

Abstract
High frequency of KRAS and TP53 mutations is a unique genetic feature of pancreatic ductal adenocarcinoma (PDAC). TP53 mutation not only renders PDAC resistance to chemotherapies but also drives PDAC invasiveness. Therapies targeting activating mutant KRAS are not available and the outcomes of current PDAC treatment are extremely poor. Here we report that MMRi62, initially identified as an MDM2-MDM4-targeting small molecule with p53-independent pro-apoptotic activity, shows anti-PDAC activity in vitro and in vivo. We show that MMRi62 inhibits proliferation, clonogenic and spheroid growth of PDAC cells by induction of cell death. MMRi62-induced cell death in PDAC is characteristic of ferroptosis which is associated with increased autophagy, increased reactive oxygen species and lysosomal degradation of NCOA4 and Ferritin Heavy Chain (FTH1). In addition to induced degradation of FTH1, MMRi62 also induces proteasomal degradation of mutant p53. Interestingly, MMRi62-induced ferroptosis occurs in PDAC cell lines harboring either KRAS and TP53 double mutations or single TP53 mutation. In orthotopic xenograft PDAC mouse models, MMRi62 was capable of inhibiting tumor growth in mice associated with downregulation of NCOA4 and mutant p53 in vivo. Strikingly, MMRi62 completely abrogated metastasis of orthotopic tumors to distant organs, which is consistent with MMRi62’s ability to inhibit cell migration and invasion in vitro. These findings identified MMRi62 as a novel ferroptosis inducer capable of suppressing PDAC growth and overcoming metastasis.
Keywords: MMRi62, mutant p53, pancreatic, ferroptosis, metastasis
Introduction
Pancreatic ductal adenocarcinoma (PDAC) ranks the fourth leading cause of cancer-related deaths in the Western world (1). The estimated new cases of pancreatic cancer were 60,430 in the US for 2021 with an estimated 48,220 deaths(1), and 90,100 in China with estimated 79,400 deaths for 2015 (2). The PDAC mortality rate has not been much improved in three decades because of late diagnosis, early metastasis, and limited response to chemotherapy and radiotherapy, and are projected to surpass breast, prostate, and colorectal cancers to become the second leading causes of cancer-related death by 2030 (3). Even for resectable tumors, surgery alone only has limited benefit, as >90% of patients relapse and then die after surgery without additional therapy (4). Surgery followed by adjuvant chemotherapy (gemcitabine plus capecitabine) is the standard of care for resectable tumors and the median survival in these patients is 26 months, with a 5-year survival of 30% (5). FOLFIRINOX regimen (5-fluorouracil/leucovorin with irinotecan and oxaliplatin) and gemcitabine/nab-paclitaxel has been used for advanced PDAC but with severe toxicity that limits patient enrollment (6). Therefore, new therapeutic strategies for PDAC are highly anticipated (5). TP53 mutation is one of the key drivers of tumorigenesis in most human cancer types (7). Mutant p53 proteins (mut-p53) often lose wild-type p53 (wt-p53) function while several hot-spot p53 mutants even acquire novel oncogenic functions, namely gain-of-function (GOF) driving aberrant cell proliferation, chemoresistance, disruption of tissue architecture, migration, invasion, and metastasis (8). It was established in mouse models that KRAS and TP53 mutations are sufficient to promote PDAC development (9). In humans, four major driver genetic mutations in PDAC includes KRAS, CDKN2A, TP53, and SMAD4 (10, 11). Genomic sequencing of primary PDAC pancreatic tumors found 60–70% bearing TP53 mutations which occur in late stage PanIN lesions, suggesting critical roles of TP53 mutation in PDAC transformation and progression (12, 13). In addition, mut-p53 drives PDAC metastasis in mouse models (14, 15) and confers PDAC resistance to chemotherapies (16).
While mutant RAS-targeted therapies are still lacking, screens of RAS-selective lethal compounds identified that RAS-driven pancreatic cancer cells are predisposed to ferroptosis, a mode of cell death involving ROS production and autophagic degradation of ferritin (17, 18). While the molecular mechanisms regulating ferroptosis is under active research, identification of novel pro-ferroptotic agents represents a new area of potential PDAC therapeutics. In this report, we describe small molecule MMRi62 as a ferroptosis inducer in pancreatic cancer cells by mechanisms involving induced degradation of mut-p53 and FTH1 that may have potential to suppress both PDAC growth and metastasis.
Materials and Methods
Chemical synthesis
Small molecule MMRi62 was synthesized in house at Chemler’s lab. The Betti reaction was for MMRi62 synthesis as described in literatures (19, 20). The structure and purity of the synthesized MMRi62 was determined by NMR and the purity was determined by high resolution mass spectra obtained at SUNY Buffalo’s mass spec facility on a ThermoFinnigan MAT XL spectrometer. Bafilomycin A1 was obtained from Sigma (Cat# 19–148) and Carfilzomib was purchased from Cayman Chemical (Cat# 17554). All the compounds were dissolved in DMSO as 10 mM stocks and stored at −20 °C.
Cell culture, proliferation assay and synergism analysis
Panc1, BxPc3, panc10.05, AsPc1, Capan2 and SW1990 pancreatic cancer cells were purchased in December 2017 from the ATCC and cultured as recommended. Cells were regularly authenticated by growth rate and morphologic observation. Panc1,BxPc3, panc10.05, AsPc1, Capan-2 and AsPc1cells were grown in a humidified incubator at 37° C under an atmosphere of 5% CO2 in air and periodically checked for mycoplasma contamination. SW1990 cells were grown in a humidified incubator at 37° C without CO2 in air. Genetic status of KRAS and TP53 were obtained from published studies (21–23). Proliferation assay was caried out with resazurin cell proliferation assay following manufacturer’s instruction (Biotium, CA, USA, Cat# 30025–1). The IC50 values and combination index were obtained by Chou-Median-Effect Equation using CompuSyn software (24) and dose-effect curves were obtained by GraphPad using affected fractions of compound-treated wells normalized against no-drug control wells using non-liner regression model.
Immunoblotting and p53 ubiquitination assay
Cells (~70% confluency) were collected via scraping on ice and lysed in a buffer containing 8M urea, 0.1M phosphate, 0.01M Tris, pH8.0, 15mM imidazole, 0.2% triton-X100. After vigorous vortex, the lysates were clarified by centrifugation at 12,000 rpm for 10 min at RT followed by protein quantification using Pierce™ BCA Kit. Equal amounts of protein (20 μg) were separated by 8 or 12% SDS-PAGE followed by transfer to nitrocellulose membranes and blotting with with primary antibodies overnight and species-specific horseradish peroxidase-conjugated secondary antibodies and chemiluminescence. The densitometry was performed using ImageJ. Primary antibodies used in this study include rabbit NCOA4 polyclonal antibody (Abclonal, cat# A5695), rabbit Anti-LC3B (Sigma, Cat# L7543), mouse mAb for p53 (DO1) and MDM2 (4B11) (Millipore-Sigma Cat#OP43 and cat# OP143), rabbit MDM4 (Proteintech cat# 17914–1-AP), rabbit mAb FTH1 (Cell Signaling #4393S) and rabbit polyclonal GAPDH (FL-335) (Santa Cruz Biotechnology, sc-25778). P53 ubiquitination assay was performed as previously described (25).
Immunofluorescence
Cells were plated in wells of 24-well plates with glass cover slips and allowed to attach to cover slips overnight. Following drug exposure, cells were fixed and permeabilized with methanol. Slides were blocked in 3% FBS and then the primary antibody was added overnight. Fluorescent secondary antibodies, goat-anti-mouse IgG (H+L)-Alexa Fluor 594 (Jackson Immunoresearch Labs, cat# 115–585-166) or Donkey Anti-Rabbit IgG (H+L)-Cy3 (Jackson Immunoresearch Labs, cat# 711–165-152) were added for 1 hour at room temperature. Nuclei were counterstained with DAPI (5mg/ml) at1:10,000 dilution and coverslips mounted using propyl gallate. Cells were imaged using a Zeiss Axioplan 2 fluorescent microscope mounted with a Hamamatsu ORCA-ER camera.
Measurement of reactive oxygen species (ROS)
Cells were plated in 6 cm dishes in ~80% confluency and allowed to attach overnight. Cells were then treated with different doses of MMRi62 for 72 hours followed by harvesting and washing with 1X PBS. Cells were stained with Cellular ROS red (0.5 μL / mL assay buffer, abcam #186027) using phenol red-free media containing 2% FBS. After 30 min of incubation in the dark at 37°C, dye-containing media was removed by centrifugation and cells were suspended in phenol red-free media containing 2% FBS. Cells were then analyzed by flow cytometry (LSR IIA, BD Biosciences) using the PE Texas Red channel and results were quantified using WinList 3D 7.1 software. Data is presented as fold change compared to control.
Knocking down p53 expression by lentiviral shRNA
Cells were infected with retrovirus containing the lentiviral vector pLKO.1-shp53 DNA (addgene #19119 (26)). Cells were cultured with puromycin for 2 weeks, and puromycin resistant clones were selected. Knockdown of p53 was detected by Western blot analysis with anti-p53 antibody.
Clonogenic assays
Two thousand cells per well were played on 6-well plates one day before and the cells were treated with MMRi62 at different concentrations for 24 hours. Then, cells were replenished with drug-free complete medium and cultured for 2 weeks during which the cell culture was replenished with fresh medium every 3 days. Cells were fixed with cold methanol and stained with 0.05% (w/v) crystal violet solution at the end of 2-week period of cell culture.
Spheroid assay
Dissociated single cells (1000 cells/well) into ultra-low attachment 96 well plates (Fisher, Cat# 07200603) were cultured in 50 μl medium mixed with 3% Matrigel for 72 h. The spheroids were treated by adding 100 μl of DMSO or 4μM MMRi62-containing 3%-matrigel in medium continuously. The plates with spheroids were then scanned on different days and the resulting images were stored as high resolution (1,200 dpi) grayscale TIFF files. Signal intensity and density were quantified using Image-Pro Plus (Media Cybernetics).
Orthotopic pancreatic tumor implantation
All animal experiments have been conducted in accordance with an IACUC protocol approved by Institutional Animal Care and Use Committee (IACUC) of Roswell Park Comprehensive Cancer Center. Orthotopic tumors were established in female SCID mice at 4–6 weeks of age. While under Isoflurane anesthetization, a 1 cm incision was made in the left abdomen of the animal to exteriorize the pancreas. Longitudinal treatment response studies were carried out with an established orthotopic model of pancreatic cancer by implanting 0.5 × 106 /50μl of Panc1luc or BxPc3luc cells in a PBS-Matrigel mixture. After injection into the pancreas, the incision was sutured aseptically. Mouse body weight was measured on a balance and tumor size was measured by bioluminescence imaging two to three times per week. Once the tumor became well-established 8 days after tumor implantation, mice were randomized according to tumor size and treatment with MMRi62 was started. The mice were injected with vehicle or MMRi62 at a dose of 25 mg/kg in a formulation of 1.25% MMRi62–10 % N-Methyl-2-pyrrolidone-10% Cremphor-EL-78.75% PBS intraperitoneally twice per week. The tumor growth and body weight were measured every week for 31 days.
Cell migration and invasion assay
The transwell chamber (pore size, 8.0 μm; Millipore, USA) with Matrigel (for invasion assay) coating was inserted into a 24-well culture plate. For the invasion assay, 8 μm pore inserts were coated with Matrigel (BD Biosciences). PDAC cells in log phase growth were cultured in 6-well plates in medium containing 1% FBS for 24 h before drug treatment. The PDAC cells (100 μl, 1×105 ) suspended in DMEM containing 1% FBS with or without 4μM MMRi62 were seeded in the top chamber, and 500 μl of medium containing 20% FBS was placed in the lower chamber as a chemoattractant. The transwell chamber was incubated for 24 h. The invaded cells on the bottom surface of filter were fixed in methanol and stained with 0.05% w/v crystal violet solution for 20 min following by wash with running water in a bucket. Cell migration and invasion was determined by counting the stained cells under a light microscope in 10 randomly selected fields.
Wound healing assay.
Pancreatic cancer cells were grown to confluence in 24-well plates. The monolayer was then artificially wounded using a sterile 200-μl pipette tip. Cell debris was removed by washing the monolayer with PBS. The cells were then incubated with medium containing 4 μM MMRi62. Wound closure was monitored by images taken at various time points at the same spot with an inverted microscope equipped with a digital camera showing cell migrated into the wound. The extent of healing was calculated as the ratio of the remaining wound areas to the original wound area.
Statistical analysis
All the IC50 results were obtained from triplicate experiments and each individual experiment had triplicate at each dose point. The immuno-blotting experiments were repeated at least three times and the representative data were presented. Statistical analyses were performed using PRISM6 Software (GraphPad Software, Inc.), ANOVA, and the Student t test. Statistical significances indicated with * were determined by P < 0.05.
Results
MMRi62 inhibits PDAC proliferation, clonogenic and spheroid growth by cell death induction in vitro
We previously reported identification of RING-domain inhibitors of MDM2-MDM4 E3 complex in a high-throughput screen. We identified several hits which we called MMRi which stands for MDM2-MDM4 RING domain inhibitors (27). MMRi6 as one of the primary hits belongs to quinolinol class which is drawing attention in drug development (28, 29). MMRi62 was identified in a cell-based screen using leukemia/lymphoma cell lines as a potent apoptosis inducer among MMRi6 analogs (Fig.1A top for MMRi62 structure). Mechanism of action studies uncovered that MMRi62 promotes MDM4 degradation and p53-independent apoptosis in leukemia cells (Lama R et al, unpublished observation). To explore whether MMRi62 has antitumor activity in solid tumors, we included MMRi62 in a panel of therapeutics in a Panc1 cell-based screen. We observed that MMRi62 did inhibit Panc1 cell proliferation in addition to gemcitabine. We then tested MMRi62 in multiple PDAC cell lines for which their KRAS and TP53 status could be retrieved from literature. Our result showed that four of six PDAC cell lines were sensitive to MMRi62 with an IC50 ranging from 0.59 to 1.65 μM in a 72h proliferation assay (Fig.1A, middle) and three lines showed intrinsic resistance to MMRi62 with IC50 of ~ 10μM. Although our cell line panel is small, it appeared that transcriptional subtype of PDAC determines MMRi62 sensitivity since three of the four MMRi62-sensitive cell lines fell into quasimesenchymal (QM-PDA) subtypes and the three resistant cell lines fell into classical/epithelial subtype as defined by transcriptome profiling (30, 31). Given the MMRi62 sensitivity is not dependent of p53 status, we focused on cell lines Panc1 (KRASG12D:TP53R273) and BxPc3 (TP53Y220) bearing mutant p53 since it occurs in 60–70% PDAC (12, 13). To better understand how MMRi62 inhibit PDAC proliferation, we performed trypan blue exclusion assay and found that MMRi62 treatment for 24h induced loss of cell viability with increased trypan blue-positive cells in MMRi62 concentration-dependent manner with significant difference as compared with non-treated cells (Fig.1 B, top, representative images, bottom quantification of live cells). We then performed clonogenic assay with three cell lines including Panc1, BxPc3 and HPAFII. HPAFII was included because it bears a TP53P151S mutant that confers resistance to cell death by anoikis in head-and neck cancer cell lines (32). Pretreatment with MMRi62 for 24h followed by clonogenic growth in the absence of the compound effectively inhibited PDAC cell colony formation in all three cell lines in a MMRi62-concentration dependent manner (Fig. 1C). To investigate whether the prodeath effect of MMRi62 goes beyond 2-dimensional culture system, we performed experiments with PDAC spheroids. The Panc1 and BxPc3 spheroids were treated with solvent DMSO or 4μM MMRi62-containing medium and spheroid images were taken on days 1, 4, 7 and 10. As shown in Fig.1D for the representative spheroid images and Fig.1E for the median diameter of spheroids, MMRi62 significantly inhibited spheroid growth as the diameter of spheroids in DMSO control group increased from 570 μm on day 1 to 720 μm on day 10 whereas the diameter of spheroids decreased from 505 μm on day 1 to 287.5 μm on day 10 in the MMRi62 group (Fig.1D). The difference between DMSO and MMRi62-treated group was statistically significant (Fig.1E) for both Panc1 and BxPc3 cells. Hematoxylin and eosin (HE) stain of the spheroids collected on 10 days showed Panc1 formed spheroid structure with irregular edges and BxPC-3 formed compact spheroid structures with vacuole inside the spheroids (Fig.1D, HE).
Fig.1. selectively inhibits proliferation and induces cell death in (QM-PDA) subtypes pancreatic cancer lines.

A, Top, chemical structure of MMRi62. Middle, growth curves of pancreatic cell lines in the presence of MMRi62 in 72h proliferation assay. Bottom, the IC 50 concentrations for each cell line and their RAS and TP53 status of each cell line was shown. B, Top, MMRi62 induces pancreatic cancer cell death. Representative images of trypan blue exclusion assay in MMRi62-treated Panc1 and BxPc3 cells at indicated concentrations for 24h. C, nontreated control. Bottom, Quantitative results of live cell fractions for Panc1 (left) and BxPc3 (right). *P < 0.05. C, Effect of MMRi62 on clonogenic growth. Cells were treated with indicated concentrations of MMRi62 for 24h followed culturing in the absence of the drug for 14 days before violet blue staining. D, Left, the shapes of spheroids of Panc1 and BxPc3 after 4mM MMRi62 for continuous treatment under light microscope at various days (D1 to D10) d and the HE staining of the final day cultures were shown. E, Quantification of the average spheroid diameter (SD in mm) at various time points for Panc1 (upper) and BxPC-3 (lower)
MMRi62 induces ferroptosis and p53 downregulation
To understand the molecular events involved in MMRi62 induced PDAC cell death, we first investigated how MMRi62 affect expression of MDM4, MDM2 and p53 since they were the initially identified targets of MMRi62. While MMRi62 treatment at low μM range for 24h did not induce measurable downregulation of MDM4 or MDM2 in Panc1 cells by Western blotting analysis, it did consistently downregulate MDM2 in BxPc3 cells (Fig.2A). However, MMRi62 does not downregulate MDM2 and MDM4 in PDAC cells as effectively as in leukemia/lymphoma cells (Lama, et al unpublished data). Interestingly, we observed a moderate downregulation of p53R273H in Panc1 and significant downregulation of p53Y220C in BxPc3 after 24h treatment (Fig.2A).
Fig.2. induces autophagic cell death associated with downregulation of p53.
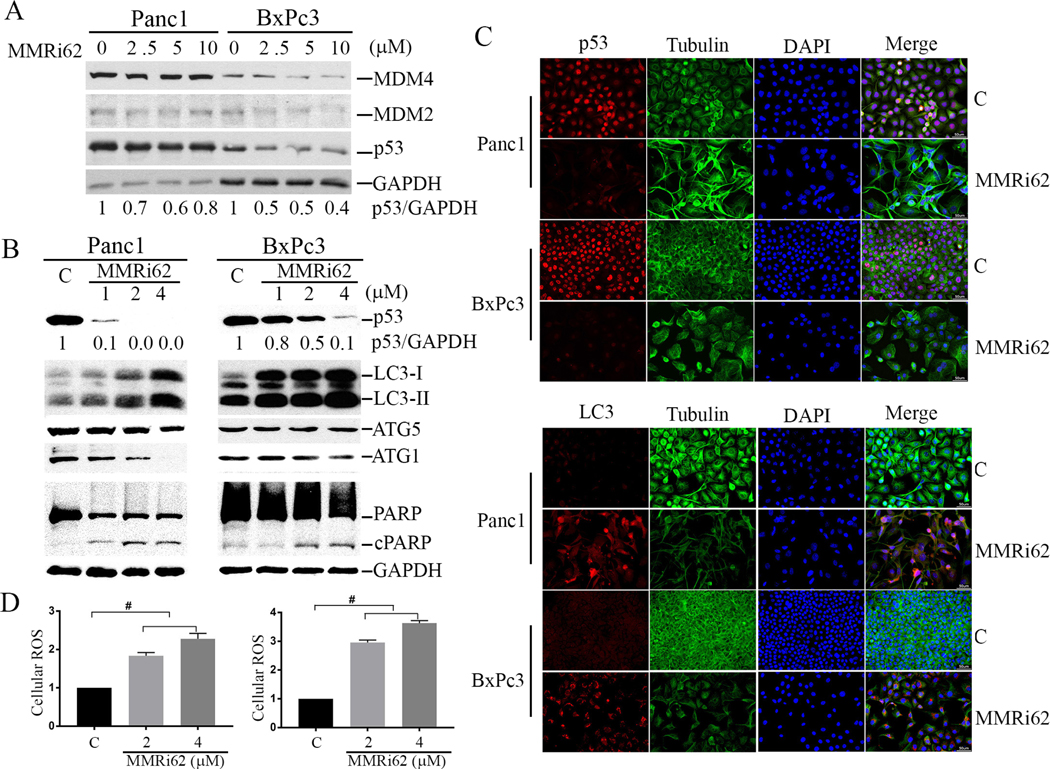
A, MMRi62 induces downregulation of p53 expression in pancreatic cancer cells. WB analysis of p53, MDM2 and MDM4 in Panc1 and BxPc3 treated for 24h with indicated concentrations of MMRi62 (μM). B, MMRi62 induces increased autophagy and apoptosis. WB analysis of p53, cleaved PARP and LC3 in Panc1 or BxPC3 cells treated with MMRi62 at indicated concentrations for 72h. C, IF staining of p53 and LC3 in Panc1 and BxPc3 after 72h treatment with 2mM MMRi62. D. MMRi62 treatment induces increase in cellular ROS after MMRi62 treatment at two concentrations for 72h. Left, Panc1, p=0.0003 and <0.0001 for C vs 2 and 4 μM MMRi62 respectively; right, BxPc3, p=<0.0001 for C vs 2 or 4 μM MMRi62 respectively.
Although MMRi62 also downregulates wt-p53 in Capan2 cells (Supplementary Fig 1), MMRi62 effect on mut-p53 downregulation in Panc1 and BxPc3 cells is significantly stronger. Of note, mut-p53 downregulation by MMRi62 in PDAC cells is of slow kinetics: extended treatment with MMRi62 for 72h caused more significantly downregulation than 24h treatment (compare Figure 2A and 2B). We then looked at the molecular events involved in apoptosis, i. e. increased caspase 3-mediated PARP cleavage (cPARP) in a prolonged treatment of 72h. Our results indicated that MMRi62 only slightly increased the levels of cPARP in the two cell lines, indicating that apoptosis was induced but was not likely the leading cause of cell death. Since PDAC cells are predisposed to ferroptosis, a mode of cell death initiated by dysregulation of iron metabolism(17, 18), we then investigated the hallmarks of ferroptosis, increased autophagy activity using LC3-phosphatidylethanolamine conjugate (LC3-II) as a marker (18). We found that MMRi62 significantly increased LC3B-II levels in a dose-dependent manner, which is in sharp contrast to a significant decrease in p53 levels in 72h-treated cells (Fig.2B). Interestingly, phospholipid-unconjugated LC3-I was also increased (Fig.2B, LC3-I). This increased autophagy activity was not associated with changes in ATG5 expression and occurred even in the presence of a slight decrease in ATG1 in Panc1 cells. Immunofluorescence (IF) microscopy confirmed that MMRi62 treatment downregulated p53 expression and increased LC3 expression in both cell lines (Fig.2C). Similarly, p53 downregulation was also confirmed by IF in MMRi62-treated spheroids of Panc1 and BxPc3 cells (Supplementary Fig 2A). Then we measured the production of cellular reactive oxygen species (ROS), another feature of ferroptosis. Our results showed that MMRi62 significantly increased cellular ROS in a dose-dependent manner as compared to non-treated cells (Fig.2D). Taken together, these results suggest that MMRi62 induces ferroptosis in PDAC cells associated with p53 downregulation.
MMRi62 induces lysosomal degradation of FTH1 and proteasomal degradation of p53 in PDAC cells
To confirm that MMRi62 induces ferroptosis, we determined the expression of ferritin heavy chain (FTH1) and ferritin receptor NCOA4, two negative regulators of ferroptosis that are downregulated during ferroptosis (33, 34). Our results showed that MMRi62 treatment induced significant decrease in NCOA4 and FTH1 protein expression in both Panc1 and BxPc3 cells (Fig.3A). We confirmed with IF microscopy that the MMRi62-induced NCOA4 downregulation also occurred in the spheroids of Panc1 and BxPc3 cells (Supplementary Fig 2B). To understand how MMRi62 induces downregulation of p53 and FTH1 in PDAC cells, we performed rescue expression using proteasomal inhibitor Carfilzomib (CFZ) and lysosomal inhibitor Bafilomycin A1 (BAF). Our results showed that CFZ nearly fully rescued p53, a minor rescue of NCOA4 but not FTH1, while BAF1 nearly fully rescued both FTH1 and NCOA4 downregulation in Panc1 cells and partially in BxPc3 cells but showed no effect on mut-p53 downregulation in both cell lines (Fig.3B). BAF rescue of FTH1 and NCOA4 degradation also suggests that MMRi62-induced increase of LC3-I and LC3-II is due to activation of autophagy rather than inhibition of lysosomal function. The partial rescue of NCOA4 and FTH1 downregulation in BxPc3 cells suggests possible transcriptional mechanisms might be involved in addition to the lysosomal degradation mechanism. As expected, MMRi62 treatment increased ubiquitinated mut-p53 in Panc1 cells in in vivo ubiquitination assays in which His-ubiquitinated proteins were brought down by nickel beads followed by WB of mut-p53 in Panc1 cells (Fig.3C). These results suggest that MMRi62 promotes proteasomal degradation of mut-p53 but lysosomal degradation of NCOA4 and FTH1.
Fig.3. induces lysosomal degradation of FTH1 and NCOA4 and proteasomal degradation of mut-p53 in PDAC cells.

A, WB analysis of NCOA4 and FTH1in Panc1 and BxPc3 cells treated with indicated concentrations of MMRi62 (μM) for 24h. Tubulin and GAPDH were used as loading controls. B, MMRi62 promotes lysosomal degradation of FTH1and NcoA4 and proteasomal degradation of mut-p53. Upper, WB analysis of the indicated proteins in Panc1 treated with indicated concentrations of MMRi62 for 24h in the presence of either 50nM of Bafilomycin B1 (BAF) or 200 nM Carfizomib (CFZ). Lower, the same as in Upper except for using BxPC3 cells., C. In vivo ubiquitnation of mut-p53 in MMRi62-treated Panc1 cells. Panc1 cells were transfected with His-tag-ubiquitin expression plasmid followed by MMRi62 treatment and his-tag pulldown and WB for p53 and polyubiquitin. Ub-p53 and polyubiquitin adducts were indicated. D, Knockdown of mut-p53 increases FTH1 expression. WB analysis of indicated proteins in BxPc3 or HPAFII cells after infection with lentiviral expressing control shRNA (shSer) or shp53. GAPDH serves as loading control. E, IC50 changes in the indicated cells.
Downregulation of mut-p53 minimally contributes to MMRi62 anti-PDAC activity
To understand whether p53 expression has direct effect on expression of FTH1 and NCOA4 and how mut-p53 is involved in MMRi62 anti-PDAC activity, we performed experiments with p53 knockdown cells. We obtained efficient p53 knockdown in BxPc3 and HPAFII, but not in Panc1 cells. Knockdown of mut-p53 increased expression of NCOA4 and FTH1, the two negative regulators of ferroptosis, suggesting that high expression of mut-p53 predisposes PDAC cells to ferroptosis (Fig. 3D). To understand how mut-p53 knockdown affects MMRi62 sensitivity, we performed proliferation assay in the presence of MMRi62 with the p53 knockdown cells. The IC50 of MMRi62 was slightly increased in both shp53-BxPc3 cells and shp53-HPAFII cells compared shRNA control cells (Fig. 3E), suggesting a minor contribution of mut-p53 status to MMRi62 induced anti-growth effect. Since mut-p53 knockdown increases NCOA4 and FTH1, these results suggest that MMRi62 induced downregulation of FTH1/NCOA4 and mut-p53 are independent effects of MMRi62, and MMRi62-induced cell death is independent of p53 downregulation and p53’s role in regulating the expression of NCOA4 and FTH1 (Fig. 3D-E).
MMRi62 inhibits pancreatic tumor growth in orthotopic xenograft mouse models
We next examined MMRi62’s in vivo efficacy in orthotopic tumor models with Panc1 and BxPc3 cells stably expressing luciferase for bioluminescence imaging (BLI). The experiments were performed by treating the nude mice bearing the Panc1luc or BxPc3luc orthotopic tumors with vehicle or MMRi62 (25 mg/kg/day, 2 days/week for 2 weeks) by intraperitoneal injection. Mouse body weight was weighed, and tumor growth was measured by BLI on days 9, 18, 28 and 37 after tumor implantation. Our results showed that MMRi62 treatment caused body weight loss at day18 but within 20% range and started to recover by day 28 and 37 (Fig.4A & 4B, left middle). MMRi62 significantly suppressed the growth of Panc1luc by 52.71% (P < 0.05) and 60.77% of BxPc31luc orthotopic tumors compared with the tumors in control animals (Fig. 4A & 4B, middle right). The average wet weights of tumor collected at the end of the experiment were 2.85g (Panc1luc) and 2.81g (BxPc3luc), respectively in MMRi62 group compared with 3.99g (Panc1luc) and 4.29g (BxPc3luc) in the vehicle-treated control group with significant differences (P=0.0002 and, 0.001 for Panc1luc and BxPc3luc respectively, Fig. 4A & 4B, bottom left). The average long diameters of the tumors were 1.36cm (Panc1luc) and 1.48cm (BxPc3luc), respectively in MMRi62 group compared with 2.29cm (Panc1luc) and 2.47 cm3 (BxPc3luc), respectively in the control group with significant differences (P=<0.0001 for both Panc1luc and BxPc3luc, Fig. 4A & 4B, bottom right).
Fig.4. inhibits growth of pancreatic tumor in orthotopic xenograft mouse models associated with downregulation of p53 and NCOA4 expression.
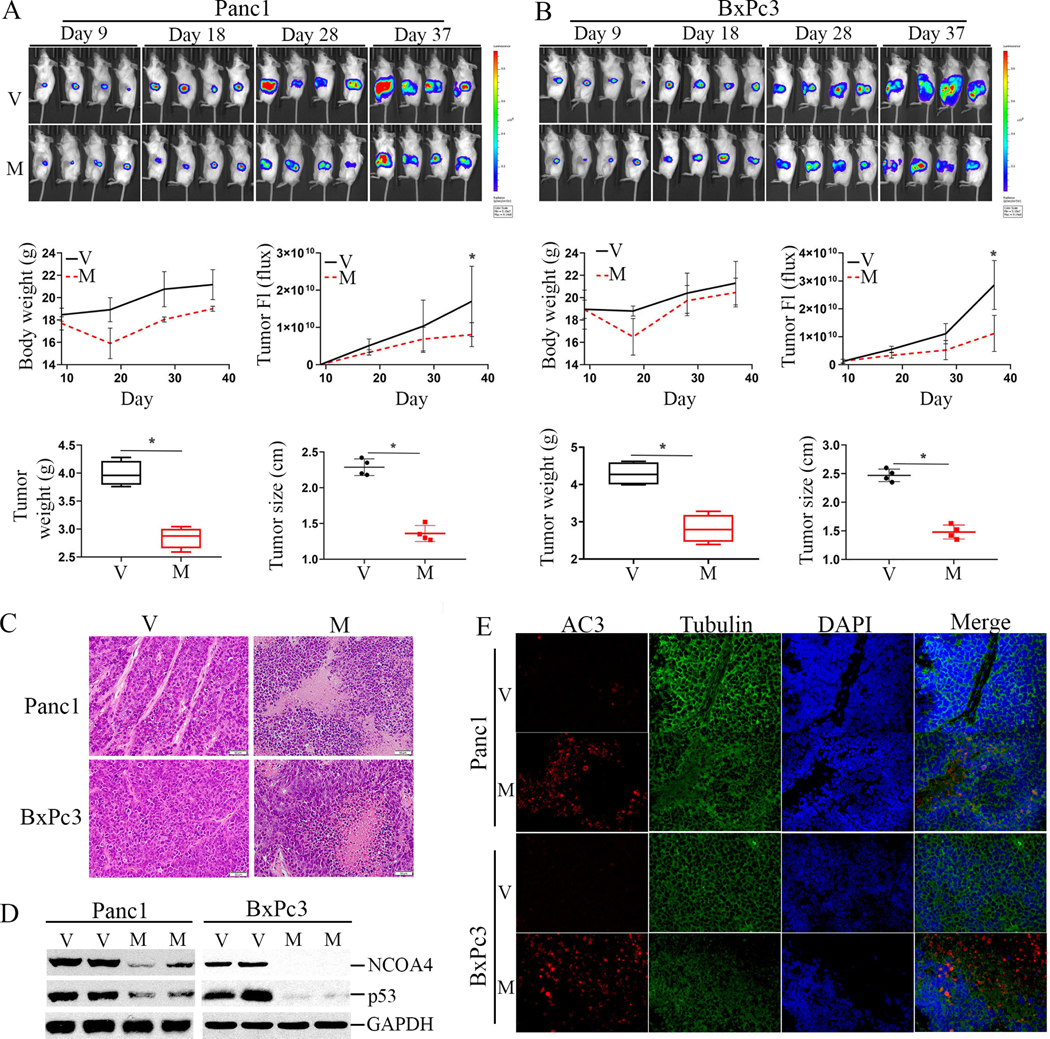
A, Top, Panc1luc cells were implanted orthotopically into the pancreas of SCID mice followed by MMRi62 treatment via intraperitoneal injection at doses of 25 mg/kg at every other day for 5 injections, 2 days/week for 2 weeks. The dorsal luciferase signals in the mice bearing Panc1luc orthotopic tumors were captured in different days during and after treatment. Middle, mouse body weight (left) and tumor BLI signal in Flux (Right) measured in indicated days in vehicle-control (V) mice and MMRi62-treated (M) mice were shown. Bottom, At the termination day of the experiments, median tumor weight in gram and median tumor size in cm in the two groups of mice were shown. *P =0.0002 and <0.00001 for Panc1 tumor weight and tumor size between V and M groups; and *P =0.001 and <0.0001 for BxPc3 tumor weight and tumor size between V and M groups. B, the same in A except for BxPc3luc cells. C, HE staining of Panc1luc and BxPc3luc orthotopic tumors collected on Day 37 in MMRi62-treated (M) as compared with vehicle-treated mice (V). D, WB analysis of the resected tumor tissues on Day 37 for the expression of p53 and NCOA4 in V and M group of mice. E, Immunofluorescence staining of activate caspase 3 (AC3) for in Panc1luc and BxPc3luc orthotopic tumors collected on Day 37 in MMRi62-treated (M) as compared with vehicle-treated mice (V).
We validated the death mode and changes in p53 and NCOA4 in these orthotopic tumors. Results of HE staining showed Panc1luc and BxPc3luc orthotopic tumors manifested necrotic tissue structures in MMRi62 group (Fig.4C). MMRi62 treatment reduced the expression of p53 and NCOA4 in both Panc1luc and BxPc3luc orthotopic tumors compared with vehicle groups (Fig.4D, compare V with M in duplicates). Results from IF microscopy for activated caspase 3 showed increase AC3 staining, suggesting a death mode composed of apoptosis and necrosis (Fig.4E).
MMRi62 prevents pancreatic cancer metastasis in vivo and invasion/migration in vitro
Early metastasis is one of the major reasons for low 5-year survival rate of PDAC patients in which mut-p53 plays a critical role (14, 15). Owing to MMRi62’s activity to downregulate mut-p53, we determined whether MMRi62 inhibits cell migration and invasion in PDAC cells. Our results from wound healing assays showed that non-treated Panc1 cells migrated into the entire wounded area by 72 hours, whereas treatment with 4 μM MMRi62 significantly inhibited Panc1 cell migration (Fig. 1A, left, compare C with M). Similarly, non-treated BxPc3 cell migrated into the entire wounded area by 48 hours, whereas treatment with 4 μM MMRi62 significantly inhibited BxPc3 cell migration (Fig. 1A, right, compare C with M). We used transwell assay to test the effect of MMRi62 on cell invasion, MMRi62 (4 μM) reduced the invasion of Panc-1 cells (Fig. 1B, left) by 46.5% (Fig. 1B, right) and of BxPc-3 cells (Fig. 1C, left) by 70.0% (Fig. 1C, right) as compared with the non-treated controls. We next examined the effect of MMRi62 on PDAC metastasis in mice bearing Panc1luc and BxPc3luc orthotopic tumors in the experiment described in Fig.4. Necropsy showed that 3 out of 4 of the Panc1luc-bearing mice and 2 out of 4 of BxPc3luc-bearing mice in the vehicle group developed metastatic lesions in the peritoneum, whereas none of the mice treated with MMRi62 had peritoneal dissemination in both Panc1luc and BxPc3luc models (Fig. 1D, small red dot). Liver metastasis occurred in 1 out of 4 mice in the vehicle-treated mice in both Panc1luc and BxPc3luc models. However, none of the MMRi62-treated mice in both models developed liver or lung metastasis (Fig. 1D, red cube & large red dot). These results were confirmed by histopathologic examinations of the liver tissues from mice in the control and treatment groups (Fig. 1E). These data suggest that MMRi62 abrogated PDAC metastasis in these two PDAC models.
Fig.5. prevents invasion in vitro and metastasis of pancreatic cancer cells in vivo.

A, MMRi62 treatment inhibits cell migration in wound-healing assay. Confluent monolayers of Panc1 (Left) and BxPc3 cells (right) were scratched with a 1mL pipette tip at experimental time zero and then were cultured with or without presence of MMRi62 (4mmol/L). The images of cell monolayers were taken at 24, 48 and 72h after the scratch and representative images were shown. Quantification of migration distance (MD in μm) were shown in histograms on left. B, MMRi62 inhibits Panc1 cell invasion in transwell assay. Left, a representative image of Panc1 cells transpassed to the other side of membrane when cells are cultured in medium without (C) or with 4mM MMRi62 (M) for 24h. Right, quantitative analysis of the transpassed cell numbers in this assay. C, MMRi62 inhibits BxPc3 cell invasion in transwell assay. The experiment was done similarly as in B except with BxPc3 cells. D, MMRi62 inhibits metastasis in orthotopic PDAC tumors. On the termination day of the in vivo efficacy experiments, mice bearing orthotopic tumors of Panc1 (left) and BxPc3 (Right) were examined for tumor metastasis to various organs. The numbers of mice with metastasis to the peritoneum, liver, and lungs were counted and presented. E, HE staining of liver metastasis in mice bearing Panc1 and BxPc3 metastasis orthotopic tumors in vehicle control (C) and MMRi62-treated (M) mice were shown. Dark and dense areas are metastatic tumors in C group.
MMRi62 inhibits proliferation of gemcitabine resistant PDAC cells
We investigated whether combination of MMRi62 and current chemotherapy gemcitabine could synergistically inhibit PDAC proliferation. Our synergism analysis indicated a weak synergism between 30–75% inhibition effect levels with combination index (CI) being about 0.7 to 0.5 (Fig.6A), suggesting that simultaneous administration of MMRi62 and gemcitabine may have synergism in low effect range but risks antagonism in high effect range. We further tested MMRi62 sensitivity in established gemcitabine-resistant BxPc3 cells (Gem-R-BxPc3 and Gem-R-HPAFII). Our results indicated that Gem-R-BxPc3 and Gem-R-HPAFII were extremely resistant to gemcitabine with IC50> 100μM while BxPc3 is extremely sensitive to gemcitabine (IC50, 0.047μM) and HPAFII is relatively resistant to gemcitabine (IC50, 8.06μM). However, MMRi62 sensitivities of Gem-R-BxPc3 and Gem-R-HPAFII were slightly reduced compared to parental cells: IC50=2.38±0.88 and 3.72±0.36μM, respectively for BxPc3 and Gem-R-BxPc3 and IC50=4.04±0.59 and 6.62±0.52μM, respectively for HPAFII and Gem-R-HPAFII) (Fig.6B & C). These results suggest that MMRi62 if used in sequential combination might be a strategy for killing gemcitabine-resistant cells.
Fig.6. Gemcitabine resistant PDAC cells remain sensitive to MMRi62 in vitro.

A, Combination index (CI) plots for Panc1 cells (top) and BxPc3 cells (bottom) derived from dose-effect data of MMRi62 combination with gemcitabine in 72h proliferation assay. Gem, gemcitabine. CI <1, synergism, CI=1, additive, CI>1, antagonism. B, drug response curves of BxPc3 and Gem-R-BxPc3 cells to gemcitabine (top) and to MMRi62 (bottom, IC50=2.38±0.88 and 3.72±0.36μM, respectively for BxPc3 and Gem-R-BxPc3). C, drug response curves of HPAFII and Gem-R-HPAFII cells to gemcitabine (top) and to MMRi62 (bottom, IC50=4.04±0.59 and 6.62±0.52μM, respectively for HPAFII and Gem-R-HPAFII).
Discussion
PDAC incidence has been on the rise and chemoresistance, both intrinsic and acquired, poses significant challenges to current combination therapies (35). Active autophagy plays an essential role in PDAC development since mice lacking the autophagy genes Atg5 or Atg7 accumulate low-grade, pre-malignant pancreatic intraepithelial neoplasia lesions without progressing to high grade pancreatic intraepithelial neoplasia and PDAC (36). Elevated autophagy in PDAC provides a surviving mechanism to resist to die under nutrient limitation of tumor microenvironment or therapeutic treatment (37). Consequently, depletion of autophagic machinery components, or hydroxychloroquine (HCQ) inhibition of autophagy, suppressed PDAC proliferation and decreased tumorigenicity in vivo (38). Autophagy is bulk degradation of proteins and other macromolecules for recycling building blocks and survival of nutrient-scarce conditions. However, paradoxically, ferritinophagy is an autophagic degradation of ferritin triggers cell death called ferroptosis (18). Ferroptosis as a preferred mode of cell death for KRAS mutant PDAC cells was discovered in studies with a RAS-selective lethal compounds like erastin and RSL3 (17). Cellular targets identified with different ferroptosis inducers include voltage-dependent anion channel (VDAC), cystine/glutamate antiporter system, GPX4 and GSH as cellular targets in ferroptosis induction (39). Ferroptosis is associated with NCOA4-mediated ferritinophagy, increased ROS and deregulation of cellular iron (18). Currently, no ferroptosis inducer agents for PDAC therapy are available in clinic. In this report, we described MMRi62 as a new ferroptosis inducer in PDAC cells that inhibited PDAC growth, cell migration in vitro and metastasis in vivo.
MMMRi62 is small molecule quinolinol initially identified as a RING domain modifier compound that induces preferential MDM4 degradation by MDM2-MDM4 E3 complex and p53-independent apoptosis in leukemia cells (Lama R et al, unpublished observation). Except for targeting MDM2-MDM4, whether MMRi62 has other cellular targets is currently unknown. This study discovers a novel pro-ferroptosis activity of MMRi62 associated with induced mut-p53 and FTH1 degradation. Our data suggest that the anti-PDAC effect of MMRi62 involves a mixture of death modes of apoptosis and ferroptosis, although necrosis might also be involved. The contribution of apoptosis may be limited since caspase 3 activation (Fig.4) is associated with weak downstream effect of PARP cleavage (Fig.2). In contrast, ferroptosis appears to be the dominating death mode since MMRi62 treatment caused robust lysosome-dependent downregulation of NCOA4 and FTH1 associated with significantly increased autophagy and ROS production (Fig.2, Fig.3, Fig.4). MMRi62 induced ferroptosis is not strictly dependent on KRAS mutation since it also occurs in wt-KRAS BxPc3 cells. This is in line with literature showing that KRAS mutation is not a prerequisite in ferroptosis since erastin-induced ferroptosis requires KRAS mutation in PDAC but is antagonized by mutant KRAS in RMS13 rhabdomyosarcoma cells (40). Our finding that MMRi62-induced ferroptosis is mutant-KRAS-independent provides a useful scaffold structure for developing new anti-PDAC ferroptosis inducers. NCOA4/FTH1 downregulation in ferritinophagy is a hallmark of ferroptosis reported with erastin (18). Consistent with reports that cancer cells with EMT phenotype are vulnerable to ferroptotic cell death due to their dependence on a lipid peroxidase pathway (41), we found that QM-PDA cells were more sensitive to MMRi62-induced cell death (Fig.1A). However, 5μM ferrostatin-1 failed to rescue MMRi62-induced ferroptosis or FTH1 degradation in Panc1 and BxPc3 cells, suggesting that MMRi62-induced ferroptosis does not involve lipid oxidation at which ferrostatin-1 inhibits and rescues erastininduced ferroptosis (42). We propose that MMRi62-induced FTH1 degradation triggers ferroptosis, since FTH1 has been validated genetically as a key target in ferroptosis in a report showing FTH1-loss causes massive ferroptosis in the developing wing of Drosophila associated with production of reactive oxygen species (33). Currently, how MMRi62 induces lysosomal degradation of NCOA4 and FTH1 and whether NCOA4 and FTH1 are direct targets of MMRi62 is under investigation. Nevertheless, this study identified MMRi62 as a potent FTH1 degradation inducer useful for FTH1 targeting.
Role of p53 in ferroptosis regulation is cell type and ferroptosis inducer dependent (43). Wt-p53 positively regulates ferroptosis by transcriptionally repressing SLC7A11, a component of anti-oxidant system Xc- in U2OS and MCF7 cells (44) but it inhibits ferroptosis by inhibiting dipeptidyl-peptidase-4 (DPP4)-dependent lipid peroxidation in colon cancer cells (45). Mutant p53 was reported to sensitize cancer cells to ferroptosis involving reduced expression of SLC7A11(43, 46). We found that mut-p53 might predispose PDAC cells to ferroptosis by reduced expression of FTH1/NCOA4 (Fig.3D). This projection was confirmed by a slightly decreased MMRi62 sensitivity in shp53 PDAC cell lines (Fig.3E). Therefore, although MMRi62 promotes mut-p53 degradation (Fig.2 & 3), MMRi62-induced ferroptosis is likely mediated by FTH1 downregulation. A surprising in vivo result was that MMRi62 completely suppressed PDAC metastasis in two orthotopic mouse models. This effect is likely mediated at least partially by mut-p53 downregulation which occurred in those tumor tissues (Fig.5D), since mutant p53 is an established metastasis driver in PDAC mouse models (14). Due to limited number of animals in this study, the MMRi62 effect on metastasis warrants further study using a larger cohort of animals.
TP53 mutation occurs in 75% of invasive pancreatic adenocarcinomas (12) and mut-p53 proteins are often stable and accumulate at high levels in cancer cells in contrast to the fast turnover and low expression of wt-p53 protein in normal cells. Strategy to reactivate mut-p53 for cancer therapy (47) may be challenged with limited efficacy since mut-p53 is often found in late stage cancers in which the p53 downstream pathways are often disrupted together with other oncogenic mutations such as MYC, RAS and/or PI3K(48). MMRi62 might be an alternative strategy to eliminate mut-p53mediated oncogenic activities including metastasis. In this sense, our finding places MMRi62 in a category of mut-p53-targeting agents. Taken together, this study identified MMRi62 as a new class of ferroptosis inducer with distinct mechanisms of action than the reported ferroptosis inducers (39). The dual targeting activity for mutp53 and FTH1 renders MMRi62 an advantage over other ferroptosis inducers. Further studies on underlying mechanisms of ferroptosis induction by MMRi62 and development of better MMRi62-like small molecules may bring about promising new therapies for recalcitrant cancers such as PDAC.
Supplementary Material
Significance:
These findings discover a new class of ferroptosis inducer for suppression of PDAC growth and metastasis.
Acknowledgments
This work was supported in part by grants from R01CA208352 (XW), Roswell Park Alliance Foundation (XW and SRC) and by the National Natural Scientific Foundation of China (JL, No. 81472246). The Roswell Park Comprehensive Cancer Center Core Grant CA16056 from NCI is gratefully acknowledged. The authors also thank Roswell Park Small Molecule Screening Shared Resource for the initial high throughput screening of MMRi preliminary hits and Roswell Park’s Translational Imaging Shared Resource (TISR) for BLI imaging. We thank Ms. Yuping Wang for her technical assistance.
Financial support:
R01CA208352 grant from National Cancer Institute, NIH, USA to XW; Roswell Park Alliance Foundation grant to XW and SRC, and a grant (No. 81472246) from National Natural Scientific Foundation of China to JL.
Footnotes
Conflict of interest statement: The authors declare no potential conflicts of interest.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest are declared by authors.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7–33. [DOI] [PubMed] [Google Scholar]
- 2.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–32. [DOI] [PubMed] [Google Scholar]
- 3.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–21. [DOI] [PubMed] [Google Scholar]
- 4.Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 5.Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol. 2018;15:333–48. [DOI] [PubMed] [Google Scholar]
- 6.Zeng S, Pottler M, Lan B, Grutzmann R, Pilarsky C, Yang H. Chemoresistance in Pancreatic Cancer. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nature reviews Cancer. 2009;9:749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Agostino S, Fontemaggi G, Strano S, Blandino G, D’Orazi G. Targeting mutant p53 in cancer: the latest insights. J Exp Clin Cancer Res. 2019;38:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. [DOI] [PubMed] [Google Scholar]
- 10.Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388:73–85. [DOI] [PubMed] [Google Scholar]
- 11.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 12.Kanda M, Sadakari Y, Borges M, Topazian M, Farrell J, Syngal S, et al. Mutant TP53 in duodenal samples of pancreatic juice from patients with pancreatic cancer or high-grade dysplasia. Clin Gastroenterol Hepatol. 2013;11:719–30 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Polireddy K, Singh K, Pruski M, Jones NC, Manisundaram NV, Ponnela P, et al. Mutant p53(R175H) promotes cancer initiation in the pancreas by stabilizing HSP70. Cancer Lett. 2019;453:122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weissmueller S, Manchado E, Saborowski M, Morris JPt, Wagenblast E, Davis CA, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell. 2014;157:382–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107:246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fiorini C, Cordani M, Padroni C, Blandino G, Di Agostino S, Donadelli M. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim Biophys Acta. 2015;1853:89–100. [DOI] [PubMed] [Google Scholar]
- 17.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanizsai I, Madacsi R, Hackler L Jr., Gyuris M, Szebeni GJ, Huzian O, et al. Synthesis and Cytoprotective Characterization of 8-Hydroxyquinoline Betti Products. Molecules. 2018;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joseph P. Errico IT, Matthew Sills, Jane Ong, Franklin Park, John Alloccoand Pam Wines. METHODS AND COMPOSITIONS OF TARGETED DRUG DEVELOPMENT. US patent. 2013. [Google Scholar]
- 21.Moore PS, Sipos B, Orlandini S, Sorio C, Real FX, Lemoine NR, et al. Genetic profile of 22 pancreatic carcinoma cell lines. Analysis of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch. 2001;439:798–802. [DOI] [PubMed] [Google Scholar]
- 22.Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ, et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994;54:3025–33. [PubMed] [Google Scholar]
- 23.Dalin S, Sullivan MR, Lau AN, Grauman-Boss B, Mueller HS, Kreidl E, et al. Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine Resistance. Cancer Res. 2019;79:5723–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81. [DOI] [PubMed] [Google Scholar]
- 25.Xu C, Fan CD, Wang X. Regulation of Mdm2 protein stability and the p53 response by NEDD4–1 E3 ligase. Oncogene. 2015;34:281–9. [DOI] [PubMed] [Google Scholar]
- 26.Godar S, Ince TA, Bell GW, Feldser D, Donaher JL, Bergh J, et al. Growthinhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu W, Xu C, Ling X, Fan C, Buckley BP, Chernov MV, et al. Targeting RING domains of Mdm2-MdmX E3 complex activates apoptotic arm of the p53 pathway in leukemia/lymphoma cells. Cell Death Dis. 2015;6:e2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thinnes CC, Tumber A, Yapp C, Scozzafava G, Yeh T, Chan MC, et al. Betti reaction enables efficient synthesis of 8-hydroxyquinoline inhibitors of 2-oxoglutarate oxygenases. Chem Commun (Camb). 2015;51:15458–61. [DOI] [PubMed] [Google Scholar]
- 29.Mohammed I, Hampton SE, Ashall L, Hildebrandt ER, Kutlik RA, Manandhar SP, et al. 8-Hydroxyquinoline-based inhibitors of the Rce1 protease disrupt Ras membrane localization in human cells. Bioorg Med Chem. 2016;24:160–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci U S A. 2015;112:E4410–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie TX, Zhou G, Zhao M, Sano D, Jasser SA, Brennan RG, et al. Serine substitution of proline at codon 151 of TP53 confers gain of function activity leading to anoikis resistance and tumor progression of head and neck cancer cells. Laryngoscope. 2013;123:1416–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mumbauer S, Pascual J, Kolotuev I, Hamaratoglu F. Ferritin heavy chain protects the developing wing from reactive oxygen species and ferroptosis. PLoS Genet. 2019;15:e1008396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarvepalli D, Rashid MU, Rahman AU, Ullah W, Hussain I, Hasan B, et al. Gemcitabine: A Review of Chemoresistance in Pancreatic Cancer. Crit Rev Oncog. 2019;24:199–212. [DOI] [PubMed] [Google Scholar]
- 36.Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. [DOI] [PubMed] [Google Scholar]
- 37.Boone BA, Zeh HJ 3rd, Bahary N. Autophagy Inhibition in Pancreatic Adenocarcinoma. Clin Colorectal Cancer. 2018;17:25–31. [DOI] [PubMed] [Google Scholar]
- 38.Bryant KL, Der CJ. Blocking autophagy to starve pancreatic cancer. Nat Rev Mol Cell Biol. 2019;20:265. [DOI] [PubMed] [Google Scholar]
- 39.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu J, Zhang C, Wang J, Hu W, Feng Z. The Regulation of Ferroptosis by Tumor Suppressor p53 and its Pathway. Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017;20:1692–704. [DOI] [PubMed] [Google Scholar]
- 46.Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, et al. Inhibiting the system xC(−)/glutathione axis selectively targets cancers with mutantp53 accumulation. Nat Commun. 2017;8:14844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duffy MJ, Synnott NC, Crown J. Mutant p53 as a target for cancer treatment. Eur J Cancer. 2017;83:258–65. [DOI] [PubMed] [Google Scholar]
- 48.Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nature reviews Cancer. 2018;18:89–102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.