

Abstract
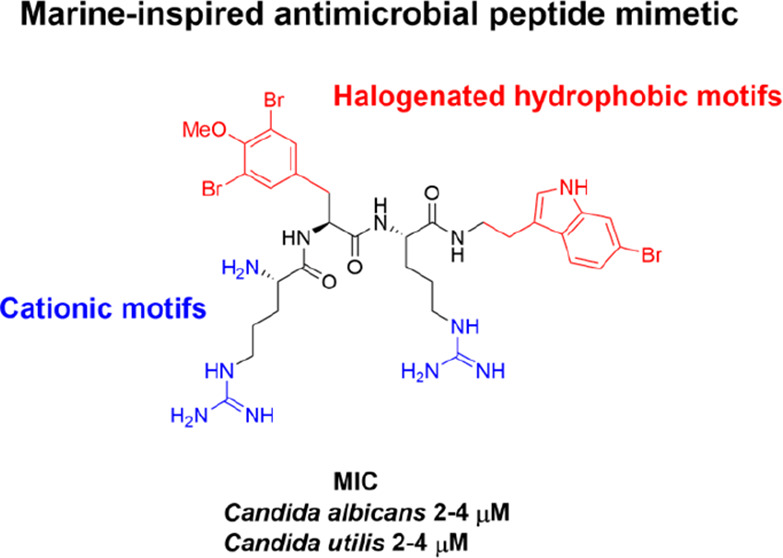
Small synthetic mimics of cationic antimicrobial peptides represent a promising class of compounds with leads in clinical development for the treatment of persistent microbial infections. The activity and selectivity of these compounds rely on a balance between hydrophobic and cationic components, and here, we explore the activity of 19 linear cationic tripeptides against five different pathogenic bacteria and fungi, including clinical isolates. The compounds incorporated modified hydrophobic amino acids inspired by motifs often found in bioactive marine secondary metabolites in combination with different cationic residues to probe the possibility of generating active compounds with improved safety profiles. Several of the compounds displayed high activity (low μM concentrations), comparable with the positive controls AMC-109, amoxicillin, and amphotericin B. A higher activity was observed against the fungal strains, and a low in vitro off-target toxicity was observed against erythrocytes and HeLa cells, thereby illustrating effective means for tuning the activity and selectivity of short antimicrobial peptides.
Keywords: Halogenated, Synthesis, Antimicrobial Peptide, Marine Natural Products, Bromotyrosine
The alarming development of antimicrobial resistance (AMR) toward our traditionally employed antibiotics represents an urgent and global challenge.1 The continued excessive and incorrect use of antibiotics, and insufficient progress in the anti-infective drug development space, causes human suffering and the associated economic burden of AMR.2 Infections and complications associated with multidrug-resistant pathogens are expected to cause the deaths of nearly 2.5 million people in Europe, Australia, and North America over the next 30 years alone, with an estimated added yearly cost as high as US$3.5 billion.2 In light of this somber scenario, it is clear that intensified efforts to find effective novel antimicrobial solutions that can exert their activity without easily inducing AMR remains a critical task.3−5
About half of the antibiotics in clinical use are derived from or inspired by natural products.6 Microorganisms, in particular, are efficient producers of potent antimicrobial secondary metabolites.6 Such compounds provide the producing organism with a chemical defense against intruders and competitors. Higher organisms are also armed with an innate chemical defense that enable a rapid response against intruding pathogens before the onset of the adaptive immune system.7 A key component of the innate defense system is antimicrobial peptides (AMPs), and more than 3000 different natural antimicrobial peptides are reported in the antimicrobial peptide database.8 Several of these, such as vancomycin, gramicidin D, colistin, and daptomycin, have made it into clinical use, thereby illustrating the increasing importance of AMPs for targeting multidrug-resistant bacteria.9 The currently approved AMPs are all of microbial origin, but several late-stage leads originating from higher mammalian sources are in late-stage clinical development.4 While these AMPs are diverse, they often share common structural motifs, such as a net positive charge (+2 to +11) and a pronounced amphiphilic structure that enables interactions with the anionic bacterial membrane, which is often the molecular target.10 These interactions lead to membrane disruption via several mechanisms, which are extensively discussed elsewhere.5,11,12 In addition, numerous AMPs also display immunomodulatory activities, which enables dual routes for bacterial destruction.12−14
Several innate mammalian AMPs are under clinical development, but a common challenge is that they are all generally poor drug leads because of their size and peptidic nature, which leads to low metabolic stability, challenging pharmacokinetics, and poor oral bioavailability.4,15 The highly elaborate amphiphilic secondary structures of several native AMPs lead to high production costs, which contributes to the delayed anticipated impact of this class of compounds.9 Attempts to generate more “druglike,” shorter AMP mimics have revealed ample room for development permitting that the balance between hydrophobicity and cationic charge is maintained.4,16,17 Compounds as small as di- and tripeptidomimetics can be prepared by the incorporation of unnatural bulky and cationic residues on different scaffold backbones, as recently extensively reviewed.4,17,18 These simplified synthetic mimics of AMPs (SMAMPs) come with several benefits, including improved metabolic stability,19 increased potential for oral bioavailability,20 and lowered production costs.16 A range of different backbones has been evaluated, and potent compounds such as AMC-109 (linear tripeptide),16 CSA-13 and CSA-131 (cationic bile acid derivatives),21,22 and brilacidin (arylamide SMAMP with a central pyrimidine)23,24 are in clinical development by Amicoat AS (NO), N8 Medical (USA), and Innovation Pharmaceuticals Inc. (USA), respectively (Figure 1).
Figure 1.

Short synthetic mimics of innate defense peptides in clinical development and structures of bioactive natural halogenated marine secondary metabolites
The success of these smaller mimics lies mainly in the ability to balance and control amphiphilicity by incorporating different hydrophobic and basic residues.16,25 Going beyond the natural hydrophobic building blocks enables the design of much smaller active structures.4
In the marine environment, several small potent bioactive secondary metabolites containing halogenated hydrophobic amino acids have been reported,26 and antibacterial brominated marine alkaloids were recently effectively identified using a virtual screening method.27 Different brominated tyrosine and tryptophan analogues are often encountered (Figure 1),27 and halogenation offers a way for nature to increase the hydrophobic volume of these residues.28 Structure–activity relationship (SAR) studies have linked much of activity of these compounds to a minimum hydrophobic volume provided by the halogens,29,30 in analogy to the SAR of AMP mimics.31,32 Furthermore, it was recently illustrated in a series of studies that inactive peptoids can be converted into antibacterials by halogenation of key residues without also generating increased hemolytic activity or in vitro cytotoxicity.33 Halogenated residues have been incorporated to generate effective antimicrobial barbiturate mimics of the eusynstyelamide marine natural products (MNPs),34 and potent cyclic peptide antifoulants have been prepared using a brominated tyrosine motif.35 On the basis of these observations, it is clear that halogenation can be used as a versatile tool for tuning the activity of small antimicrobial compounds.
Continuing to explore the beneficial effects of halogenation on the activity and safety profiles of small AMPs mimics, the current study investigates the effect of incorporating natural marine-inspired halogenated hydrophobic amino acids. A series of naturally inspired halogenated building blocks were designed, prepared, and incorporated into a series of modified linear cationic tripeptides (Figure 2).
Figure 2.

Structure of the C-terminally capped tripeptide scaffold and hydrophobic and cationic residues used to generate the currently studied library of compounds.
The biological effects of the novel peptides were evaluated against five human pathogenic bacteria and fungi (including clinical isolates), as well as against human erythrocytes and HeLa cells, to probe off-target toxicity.
The compounds were assembled to investigate the potential of steering the bioactivity and cellular selectivity of a tripeptidic cationic AMP lead by incorporation of modified halogenated hydrophobic residues, as found in several marine natural products. The employed short AMP scaffold has been optimized by incorporating synthetic cationic and hydrophobic residues, which provides high resilience against enzymatic degradation;36−38 the possibility for oral uptake; and an activity superior to several commercially employed antimicrobial drugs, such as amorolfine and terbinafine ex vivo.39 The incorporation of different halogen atoms in drug leads is a common strategy to tune and improve their bioactivities and specificities and it has successfully been used in longer peptoids.33 The halogenation of hydrophobic amino acids in nature is generally enzymatically catalyzed by substrate-specific halogenases or less specific haloperoxidases.28 These enzymes efficiently incorporate halogen atoms at phenolic and indolic moieties that are challenging to mimic in the synthetic laboratory. Basing our study on a highly active tripeptide scaffold, the most feasible way to control the incorporation of the halogenated moieties was via the design of a series of Fmoc-protected halogenated building blocks, as shown in Figure 3.
Figure 3.

Structures of modified unnatural amino acids and amine building blocks designed and prepared in the current study.
All the building block were prepared in high yields and purities, and the methods developed offer ready access to a range of useful building blocks for the preparation of modified peptides. To probe the effect of stereochemistry on structure and activity,25,40 both enantiomers of BrW were prepared using a stereoselective method and incorporated in peptides 3 and 4. None of the designed halogenated building blocks presented any synthetic challenges when incorporated into the peptides and the tripeptide library (Table 1), which was assembled according to the outlined procedure employing standard Fmoc SPPS chemistry (Figure 4).
Table 1. Composition and HPLC-Retention Times of the Prepared Peptides.
| peptide | P1 | P2 | P3 | P4 | Mw (g/mol) | retention time (min) |
|---|---|---|---|---|---|---|
| 1 | Ph | R | Tbta | R | 788.10 | 9.23 |
| 2 | Ph | R | Ph | R | 580.74 | 7.61 |
| 3 | Ph | R | S-BrW | R | 698.67 | 7.98 |
| 4 | Ph | R | R-BrW | R | 698.67 | 7.62 |
| 5 | Ph | R | diBrOMe | R | 768.56 | 8.11 |
| 6 | Ph | R | OMeBip | R | 686.86 | 8.18 |
| 7 | Ph | R | diBrOMeBip | R | 844.65 | 8.68 |
| 8 | diBrOMe | R | diBrOMe | R | 956.37 | 8.56 |
| 9 | BrOMe | R | BrOMe | R | 798.58 | 8.09 |
| 10 | diBrOH | R | diBrOH | R | 928.32 | 7.77 |
| 11 | diBrOMe | R | W | R | 807.59 | 8.18 |
| 12 | diBrOMe | R | BrW | R | 886.49 | 8.43 |
| 13 | Br-Ind | R | diBrOMe | R | 886.49 | 8.47 |
| 14 | Ph | R | Bip | KN(Me)3 | 671.91 | 8.19 |
| 15 | Ph | R | Bip | C5N(Me)3 | 656.90 | 8.55 |
| 16 | Ph | KN(Me)3 | Bip | R | 671.91 | 8.20 |
| 17 | Ph | KN(Me)3 | Bip | KN(Me)3 | 686.98 | 8.15 |
| 18 | Ph | R | diBrOMe | C5N(Me)3 | 768.62 | 8.45 |
| 19 | diBrOMe | K | diBrOMe | K | 900.35 | 8.47 |
Tri-tert-butyl tryptophan.
Figure 4.

General synthetic Fmoc protocol employed for library generation.
The bioactivity of the assembled peptides was assessed against Escherichia coli and Staphylococcus aureus, the yeasts Candida albicans and Candida utilis (clinical isolate, subcultured from Auckland City Hospital), and filamentous fungus Aspergillus fumigatus (clinical isolate subcultured from Auckland City Hospital) using Clinical and Laboratory Standards Institute (CLSI) recommended protocols. Several of the compounds displayed high bioactivities comparable with the positive control peptide AMC-109 (1) and clinically employed anti-infectives, such as amoxicillin, polymyxin B, and amphotericin B, as presented in Table 2. Compound 1 is a highly potent linear AMP mimic derived from lactoferricin, which is structurally closely related (Figure 1) to the developed compounds and is in clinical development by Amicoat AS. Antifungal activities superior to both terbinafine and amorolfine39 have been reported for 1, and its development has been recently reviewed.16
Table 2. Antimicrobial Activity of the Prepared Peptides.
| peptide | MIC (μM) |
||||
|---|---|---|---|---|---|
| E. colia | S. aureusb | C. albicansc | C. utilisd | A. fumigatuse | |
| 1 | 2–4 | 2–4 | 4 | 2 | 8 |
| 2 | >64 | >64 | >64 | >64 | >64 |
| 3 | >64 | >64 | 16–32 | 8–16 | >64 |
| 4 | >64 | >64 | >64 | 16–32 | >64 |
| 5 | >64 | >64 | 16 | 8 | 64 |
| 6 | >64 | 32–64 | 2 | 2–4 | >64 |
| 7 | 16 | 8 | 4 | 4 | 16–32 |
| 8 | 8 | 4 | 4 | 4 | 16 |
| 9 | >64 | >64 | 32 | 4 | >64 |
| 10 | >64 | >64 | >64 | 64 | 64 to >64 |
| 11 | >64 | 64 | 8–16 | 8 | 64 |
| 12 | 16 | 8 | 2–4 | 2–4 | 16 |
| 13 | >64 | >64 | >64 | >64 | >64 |
| 14 | >64 | >64 | >64 | >64 | >64 |
| 15 | >64 | >64 | >64 | >64 | >64 |
| 16 | >64 | >64 | >64 | >64 | >64 |
| 17 | >64 | >64 | >64 | 64 | >64 |
| 18 | >64 | >64 | >64 | >64 | >64 |
| 19 | >64 | 64 | >64 | 32 | >64 |
| amoxicillin | 16 | 4 | f | ||
| polymyxin B | 0.0625–0.125 | >64 | |||
| amphotericin B | 1 | 2 | 4 | ||
ATCC 25922.
ATCC 29213.
SC5314.
SVB-Y1.
SVB-F136.
Not tested.
In comparison with the highly active 1, the majority of the prepared compounds were generally inferior antibacterials, with only compounds 7, 8, and 12 displaying strong antibacterial activity comparable with amoxicillin. A higher activity was observed against the Gram-positive S. aureus, which is in accordance with the literature.16 Because of material availability, the compounds were only tested up to 64 μM, and it is likely that several of the compounds are active at higher concentrations, although this is of less clinical relevance. The active compounds contained combinations of diBrOMeBip, diBrOMe, and BrW as central P3 side chains and C-terminal P1 capping groups balanced by two cationic R side chains as the P2 and P4 units. This combination provides the active compounds with sufficient hydrophobicity for bioactivity (retention time ranging from 8.43 to 8.68 min). This is in good correlation with earlier studies indicating that an amphiphilic solution structure40 and a threshold hydrophobicity is key for antibacterial activity.20 Compound 19 displays a weak activity linked to its two diBrOMe units that is still significantly lower than the activity of 8, which incorporates two R instead of K, thereby reducing the hydrogen bonding and membrane-interacting potential of 19.16,41 This reduced ability for hydrogen bonding is believed to be the main driver behind the low antibacterial activity observed for compounds 14–19.16
The antifungal activity was more pronounced, with several compounds displaying equivalent and better potency compared with 1 and amphotericin B against the two yeasts C. albicans and C. utilis. Compounds 6–8 and 12 were again the most active ones with low μM MICs. The diBrOMe units were shown to be effective hydrophobic elements that confer a high antifungal activity, while the more polar diBrOH units yielded inactive compounds. This is an interesting observation because these active leads are less polar than 1, which could lead to a lower off-target cellular toxicity. As observed against the bacteria, compounds 14–19, which display less basic cationic side chains, were also typically inactive against tested yeast strains. The ability to form stable amphiphilic solution structures is key for these types of short antimicrobials.4 On the basis of the observed retention times, nothing suggests that the novel building blocks prevent this with a good linear relationship between retention time and the theoretical solvent-excluded volume (R2 = 0.83, calculated using ChemBioOffice), thereby indicating no major barriers for the formation of bioactive structures, which often leads to significantly lower retention times.25,40 To further probe the importance of solution structure, compound 4 was prepared by incorporation of d-BrW as the hydrophobic P3 substituent. Compound 4 was much less active than the all-l isomer 3, and this was also reflected in the shorter retention time of 4, which supports that the mixed stereochemistry often reduces the bioactivity by preventing the formation of the bioactive solution conformers.25 A similar trend in antifungal bioactivity was observed against filamentous fungus A. fumigatus. The clinical Aspergillus isolate is a robust pathogen (Table 2), and the recorded antifungal MICs were approximately 4 times higher in comparison with the yeast. Compounds 7, 8, and 12 were once again the most active leads, thereby supporting earlier observations of high antifungal potential.39 The activity of most highly active membrane-active compounds are in the low micromolar range, and the observed bioactivities thus indicate that the leads from the current study are potent antimicrobials.4 The mode of action of the compounds was not studied, but it is expected, on the basis of structural similarity and observed trends in bioactivity, that they are membrane active compounds.4,16,25
A common challenge for cationic AMPs and their related smaller mimics is the therapeutic window.4,42 The balance between charge and hydrophobicity dictates the microbial bioactivity, but the hydrophobic component is also the major molecular driver behind off-target toxicity.43 As such, one of the hypotheses of the current study was to examine if the marine-inspired hydrophobic units could confer a high bioactivity with a lower toxicity. To investigate the toxicity, a selection of compounds was first screened against human erythrocytes using established protocols.44 Erythrocytes are often used to assess toxicity of AMPs. However, the range of different assay conditions and cell types used makes comparison of absolute hemolysis values between studies difficult,43 and optimized and standardized methods are needed for in vitro determination of hemolytic properties.45 Our employed method assessed the hemolysis (%) of the compounds at 100 μM, and the results are presented in Table 3.
Table 3. Hemolytic Activity of Selected Prepared Compounds.
| peptide | hemolytic activity (%) | standard deviation |
|---|---|---|
| 1 | 4.23 | 0.12 |
| 2 | 0.33 | 0.03 |
| 3 | 0.29 | 0.02 |
| 4 | 0.27 | 0.00 |
| 5 | 0.34 | 0.02 |
| 6 | 0.40 | 0.03 |
| 7 | 1.38 | 0.48 |
| 8 | 1.62 | 0.04 |
| 9 | 0.29 | 0.01 |
| 11 | 0.35 | 0.01 |
| 12 | 1.19 | 0.16 |
| 1% DMSO | 0 | 0.02 |
| 1% Triton | 100 | 10 |
The hemolysis results indicate a low hemolytic activity for all the prepared compounds compared with 1 at 100 μM. Although not directly comparable, 1 has previously been reported with an EC50 of 222 μM against human erythrocytes, which implies significant therapeutic indexes for the prepared compounds.39 Nonoptimized hemolysis experiments using isolated and washed erythrocytes for initial toxicity assessment can both under- and overestimate systemic toxicity, and the values should only be seen as a relative comparison between the currently evaluated compounds.43 To obtain a more accurate assessment of cellular toxicity, a selection of compounds with ranging bioactivity and hemolytic activity was also evaluated against human epithelial cells (HeLa cells)46 up to 87.5 μM, as shown in Figure 5. The results from the HeLa cytotoxicity experiment indicate a high cellular tolerance for all the prepared compounds in the given concentration range. IC50 values could only be obtained for the positive control 1, which illustrates the large differences in toxicity. In fact, only compound 8 displayed a reduced viability at the highest concentration, while highly active 7 and 12 did not induce a reduction in cell viability over 24 h, thereby illustrating a higher tolerance in comparison with 1.
Figure 5.

Cytotoxicity dose–response curves for compounds (A) 1, (B) 3, (C) 6, (D) 7, (E) 8 and (F) 12. HeLa cells were treated with serial dilutions of peptides (1.4–87.5 μM), and cell viability was measured by PrestoBlue assays after 24 h incubation at 37 °C. An IC50 value of 46.6 μM was determined for 1 (A), while all remaining compounds (B–G) had IC50 values above 87.5 μM. Data is shown as mean ± SD of two independent experiments, each performed in triplicate.
Taken together, both the hemolysis and cytotoxicity evaluations indicate that the compounds with halogenated hydrophobic residues are much less toxic than the positive control AMC-109. This is likely linked to the reduced hydrophobicity of the P1 and P3 side chains.
In the current study, we have prepared a series of 19 amphiphilic tripeptides and evaluated their antimicrobial activity against five different strains of bacteria and fungi, including clinical isolates. The compounds were designed to incorporate unnatural halogenated residues inspired by bioactive marine secondary metabolites with the aim of conferring high antimicrobial activity but with a lower off-target toxicity. The prepared compounds were highly active against tested fungi, and the most active compounds showed promising antibacterial activities compared with amphotericin B and amoxicillin. The higher activity against the fungal strains suggests subtle differences in the structural requirements for these compounds against the different types of microbes. Significantly lower toxicity against both human erythrocytes and human HeLa cells were observed in comparison with the related clinical AMP lead AMC-109 (1) with a maintained high bioactivity. Our study provides insight for improving the therapeutic window of this promising class of compounds, which may find additional uses for peptide drug development. The incorporation of the developed marine-inspired halogenated building blocks may also impact the metabolic half-life and protein binding in a beneficial way, which warrants further investigation.
Acknowledgments
L.M. and A.J.C. are grateful to Carl Tryggers Stiftelse for a postdoctoral fellowship (A.J.C.). M.H. and A.R. acknowledge the Åke Wibergs Stiftelse for a postdoctoral fellowship (A.R.). L.M. wishes to thank the Uppsala Antibiotic Centre for support. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00093.
Synthesis, 1H and 13C NMR spectra, LC traces for all compounds, and additional experimental details (PDF)
Author Contributions
° A.J.C. and Y.E. contributed equally to this work. Conceptualization, J.S., Y.E., and N.M.; Methodology, A.J.C., Y.E., M.A.B., L.W.K.M., and J.S.; Investigation, A.J.C., N.M., Y.E., H.W., A.C., A.R., J.A.H., and M.H.; Data curation, A.J.C., M.H., L.W.K.M., and J.S.; Writing—original draft preparation, J.S., Y.E., A.J.C., and L.W.K.M.; Writing—review and editing, J.S., N.M., Y.E., A.J.C., and L.W.K.M., Visualization, M.H. and J.S.; Supervision, J.S. and L.W.K.M.; Project administration, J.S., A.J.C., Y.E., and L.W.K.M.; Funding acquisition, M.A.B., F.B., and L.W.K.M. All authors have read and agreed to the published version of the manuscript.
The authors are grateful for support from the NZ Ministry of Business, Innovation & Employment via Smart Idea Grant CAWX1805 (M.A.B.) and Endeavor Grant UOAX2010 (M.A.B., A.C.).
The authors declare no competing financial interest.
Supplementary Material
References
- Dadgostar P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. 10.2147/IDR.S234610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer U. The cost of antimicrobial resistance. Nat. Rev. Microbiol. 2019, 17 (1), 3–3. 10.1038/s41579-018-0125-x. [DOI] [PubMed] [Google Scholar]
- Yoneyama H.; Katsumata R. Antibiotic resistance in bacteria and its future for novel antibiotic development. Biosci. Biotechnol. Biochem. 2006, 70 (5), 1060–1075. 10.1271/bbb.70.1060. [DOI] [PubMed] [Google Scholar]
- Svenson J.; Molchanova N.; Schroeder C. I. Antimicrobial Peptide Mimics for Clinical Use: Does Size Matter?. Front. Immunol. 2022, 13, 915368. 10.3389/fimmu.2022.915368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magana M.; Pushpanathan M.; Santos A. L.; Leanse L.; Fernandez M.; Ioannidis A.; Giulianotti M. A.; Apidianakis Y.; Bradfute S.; Ferguson A. L.; Cherkasov A.; Seleem M. N.; Pinilla C.; de la Fuente-Nunez C.; Lazaridis T.; Dai T.; Houghten R. A.; Hancock R. E. W.; Tegos G. P. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20 (9), e216–e230. 10.1016/S1473-3099(20)30327-3. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83 (3), 770–803. 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- Iwasaki A.; Medzhitov R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16 (4), 343–353. 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasloff M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415 (6870), 389–395. 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- Chen C. H.; Lu T. K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9 (1), 24. 10.3390/antibiotics9010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaman M. R.; Yount N. Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55 (1), 27–55. 10.1124/pr.55.1.2. [DOI] [PubMed] [Google Scholar]
- Mahlapuu M.; Hakansson J.; Ringstad L.; Bjorn C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell Infect. Microbiol. 2016, 6, 194. 10.3389/fcimb.2016.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlapuu M.; Björn C.; Ekblom J. Antimicrobial peptides as therapeutic agents: Opportunities and challenges. Crit. Rev. Biotechnol. 2020, 40 (7), 978–992. 10.1080/07388551.2020.1796576. [DOI] [PubMed] [Google Scholar]
- Jenssen H.; Hancock R. E. Therapeutic potential of HDPs as immunomodulatory agents. Methods Mol. Biol. 2010, 618, 329–347. 10.1007/978-1-60761-594-1_20. [DOI] [PubMed] [Google Scholar]
- Bowdish D. M.; Davidson D. J.; Scott M. G.; Hancock R. E. Immunomodulatory activities of small host defense peptides. Antimicrob. Agents Chemother. 2005, 49 (5), 1727–1732. 10.1128/AAC.49.5.1727-1732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar T.; Chetia M.; Chatterjee S. Antimicrobial Peptides and Proteins: From Nature’s Reservoir to the Laboratory and Beyond. Front. Chem. 2021, 9, 691532. 10.3389/fchem.2021.691532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen J. S. M.; Grant T. M.; Rennison D.; Brimble M. A.; Svenson J. Very short and stable lactoferricin-derived antimicrobial peptides: design principles and potential uses. Acc. Chem. Res. 2019, 52 (3), 749–759. 10.1021/acs.accounts.8b00624. [DOI] [PubMed] [Google Scholar]
- W Scott R.; N Tew G. Mimics of host defense proteins; strategies for translation to therapeutic applications. Curr. Top. Med. Chem. 2016, 17 (5), 576–589. 10.2174/1568026616666160713130452. [DOI] [PubMed] [Google Scholar]
- Hellewell L.; Gilani N. M.; Stanton C. J.; Pelligand L.; Franzyk H.; Guardabassi L.; Good L. Efficacy of natural antimicrobial peptides versus peptidomimetic analogues: a systematic review. Future Med. Chem. 2022, 14 (24), 1899–1921. 10.4155/fmc-2022-0160. [DOI] [PubMed] [Google Scholar]
- Karstad R.; Isaksen G.; Wynendaele E.; Guttormsen Y.; De Spiegeleer B.; Brandsdal B. O.; Svendsen J. S.; Svenson J. Targeting the S1 and S3 subsite of trypsin with unnatural cationic amino acids generates antimicrobial peptides with potential for oral administration. J. Med. Chem. 2012, 55 (14), 6294–6305. 10.1021/jm3002058. [DOI] [PubMed] [Google Scholar]
- Flaten G. E.; Kottra G.; Stensen W.; Isaksen G.; Karstad R.; Svendsen J. S.; Daniel H.; Svenson J. In vitro characterization of human peptide transporter hPEPT1 interactions and passive permeation studies of short cationic antimicrobial peptides. J. Med. Chem. 2011, 54 (7), 2422–2432. 10.1021/jm1015704. [DOI] [PubMed] [Google Scholar]
- Lai X. Z.; Feng Y.; Pollard J.; Chin J. N.; Rybak M. J.; Bucki R.; Epand R. F.; Epand R. M.; Savage P. B. Ceragenins: cholic acid-based mimics of antimicrobial peptides. Acc. Chem. Res. 2008, 41 (10), 1233–1240. 10.1021/ar700270t. [DOI] [PubMed] [Google Scholar]
- Hashemi M. M.; Rovig J.; Bateman J.; Holden B. S.; Modelzelewski T.; Gueorguieva I.; von Dyck M.; Bracken R.; Genberg C.; Deng S.; Savage P. B. Preclinical testing of a broad-spectrum antimicrobial endotracheal tube coated with an innate immune synthetic mimic. J. Antimicrob. Chemother. 2018, 73 (1), 143–150. 10.1093/jac/dkx347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H.; Doerksen R. J.; Jones T. V.; Klein M. L.; Tew G. N. Biomimetic facially amphiphilic antibacterial oligomers with conformationally stiff backbones. Chem. Biol. 2006, 13 (4), 427–435. 10.1016/j.chembiol.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Mensa B.; Howell G. L.; Scott R.; DeGrado W. F. Comparative mechanistic studies of brilacidin, daptomycin, and the antimicrobial peptide LL16. Antimicrob. Agents Chemother. 2014, 58 (9), 5136–5145. 10.1128/AAC.02955-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T. M.; Rennison D.; Krause A. L.; Mros S.; Ferguson S. A.; Cook G. M.; Cameron A.; Arabshahi H. J.; Brimble M. A.; Cahill P.; Svenson J. Stereochemical Effects on the Antimicrobial Properties of Tetrasubstituted 2,5-Diketopiperazines. ACS Med. Chem. Lett. 2022, 13 (4), 632–640. 10.1021/acsmedchemlett.1c00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble G. W. Biological activity of recently discovered halogenated marine natural products. Mar. Drugs 2015, 13 (7), 4044–4136. 10.3390/md13074044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever J.; Kreuder F.; Henry J.; Hung A.; Allard P.-M.; Brkljača R.; Rix C.; Taki A. C.; Gasser R. B.; Kaslin J.; et al. Targeted Isolation of Antibiotic Brominated Alkaloids from the Marine Sponge Pseudoceratina durissima Using Virtual Screening and Molecular Networking. Mar. Drugs 2022, 20 (9), 554. 10.3390/md20090554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner C.; El Omari M.; König G. M. Biohalogenation: nature’s way to synthesize halogenated metabolites. J. Nat. Prod. 2009, 72 (3), 540–553. 10.1021/np800651m. [DOI] [PubMed] [Google Scholar]
- Olsen E. K.; Hansen E.; Moodie L. W. K.; Isaksson J.; Sepčić K.; Cergolj M.; Svenson J.; Andersen J. H. Marine AChE inhibitors isolated from Geodia barretti: natural compounds and their synthetic analogs. Org. Biomol. Chem. 2016, 14 (5), 1629–1640. 10.1039/C5OB02416A. [DOI] [PubMed] [Google Scholar]
- Moodie L. W.; Žužek M. C.; Frangež R.; Andersen J. H.; Hansen E.; Olsen E. K.; Cergolj M.; Sepčić K.; Hansen K. Ø.; Svenson J. Synthetic analogs of stryphnusin isolated from the marine sponge Stryphnus fortis inhibit acetylcholinesterase with no effect on muscle function or neuromuscular transmission. Org. Biomol. Chem. 2016, 14 (47), 11220–11229. 10.1039/C6OB02120D. [DOI] [PubMed] [Google Scholar]
- Labriere C.; Cervin G.; Pavia H.; Hansen J. H.; Svenson J. Structure-Activity Relationship Probing of the Natural Marine Antifoulant Barettin. Mar. Biotechnol. 2021, 23 (6), 904–916. 10.1007/s10126-021-10074-z. [DOI] [PubMed] [Google Scholar]
- Trepos R.; Cervin G.; Hellio C.; Pavia H.; Stensen W.; Stensvåg K.; Svendsen J.-S.; Haug T.; Svenson J. Antifouling compounds from the sub-arctic ascidian Synoicum pulmonaria: Synoxazolidinones A and C, pulmonarins A and B, and synthetic analogues. J. Nat. Prod. 2014, 77 (9), 2105–2113. 10.1021/np5005032. [DOI] [PubMed] [Google Scholar]
- Molchanova N.; Nielsen J. E.; Sorensen K. B.; Prabhala B. K.; Hansen P. R.; Lund R.; Barron A. E.; Jenssen H. Halogenation as a tool to tune antimicrobial activity of peptoids. Sci. Rep. 2020, 10 (1), 14805. 10.1038/s41598-020-71771-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen M. H.; Engqvist M.; Ausbacher D.; Anderssen T.; Langer M. K.; Haug T.; Morello G. R.; Liikanen L. E.; Blencke H.-M.; Isaksson J.; et al. Amphipathic barbiturates as mimics of antimicrobial peptides and the marine natural products eusynstyelamides with activity against multi-resistant clinical isolates. J. Med. Chem. 2021, 64 (15), 11395–11417. 10.1021/acs.jmedchem.1c00734. [DOI] [PubMed] [Google Scholar]
- Grant T. M.; Rennison D.; Cervin G.; Pavia H.; Hellio C.; Foulon V.; Brimble M. A.; Cahill P.; Svenson J. Towards eco-friendly marine antifouling biocides-Nature inspired tetrasubstituted 2,5-diketopiperazines. Sci. Total Environ. 2022, 812, 152487. 10.1016/j.scitotenv.2021.152487. [DOI] [PubMed] [Google Scholar]
- Svenson J.; Stensen W.; Brandsdal B. O.; Haug B. E.; Monrad J.; Svendsen J. S. Antimicrobial peptides with stability toward tryptic degradation. Biochemistry 2008, 47 (12), 3777–3788. 10.1021/bi7019904. [DOI] [PubMed] [Google Scholar]
- Karstad R.; Isaksen G.; Brandsdal B. O.; Svendsen J. S.; Svenson J. Unnatural amino acid side chains as S1, S1′, and S2′ probes yield cationic antimicrobial peptides with stability toward chymotryptic degradation. J. Med. Chem. 2010, 53 (15), 5558–5566. 10.1021/jm1006337. [DOI] [PubMed] [Google Scholar]
- Svenson J.; Vergote V.; Karstad R.; Burvenich C.; Svendsen J. S.; De Spiegeleer B. Metabolic fate of lactoferricin-based antimicrobial peptides: effect of truncation and incorporation of amino acid analogs on the in vitro metabolic stability. J. Pharm. Exp. Ther. 2010, 332 (3), 1032–1039. 10.1124/jpet.109.162826. [DOI] [PubMed] [Google Scholar]
- Stensen W.; Turner R.; Brown M.; Kondori N.; Svendsen J. S.; Svenson J. Short Cationic Antimicrobial Peptides Display Superior Antifungal Activities toward Candidiasis and Onychomycosis in Comparison with Terbinafine and Amorolfine. Mol. Pharmaceutics 2016, 13 (10), 3595–3600. 10.1021/acs.molpharmaceut.6b00654. [DOI] [PubMed] [Google Scholar]
- Isaksson J.; Brandsdal B. O.; Engqvist M.; Flaten G. E.; Svendsen J. S.; Stensen W. A synthetic antimicrobial peptidomimetic (LTX 109): stereochemical impact on membrane disruption. J. Med. Chem. 2011, 54 (16), 5786–5795. 10.1021/jm200450h. [DOI] [PubMed] [Google Scholar]
- Svenson J.; Karstad R.; Flaten G. E.; Brandsdal B. O.; Brandl M.; Svendsen J. S. Altered activity and physicochemical properties of short cationic antimicrobial peptides by incorporation of arginine analogues. Mol. Pharmaceutics 2009, 6 (3), 996–1005. 10.1021/mp900057k. [DOI] [PubMed] [Google Scholar]
- Haug B. E.; Stensen W.; Kalaaji M.; Rekdal O.; Svendsen J. S. Synthetic antimicrobial peptidomimetics with therapeutic potential. J. Med. Chem. 2008, 51 (14), 4306–4314. 10.1021/jm701600a. [DOI] [PubMed] [Google Scholar]
- Greco I.; Molchanova N.; Holmedal E.; Jenssen H.; Hummel B. D.; Watts J. L.; Håkansson J.; Hansen P. R.; Svenson J. Correlation between hemolytic activity, cytotoxicity and systemic in vivo toxicity of synthetic antimicrobial peptides. Sci. Rep. 2020, 10 (1), 1–13. 10.1038/s41598-020-69995-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szałaj N.; Lu L.; Benediktsdottir A.; Zamaratski E.; Cao S.; Olanders G.; Hedgecock C.; Karlén A.; Erdélyi M.; Hughes D.; Mowbray S. L.; Brandt P. Boronic ester-linked macrocyclic lipopeptides as serine protease inhibitors targeting Escherichia coli type I signal peptidase. Eur. J. Med. Chem. 2018, 157, 1346–1360. 10.1016/j.ejmech.2018.08.086. [DOI] [PubMed] [Google Scholar]
- Sæbø I. P.; Bjørås M.; Franzyk H.; Helgesen E.; Booth J. A. Optimization of the Hemolysis Assay for the Assessment of Cytotoxicity. Int. J. Mol. Sci. 2023, 24 (3), 2914. 10.3390/ijms24032914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palica K.; Vorácová M.; Skagseth S.; Andersson Rasmussen A.; Allander L.; Hubert M.; Sandegren L.; Schrøder Leiros H.-K.; Andersson H.; Erdélyi M. Metallo-β-Lactamase Inhibitor Phosphonamidate Monoesters. ACS Omega 2022, 7 (5), 4550–4562. 10.1021/acsomega.1c06527. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.