

Abstract

A novel class of potent NaV1.7 inhibitors has been discovered. The replacement of diaryl ether in compound I was investigated to enhance mouse NaV1.7 inhibitory activity, which resulted in the discovery of N-aryl indoles. The introduction of the 3-methyl group is crucial for high NaV1.7 in vitro potency. The adjustment of lipophilicity led to the discovery of 2e. Compound 2e (DS43260857) demonstrated high in vitro potencies against both human and mouse NaV1.7 with high selectivity over NaV1.1, NaV1.5, and hERG. In vivo evaluations revealed 2e demonstrating potent efficacy in PSL mice with excellent pharmacokinetics.
Keywords: voltage-gated sodium channel (NaV), pain, acyl sulfonamide, indole
The inhibition of voltage-gated sodium channel 1.7 (NaV1.7) is a promising therapeutic target for treating various pain disorders,1,2 while it is also true that multiple selective NaV1.7 inhibitors failed to prove their utility in clinical trials. The pioneering molecule, PF-05089771, is a highly potent selective NaV1.7 inhibitor, which showed limited efficacy against patients with diabetic peripheral neuropathy. Although PF-05089771 was well tolerated, the elevation of cholesterol levels was reported in phase 2 clinical trial.3 Thus, further R&D activities are essential for acquiring novel analgesic agents.4−7
In the course of our research dedicated to acquiring potent selective NaV1.7 inhibitors,8−10 we focused on the subtype-selective acyl sulfonamide derivative I reported by the Pfizer group (Figure 1).11−14 Compound I exerted potent human NaV1.7 inhibitory activity, whereas modest mouse NaV1.7 potency was observed (hNaV1.7 IC50: 0.052 μM, mNaV1.7 IC50: 1.7 μM). As we believe our preclinical evaluation process including in vivo efficacy evaluation with neuropathic pain model mice enables the extrapolation to human efficacy, improving in vitro potency against mice is an essential task. Our strategy consisted of addressing such issues through the modifications of compound I. In this paper, we report the discovery and structure–activity relationship (SAR) of N-aryl indole derivatives with improved mouse NaV1.7 activity in vitro. The high mouse NaV1.7 activity with excellent PK profiles warranted in vivo evaluations. The optimized compounds demonstrated potent analgesic efficacy in vivo.
Figure 1.

NaV1.7 inhibitor I reported by the Pfizer group with IC50 values and molecular design of novel indole derivatives A and B. Data were obtained with a high-throughput electrophysiology system (IonWorks Quattro) in-house. Values are from a single experiment run in quadruplicate.
Initial efforts focused on converting the acyl sulfonamide moiety without success. Consequently, the modification of diaryl ether was investigated. The existence of a chloro substituent in the central benzene ring suggested that there was sufficient space to accommodate a bicyclic ring such as compound A (Figure 1). At the same time, another type of annulated analog B was designed. As compound B and its analogs did not show potent NaV1.7 activities, the derivatization of compound A was commenced.
Initial SAR results of compound A are summarized in Table 1. Neither N-phenyl indole 1a nor N-benzyl indole 1b showed NaV1.7 activity. The same poor results were obtained in regioisomer 1c. To our surprise, 3-methyl substitution is critical for the activity because 3-methyl derivative 1d inhibited human NaV1.7 modestly, whereas 3-unsubstituted derivative 1a was inactive. Compound 1d exhibited a 10-fold reduction in mouse NaV1.7 potency compared with the human ortholog. The introduction of a 1-methyl group in 3-phenyl indole 1c did not lead to any inhibitory potency (compound 1e).
Table 1. IC50 Values of NaV1.7 Inhibitors 1a.

| X | Y | hNaV1.7 IC50 (μM) | mNaV1.7 IC50 (μM) | |
|---|---|---|---|---|
| I | 0.052 | 1.7 | ||
| 1a | Ph | H | >10 | >10 |
| 1b | CH2Ph | H | >10 | >10 |
| 1c | H | Ph | >10 | >10 |
| 1d | Ph | Me | 0.33 | 2.7 |
| 1e | Me | Ph | >10 | >10 |
Data were obtained with a high-throughput electrophysiology system (IonWorks Quattro). Values are from a single experiment run in quadruplicate.
Accordingly, further optimization of compound 1d was performed, as shown in Table 2. The inhibitory activities of hNaV1.1, hNaV1.5, and hERG were assessed, because NaV1.1 is expressed in the brain and the inhibition of NaV1.1 leads CNS adverse effects, whereas the inhibition of NaV1.5 or hERG expressed in cardiac muscle causes cardiac adverse effect. Compound 1d showed good subtype selectivity over hNaV1.1 and hNaV1.5 (both 240-fold) without affecting hERG activity at 100 μM (less than 20%). Compound 1d also exerted high solubility, whereas 1d showed high protein binding (PB) ability owing to its acidity. Consequently, focus was placed on the enhancement of human and mouse NaV1.7 potencies by the introduction of substituents on the phenyl ring. When a fluoro group was incorporated, 3-isomer 1g boosted hNaV1.7 potency by 10-fold, whereas attenuation of the activity was observed in 2-isomer 1f. 4-Substitution maintained the in vitro profile (compound 1h). 3-Substituted analog 1g also showed improved mNaV1.7 activity by 3-fold. No hERG liability was confirmed for 1g and 1h. Although the introduction of the 3-chloro group enhanced hNaV1.7 activity, mNaV1.7 potency of 1i was still in the micromolar range. Chloro derivatives 1i and 1j exhibited enhanced hERG inhibitory potency. The introduction of a methyl or trifluoromethyl group also increased hERG liability (1k and 1m), whereas methoxy analog 1l did not. Although the introduction of a lipophilic substituent at the 3-position improved NaV1.7 potencies against both species, further enhancement of mNaV1.7 activity was required.
Table 2. In Vitro Profiles of Compound 1.
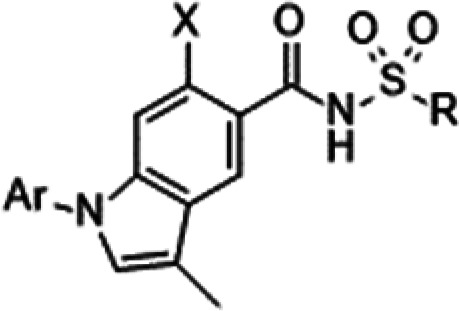
| Ar | X | R | hNaV1.1 IC50 (μM)a | hNaV1.5 IC50 (μM)a | hNaV1.7 IC50 (μM)a | mNaV1.7 IC50 (μM)a | hERG inhibition at 100 μMa,b | Log Dc | Solubility (μM)d | Mouse PB free (%)d | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1d | Ph | H | Me | 80 | 79 | 0.33 | 2.7 | 17.1 ± 3.8 | 1.1 | >660 | <0.20 |
| 1f | 2-F-C6H4 | H | Me | NTe | 100 | 5.9 | 12 | NTf | NTf | NTf | NTf |
| 1g | 3-F-C6H4 | H | Me | 12 | 35 | 0.038 | 0.90 | 13.2 ± 6.0 | 1.2 | 600 | <0.20 |
| 1h | 4-F-C6H4 | H | Me | 52 | 42 | 0.19 | 2.8 | 5.6 ± 2.3 | 1.1 | 140 | <0.20 |
| 1i | 3-Cl-C6H4 | H | Me | 20 | 34 | 0.087 | 1.7 | 43.5 ± 11.9 | 1.8 | 620 | <0.20 |
| 1j | 4-Cl-C6H4 | H | Me | >100 | 23 | 0.26 | 7.9 | 51.1 ± 7.6 | 1.8 | 26 | NTf |
| 1k | 3-Me-C6H4 | H | Me | 39 | NTf | 0.058 | 1.6 | 51.0 ± 10.0 | 1.6 | 550 | NTf |
| 1l | 3-MeO-C6H4 | H | Me | 79 | 71 | 0.19 | 2.1 | 18.9 ± 1.6 | 1.2 | 580 | <0.20 |
| 1m | 3-CF3-C6H4 | H | Me | 20 | 27 | 0.034 | 0.68 | 49.8 ± 8.2 | 1.7 | >820 | 0.20 |
| 1n | 3-F-C6H4 | F | Me | >100 | 31 | 0.014 | 0.98 | 15.6 ± 2.6 | 0.8 | 430 | <0.20 |
| 1o | 3-CF3-C6H4 | F | Me | >100 | 14 | 0.019 | 0.016 | 54.8 ± 15.2 | 1.5 | 230 | 0.40 |
| 1p | 3-F-C6H4 | H | c-Pr | 4.8 | 14 | 0.032 | 0.81 | 42.1 ± 5.0 | 1.8 | 440 | <0.20 |
Data were obtained with a high-throughput electrophysiology system (IonWorks Quattro). Values are from a single experiment run in quadruplicate.
hERG inhibitory activity at 10, 30, and 100 μM was assessed, and the dose response was confirmed. Each value represents the mean ± SD.
Distribution coefficients (log D) were measured after partition between 1-octanol and PBS (pH = 7.4).
Unbound fractions (%) in mouse plasma.
Aqueous thermodynamic solubility at pH 6.8.
Not tested.
The enhancement of hERG liability can be explained by the lipophilicity of molecules. Phenyl (1d), fluoro (1f and 1g), and methoxy analogs (1l) showed log D values of 1.1–1.2, which inhibited hERG activity by less than 20% at 100 μM. In contrast, chloro (1i and 1j), methyl (1k), and trifluoromethyl analogs (1m) showed log D values of 1.6–1.8, which resulted in the inhibition of hERG by more than 20% at 100 μM. As such, to avoid hERG liability, compounds need to be derivatized with a log D value of less than 1.5.
The introduction of a 6-fluoro substituent on the indole ring resulted in the expansion of hNaV1.1 selectivity (compound 1n). Less lipophilicity of 1n led to less hERG liability. Excellent mNaV1.7 activity was achieved in 1o. Thus, 3-trifluoromethyl derivative 1o successfully demonstrated excellent human and mouse NaV1.7 activities with excellent subtype selectivity (over 5000-fold and 740-fold against human NaV1.1 and NaV1.5, respectively).19 However, less hERG inhibition is preferable.20
When a cyclopropyl group was introduced on acyl sulfonamide, 1p retained high human and mouse NaV1.7 activities. Enhanced hERG inhibition was observed for 1p owing to its elevated lipophilicity (log D: 1.8). Furthermore, compound 1p lost high hNaV1.1 selectivity.
As described above, the adjustment of lipophilicity was essential to avoid hERG liability. The replacement of the phenyl group with a pyridine ring was then explored because the derivatization of the phenyl analog would increase hERG liability. The compound with less lipophilicity was also expected to cause less PB ability. Although the 2- or 4-pyridine derivative lost hNaV1.7 potency, 3-pyridine 2a exhibited diminished hNaV1.7 activity (Table 3). The substituent on the acyl sulfonamide moiety was fixed to a cyclopropyl group owing to methyl analog 2b showing no NaV1.7 potency. The introduction of a fluoro group on the pyridine ring regained NaV1.7 activities against both species (compound 2c). More lipophilic compounds 2d and 2e showed high NaV1.7 potencies, with 5-trifluoromethyl derivative 2e in particular exhibiting excellent human and mouse NaV1.7 activities without hERG liability.21 Compound 2e exhibited good subtype selectivity (440-fold and 930-fold against hNaV1.1 and hNaV1.5, respectively). 4-Trifluoromethyl substitution resulted in the reduction of mNaV1.7 activity (compound 2f). The introduction of a 6-fluoro substituent on the indole ring led to the discovery of 2g, which showed high human and mouse NaV1.7 activities without hREG liability. Compound 2g showed diminished subtype selectivity over hNaV1.1 (30-fold).
Table 3. In Vitro Profiles of Compound 2.

| X1 | X2 | X3 | R | hNaV1.1 IC50 (μM)a | hNaV1.5 IC50 (μM)a | hNaV1.7 IC50 (μM)a | mNaV1.7 IC50 (μM)a | hERG inhibition at 100 μMa,b | Log Dc | Solubility (μM)d | Mouse PB free (%)d | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2a | H | H | H | c-Pr | >100 | >300 | 1.6 | >10 | –8.8 ± 3.1 | NTe | NTe | NTf |
| 2b | H | H | H | Me | NTf | >300 | >10 | NTf | –0.1 ± 11.6 | NTe | NTe | NTf |
| 2c | H | F | H | c-Pr | >100 | >300 | 0.86 | 2.7 | –1.1 ± 5.8 | NTe | NTe | NTf |
| 2d | H | Cl | H | c-Pr | 9.9 | 107 | 0.085 | 0.46 | 34.1 ± 2.1 | 0.80 | 35 | NTf |
| 2e | H | CF3 | H | c-Pr | 6.6 | 14 | 0.015 | 0.061 | 6.9 ± 9.0 | 1.0 | 130 | 0.50 |
| 2f | CF3 | H | H | c-Pr | >100 | 210 | 0.059 | 1.6 | 21.0 ± 16.3 | 1.0 | 29 | 1.4 |
| 2g | H | CF3 | F | c-Pr | 1.2 | 32 | 0.040 | 0.015 | –2.9 ± 0.0 | 0.60 | 25 | 0.7 |
Data were obtained with a high-throughput electrophysiology system (IonWorks Quattro). Values are from a single experiment run in quadruplicate.
hERG inhibitory activity at 10, 30, and 100 μM was assessed, and the dose response was confirmed. Each value represents the mean ± SD.
Distribution coefficients (log D) were measured after partition between 1-octanol and PBS (pH = 7.4).
Unbound fractions (%) in mouse plasma.
Aqueous thermodynamic solubility at pH 6.8.
Not tested.
Compounds 2e and 2g were advanced to in vivo efficacy evaluation and PK study in mice. Compound 1p with poor mouse in vitro potency was also selected. The in vivo efficacy was assessed in mice with thermal hyperalgesia reduction by partial sciatic nerve ligation (PSL, Seltzer), a model of neuropathic pain at 1 mg/kg p.o.22 The paw withdrawal latency (PWL, s) was assessed at 30, 60, 120, 180, and 240 min after oral administration of the test compound (Plantar test). The time course of PWL is depicted in Figure 2. The development of hyperalgesia was confirmed in the model control group because the pain threshold was significantly lower in that group than in the normal control group. For compound 1p, the analgesic efficacy peaked at 30 min (73% compared with the normal group), after which this modest efficacy gradually disappeared. Compound 2g induced analgesic efficacy (64% compared with the normal group) comparable to that of 1p at 30 min. This antithermal efficacy lasted until 60 min, after which it diminished until disappearing at 180 min. When 2e was administered at 1 mg/kg, the onset of the analgesic effect was delayed compared with that of other compounds. Although compound 2e did not show potent analgesic efficacy at 30 min, it demonstrated superior efficacy at 60 and 120 min, which means that it reversed the thermal hyperalgesia to normal levels (94% and 85% compared with the normal group, respectively). This in vivo efficacy assessment revealed that compound 2e is superior in terms of both intensity and duration of efficacy.
Figure 2.

Time course of PWL (s) induced by compounds 1p, 2e, and 2g. The effect of compounds on thermal hyperalgesia in PSL mice. The compound was administered p.o. in 0.5% MC in three experiments (n = 4). Normal and vehicle groups are indicated based on three pooled experiments (n = 12), whereas normal and vehicle groups at 240 min are indicated based on two pooled experiments (n = 8). The analgesic efficacy of 1p at 240 min was not measured. Each value is the mean ± SEM. Statistical significance compared with vehicle treatment is denoted by *(p < 0.05), **(p < 0.01), as determined by the Aspin–Welch’s or Student’s t-test.
Mouse PK parameters were acquired after oral administration of the test compound at 1 mg/kg (Table 4). Total body clearance (CL), distribution volume at a steady state (Vss), and F value (%) were calculated after intravenous administration (1 mg/kg) of the compounds to mice (n = 2–3). All the three compounds exhibited high plasma exposure after oral administration at 1 mg/kg. Compounds 2e and 2g possessed higher plasma exposure than 1p, and showed suitable CL, Vss, and F (%) for clinical candidate selection.23
Table 4. PK Parameters and NaV1.7 Coverage in Mice.
| PK parameters
in ddY mice at 1 mg/kga |
||||||||
|---|---|---|---|---|---|---|---|---|
| Cmax (μM) | Tmax (h) | AUC0–24h (h·μM) | T1/2 (h) | CL (mL/min/kg) | Vss (L/kg) | F (%) | Mouse NaV1.7 coverage (Fold)b | |
| 1p | 1.7 ± 0.39c | 1.0 ± 0.87c | 12.1 ± 2.1c | 5.0 ± 2.6c | 2.58 ± 0.33c | 0.57 ± 0.03c | 65 ± 12c | <0.0042 |
| 2e | 2.3 ± 0.14c | 3.3 ± 4.0c | 41.5 ± 2.4c | 17.0 ± 6.1c | 1.01 ± 0.15c | 0.81 ± 0.05c | 117 ± 23c | 0.20 |
| 2g | 1.4 (1.5, 1.4)d | 6.0 (6.0, 6.0)d | 23.6 (24.0, 23.3)d | 13.5 (11.6, 15.5)d | NTe | NTe | NTe | 0.65 |
The test compounds in 0.5% MC were administered to ddY mice at 1 mg/kg (p.o.). Total body clearance (CL), distribution volume at a steady state (Vss), and F value were calculated after intravenous (1 mg/kg) administration of the test compounds. Each value represents the mean ± SD.
Mouse NaV1.7 coverage = plasma Cmax_free/mNaV1.7 IC50.
n = 3.
Individual data (n = 2).
Not tested.
Mouse NaV1.7 coverage was calculated from free plasma Cmax. As the sodium currents directly affect pain signaling, pain signaling would be governed by NaV1.7 coverage at each time. At the same time, we believe the duration of the compound’s plasma exposure, which correlates with AUC is also an important factor for the strength of efficacy.24 The lower efficacy of 1p can be rationalized by very low NaV1.7 coverage (<0.0042-fold). The target coverage of compound 2e was 0.20-fold, which is comparable to those of other in vivo efficacious acyl sulfonamides.25 Thus, it is fair to conclude that compound 2e caused robust analgesic efficacy through NaV1.7 inhibition, whereas in vivo efficacy of compound 2g is controversial. Although the target coverage of 2g is the highest among the three compounds, its efficacy is comparable to 1p, whose target coverage is very low.
Time course of the PK study after oral administration of the compounds is shown in Figure 3. Compound 2e retained high plasma concentration until 8 h, which may contribute to the potent efficacy as well as the duration of efficacy.24 However, the duration of high plasma concentration of 2e did not correlate with the duration of efficacy, because the efficacy of 2e in the Plantar test disappeared at 4 h. In a similar manner, the plasma concentration of 2g peaked at 6 h, whereas its efficacy in the Plantar test peaked at 1 h and disappeared at 3 h. Although T1/2 values of 2e and 2g in the mice PK study could have the potential to extrapolate once-daily administration in clinical settings, the duration of in vivo efficacy is much shorter than expected in the PK study. The reasons for such PK and PD discrepancies remain unclear, on which further research including the evaluation of DRG concentration or residence time will be required.24
Figure 3.

Time course of PK study after oral administration 1p (n = 3), 2e (n = 3), and 2g (n = 2). The compounds in 0.5% MC were administered to ddY mice at 1 mg/kg (p.o.). Each value is the mean ± SD.
In summary, a novel class of potent NaV1.7 inhibitors has been discovered. Diaryl ether of compound I can be replaced with N-aryl indole. The 3-methyl group is essential for reaching high potency in vitro. For the avoidance of hERG liability, the phenyl group was replaced with a 3-pyridine ring, followed by optimization leading to the discovery of 2e. Compound 2e (DS43260857) demonstrated highly potent in vitro activities against human and mouse NaV1.7 with an excellent PK profile and demonstrated potent in vivo efficacy in PSL mice at 1 mg/kg. The PD duration was not correlated with T1/2 in the PK study, which needs to be clarified by further research.
Acknowledgments
We would like to thank Dr. Kazunori Fujimoto, R&D Division, Daiichi Sankyo Co., Ltd., for his kind support of this research.
Glossary
Abbreviations
- NaV
voltage-gated sodium channel
- SAR
structure–activity relationship
- PB
protein binding
- PSL
partial spinal nerve ligation
- PWL
paw withdrawal latency
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00079.
Experimental procedures and characterization data of key synthesized compounds; procedures for pharmacological activities (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Cox J. J.; Reimann F.; Nicholas A. K.; Thornton G.; Roberts E.; Springell K.; Karbani G.; Jafri H.; Mannan J.; Raashid Y.; Al-Gazali L.; Hamamy H.; Valente E. M.; Gorman S.; Williams R.; McHale D. P.; Wood J. N.; Gribble F. M.; Woods C. G. An SCN9A Channelopathy Causes Congenital Inability to Experience Pain. Nature 2006, 444, 894–898. 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minett M. S.; Nassar M. A.; Clark A. K.; Passmore G.; Dickenson A. H.; Wang F.; Malcangio M.; Wood J. N. Distinct Nav1.7-Dependent Pain Sensations Require Different Sets of Sensory and Sympathetic Neurons. Nat. Commun. 2012, 3, 791. 10.1038/ncomms1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell A.; Collins S.; Ali Z.; Iavarone L.; Surujbally R.; Kirby S.; Butt R. P. Efficacy of the Nav1.7 blocker PF-05089771 in a randomised, placebo-controlled, double-blind clinical study in subjects with painful diabetic peripheral neuropathy. Pain 2018, 159, 1465–1476. 10.1097/j.pain.0000000000001227. [DOI] [PubMed] [Google Scholar]
- For a recent review of NaV1.7 inhibitors with the challenges, see references (5−7).
- Kitano Y.; Shinozuka T. Inhibition of NaV1.7: The Possibility of Ideal Analgesics. RSC Med. Chem. 2022, 13, 895–920. 10.1039/D2MD00081D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy J. V.; Pajouhesh H.; Beckley J. T.; Delwig A.; Du Bois J.; Hunter J. C. Challenges and Opportunities for Therapeutics Targeting the Voltage-Gated Sodium Channel Isoform NaV1.7. J. Med. Chem. 2019, 62, 8695–8710. 10.1021/acs.jmedchem.8b01906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerrall S. J.; Sutherlin D. P. NaV1.7 inhibitors for the treatment of chronic pain. Bioorg. Med. Chem. Lett. 2018, 28 (19), 3141–3149. 10.1016/j.bmcl.2018.08.007. [DOI] [PubMed] [Google Scholar]
- Suzuki S.; Kuroda T.; Kimoto H.; Domon Y.; Kubota K.; Kitano Y.; Yokoyama T.; Shimizugawa A.; Sugita R.; Koishi R.; Asano D.; Tamaki K.; Shinozuka T.; Kobayashi H. Discovery of (Phenoxy-2-hydroxypropyl)piperidines as a Novel Class of Voltage-Gated Sodium Channel 1.7 Inhibitors. Bioorg. Med. Chem. Lett. 2015, 25 (22), 5419–5423. 10.1016/j.bmcl.2015.09.005. [DOI] [PubMed] [Google Scholar]
- Tanaka K.; Suzuki S.; Kobayashi H.; Shibuya S.; Naito S.; Kimoto H.; Domon Y.; Kubota K.; Kitano Y.; Yokoyama T.; Shimizugawa A.; Koishi R.; Asano D.; Takasuna K.; Shinozuka T. Discovery of a Novel Class of State-Selective NaV1.7 Inhibitors for the Treatment of Neuropathic Pain. Chem. Pharm. Bull. 2020, 68, 653–663. 10.1248/cpb.c20-00126. [DOI] [PubMed] [Google Scholar]
- Shinozuka T.; Kobayashi H.; Suzuki S.; Tanaka K.; Karanjule N.; Hayashi N.; Tsuda T.; Tokumaru E.; Inoue M.; Ueda K.; Kimoto H.; Domon Y.; Takahashi S.; Kubota K.; Yokoyama T.; Shimizugawa A.; Koishi R.; Asano D.; Sakakura T.; Takasuna K.; Abe Y.; Watanabe T.; Kitano Y. Discovery of DS-1971a, a Potent Selective NaV1.7 Inhibitor. J. Med. Chem. 2020, 63, 10204–10220. 10.1021/acs.jmedchem.0c00259. [DOI] [PubMed] [Google Scholar]
- Bell A. S.; Brown A. D.; De Groot M. J.; Lewthwaite R. A.; Marsh I. R.; Millan D. S.; Pacheco M. P.; Rawson D. J.; Sciammetta N.; Storer R. I.; Swain N. A.; Gaulier S. M.. Preparation of Heteroaryl Sulfonamide Derivatives as Nav1.7 Inhibitors. PCT Int. Appl. WO2012007861, Jan 19, 2012.
- Xenon/Genentech disclosed the
structure of acyl sulfonamides that progressed to clinical trials.
Structures of GDC-0276 and GDC-0310 with reported in vitro IC50 values.13

- Safina B. S.; McKerrall S. J.; Sun S.; Chen C.-A.; Chowdhury S.; Jia Q.; Li J.; Zenova A. Y.; Andrez J.-C.; Bankar G.; Bergeron P.; Chang J. H.; Chang E.; Chen J.; Dean R.; Decker S. M.; DiPasquale A.; Focken T.; Hemeon I.; Khakh K.; Kim A.; Kwan R.; Lindgren A.; Lin S.; Maher J.; Mezeyova J.; Misner D.; Nelkenbrecher K.; Pang J.; Reese R.; Shields S. D.; Sojo L.; Sheng T.; Verschoof H.; Waldbrook M.; Wilson M. S.; Xie Z.; Young C.; Zabka T. S.; Hackos D. H.; Ortwine D. F.; White A. D.; Johnson J. P. Jr; Robinette C. L.; Dehnhardt C. M.; Cohen C. J.; Sutherlin D. P. Discovery of acyl-sulfonamide inhibitors GDC-0276 and GDC-310. J. Med. Chem. 2021, 64, 2953–2966. 10.1021/acs.jmedchem.1c00049. [DOI] [PubMed] [Google Scholar]
- Close indole and indazole-based analogs were reported, see references15−18.
- Luo G.; Chen L.; Easton A.; Newton A.; Bourin C.; Shields E.; Mosure K.; Soars M. G.; Knox R. J.; Matchett M.; Pieschl R. L.; Post-Munson D.; Wang S.; Herrington J.; Graef J.; Newberry K.; Sivarao D. V.; Senapati A.; Bristow L.; Meanwell N. A.; Thompson L. A.; Dzierba C. D. Discovery of Indole- and Indazole-acylsulfonamides as Potent and Selective NaV1.7 Inhibitors for the Treatment of Pain. J. Med. Chem. 2019, 62, 831–856. 10.1021/acs.jmedchem.8b01550. [DOI] [PubMed] [Google Scholar]
- Jones H. M.; Butt R. P.; Webster R. W.; Gurrell I.; Dzygiel P.; Flanagan N.; Fraier D.; Hay T.; Iavarone L. E.; Luckwell J.; Pearce H.; Phipps A.; Segelbacher J.; Speed B.; Beaumont K. Clinical micro-dose studies to explore the human pharmacokinetics of four selective inhibitors of human NaV1.7 voltage-dependent sodium channels. Clin. Pharmacokinet. 2016, 55, 875–887. 10.1007/s40262-015-0365-0. [DOI] [PubMed] [Google Scholar]
- Bankar G.; Goodchild S. J.; Howard S.; Nelkenbrecher K.; Waldbrook M.; Dourado M.; Shuart N. G.; Lin S.; Young C.; Xie Z.; Khakh K.; Chang E.; Sojo L. E.; Lindgren A.; Chowdhury S.; Decker S.; Grimwood M.; Andrez J. C.; Dehnhardt C. M.; Pang J.; Chang J. H.; Safina B. S.; Sutherlin D. P.; Johnson J. P. Jr; Hackos D. H.; Robinette C. L.; Cohen C. J. Selective NaV1.7 Antagonists with Long Residence Time Show Improved Efficacy against Inflammatory and Neuropathic Pain. Cell Rep. 2018, 24, 3133–3145. 10.1016/j.celrep.2018.08.063. [DOI] [PubMed] [Google Scholar]
- Wu Y.-J.; Venables B.; Guernon J.; Chen J.; Sit S.-Y.; Rajamani R.; Knox R. J.; Matchett M.; Pieschl R. L.; Herrington J.; Bristow L. J.; Meanwell N. A.; Thompson L. A.; Dzierba C. Discovery of New Indole-based Acylsulfonamide NaV1.7 Inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 659–663. 10.1016/j.bmcl.2018.12.013. [DOI] [PubMed] [Google Scholar]
- As the preliminary in vivo assessment of 1o at 3 mg/kg with PSL mice revealed modest efficacy, compound 1o was deprioritized.
- There is a possibility that such a high safety margin against hERG is not necessarily required.
- The modifications of substituents on the acyl sulfonamide group in 2e gave poor results, see: Table S2 in the Supporting Information.
- Seltzer Z.; Dubner R.; Shir Y. A Novel Behavioral Model of Neuropathic Pain Disorders Produced in Rats by Partial Sciatic Nerve Injury. Pain 1990, 43 (2), 205–218. 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- Compound 2e did not inhibit CYP enzymes (1A2, 2C9, 2D6, and 3A4) at 10 μM, and was negative for the mechanism-based inhibition of CYP3A4 enzyme. GSH trapping assay in human liver microsomes showed no GSH adduct formation of compound 2e.
- We are proposing high DRG concentration with a long residence time, which might reflect time above IC50 considering the unbound fraction in the PK study could be an important factor for the in vivo efficacy, see reference (5).
- NaV1.7 coverage of in vivo efficacious acyl sulfonamides is from 0.17 to 36-fold, see reference (5), Table 8.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.