6 was prepared in 11 steps that only involved five column chromatography purifications from a penta-acetylated mannose starting material (Scheme 1A). The first 6 steps of the synthesis follow those previously reported by Bertozzi to achieve the phosphonate modified intermediate 1.8 From there, a hydrogenation reaction was performed to reduce the double bond, as well as to remove the benzyl group and TMS groups to yield intermediate 2. Next, the unprotected hydroxy groups were acetylated to give compound 3, which was then reacted with allyl alcohol to yield glycoside 4. The ethoxy protecting groups on the phosphonate were then removed to give precursor 5, and the acyl groups were removed to give the final M6Pn 6. The allyl conjugated mannose-6-phosphonate was produced with an overall yield of ∼10% from commercially available starting material.
Scheme 1. Synthesis and Incorporation of a Mannose-6-phosphonate (M6Pn) onto a Peptide Backbone. A) Synthesis of M6Pn with Anomeric Alkene Group: a) Pd–C, H2, MeOH/AcOH, 73%; b) Pyr, Ac2O, DMAP, 93%; c) Ally Alcohol, BF3*OEt2, 0°C → 25°C, 54%; d) Pyr, TMSBr, 90%; e) 0.5 M NaOMe in MeOH, Quantitative. B) Incorporation of M6Pn to Peptide Backbone via the Thiol–ene Reaction. For Each Cysteine Residue on the Backbone, 2 equiv of M6Pn Were Used. DPAP Served as the Radical Initiator, and UV at 365 nm Was Used to Activate.
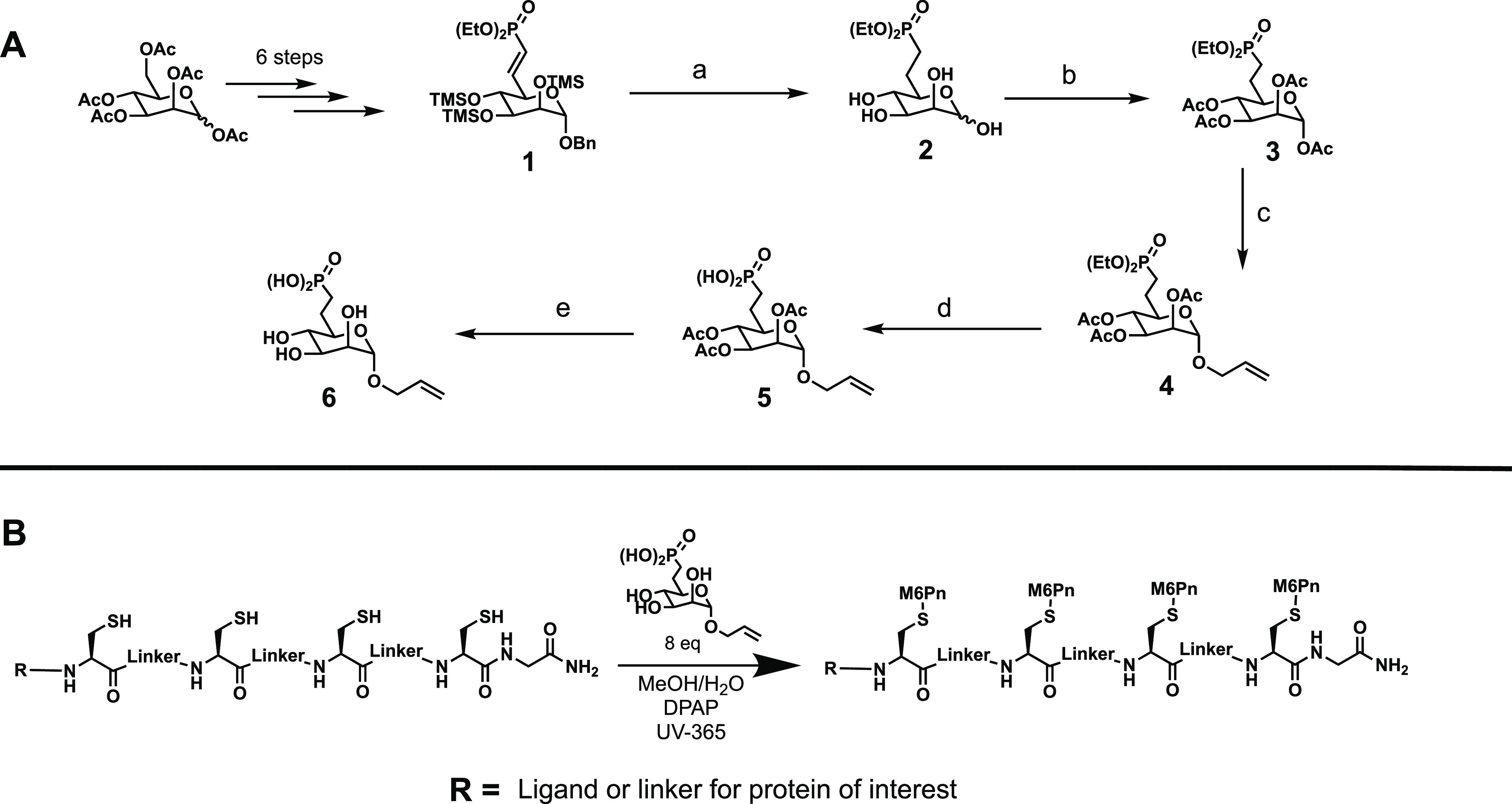
Next, conjugation of the M6Pn to the peptide backbone was achieved via the thiol–ene reaction (Scheme 1B). The peptide backbones were produced first via standard Fmoc solid phase peptide synthesis, then cleaved from the solid support resin followed by deprotection. A number of these peptides were synthesized with different structures for testing uptake and degradation. The deprotected peptides were then combined with the M6Pn in a mixed solvent of water and methanol, and reacted with the photoinitiator 2,2-dimethoxy-2-phenylacetophenone under an ultraviolet lamp set to 365 nm.26 It was found that this reaction proceeded quickly and in near quantitative yields as long as the reaction is kept under inert atmosphere with a proper mixture of water and methanol. Fine-tuning of specific conditions was needed for each peptide backbone to achieve high conversion. After lyophilization, the biotinylated M6Pn-peptide conjugates were found by LCMS (liquid chromatography mass spectrometry) analysis to have high purities, and could be used directly for cellular uptake of a model protein target. Our strategy provides an efficacious method for creating controlled and specifically designed M6Pn-peptides that are capable of linking to a variety of targeting ligands for different POIs after replacing the biotin with an azide, which can couple with alkyne-labeled antibodies or other types of binders.
A series of M6Pn-peptide conjugates were created to test the uptake requirements of the M6PR. Based on the previously reported M6Pn-based ligands for drug delivery through M6PR, adequate binding can be achieved with a multimeric display of around three M6Pn units.15 In order to test the multivalent requirements in binding M6PR for LYTACs, five initial M6Pn-peptide conjugates were prepared with varying multivalency (Figure 2A). Conjugate 7 consisted of a monomeric M6Pn presenting ligand, conjugate 8 consisted of a dimeric ligand, conjugates 9 and 10 consisted of trimeric ligands, and conjugate 11 consisted of a tetrameric ligand. In order to test for uptake, each of the five conjugates contained a terminal biotin moiety that can bind to a fluorescent neutravidin (NA-650), a model POI. To test the uptake of NA-650, huh7 (human hepatoma-derived 7) cells were incubated with NA-650 and 10 uM of M6Pn-biotin conjugate for 24 h. The uptake of NA-650 was then measured by examining the fluorescent intensity via plate reader. It was found that the monomeric and dimeric M6Pn-biotin conjugates 7 and 8 were unable to mediate any uptake, and the trimeric conjugates 9 and 10 offered minimal uptake when compared to the tetrameric conjugate 11 (Figure 2B). This supports the idea of specific multivalent requirements for efficient uptake via the M6PR using M6Pn derivatives.
Figure 2.

Initial M6Pn-conjugate uptake studies using biotin/neutravidin assay. A) Structure of conjugates 7 through 11, ranging from monomeric to tetrameric. All conjugates contain a terminal biotin moiety. B) Fold change in mean fluorescent intensity (MFI) data from uptake studies of conjugates 7 through 11. Huh7 cells were incubated with 500 nM of neutravidin-650 (NA-650) and 10 uM of M6Pn-biotin at 37 °C for 24 h. The cells were then washed with PBS, and the uptake was determined by measuring the fluorescent intensity at 650 nm excitation/680 nm emission. Data is shown in relative fluorescent units.
It is interesting to note, however, the large increase in uptake efficacy when going from the trimeric conjugates (9 and 10) to the tetrameric conjugate (11), when compared to the lesser increase when moving from dimeric to trimeric (Figure 2B). It is possible that this preference of the M6PR for tetrameric M6Pn ligands can be explained using known structure data on the receptor. The M6PR has been found to contain four total binding sites for M6P, two of which are considered high affinity binding sites for M6P that contain essential residues for binding.27−29 In addition, several reports indicate that M6PR forms receptor dimers as part of the internalization process.30−33 Together, these pieces of information indicated that an effective ligand would consist of a tetrameric M6Pn conjugate composed of two sets of M6Pn dimers separated by a linker, which would be able to bind the two high affinity M6P binding sites on two different M6PR monomers, leading to M6PR dimerization and internalization into the cell. The large increase in uptake efficacy supports this hypothesis and is consistent with previous reports of M6PR dimerization as a required event for internalization, though more evidence that is beyond the scope of this study will be necessary to validate this mechanism. In addition to providing useful structure activity relationship (SAR) data, these results also show that a streamlined and specifically designed M6Pn based LYTAC is capable of recruiting an extracellular protein and bringing it into the cell via the M6PR.
With the results from the NA-650 uptake experiments in hand, conjugate 11 was chosen to test the ability of the designed M6Pn-LYTAC to degrade a target POI by replacing the biotin motif with a binder of POI. Epidermal growth factor receptor (EGFR), which is overexpressed in many tumors,34−37 was chosen as the POI to test the M6Pn-LYTACs. First, the terminal biotin in 11 moiety was replaced with an azido group to create conjugate 12 (Figure 3A). The anti-EGFR antibody Cetuximab (Ctx) was then reacted with commercially available DBCO-NHS activated ester (dibenzo cyclooctyne-N-hydroxysuccinimidyl ester), which contains a strained alkyne group, to create Ctx-DBCO (Figure 3B). The Ctx-DBCO was then reacted with conjugate 12 through a copper free click reaction to yield the antibody conjugated LYTAC, Ctx-DBCO-12. MALDI analysis revealed that each Ctx was labeled with ∼2 ligand 12 on average (Figure S1). This full conjugate was then incubated at various concentrations with HeLa cells expressing EGFR for 24 h to establish the dose-dependent degradation by Western blot analysis (Figure 3C).
Figure 3.

Design, synthesis, and degradation study of an antibody conjugated M6Pn LYTAC. A) Structure of conjugate 12, similar to that of conjugate 11 but with the terminal biotin moiety replaced by an azido group. This modification was made during SPPS of the peptide backbone. B) Linkage of conjugate 12 to the EGFR antibody Cetuximab (Ctx). First, Ctx was mixed with DBCO-NHS at a molar ratio of 1:25 and incubated overnight to produce Ctx-DBCO. Next, the Ctx-DBCO was mixed with 25 equiv of conjugate 12 and incubated overnight to yield Ctx-DBCO-12, which was characterized by MALDI-TOF-MS. C) Dose response study of Ctx-DBCO-12 on Hela cells with EGFR. Ctx-DBCO-12 in concentrations of 0, 0.1, 1, 10, and 100 nM were incubated with Hela cells for 24 h. Next, the cells were lysed and EGFR degradation was measured using Western blot analysis. D) Time course study of Ctx-DBCO-12 on Hela cells with EGFR. Hela cells were incubated with 10 nM of Ctx-DBCO-12, and samples were collected at various time points and analyzed by Western blot to determine EGFR degradation.
It was found that Ctx-DBCO-12 was able to induce degradation with as low as 0.1 nM concentration, with the maximum EGFR degradation achieved at 100 nM with ∼40% degradation. Following the dose–response, a time-course study was done to determine the degradation effect over time (Figure 3D). The study was done using 10 nM of Ctx-DBCO-12, and showed that significant levels of degradation were achieved as early as 2–4 h, and the effect lasted through the last time point taken at 48 h. This suggests that treatment with the M6Pn based LYTAC may offer a sustained level of protein degradation. Overall, the degradation studies show that our specifically designed M6Pn-LYTAC is capable of targeting and degrading a cell membrane associated protein.
Following the successful degradation of EGFR using Ctx-DBCO-12, we decided to expand the variety of linkers and multivalency of the conjugates for further improvements. A second series of conjugates were prepared with a terminal biotin moiety for testing uptake using NA-650 as the model target (Figure 4A). These conjugates, 13 through 24, were designed to expand the scope and variety of conjugate structures. Conjugates 13 through 23 are all tetrameric presenting M6Pn conjugates which follow the outlined design of two sets of M6Pn dimers separated by a linker (linker A). The linker A is composed of an amino acid with increasing degrees of separation, ranging from one atom separation to seven atom separation, and varying in composition. Each tetrameric conjugate also contains a second linker (linker B) which separates the M6Pn of the dimers. These were varied as well in length and composition. Conjugates 13 through 19 have different linker A in terms of composition and length while keeping linker B as a single lysine residue. Conjugates 20 through 23 have the same C3 linker A used in conjugate 11, but different linker B. In addition to lysine in each linker B, an additional lysine, glycine, serine, or phenylalanine was incorporated to change the length and composition. Lastly, conjugate 24 expanded the multivalency to a hexameric presenting M6Pn conjugate to further test the effects of multivalency on uptake efficacy. These 12 conjugates were tested for cellular uptake of NA-650, and compared to conjugate 11 (Figure 4B).
Figure 4.

Additional M6Pn-conjugate uptake studies using the biotin/neutravidin system. A) Structure of conjugates 13 through 24. Conjugates 13 through 23 use the general tetrameric structure shown above the table, modifying both linker A and linker B for structure and composition. Conjugate 24 changes the multivalency to a hexameric structure. All conjugates contain a terminal biotin moiety. B) Fold change in MFI data from uptake studies of conjugates 17 through 24. Huh7 cells were incubated with 500 nM of neutravidin-650 (NA-650) and 10 uM of M6Pn-biotin at 37 °C for 24 h. The cells were then washed with PBS, and the uptake was determined by measuring the fluorescent intensity at 650 nm excitation/680 nm emission. Data is shown in relative fluorescent units.
While conjugates 13 through 16, 18, 22, and 23 all performed similarly to conjugate 11, there was more significant difference in the performance of the other conjugates (Figure 4B). Compared to 11, conjugates 20 and 21 showed obvious less uptake. This is interesting, as the other two conjugates with different linker B, conjugates 22 and 23, had similar uptake as 11. It is possible that the composition of the linker as opposed to linker length matters more, as 20 contains two charged lysines, while 22 and 23 contain a lysine and an uncharged residue. Conjugate 21 is a more unique case, as it contains glycine, which would possibly add more flexibility to the conjugate backbone. Conjugate 15 also contained glycine residues, this time for linker A, but showed effective uptake. It is possible that for these M6Pn-LYTACs, flexibility is desirable between the two sets of dimers (in linker A), but less desirable between the M6Pns that target the binding domains on one M6PR (in linker B). In addition to the effect of linker B, conjugates 17 and 19 had increased uptake compared to 11 (Figure 4B). These conjugates contain either a C6 or a C7 linker respectively, and while they are not largely different in terms of linker length compared to the other conjugates (e.g., 18), their composition of only carbon may offer advantages that lead to more efficacious uptake.
Of particular note is 24, the hexameric M6Pn conjugate. This conjugate showed the most efficacious uptake of all those tested; however, it did not offer the same increase in uptake over the best performing tetrameric conjugate, 19, compared to the increase in uptake from the trimeric conjugate to the tetrameric. Conjugate 19, the most efficacious tetrameric conjugate, offered a 3.5-fold increase in uptake over the trimeric conjugate 10, while the hexameric conjugate 24 only offered a 1.5-fold increase in uptake compared to 19. While the hexameric conjugate did show the highest uptake, it takes more steps to prepare the peptide backbone and more M6Pn units for the thiol–ene coupling. This suggests that tetrameric conjugates offer the greatest balance of preparation and uptake efficacy in the performed study.
As previously mentioned, all of the conjugates tested (7 through 24) were not purified following the thiol–ene reaction to conjugate the M6Pn to the peptide backbone. While this reaction is highly efficient, it does involve an excess of the M6Pn 6. To test the effect this excess reagent had on the uptake data shown in Figure 2B and 4B, the most representative conjugates 7, 8, 9, 19, and 24 were purified via HPLC and tested for uptake of the NA-650 in comparison to the corresponding nonpurified conjugates (Figure S2). This represents the best performing monomeric, dimeric, trimeric, tetrameric, and hexameric conjugates. For all conjugates except 24, the pure and nonpure had very similar uptake efficiency. For conjugate 24, the pure compound had noticeable higher uptake than the crude, which is in line with previous findings.8 We can conclude that the impact of the excess M6Pn presented in the system after the thiol–ene reaction on the uptake in the NA-650 assay is minimal and the overall SAR trend remains the same following purification.
Herein, we have shown the synthesis and successful uptake and degradation of a specifically designed M6Pn-LYTAC. This LYTAC builds on those previously reported, with a simplified synthetic pathway due to the efficient thiol–ene coupling chemistry and a well-defined modular structure. These M6Pn-peptide conjugates allow for the incorporation of a variety of ligands or linkers for targeted POIs. Showcased here, the conjugates were able to be linked to a small molecule ligand, biotin, for binding a fluorescent neutravidin protein for studying uptake efficacy. In addition, the conjugates could be designed to contain an azido moiety for click-chemistry linkage to an antibody ligand, cetuximab, for EGFR. This cetuximab-M6Pn conjugate was able to successfully internalize and degrade EGFR. While both the neutravidin and EGFR served as test systems for the LYTACs, they could readily be adapted for linking to any variety of small molecule, peptide, or antibody ligands in relevant therapeutic applications. While this degrader (Ctx-DBCO-12) had decreased degradation efficacy compared to the first generation M6Pn based LYTACs,8 the advances in being able to use well-defined ligands with the flexibility for further improvement should increase its utility for the community.
In addition to showing the degradation capabilities of the designed M6Pn-LYTACs, the uptake studies enabled the further investigation of the M6PR recruiting requirements. In total, 17 different M6Pn-peptide conjugates were synthesized and tested for uptake efficacy. These included conjugates that tested the multivalency requirements for recruiting M6PR, as well as conjugates that tested the effect of size and composition of the peptide backbone on uptake efficacy. We found that tetrameric M6Pn-conjugates offered the greatest balance of synthetic simplicity and uptake efficacy. We also found that while conjugate length did not seem to be a significant factor in uptake, composition may have an impact. Further studies will continue to expand the variety of conjugate structures to further elucidate the M6PR recruiting requirements for an effective M6Pn-LYTAC, as well as utilize the existing conjugate designs to target and degrade therapeutically relevant proteins with different ligand types.
Acknowledgments
Support for this research was provided by the University of Wisconsin – Madison Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation through a UW2020 award (before July 1st, 2022) and National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM148266. C.M.S. thanks fellowship provided by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number T32HL007936. This study made use of the Medicinal Chemistry Center at UW-Madison instrumentation, supported by the Lachman Institute for Pharmaceutical Development, University of Wisconsin School of Pharmacy and the University of Wisconsin Carbone Cancer Center Support Grant NIH P30 CA014520.
Glossary
Abbreviations
- LYTAC
lysosome targeting chimera
- M6PR
mannose-6-phosphate receptor
- M6Pn
mannose-6-phosphonate
- POI
protein of interest
- PROTAC
proteolysis targeting chimera
- RIPK2
receptor-interacting protein kinase 2
- BRD4
bromodomain-containing protein 4
- ASGPR
asialoglycoprotein receptor
- M6P
mannose-6-phosphate
- LCMS
liquid chromatography mass spectrometry
- NA-650
fluorescent streptavidin
- Huh7
human hepatoma derived 7 cells
- MFI
mean fluorescent intensity
- SAR
structure activity relationship
- Ctx
cetuximab
- EGFR
epidermal growth factor receptor
- DBCO-NHS
dibenzo cyclooctyne-N-hydroxysuccinimidyl ester
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00479.
Supplemental figures detailing full LYTAC MALDI analysis and uptake comparisons of crude and pure conjugates; Additional experimental details; Copies of 1H NMR data; Copies of 13C{1H}NMR data; Copies of HRMS data; Copies of HPLC data (PDF)
Author Contributions
§ (C.M.S., Y.Z., P.T.) These authors contributed equally.
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Medicinal Chemistry Letters virtual special issue “New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science”.
Supplementary Material
References
- Valeur E.; Narjes F.; Ottmann C.; Plowright A. T. Emerging Modes-of-Action in Drug Discovery. MedChemComm 2019, 10 (9), 1550–1568. 10.1039/C9MD00263D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 2015, 11 (8), 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348 (6241), 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22 (6), 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K. H.; Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10 (8), 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K. M.; Kim K. B.; Verma R.; Ransick A.; Stein B.; Crews C. M.; Deshaies R. J. Development of Protacs to Target Cancer-Promoting Proteins for Ubiquitination and Degradation. Mol. Cell Proteomics 2003, 2 (12), 1350–1358. 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- Uhlén M.; Fagerberg L.; Hallström B. M.; Lindskog C.; Oksvold P.; Mardinoglu A.; Sivertsson A.; Kampf C.; Sjöstedt E.; Asplund A.; Olsson I. M.; Edlund K.; Lundberg E.; Navani S.; Szigyarto C. A. K.; Odeberg J.; Djureinovic D.; Takanen J. O.; Hober S.; Alm T.; Edqvist P. H.; Berling H.; Tegel H.; Mulder J.; Rockberg J.; Nilsson P.; Schwenk J. M.; Hamsten M.; von Feilitzen K.; Forsberg M.; Persson L.; Johansson F.; Zwahlen M.; von Heijne G.; Nielsen J.; Pontén F. Tissue-Based Map of the Human Proteome. Science 2015, 347 (6220), 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Banik S. M.; Pedram K.; Wisnovsky S.; Ahn G.; Riley N. M.; Bertozzi C. R. Lysosome-Targeting Chimaeras for Degradation of Extracellular Proteins. Nature 2020, 584 (7820), 291–297. 10.1038/s41586-020-2545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Leroux J. C.; Castagner B. Well-Defined Multivalent Ligands for Hepatocytes Targeting via Asialoglycoprotein Receptor. Bioconjug Chem. 2017, 28 (2), 283–295. 10.1021/acs.bioconjchem.6b00651. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Teng P.; Montgomery N. T.; Li X.; Tang W. Development of Triantennary N-Acetylgalactosamine Conjugates as Degraders for Extracellular Proteins. ACS Cent Sci. 2021, 7 (3), 499–506. 10.1021/acscentsci.1c00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn G.; Banik S. M.; Miller C. L.; Riley N. M.; Cochran J. R.; Bertozzi C. R. LYTACs That Engage the Asialoglycoprotein Receptor for Targeted Protein Degradation. Nat. Chem. Biol. 2021, 17 (9), 937–946. 10.1038/s41589-021-00770-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caianiello D. F.; Zhang M.; Ray J. D.; Howell R. A.; Swartzel J. C.; Branham E. M. J.; Chirkin E.; Sabbasani V. R.; Gong A. Z.; McDonald D. M.; Muthusamy V.; Spiegel D. A. Bifunctional Small Molecules That Mediate the Degradation of Extracellular Proteins. Nat. Chem. Biol. 2021, 17 (9), 947–953. 10.1038/s41589-021-00851-1. [DOI] [PubMed] [Google Scholar]
- Hoogendoorn S.; van Puijvelde G. H. M.; Kuiper J.; van der Marel G. A.; Overkleeft H. S. A Multivalent Ligand for the Mannose-6-Phosphate Receptor for Endolysosomal Targeting of an Activity-Based Probe. Angew. Chem. Int. Ed 2014, 53 (41), 10975–10978. 10.1002/anie.201406842. [DOI] [PubMed] [Google Scholar]
- Ali L. M. A.; Simon M.; el Cheikh K.; Aguesseau-Kondrotas J.; Godefroy A.; Nguyen C.; Garcia M.; Morère A.; Gary-Bobo M.; Maillard L. Topological Requirements for CI-M6PR-Mediated Cell Uptake. Bioconjug Chem. 2019, 30 (10), 2533–2538. 10.1021/acs.bioconjchem.9b00590. [DOI] [PubMed] [Google Scholar]
- Hyun J. Y.; Kim S.; Lee H. S.; Shin I. A Glycoengineered Enzyme with Multiple Mannose-6-Phosphates Is Internalized into Diseased Cells to Restore Its Activity in Lysosomes. Cell Chem. Biol. 2018, 25 (10), 1255–1267. 10.1016/j.chembiol.2018.07.011. [DOI] [PubMed] [Google Scholar]
- Reintjens N. R. M.; Tondini E.; Vis C.; McGlinn T.; Meeuwenoord N. J.; Hogervorst T. P.; Overkleeft H. S.; Filippov D. v.; van der Marel G. A.; Ossendorp F.; Codée J. D. C. Multivalent, Stabilized Mannose-6-Phosphates for the Targeted Delivery of Toll-Like Receptor Ligands and Peptide Antigens. ChemBioChem. 2021, 22 (2), 434–440. 10.1002/cbic.202000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Liu H.; He J.; Ou C.; Donahue T. C.; Muthana M. M.; Su L.; Wang L. X. Site-Specific Chemoenzymatic Conjugation of High-Affinity M6P Glycan Ligands to Antibodies for Targeted Protein Degradation. ACS Chem. Biol. 2022, 17 (11), 3013–3023. 10.1021/acschembio.1c00751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanjean A.; Garcia M.; Leydet A.; Montero J. L.; Morère A. Synthesis and Receptor Binding Affinity of Carboxylate Analogues of the Mannose 6-Phosphate Recognition Marker. Bioorg. Med. Chem. 2006, 14 (10), 3575–3582. 10.1016/j.bmc.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Sly W. S.; Vogler C.; Grubb J. H.; Levy B.; Galvin N.; Tan Y.; Nishioka T.; Tomatsu S. Enzyme Therapy in Mannose Receptor-Null Mucopolysaccharidosis VII Mice Defines Roles for the Mannose 6-Phosphate and Mannose Receptors. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (41), 15172–15177. 10.1073/pnas.0607053103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gary-Bobo M.; Nirde P.; Jeanjean A.; Morere A.; Garcia M. Mannose 6-Phosphate Receptor Targeting and Its Applications in Human Diseases. Curr. Med. Chem. 2007, 14 (28), 2945–2953. 10.2174/092986707782794005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe A. B. Thiol-Ene “Click” Reactions and Recent Applications in Polymer and Materials Synthesis. Polym. Chem. 2010, 1 (1), 17–36. 10.1039/B9PY00216B. [DOI] [Google Scholar]
- Guo L.; Zhou Y.; Nie X.; Zhang Z.; Zhang Z.; Li C.; Wang T.; Tang W. A Platform for the Rapid Synthesis of Proteolysis Targeting Chimeras (Rapid-TAC) under Miniaturized Conditions. Eur. J. Med. Chem. 2022, 236, 114317. 10.1016/j.ejmech.2022.114317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts B. L.; Ma Z. X.; Gao A.; Leisten E. D.; Yin D.; Xu W.; Tang W. Two-Stage Strategy for Development of Proteolysis Targeting Chimeras and Its Application for Estrogen Receptor Degraders. ACS Chem. Biol. 2020, 15 (6), 1487–1496. 10.1021/acschembio.0c00140. [DOI] [PubMed] [Google Scholar]
- Hendrick C. E.; Jorgensen J. R.; Chaudhry C.; Strambeanu I. I.; Brazeau J. F.; Schiffer J.; Shi Z.; Venable J. D.; Wolkenberg S. E. Direct-to-Biology Accelerates PROTAC Synthesis and the Evaluation of Linker Effects on Permeability and Degradation. ACS Med. Chem. Lett. 2022, 13 (7), 1182–1190. 10.1021/acsmedchemlett.2c00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd N.; Vijayakrishnan B.; Koeppe J. R.; Davis B. G. Thiyl Glycosylation of Olefinic Proteins: S-Linked Glycoconjugate Synthesis. Angew. Chem. Int. Ed 2009, 48 (42), 7798–7802. 10.1002/anie.200903135. [DOI] [PubMed] [Google Scholar]
- Olson L. J.; Castonguay A. C.; Lasanajak Y.; Peterson F. C.; Cummings R. D.; Smith D. F.; Dahms N. M. Identification of a Fourth Mannose 6-Phosphate Binding Site in the Cation-Independent Mannose 6-Phosphate Receptor. Glycobiology 2015, 25 (6), 591. 10.1093/glycob/cwv001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson L. J.; Misra S. K.; Ishihara M.; Battaile K. P.; Grant O. C.; Sood A.; Woods R. J.; Kim J. J. P.; Tiemeyer M.; Ren G.; Sharp J. S.; Dahms N. M. Allosteric Regulation of Lysosomal Enzyme Recognition by the Cation-Independent Mannose 6-Phosphate Receptor. Commun. Biol. 2020, 3 (1), 1–15. 10.1038/s42003-020-01211-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong P. Y.; Kornfeld S. Ligand Interactions of the Cation-Dependent Mannose 6-Phosphate Receptor: Comparison with the Cation-Independent Mannose 6-Phosphate Receptor. J. Biol. Chem. 1989, 264 (14), 7970–7975. 10.1016/S0021-9258(18)83137-4. [DOI] [PubMed] [Google Scholar]
- Byrd J. C.; Park J. H. Y.; Schaffer B. S.; Garmroudi F.; MacDonald R. G. Dimerization of the Insulin-like Growth Factor II/Mannose 6-Phosphate Receptor. J. Biol. Chem. 2000, 275 (25), 18647–18656. 10.1074/jbc.M001273200. [DOI] [PubMed] [Google Scholar]
- Kreiling J. L.; Byrd J. C.; MacDonald R. G. Domain Interactions of the Mannose 6-Phosphate/Insulin-like Growth Factor II Receptor. J. Biol. Chem. 2005, 280 (22), 21067–21077. 10.1074/jbc.M412971200. [DOI] [PubMed] [Google Scholar]
- York S. J.; Arneson L. S.; Gregory W. T.; Dahms N. M.; Kornfeld S. The Rate of Internalization of the Mannose 6-Phosphate/Insulin-like Growth Factor II Receptor Is Enhanced by Multivalent Ligand Binding. J. Biol. Chem. 1999, 274 (2), 1164–1171. 10.1074/jbc.274.2.1164. [DOI] [PubMed] [Google Scholar]
- Ali L. M. A.; Simon M.; el Cheikh K.; Aguesseau-Kondrotas J.; Godefroy A.; Nguyen C.; Garcia M.; Morère A.; Gary-Bobo M.; Maillard L. Topological Requirements for CI-M6PR-Mediated Cell Uptake. Bioconjug Chem. 2019, 30 (10), 2533–2538. 10.1021/acs.bioconjchem.9b00590. [DOI] [PubMed] [Google Scholar]
- Pabla B.; Bissonnette M.; Konda V. J. Colon Cancer and the Epidermal Growth Factor Receptor: Current Treatment Paradigms, the Importance of Diet, and the Role of Chemoprevention. World J. Clin Oncol 2015, 6 (5), 133–141. 10.5306/wjco.v6.i5.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M.; Zouhair A.; Azria D.; Ozsahin M. The Epidermal Growth Factor Receptor (EGFR) in Head and Neck Cancer: Its Role and Treatment Implications. Radiat Oncol 2006, 1, 11. 10.1186/1748-717X-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell R.; Felip E.; Garcia-Campelo R.; Balaña C. The Biology of Non-Small-Cell Lung Cancer: Identifying New Targets for Rational Therapy. Lung Cancer 2004, 46 (2), 135–148. 10.1016/j.lungcan.2004.04.031. [DOI] [PubMed] [Google Scholar]
- Hashmi A. A.; Naz S.; Hashmi S. K.; Irfan M.; Hussain Z. F.; Khan E. Y.; Asif H.; Faridi N. Epidermal Growth Factor Receptor (EGFR) Overexpression in Triple-Negative Breast Cancer: Association with Clinicopathologic Features and Prognostic Parameters. Surg Exp Pathol 2019, 2 (1), 6. 10.1186/s42047-018-0029-0. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.