

Abstract

The role of peroxisome proliferator-activated receptor alpha (PPARα) in retinal biology is clarifying, and evidence demonstrates that novel PPARα agonists hold promising therapeutic utility for diseases like diabetic retinopathy and age-related macular degeneration. Herein, we disclose the design and initial structure–activity relationships for a new biaryl aniline PPARα agonistic chemotype. Notably, this series exhibits subtype selectivity for PPARα over other isoforms, a phenomenon postulated to be due to the unique benzoic acid headgroup. This biphenyl aniline series is sensitive to B-ring functionalization but allows isosteric replacement, and provides an opportunity for C-ring extension. From this series, 3g, 6j, and 6d were identified as leads with <90 nM potency in a cell-based luciferase assay cell and exhibited efficacy in various disease-relevant cell contexts, thereby setting the stage for further characterization in more advanced in vitro and in vivo models.
Keywords: Diabetic Retinopathy, PPARα, Structure−Activity Relationships, Retina
Diabetic retinopathy (DR) is a common microvascular complication of diabetes and the leading cause of blindness in the working-age population.1 Although several treatment options are available,2 addressing the complex nature of DR remains a significant challenge.3 Direct intraocular injection of anti-VEGF antibodies has emerged as the standard of care; however, this treatment suffers from the requirement of recurrent injections, and ∼40% of patients fail to respond to this therapy.4 A large part of unresponsiveness is likely due to disease heterogeneity, with other pathological features of DR (e.g., inflammation, fibrosis, ischemia, and vascular leakage) being more influential in certain patient subsets, rather than VEGF-mediated processes (e.g., neovascularization).5 New therapies that are superior or complementary to current approaches would be valuable to patients, especially for those that remain refractory to anti-VEGF options. Ideally, unlike current treatment choices, new approaches would be noninvasive (to the eye), address other pathological drivers, and be noncontingent on specialized facilities.
One target that is gaining popularity for DR is peroxisome proliferator-activated receptor alpha (PPARα). PPARα is a ligand-activated transcription factor with known endogenous (e.g., fatty acids) and synthetic ligands.6 Two additional PPAR members (PPARγ and PPARβ/δ) exist, and each PPAR subtype exhibits a unique tissue distribution, maintains diverse functions, and can be selectively targeted.7 Specifically, it has been shown that PPARα is expressed at high levels in all types of retinal cells and plays fundamental roles not only in regulating VEGF but also in mitochondrial function, inflammation, apoptosis, and angiogenesis in the retina.8 PPARα levels in the retina are significantly decreased in diabetic animal models and humans, and PPARα ablation results in retinal mitochondrial dysfunction, a declined electroretinogram (ERG) response, and retinal cell apoptosis.8−10
Two large, prospective, independent clinical trials (FIELD and ACCORD) have demonstrated that fenofibrate, an orally administered medication used to treat dyslipidemia, significantly reduced the progression of DR and other microvascular end points in patients with type 2 diabetes.11,12 The therapeutic benefits are not related to its lipid-lowering activity but rather result from the agonism of PPARα by the major metabolite, fenofibric acid (FA).9,13 FA, however, suffers from a low affinity for PPARα, lack of selectivity among PPAR subtypes, dose-limiting toxicities, and is no longer protected by composition of matter patents, all of which will likely limit its development and use as a DR therapy.14 Mechanistic and clinical results, however, paint a compelling picture for developing novel PPARα agonists as mechanistically differentiated therapeutics for DR. A large body of work, primarily from the 1990s, exists for the development of PPARα agonists for the treatment of dyslipidemia.15 While these efforts produced potent and subtype-selective PPARα agonists, the link between PPARα and retinal diseases had not been established at the time, and thus, no ocular safety or efficacy data is reported for these compounds. Rather than repurposing reported chemotypes, most of which lack disclosure of comprehensive PK/PD profiles, we opted to develop novel PPARα agonists.
Recently, we published structure–activity relationship (SAR) studies on a new PPARα agonist, 7-chloro-8-methyl-2-phenylquinoline-4-carboxylic acid (Y-0452), which led to the 4-benzyloxy-benzylamino chemotype, exemplified by A190 (Figure 1A), that exhibits improved potency and selectivity over FA.16,17 In that study, we capitalized on (1) computationally guided design to develop compounds more compatible with the U-shaped PPARα binding pocket and (2) installation of a benzoic acid motif, which provided subtype selectivity. Upon inspection of the 4-benzyloxy-benzylamino SAR data, we recognized that the area around the C-ring was restricted to small substituents, and we hypothesized that removal of the linker between the B- and C-ring to provide a biaryl tail would reduce the number of rotatable bonds and allow for more diverse C-ring functionalization. If successful, this would provide a novel biaryl chemotype as an additional series for exploration and development.
Figure 1.

Structural derivation of A229 and evidence of on-target activity. (A) Structure of A229. (B) Dose–response for PPARα agonism as detected in a luciferase-based cell assay (IB0011, Indigo Biosciences). (C) Subtype activation of PPAR isoforms as detected in luciferase-based cell assays (IB0010 and IB0012, Indigo Biosciences). *Percent activation in comparison with maximum observed signal for the respective positive control [PPARα = GW7647 (90 nM), PPARγ = rosiglitazone (2.5 μM), and PPARδ = GW0742 (7.5 nM)]. (D) mRNA levels of PPARα target genes essential for mitochondrial function and (E) mRNA levels of PPARα target genes involved in mitochondrial biogenesis observed following RT-PCR analysis of primary human RPE (hRPE) cells after 24 h (n = 6) treatment with 25 μM FA or A229.
To test the hypothesis that the linker between the B- and C-rings was dispensable, we synthesized A229 (Figure 1A). As shown in Figure 1B,C, A229 exhibits good potency and subtype selectivity for PPARα when assessed in a commercial primary luciferase-based cell assay. In human retinal pigment epithelial (hRPE) cells, A229 upregulates PPARα target genes to a larger extent than FA (Figure 1D,E).
To confirm PPARα agonism, we assessed A229 in a variety of cell-based phenotypic assays, which were established to provide outputs indicative of PPARα agonism. These studies revealed that at 25 μM A229 (1) exhibited cell protection upon palmitate challenge in ARPE19 cells (a cell line derived from human RPE), 661W cells (a murine photoreceptor precursor cell line), and MIO-M1 cells (a cell line derived from human Müller cells) (Figure 2A and Supplementary Figure S1); (2) reduced VEGF secretion in MIO-M1 and ARPE19 cells following hypoxic challenge induced by CoCl2 (Figure 2B and Supplementary Figure S2); and (3) reduced the production of reactive oxygen species (ROS) in 661W, primary human RPE (hRPE), and MIO-M1 cells following palmitate challenge (Figure 2C and Supplementary Figure S3).
Figure 2.

Cell-based bioactivity of A229. (A) hRPE cells were treated with 25 μM of A229 or FA followed by coincubation with palmitate (PA, 150 μM). Cell viability was determined by counting viable cells. (B) MIO-M1 cells were treated with 25 μM of A229 or FA followed by coincubation with CoCl2 (200 μM). Soluble VEGF (sVEGF) levels were measured via ELISA. (C) 661W cells were treated with 25 μM of A229 or FA followed by coincubation with PA (150 μM). ROS production was measured through the production and detection of fluorescent dichlorofluorescein from 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA). *P < 0.05, **P < 0.01.
These promising results led us to classify this diphenyl aniline scaffold as a new PPARα agonistic chemotype, and a preliminary SAR of this chemotype was conducted and is reported herein.
To probe the chemotype SAR, we set out to synthesize a small library of <50 compounds. These initial efforts mainly focused on determining the impact of A-ring fluorination, B-ring functionalization and isosteric replacement, and C-ring substitution (Figure 3). Analogues were inspired by what we have learned about the 4-benzyloxy-benzylamino chemotype, and our objective was to determine how amenable this new scaffold is to modification while retaining subtype selectivity and potency.
Figure 3.

Approaches to explore the PPARα binding pocket.
For the initial assessment of the synthesized library, we performed a three-concentration (0.5, 5, and 50 μM) dose–response assessment of derivatives for in vitro human PPARα (hPPARα) agonism (Supplementary Figures S4–S7) utilizing a commercially available hPPARα luciferase cell reporter assay (Indigo Biosciences). As per manufacturer’s protocol, GW7647, a well-established potent and selective PPARα full-agonist, served as the positive control.18 This three-concentration approach provides a first-pass readout on potency, efficacy, and toxicity/off-target effects (PET profile) and is a cost-effective strategy to prioritize compounds for further assessment. A colorimetric activity code was utilized to more easily visualize the PPARα PET profile of each compound, and Table 1 presents the outcomes from the three-dose evaluation. Low PPARα agonism is represented as a light green box, moderate agonism as a medium green box, and high agonism as a dark green box. The descriptor for the level of agonism is respective to GW7647.
Table 1. Three-Dose Luciferase PET Profiles.

Detailed levels of agonism are shown in the Supporting Information. Data are based on at least two independent experiments run in triplicate.
We began with the structural interrogation with the C-ring. Compounds 3g, 6a, and 6g (Table 1) were designed using information learned from the 4-benzyloxy chemotype. On the basis of the apparent high potency (i.e., high agonism at 0.5 μM) and no evidence of cytotoxicity or luciferase inhibition (i.e., decreased luciferin production) until 50 μM compound concentration, compound 3g with the trifluoromethyl motif was selected for a more refined dose–response determination. The EC50 value of compound 3g was determined to be 83 nM, a >7-fold improvement over A229 (Table 2). This is contrary to the effect of fluoro- to trifluoromethyl substitution on the 4-benzyloxy chemotype, which negatively affects activity.16 The location of the trifluoromethyl motif was then assessed. The outcome of the 3g (para), 6g (ortho), and 6h (meta) PET profiles, clearly show that the p-CF3 (3g) is optimal (Table 1), as it is the only analogue that exhibits high agonism over the 100-fold dose range. To aid in our understanding of the effect trifluoromethyl location has on target binding, we performed a docking simulation of 3g, 6g, and 6h. As presented in Figure 4A, a significantly different binding mode is predicted for 6g, thereby suggesting binding pocket incompatibility with the ortho-CF3 placement. Because of the higher agonism level elicited by the para-analogue (vs the meta) and the steric discord of the ortho placement, we investigated substitution at the para position of the C-ring to generate 6i–6o. It was noted that installation of a trifluoromethoxy group (6j) produced significant hPPARα agonism at 0.5 μM and 5 μM without observed evidence of off-target effects at 50 μM (also see Supplementary Figure S10), thereby suggesting an improvement from 3g. Compound 6j was selected for a more detailed dose–response assessment and revealed an EC50 of 64 nM (Table 2). It is worth noting that for 3g, 6k, and A229 (6l), we also assessed the corresponding A-ring des-fluoro derivatives (i.e., 3c, 3d, and 3b, respectively), and in each case, the potency, level of agonism, and/or evidence of toxicity worsened (Table 1), which alludes to the importance of the A-ring fluorine.
Table 2. In Vitro Activity of Selected Compoundsa.
| compound | EC50 hPPARα (μM ± SEM)b | hill slope | maximal relative activation of hPPARβ/δ (% ± SEM)c,e | maximal relative activation of hPPARγ (% ± SEM)d,e | clogPf | ligand efficiencyg |
|---|---|---|---|---|---|---|
| A229 (6l) | 0.59 ± 0.051 | 1.4 | 12.1 ± 1.0 | 11.2 ± 3.0 | 4.8 | 0.341 |
| 3g | 0.083 ± 0.011 | 2.2 | 31.1 ± 1.0 | 22.5 ± 6.7 | 5.4 | 0.347 |
| 6d | 0.080 ± 0.032 | 1.0 | 25.1 ± 4.1 | 35.7 ± 4.2 | 5.7 | 0.343 |
| 6j | 0.064 ± 0.018 | 1.8 | 6.1 ± 3.6 | 9.3 ± 2.3 | 5.7 | 0.337 |
| 8b | 0.30 ± 0.096 | 1.5 | 21.1 ± 6.2 | 19.9 ± 10.3 | 4.6 | 0.317 |
| GW7647 | 0.0057 ± 0.00029 | 1.8 | N.D. | N.D. | 7.0 | 0.340 |
EC50 values and maximal relative activation were calculated as mean ± SEM of at least two independent experiments performed in triplicate.
Full dose–response curves are shown in the Supporting Information (Figure S9).
Maximal relative activation of hPPARβ/δ refers to the activity of known PPARβ/δ agonist GW0742 at 0.0075 μM concentration, which was defined as 100% activation.
Maximal relative activation of hPPARγ refers to the activity of known PPARγ agonist rosiglitazone at 2.5 μM concentration, which was defined as 100% activation.
For PPARβ/δ and PPARγ assays, a six-point dose–response range was completed for each compound ranging from 13 nM to 100 μM (13 nM, 77 nM, 463 nM, 3 μM, 17 μM, and 100 μM). Because of observed cytotoxicity at 100 μM for several compounds, the agonism level at 17 μM was selected as the point of comparison. N.D. = not determined.
Calculated logP (clogP) values were predicted using ADMETlab 2.0.19
Ligand efficiency (LE) values were calculated using the equation: LE = (1.37/the number of heavy atoms) × pEC50.20
Figure 4.

Predicted binding mode of select biaryl anilnes. (A) Overlay of predicted binding poses for 3g (blue), 6g (magenta), and 6h (yellow). (B) Predicted binding poses of A190 (blue) and 6j (yellow) within the binding pocket of PPARα. (C) Predicted binding poses of GW0742 (green) and 6j (yellow) within the binding pocket of PPARβ/δ. (D) Predicted binding pose of rosiglitazone (green) and 6j (yellow) within the binding pocket of PPARγ. Described hydrogen bonds are indicated with a yellow dashed line.
In the 4-benxyloxy chemotype, fluorination of the A-ring was initially pursued to block a predicted site of metabolism. Because A229 was found to exhibit rather poor aqueous solubility in the cell-based assays (clogP = 4.8, tPSA = 49.3), we recognized that a meta-pyridyl nitrogen (3a) may provide a way to enhance solubility (clogP = 3.9, tPSA = 62.2) and continue to preclude metabolic liability. Unfortunately, analogue 3a lost significant potency (Table 1), as indicated by the colorimetric readout, compared with A229. This further indicates that the hydrophobic properties of the fluorine likely play a significant role in the observed activity, an effect not predicted in our molecular design. While a decrease in logP may be desirable for solubility, this seems counterproductive to target engagement. This can also be observed in Table 2, which illustrates a correlation between higher clogP and activity.
To further understand the importance of the fluorine location on the A-ring, we performed a “fluorine walk” (3e–h, Table 1). In this investigation, evidence of significant off-target effects was especially observed for 3e at 50 μM, as indicated by lower luminescence signals. This decrease in luminescence was confirmed to be due to cell cytotoxicity (Supplementary Figures S10 and S11). Compound 3g, with the 5-fluorine, was found to exhibit the highest level of hPPARα agonism at 500 nM with only a slight indication of off-target issues at 50 μM. Because of the PET profile of 3g, which showed that the 5-fluoro A-ring exhibits optimal potency, high levels of hPPARα agonism, and low evidence of off-target issues, further modification was performed retaining this fluorine location.
In the case of the 4-benzyloxy-benzylamino chemotype from our previous studies, it was noted that modifications to the B-ring were acceptable and often led to dramatic effects on the potency. One interesting observation, however, was that a “third extension” could be incorporated off of the B-ring, that was predicted to extend into an unexplored (at the time) pocket.17,21 Since our disclosure, it has been revealed that the subtype-selective PPARα agonist, pemafibrate, also exploits this pocket.21 In the case of the 4-benzyloxy series, exploitation of this additional binding pocket failed to produce improvement in potency and selectivity. To investigate if this trend held in the biphenyl series, we designed and synthesized a small subset of B-ring variants containing a hydroxy (6b), methyl (6p), benzyl (6q), 4-methylbenzyl (6r), and 4-chlorophenol (6s). Derivatives 6b and 6p–6s were assessed to obtain their PET profile, and the results are shown in Table 1. Interestingly, extension off of the B-ring in the biphenyl series was not compatible with binding, as all 5 analogues lacked significant hPPARα agonism.
A strong methyl effect was observed in 4-benzyloxy-benzylamino chemotype when a methyl group was installed at the 2-position of the B-ring. Incorporation of the methyl group significantly enhanced potency (∼21-fold improvement) and subtype selectivity (>2700-fold). This significant impact on activity inspired us to synthesize 6d, which includes a methyl group at the desired position.16 As shown in Table 1, the addition of the methyl group provides a similar PET profile as the des-methyl comparator (3g) with high agonism at 500 nM, albeit with more evidence of off-target effects at 5 and 50 μM. Other methylation or halogenation patterns were also briefly explored within this series (6c, 6e, and 6f), none of which provided improvement. Compound 6d was selected for more refined dose–response assessment. As shown in Table 2, 6d (80 nM) exhibits equipotency to the des-methyl 3g. Thus, B-ring methylation in this biphenyl series fails to produce the same “magic methyl” effect as that observed in the 4-benzyloxy-benzylamino chemotype. This was a bit surprising, given that the B-ring is predicted to bind nearly superimposable with the B-ring in the 4-benzyloxy chemotype (Figure 4B). This result, however, confirms our original suspicion that the “magic methyl” effect in the 4-benzyloxy series is due to more than simply increased hydrophobic interactions with the target and likely arises from the reorientation of the benzoic acid motif to provide T-shaped π-interactions with His-440.
Exploration of the composition of the B-ring was interrogated through bioisosteric replacement, and the results are shown in Table 1. Pyridine, thiazole, and triazole ring systems were incorporated to generate 8a–8e and 12a–12d. Within the heterocycle subsets, a similar pattern for C-ring substitution was observed (A229 vs 3g, 8d vs 8e, 12a vs 12b, and 12c vs 12d) in that the introduction of p-CF3 on C-ring showed a higher agonism and potency than p-F. Pyridine 8b exhibited low evidence of toxicity/off-target effects with consistent partial agonism across all three doses evaluated and was selected for a more detailed dose–response assessment to obtain an EC50 value of 300 nM (Table 2). Because of the lower levels of agonism for other heterocycles, no other pyridines or thiazoles were progressed further. In agreement with our previous results for the B-ring phenyl series, within the triazoles (12a–d), the presence of 5-fluorine on the A-ring (12c) proved significantly important in potency and reducing cytotoxicity at 50 μM.
Lastly, the effect of linker substitution and extension between the A- and B-rings was briefly evaluated (15a,b and 22). Unfortunately, a decline in hPPARα agonism level and potency was observed for all three derivatives (Table 1).
Compounds 3g and 6j were advanced to the same cell-based assays as A229 (Figure 2), and as shown in Figure 5, both compounds improved cell viability in hRPE cells under PA challenge (Figure 5A), reduced VEGF production in CoCl2-challenged MIO-M1 cells (Figure 5B), and protected against PA-induced ROS production (Figure 5C) in 661W cells, which are all responses expected for PPARα agonism. It should be noted that 3g and 6j outperformed FA in each study, similarly to A229.
Figure 5.

Conformation of cell-based bioactivity of 3g and 6j. (A) Human primary retinal pigment epithelial (hRPE) cells were treated with fenofibric acid (FA), A229, 3g, or 6j at a concentration of 25 μM for 4 h, followed by coincubation with 150 μM palmitate (PA) for an additional 24 h. Cells were stained with Hoechst and counted using Cytation 5 (mean ± SEM, n = 6). *P < 0.05, **P < 0.01. (B) FA, A229, 3g, and 6j reduced CoCl2-induced secretion of soluble vascular endothelial growth factor (sVEGF). Human Muller cells (MIO-M1) were treated with FA, A229, 3g, or 6j (25 μM) for 4 h and CoCl2 (200 μM) for another 24 h. The conditioned media were collected for sVEGF ELISA and normalized by total protein concentration (mean ± SEM, n = 3). *P < 0.05, **P < 0.01. (C) FA, A229, 3g, and 6j treatment decreased retinal oxidative stress. Retinal 661W cells were treated with FA, A229, 3g, or 6j (25 μM) for 4 h, followed by palmitate (PA, 150 μM) treatment and coincubation for another 24 h. ROS production was measured using H2DCFDA and normalized by total protein concentration (mean ± SEM, n = 6). *P < 0.05, **P < 0.01, ***P < 0.001.
On the basis of these first-generation SAR studies, we determined that (1) the 5-position is optimal for fluorine installation on the A-ring and proves to be significant for potency; (2) B-ring methylation at the 2-position fails to enhance the potency, contrary to the 4-benzyloxy-benzylamino chemotype; (3) extension off the 3-position of the B-ring with a third substituent to exploit an adjacent pocket is detrimental to activity, at least for the diverse groups installed; (4) isosteric replacement of the B-ring with various heterocycles is allowed but affects the level of agonism and/or produces suboptimal PET profiles; and (5) C-ring substitution shows similar trends in B-ring phenyl and heteroaryl series.
As mentioned, because of the diverse tissue distribution and function of isoforms and a desire to minimize side effects, our goal is to develop selective PPARα agonists. Derivatives 3g, 6d, 6j, and 8b were further evaluated for selectivity profiles by performing the same luciferase-based assessment with comparable commercially available hPPARβ/δ and hPPARγ cell reporter assays (Indigo Biosciences). GW0742 and rosiglitazone were used as positive controls for hPPARβ/δ and hPPARγ agonism, respectively. As seen in Table 2, all four compounds agonized the other two subtypes to a much lower extent than they do PPARα. This selectivity profile further confirms our hypothesis that the benzoic acid motif is the key contributor to the selectivity of the 4-benzyloxy-benzylmino and biphenyl chemotypes, a feature that is unique compared with the pharmacophore of other PPARα agonists, including the fibrates. On the basis of computational studies, we believe this selectivity arises from the difference in residues within the PPAR ligand binding pocket. As an example, 6j was docked into PPARβ/δ and PPARγ to provide potential insight into the selectivity of biaryl aniline within the PPAR family (Figure 4C,D). His-287, His-413, and Tyr-437, three critical residues in eliciting agonism, demonstrate critical hydrogen bonding interactions between the carboxylate of GW0742, which serves as a potent PPARβ/δ agonist.22 However, a predicted loss of hydrogen bonding between the carboxylic acid of 6j and His-413 may contribute to the weaker observed potency and efficacy against PPARβ/δ. Although 6j and GW0742 both present a carboxylate, GW0742 exhibits more structural compatibility with the cavity of PPARβ/δ and likely capitalizes on additional hydrogen bonding with residues. Rosiglitazone, which serves as a potent PPARγ agonist, exhibits hydrogen bonding with Ser-289, His-323, His-449, and Tyr-473 (Figure 4D).23 In this docking study, we observed that 6j is predicted to lack interactions between these residues that significantly contribute to the potency and efficacy of PPARγ.
The interpretation of SAR for ligand-activated nuclear transcription factors is not a trivial process, as these ligands are known to elicit changes in protein–protein interactions that comprise various coregulators and dimerization partners that exhibit varying levels of permissiveness. Recently, work was published highlighting the cooperative nature of several PPARγ ligands and the coregulator MED1.24 While PPARγ is not our isoform of interest, we realize that this concept likely holds for PPARα modulators and coregulators. One way to obtain potential insight about cooperativity is to compare the Hill slopes of the cell-based luciferase dose–response curves.
As shown in Table 2, all ligands except 6d exhibit a Hill slope > 1.0, thereby suggesting that ligand activity exhibits some type of cooperativity. This may allude to part of the reason that methylation on this scaffold fails to produce the similar jump in potency as observed in the 4-benzyloxybenzylamino chemotype on which B-ring methylation results in retention of a Hill slope > 1.5. On the contrary, in this new biaryl aniline series, B-ring methylation is detrimental to cooperativity and, thus, likely counter-productive to downstream activity, even if binding affinity is enhanced. Without detailed biochemical and biophysical assays like those described for PPARγ, however, this is merely a hypothesis that requires further interrogation.
Synthetic access to the compounds in this series was straightforward and leveraged well-established methods that relied primarily on reductive amination of requisite benzoic acids with functionalized aldehydes. Analogues 3a–3h were synthesized according to Scheme 1. Because of its low solubility in toluene, which proved problematic for the condensation reaction, the carboxylic acid present in 5-aminonicotinic acid was masked to afford ester 1, which underwent reductive amination with 4-(4-fluorophenyl)benzaldehyde to generate diphenyl aniline 2. Saponification of intermediate 2 generated carboxylic acid 3a. Analogues 3b–3h were obtained following a reductive amination reaction between biphenyl-4-carboxaldehydes and 3-aminobenzoic acids.
Scheme 1. General Synthetic Approach for Analogues 3a–3h.

Reagents and reaction conditions: (i) SOCl2, MeOH, 0 °C to reflux, 16 h, 100%; (ii) (a) toluene, 115 °C, 1–2 h; (b) HOAc, NaBH(OAc)3, THF, 0–25 °C, 14–16 h, 34–97%; (iii) LiOH·H2O, THF/MeOH/H2O (v/v/v = 3:1:1), 25 °C, 2–16 h, 90%.
The general synthetic approach for analogues 6a–6s and 8a–8e are illustrated in Scheme 2 and Scheme 3, respectively. Aldehyde intermediates 4a–4k, 4m-4o, 7a-7c were generated by Suzuki cross-coupling reactions between substituted 4-bromobenzaldehydes and aryl boronic acids. Alkylation of the hydroxy group present in intermediate 4b gave intermediates 5a–5d. Reductive amination between the substituted aldehydes (generated and commercially available) and 3-amino-5-fluorobenzoic acid yielded analogues 6a–6s and 8a–8e.
Scheme 2. General Synthetic Approach for Analogues 6a–6s.

Reagents and reaction conditions: (i) Pd(dppf)2Cl2·DCM, K2CO3, dioxane/H2O (v/v = 9:1), N2, 90 °C, 3–4 h, 27–98%; (ii) (a) Toluene, 115 °C, 1–2 h, (b) HOAc, NaBH(OAc)3, THF, 0–25 °C, 14–16 h, 40–89%; (iii) For 5a = CH3I, K2CO3, DMF, 70 °C, 16 h, 90%; for 5b = benzyl bromide, K2CO3, DMF, 80 °C, 16 h, 97%; for 5c = 4-methylbenzyl bromide, K2CO3, DMF, 80 °C, 16 h, 93%; for 5d = 4-chlorobenzyl bromide, K2CO3, DMF, 80 °C, 16 h, 86%.
Scheme 3. General Synthetic Approach for Analogues 8a–8e.

Reagents and reaction conditions: (i) Pd(dppf)2Cl2·DCM, K2CO3, dioxane/H2O (v/v = 9:1), N2, 90 °C, 3 h, 40–92%; (ii) Pd(PPh3)4, Na2CO3, EtOH/DME (v/v = 1:1), N2, 100 °C, 24 h, 30%; (iii) (a) toluene, 115 °C, 2 h, (b) HOAc, NaBH(OAc)3, THF, 0–25 °C, 14 h, 32–58%.
The general synthetic route for triazoles 12a–12d is presented in Scheme 4. Monoalkylation of methyl 3-aminobenzoates with propargyl bromide provided terminal alkynes 9a,b, which were reacted with 4-substituted azidobenzenes under Cu(I)-catalyzed click conditions to elicit an azide–alkyne cycloaddition (CuAAC) to afford triazoles 11a–11d. The methyl ester of intermediate 11a–11d was cleaved by saponification to reveal carboxylic acid 12a–12d.
Scheme 4. General Synthetic Approach for Analogues 12a–12d.

Reagents and reaction conditions: (i) for 9a = propargyl bromide, K2CO3, DMF, 25 °C, 24 h, 55%; for 9b = propargyl bromide, K2CO3, DMF, 50 °C, 15 h, 44%; (ii) CuO4·5H2O, sodium l-ascorbate, tert-butanol/H2O (v/v = 1:1), 50 °C, 16 h, 72–95%; (iii) LiOH·H2O, THF/MeOH/H2O (v/v/v = 3:1:1), 25 °C, 2 h, 78–96%.
For the last cohort of analogues, the general synthetic route for 15a,b, and 22 is shown in Scheme 5. A Suzuki reaction between phenyl bromides and (4-fluorophenyl)boronic acid provided 13a,b, which were then reacted with methyl 3-fluoro-5-hydroxybenzoate to form ether 14a,b via a Mitsunobu reaction. Removal of the methyl ether afforded final compounds 15a,b. Intermediate 17 was generated from phenyl bromide through a Suzuki reaction followed by iodination. Terminal alkyne 19 was obtained by a Sonogashira reaction between methyl 3-bromo-5-fluorobenzoate and ethynyltrimethylsilane followed by deprotection. A Sonogashira reaction between the diphenyl iodide 17 and intermediate 19 yielded internal alkyne 20, which was hydrogenated in the presence of palladium to give intermediate 21. Saponification of ester 21 gave compound 22.
Scheme 5. General Synthetic Approach for Analogues 15a,b, 22, and 24.
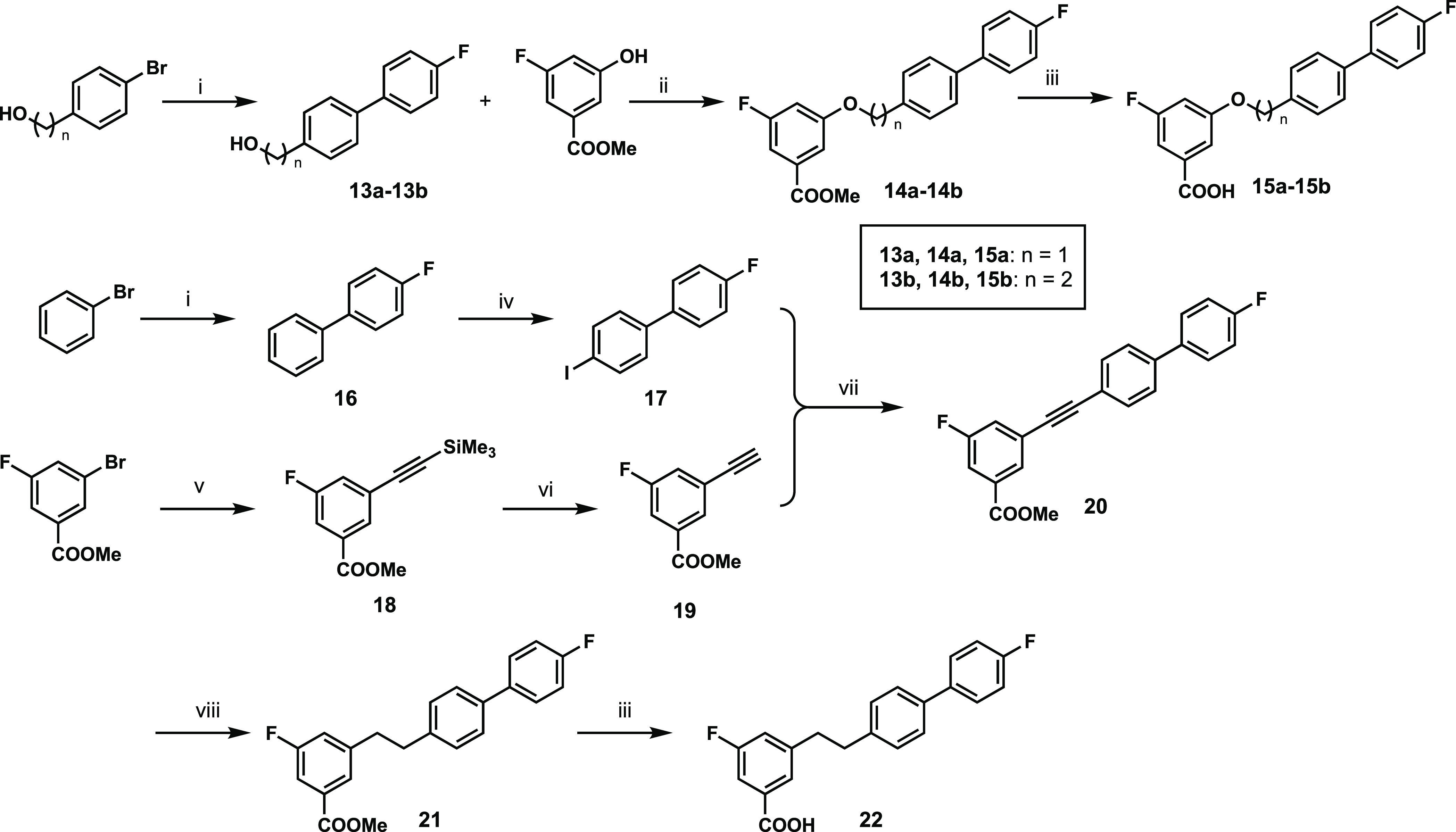
Reagents and reaction conditions: (i) 4-fluorophenylboronic acid, Pd(dppf)2Cl2·DCM, K2CO3, dioxane/H2O (v/v = 9:1), N2, 90 °C, 3 h, 78–100%; (ii) PPh3, DIAD, THF, N2, 0–25 °C, 16 h, 50–76%; (iii) LiOH·H2O, THF/MeOH/H2O (v/v/v = 3:1:1), 25 °C, 2–16 h, 85–97%; (iv) iodine, iodic acid, 80% AcOH, concd. H2SO4, 80 °C, 16 h, 88%; (v) ethynyltrimethylsilane, Pd(PPh3)2Cl2, TEA, CuI, CH3CN, N2, 60 °C, 2 h, 94%; (vi) K2CO3, MeOH, 25 °C, 20 min, 95%; (vii) Pd(PPh3)2Cl2, CuI, DMF/TEA (v/v = 2:1), N2, 25 °C, 2 h, 68%; (viii) 10% Pd/C, MeOH, 50 °C, 3 h, 100%.
In conclusion, we have designed a novel biphenyl aniline PPARα agonistic subtype-selective chemotype. The results from this initial SAR interrogation define guiding principles for the continued optimization of this scaffold including (1) the importance and location of A-ring fluorination, (2) the allowability of B-ring isosteric replacement but likely not functionalization, (3) the existence of space off the para-position of the C-ring, and (4) the necessity for the nitrogen-based linker between the A- and B-rings. This chemotype represents the second to arise from the initial hit, Y-0452, and the ability to edit the composition of the B-ring opens avenues for further pipeline diversification.
Acknowledgments
The work was supported by the National Eye Institute of the National Institutes of Health (EY030472, A.S.D. and J.X.M; EY028949, J.X.M; and EY033330, J.X.M). The content included in this manuscript does not necessarily reflect the position or the policy of the federal government, and no official endorsement should be inferred.
Glossary
Abbreviations
- CuAAC
copper catalyzed azide–alkyne coupling
- DR
diabetic retinopathy
- EC50
effective concentration for half-maximal activity
- FA or FenoFA
fenofibric acid
- hPPAR
human peroxisome proliferator-activated receptor
- hRPE
human retinal epithelium
- PA
palmitic acid
- PPAR
peroxisome proliferator-activated receptor
- PET
potency efficacy and toxicity profile
- ROS
reactive oxygen species
- RT-PCR
reverse transcription-polymerase chain reaction
- SAR
structure–activity relationship
- SEM
standard error of the mean
- VEGF
vascular endothelial growth factor.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00056.
Biological methods, cell-based assessment of A229, three-dose response results for PPARα agonism, isoform selectivity data, dose response curves for select compounds, cytotoxicity data for select compounds, synthetic procedures, characterization data, and annotated 1H and 13C NMR spectra (PDF)
Author Contributions
# J.J.L. and Z.H. contributed equally. J.J.L., Z.H., and A.S.D. designed all compounds, and J.J.L., Z.H., and D.N. synthesized all molecules. J.J.L. conducted the luciferase assessment and multiplex cytotoxicity assay. Y.A.W. performed all biology on A229. W.L. and Y.C. completed the follow-up cell-based assays for 3g and 6j. The manuscript was written by J.J.L., Z.H.. and A.S.D. and edited and approved by all authors.
The authors declare the following competing financial interest(s): A.S.D. and J.X.M. are co-founders of Excitant Therapeutics.
Special Issue
Published as part of the ACS Medicinal Chemistry Lettersvirtual special issue “Celebrating the 60th Anniversary of the MIKIW Meeting-in-Miniature”.
Supplementary Material
References
- Nentwich M. M.; Ulbig M. W. Diabetic Retinopathy - Ocular Complications of Diabetes Mellitus. World J. Diabetes 2015, 6 (3), 489–499. 10.4239/wjd.v6.i3.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd K.Diabetic Retinopathy: Causes, Symptoms, Treatment; American Academy of Ophthalmology, 2022. https://www.aao.org/eye-health/diseases/what-is-diabetic-retinopathy (accessed 2023-03-02). [Google Scholar]
- Cai X.; McGinnis J. F. Diabetic Retinopathy: Animal Models, Therapies, and Perspectives. J. Diabetes Res. 2016, 2016, 3789217. 10.1155/2016/3789217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simó R.; Hernández C. Novel Approaches for Treating Diabetic Retinopathy Based on Recent Pathogenic Evidence. Progress in Retinal and Eye Research 2015, 48, 160–180. 10.1016/j.preteyeres.2015.04.003. [DOI] [PubMed] [Google Scholar]
- Sapieha P.; Hamel D.; Shao Z.; Rivera J. C.; Zaniolo K.; Joyal J. S.; Chemtob S. Proliferative Retinopathies: Angiogenesis That Blinds. International Journal of Biochemistry & Cell Biology 2010, 42 (1), 5–12. 10.1016/j.biocel.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Berger J.; M?ller D. E. The Mechanisms of Action of PPARs. Annual Review of Medicine 2002, 53 (1), 409–435. 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- Kliewer S. A.; Forman B. M.; Blumberg B.; Ong E. S.; Borgmeyer U.; Mangelsdorf D. J.; Umesono K.; Evans R. M. Differential Expression and Activation of a Family of Murine Peroxisome Proliferator-Activated Receptors. Proc. Natl. Acad. Sci. U. S. A. 1994, 91 (15), 7355–7359. 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Chen Y.; Ding L.; He X.; Takahashi Y.; Gao Y.; Shen W.; Cheng R.; Chen Q.; Qi X.; Boulton M. E.; Ma J. Pathogenic Role of Diabetes-Induced PPAR-α down-Regulation in Microvascular Dysfunction. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (38), 15401–15406. 10.1073/pnas.1307211110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Hu Y.; Lin M.; Jenkins A. J.; Keech A. C.; Mott R.; Lyons T. J.; Ma J. Therapeutic Effects of PPARα Agonists on Diabetic Retinopathy in Type 1 Diabetes Models. Diabetes 2013, 62 (1), 261–272. 10.2337/db11-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearsall E. A.; Cheng R.; Zhou K.; Takahashi Y.; Matlock H. G.; Vadvalkar S. S.; Shin Y.; Fredrick T. W.; Gantner M. L.; Meng S.; Fu Z.; Gong Y.; Kinter M.; Humphries K. M.; Szweda L. I.; Smith L. E. H.; Ma J. PPARα Is Essential for Retinal Lipid Metabolism and Neuronal Survival. BMC Biology 2017, 15 (1), 113. 10.1186/s12915-017-0451-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keech A.; Mitchell P.; Summanen P.; O’Day J.; Davis T.; Moffitt M.; Taskinen M.-R.; Simes R.; Tse D.; Williamson E.; Merrifield A.; Laatikainen L.; d’Emden M.; Crimet D.; O’Connell R.; Colman P. Effect of Fenofibrate on the Need for Laser Treatment for Diabetic Retinopathy (FIELD Study): A Randomised Controlled Trial. Lancet 2007, 370 (9600), 1687–1697. 10.1016/S0140-6736(07)61607-9. [DOI] [PubMed] [Google Scholar]
- Effects of Medical Therapies on Retinopathy Progression in Type 2 Diabetes. N Engl J. Med. 2010, 363 (3), 233–244. 10.1056/NEJMoa1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noonan J. E.; Jenkins A. J.; Ma J.-X.; Keech A. C.; Wang J. J.; Lamoureux E. L. An Update on the Molecular Actions of Fenofibrate and Its Clinical Effects on Diabetic Retinopathy and Other Microvascular End Points in Patients With Diabetes. Diabetes 2013, 62 (12), 3968–3975. 10.2337/db13-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Kennedy L. J.; Shi Y.; Tao S.; Ye X.-Y.; Chen S. Y.; Wang Y.; Hernández A. S.; Wang W.; Devasthale P. V.; Chen S.; Lai Z.; Zhang H.; Wu S.; Smirk R. A.; Bolton S. A.; Ryono D. E.; Zhang H.; Lim N.-K.; Chen B.-C.; Locke K. T.; O’Malley K. M.; Zhang L.; Srivastava R. A.; Miao B.; Meyers D. S.; Monshizadegan H.; Search D.; Grimm D.; Zhang R.; Harrity T.; Kunselman L. K.; Cap M.; Kadiyala P.; Hosagrahara V.; Zhang L.; Xu C.; Li Y.-X.; Muckelbauer J. K.; Chang C.; An Y.; Krystek S. R.; Blanar M. A.; Zahler R.; Mukherjee R.; Cheng P. T. W.; Tino J. A. Discovery of an Oxybenzylglycine Based Peroxisome Proliferator Activated Receptor α Selective Agonist 2-((3-((2-(4-Chlorophenyl)-5-Methyloxazol-4-Yl)Methoxy)Benzyl)(Methoxycarbonyl)Amino)Acetic Acid (BMS-687453). J. Med. Chem. 2010, 53 (7), 2854–2864. 10.1021/jm9016812. [DOI] [PubMed] [Google Scholar]
- Pirat C.; Farce A.; Lebègue N.; Renault N.; Furman C.; Millet R.; Yous S.; Speca S.; Berthelot P.; Desreumaux P.; Chavatte P. Targeting Peroxisome Proliferator-Activated Receptors (PPARs): Development of Modulators. J. Med. Chem. 2012, 55 (9), 4027–4061. 10.1021/jm101360s. [DOI] [PubMed] [Google Scholar]
- Dou X.; Nath D.; Shin H.; Nurmemmedov E.; Bourne P. C.; Ma J.-X.; Duerfeldt A. S. Evolution of a 4-Benzyloxy-Benzylamino Chemotype to Provide Efficacious, Potent, and Isoform Selective PPARα Agonists as Leads for Retinal Disorders. J. Med. Chem. 2020, 63 (6), 2854–2876. 10.1021/acs.jmedchem.9b01189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou X.-Z.; Nath D.; Shin Y.; Ma J.-X.; Duerfeldt A. S. Structure-Guided Evolution of a 2-Phenyl-4-Carboxyquinoline Chemotype into PPARα Selective Agonists: New Leads for Oculovascular Conditions. Bioorg. Med. Chem. Lett. 2018, 28 (16), 2717–2722. 10.1016/j.bmcl.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P. J.; Stuart L. W.; Hurley K. P.; Lewis M. C.; Winegar D. A.; Wilson J. G.; Wilkison W. O.; Ittoop O. R.; Willson T. M. Identification of a Subtype Selective Human PPARα Agonist through Parallel-Array Synthesis. Bioorg. Med. Chem. Lett. 2001, 11 (9), 1225–1227. 10.1016/S0960-894X(01)00188-3. [DOI] [PubMed] [Google Scholar]
- ADMETlab 2.0. https://admetmesh.scbdd.com/service/evaluation/cal (accessed 2023-05-04). [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The Role of Ligand Efficiency Metrics in Drug Discovery. Nat. Rev. Drug Discovery 2014, 13 (2), 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y.; Takei K.; Arulmozhiraja S.; Sladek V.; Matsuo N.; Han S.; Matsuzaka T.; Sekiya M.; Tokiwa T.; Shoji M.; Shigeta Y.; Nakagawa Y.; Tokiwa H.; Shimano H. Molecular Association Model of PPARα and Its New Specific and Efficient Ligand, Pemafibrate: Structural Basis for SPPARMα. Biochem. Biophys. Res. Commun. 2018, 499 (2), 239–245. 10.1016/j.bbrc.2018.03.135. [DOI] [PubMed] [Google Scholar]
- Batista F. A. H.; Trivella D. B. B.; Bernardes A.; Gratieri J.; Oliveira P. S. L.; Figueira A. C. M.; Webb P.; Polikarpov I. Structural Insights into Human Peroxisome Proliferator Activated Receptor Delta (PPAR-Delta) Selective Ligand Binding. PLoS One 2012, 7 (5), e33643 10.1371/journal.pone.0033643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J.; Brust R.; Mosure S. A.; Bass J.; Munoz-Tello P.; Lin H.; Hughes T. S.; Tang M.; Ge Q.; Kamenekca T. M.; Kojetin D. J. Cooperative Cobinding of Synthetic and Natural Ligands to the Nuclear Receptor PPARγ. eLife 2018, 7, e43320 10.7554/eLife.43320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vink P. J.; Koops A. A.; D’Arrigo G.; Cruciani G.; Spyrakis F.; Brunsveld L. Cooperativity as Quantification and Optimization Paradigm for Nuclear Receptor Modulators. Chem. Sci. 2022, 13 (9), 2744–2752. 10.1039/D1SC06426F. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.