

Abstract
Background
Pancreatic adenocarcinoma (PDAC) is well known for its rapid distant metastasis and local destructive behavior. Loss of Krüppel-like factor 10 (KLF10) contributes to distant migration of PDAC. The role of KLF10 in modulating tumorigenesis and stem cell phenotypes of PDAC is unclear.
Methods
Additional depletion of KLF10 in KC (LSL: KrasG12D; Pdx1-Cre) mice, a spontaneous murine PDAC model, was established to evaluate tumorigenesis. Tumor specimens of PDAC patients were immune-stained of KLF10 to correlate with local recurrence after curative resection. Conditional overexpressing KLF10 in MiaPaCa and stably depleting KLF10 in Panc-1 (Panc-1-pLKO-shKLF10) cells were established for evaluating sphere formation, stem cell markers expression and tumor growth. The signal pathways modulated by KLF10 for PDAC stem cell phenotypes were disclosed by microarray analysis and validated by western blot, qRT-PCR, luciferase reporter assay. Candidate targets to reverse PDAC tumor growth were demonstrated in murine model.
Results
KLF10, deficient in two-thirds of 105 patients with resected pancreatic PDAC, was associated with rapid local recurrence and large tumor size. Additional KLF10 depletion in KC mice accelerated progression from pancreatic intraepithelial neoplasia to PDAC. Increased sphere formation, expression of stem cell markers, and tumor growth were observed in Panc-1-pLKO-shKLF10 compared with vector control. Genetically or pharmacologically overexpression of KLF10 reversed the stem cell phenotypes induced by KLF10 depletion. Ingenuity pathway analysis and gene set enrichment analysis showed that Notch signaling molecules, including Notch receptors 3 and 4, were over-expressed in Panc-1-pLKO-shKLF10. KLF10 transcriptionally suppressed Notch-3 and -4 by competing with E74-like ETS transcription factor 3, a positive regulator, for promoter binding. Downregulation of Notch signaling, either genetically or pharmacologically, ameliorated the stem cell phenotypes of Panc-1-pLKO-shKLF10. The combination of metformin, which upregulated KLF10 expression via phosphorylating AMPK, and evodiamine, a non-toxic Notch-3 methylation stimulator, delayed tumor growth of PDAC with KLF10 deficiency in mice without prominent toxicity.
Conclusions
These results demonstrated a novel signaling pathway by which KLF10 modulates stem cell phenotypes in PDAC through transcriptionally regulating Notch signaling pathway. The elevation of KLF10 and suppression of Notch signaling may jointly reduce PDAC tumorigenesis and malignant progression.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12929-023-00937-z.
Keywords: Krüppel-like factor 10, Notch signaling, Notch-3/4, ELF3, Pancreatic adenocarcinoma
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest malignancies, typically presents with early distant metastasis and local invasion. PDAC is resistant to most cancer therapies, and despite recent progress in early detection and therapeutic efficacy, the overall patient survival rate remains low [1]. Therefore, the development of new biologic markers and therapeutic targets for PDAC is an urgent matter.
Our previous studies have revealed that Krüppel-like factor 10 (KLF10), a zinc finger-containing transcription factor originally identified as an early response gene of TGFβ (TGFβ-inducible early gene-1; TIEG1), was downregulated in two-thirds of PDAC patients due to epigenetic and post-translational regulation [2, 3]. KLF10 was reported to positively respond to an anti-proliferative signal of TGFβ [4]. Previous studies, including our own, have reported that KLF10 deficiency activates the SDF/CXCR4 signaling pathway and transcriptionally upregulates Sirtuin 6 and Slug to modulate glycolysis and epithelial–mesenchymal transition (EMT) for distant metastasis in PDAC and lung cancer [5–7]. In contrast to TGFβ in advanced cancers, loss of KLF10 was associated with rapid distant metastasis, relatively short survival, and resistance to radiotherapy [3, 8]. Distinguishing the anti-proliferative functions from the pro-metastatic functions of TGFβ reveals KLF10 to be a potential target for the development of therapies for cancers with aberrant TGFβ pathways, such as PDAC [6]. However, the effects of KLF10 on the cancer stem cell phenotypes of PDAC are still unclear.
In this study, we found that additional deletion of KLF10 in the murine model of spontaneous pancreatic cancer accelerated PDAC tumorigenesis. KLF10 deletion transcriptionally upregulated Notch signaling molecules to promote sphere formation, and tumorigenesis of PDAC. Mechanistically, KLF10 competed with E74-like ETS transcription factor 3 (ELF3) for promoter binding of Notch-3 and -4 for transcription regulation. Upregulating KLF10 and/or suppressing Notch signaling, genetically or pharmacologically, delayed tumor growth of PDAC with KLF10 deficiency.
Materials and methods
Cell culture and chemicals
Human pancreatic cancer cell lines, Panc-1 (BCRC 60284) and MiaPaCa (BCRC 60139), were purchased from the Bioresource Collection and Research Center, Taiwan, and were authenticated by DNA fingerprinting. Panc-1 cells labeled with firefly luciferase plasmid vector (Panc-1-Luc) were kindly provided from Dr. Kelvin K. C. Tsai from Taipei Medical University. The mouse primary pancreatic adenocarcinoma (PDAC) cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 mg/mL streptomycin and 2 mM glutamine at 37 °C in a 5% CO2 incubator; maintained for < 6 passages and histo-pathologically characterized through SCID mice xenograft studies. Metformin hydrochloride (D150959,), dorsomorphin (Compound C, P5499) and evodiamine (E3531) were purchased from Sigma-Aldrich. N-[N-(3,5-Difluorophenacetyl-l-alanyl)]-(S)-phenylglycine t-butyl ester (DAPT, HY-13027) was from MedChem Express.
Flow cytometry
Cells were washed in phosphate buffered saline (PBS) and collected by centrifugation. The cells were stained with primary antibodies including CD44-FITC (560977, BD Pharmingen), CD24-Alexa 647 (56144, BD Pharmingen), c-Met-Alexa 647 (566014, BD Pharmingen), CD326-BB515 (565398, BD Pharmingen), CD133-PE (VII 70485, BD Pharmingen) at 4 °C for 30 min. After washing with PBS twice and re-suspended in 800 µL PBS, the cells were filtered through a 70 mm nylon mesh and carried out on a flow cytometer (BD Biosciences, Heidelberg, Germany). The viable and single cells were gated for analyses. BD CellQuest pro (BD Biosciences) and FlowJo (BD Bioscience) software were used to analyze the data.
Transgenic mice, animal xenograft and orthotopic tumor model
Mice were housed at the animal core facility of the National Health Research Institutes (NHRI) and the University of Kaohsiung Medical University (UKMU), Taiwan. The facilities were approved by the National Association for Accreditation of Laboratory Animal Care, Taiwan, and is maintained in accordance with the regulations and standards of the NHRI and UKMU Animal Council’s procedural and ethical guidelines. Pdx-1-Cre, LSL-KrasG12D and p53Loxp/Loxp breeder mice, generated by Drs. Andrew M. Lowy, Tyler Jacks and Anton Berns [9], were obtained from the Mouse Models of Human Cancers Consortium (MMHCC) under material transfer agreements. KLF10Loxp/Loxp (KLF10 L/L) mice were generously provided by Drs. John Hawse and Malayannan Subramaniam at the Mayo Clinic. All compound mutant mice were on a mixed genetic 129Sv × C57BL/6 background, and detailed genotyping protocols are available on request. Nonobese diabetic (NOD)/severe combined immunodeficient (SCID) mice (NOD. CB17-Prkdcscid/Jnarl) were purchased from the National Laboratory Animal Center, Taipei, Taiwan.
For the murine model of orthotopic pancreas tumor, a small left abdominal flank incision was made, and the spleen was exteriorized. A 1-mL syringe with a 29-gauge needle, in which Panc-1-Luc cells were suspended in PBS with VitroGel, (VHM01, Thewell, Bioscience) at a concentration of 1 × 106 cells/50 μL, was inserted into the tail of pancreas. To prevent tumor cell leakage and bleeding, a cotton swab was held over the injection site for 1 min. The wound was sutured with 5-0 Chromic catgut (CC125, Shin N Med. Inst. Co). For in vivo limiting dilution analysis, tumor cells of 1 × 103, 1 × 104 and 4 × 104 were mixed with VitroGel, (VHM01) at 1:1 ratio and injected subcutaneously into right flank of NOD/SCID mice. Tumor volume was measured twice every week with a caliper, and the volume was calculated using the formula of Tumor volume = 1/2 (length × width2) [10]. DAPT was prepared by dissolving in 30% dimethyl sulfoxide (DMSO) and diluted with PBS to 15 mg/kg/day and intraperitoneally injected 5 days every week for 2 weeks [11] before sacrifice. Metformin was dissolved in distilled water at dose of 120 mg/kg and intraperioneally injected for 3 weeks, 5 days a week. For evodiamine treatment, mice were given 15–30 mg/kg of evodiamine dissolved in DMSO, diluted in PBS, and intraperitoneally injected for 3 weeks, 5 days a week [12].
Mice were anesthetized by intraperitoneal administration of Attane (isoflurane, DP4900V22LA00), and tumor tissues were collected for further analysis. Pancreatic tissue samples were fixed in 10% buffered formalin overnight, washed with 1× PBS, and transferred to 70% ethanol before paraffin embedding, sectioning, and hematoxylin and eosin staining.
Immunohistochemical (IHC)
Paraffin-embedded tissue sections were deparaffinized in xylene, rehydrated through a series of ethanol dilutions, and boiled for 15 min in 10 µM citrate buffer at pH 6.0. Endogenous peroxidase activity was suppressed using a peroxidase block (RE7148, Novolink) for 5 min. The tissue sections were then blocked using protein blocks (RE-7159, Novolink) and incubated overnight at 4 °C with antibodies against KLF10 (1:400, mouse anti-human monoclonal antibody, LTK BioLaboratories), Notch-3 (1:500, ab23426, Abcam, Cambridge, MA), Notch-4 (1:200, ab199295, Abcam). Antibody detection was performed using the Novolink Max Polymer Detection System (RE7280-K, Leica Biosystems).
Immunoblotting
Cell extracts were prepared in lysis buffer (RIPA Cell Lysis Buffer 5×, RP05-100, Visual Protein, Taiwan) that contained a 1× protease inhibitor mixture (4693132001, Roche) and 1 mM phenylmethylsulfonyl fluoride. For western blot analysis, cell extracts were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The electro-transferred membrane (Protran ™ Nitrocellulose membrane NBA 085C001EA) was then incubated with the secondary antibody (92668072, IRDye® 680RD Donkey anti-Mouse IgG; 92632211 IRDye® 800CW Donkey anti-Rabbit IgG; LI-COR Biosciences) and was developed with a LI-COR Odyssey system (LI-COR). We used Notch-1 (1:1000, ab8952, Abcam), Notch-3 (1:1000, ab23426, Abcam), Notch-4 (1:1000, ab199295, Abcam), phospho-AMP-activated protein kinase (AMPK; 1:500, E-ab-21121, Elabscience), c-Myc (1:1000, E-ab-30975, Elabscience), Hes7 (1:1000, E-ab-18076, Elabscience), RBX1 (1:1000, E-ab-18881, Elabscience), FBXW7 (1:1000, E-ab-11064, Elabscience), DLL1 (1:1000, E-ab-66262, Elabscience), Hes1 (1:200, sc166410, Santa Cruz) and Hey L (1:200, sc81294, Santa Cruz) to detect Notch pathway associated protein markers. The KLF10 antibody was purchased from Abcam (1:1000, ab73537). β-Actin antibody (1:1000, E-ab-20094, Elabscience) at a 1:3000 dilution was used as control.
Transfection and transduction
The construction of KLF10 expression vectors (HA-KLF10) has been described previously [13]. The stable downregulation of scramble and KLF10 plasmids (TRCN0000318921) in Panc-1 cells (Panc-1-pLKO-shKLF10) was achieved using a retrovirus-mediated RNA interference system (pSUPER.retro.puro, VEC-PRT-0001, OligoEngine) using shRNAs purchased from the National RNAi Core Facility of Academia Sinica (Taipei, Taiwan): 5′-GAACCCTCTCAAGTGTCAAAT-3′. Infected cell populations were selected using 2 mg/mL puromycin. The effect of shRNA knockdown was rescued by HA-KLF10 to rule out off-target effect [7]. As described previously [8], the Lenti-X™ Tet-One™ Inducible Expression Systems pLVX-TetOne-puro vector plasmids (631849, Clontech) was used for generation of stable doxycycline inducible clones of KLF10 insert under the TRE3G promoter in MiaPaCa cells (MiaPaCa-pLVX-KLF10). To determine whether Notch-3 or -4 are major downstream mediators of KLF10, stable KLF10 mRNA silencing clone was transiently transduced with Notch-3 shRNA TRCN0000020236 (RNAiCORE, Sinica, Taiwan) and Notch-4 shRNA TRCN0000020271 (RNAiCORE, Sinica) using TransIT-X2® Dynamic system (MIR 600, Mirus, Bio) for 48 h. The efficacy of transfection was confirmed through western blotting.
Microarray assay and bioinformatics methods
The total RNA was extracted from Panc-1-pLKO and Panck-1-pLKO-shKLF10 cells by the RNeasy Mini Kit (74004, Qiagen, Hilden, Germany) according to the manufacturer’s instructions. RNA quality and integrity was measured by a 2100 Bioanalyzer (Agilent). Gene expression profiling was performed using the Illumina HumanHT-12 V4.0 chip (Illumina, Inc.) Microarray data normalization was performed using method of model based background correction (PMID: 20502629) in R. Differential gene expression levels between Panc-1-pLKO and Panc-1-pLKO-shKLF10 cell lines were identified by pairwise comparison analyses (≥ 1.5-fold threshold), and expression patterns were analyzed by hierarchical clustering. The canonical pathway analysis tool in Ingenuity Pathways Analysis (IPA, Ingenuity Systems) was used to identify cancer signaling pathways associated with differentially expressed genes. Gene set enrichment analysis (GSEA) was performed using the javaGSEA software developed at the Broad Institute (Cambridge, MA) and the MSigDB Hallmark gene set collection. Gene sets with a nominal of p value < 0.05 and false discovery rate (FDR) value ≤ 25% were considered to have significant enrichment. Microarray data are available in Gene Expression Omnibus (GEO) under accession number GSE218172 for gene expression.
Chromatin immunoprecipitation (CHIP), ChIP-polymerase chain reaction (PCR) and quantitative real-time PCR
The EZ ChIP chromatin immunoprecipitation kit (#17-371, Sigma-Aldrich) was used according to the manufacturer’s protocol. Briefly, the cells were treated with 1% formaldehyde to cross-link proteins to DNA. The cells were lysed with protease inhibitors, sonicated to shear DNA into fragments and incubated with antibodies against KLF10, RNA polymerase III or anti-rabbit IgG overnight. The purified DNA and input genomic DNA were analyzed using real-time PCR. The primers used are as follow: promoter PCR sequences for Notch-3: Forward, TCACAGAGGAAGTGGGTTGC; Reverse, CAGCCTCAGACCTCAGACA. Notch-4: Forward, CCCCAAAGTTGTCCTGGGTT; Reverse, TCCTTGGGATGCAGGGAATG. The results were quantitated by Quantstudio 3 (Applied biosystems).
For RT-PCR, a reverse transcription kit (RR037Q, Takara Bio, San Jose, CA) and KAPA SYBR®FAST Mix (KK4600, KAPA Biosystems) were used as recommended by the manufacturer. The primers used were listed in Additional file 6: Table S1.
Plasmid construction and promoter luciferase assay
The specific protein-1 (SP-1) binding sites over proximal promoter fragments, spanning within 1 Kb, of Notch-3 and -4 were cloned to drive the luciferase gene in the pGL4.1-based luciferase expression plasmid (#E6651, Promega; Additional file 3: Fig. S3A, B). Cells were transfected with individual reporter constructs and co-transfected with pRLTK (E2241, Promega), which constitutively expresses renilla luciferase, to normalize transfection efficiency. The ELF3/ELF3 S68Amut expression vectors were constructed in the pcDNA 3.1(+) vector (V790-20, Addgene). After transfection for 24 h of HA-KLF10 and/or ELF3/ELF3 S68Amut at various ratio, the promoter activities were determined using a dual-luciferase assay kit (E1910, Promega). Data are presented as the ratio of promoter reporter luciferase activity to control vector pGL4.1-enhancer luciferase activity.
Additional information on materials can be found in Additional file 7: Materials and methods.
Results
KLF10 deficiency correlated with accelerated pancreatic tumor growth
Reduced KLF10 expression was noted in the tumor tissues of PDAC patients compared with normal pancreas tissue in immunohistochemistry (IHC) studies. Both cytosol and nuclei in normal pancreas and pancreatic intraepithelial neoplasm (PanIN) exhibited stronger staining than did tumor cells of PDAC (Fig. 1A). The findings are consistent with those of our previous study that involved a tumor/normal tissue paired-tissue array [3]. Gene expression profiles of PDAC also showed reduced KLF10 transcripts of tumor tissue compared with normal pancreas tissue in several databases (Fig. 1B). A trend of faster local recurrence was evident in 105 patients with resected PDAC (Fig. 1C; 18.4 vs 78.2 months, p = 0.092) [ClinicalTrials.gov. identifier: NCT 00994721]. A larger tumor size (Fig. 1D, p = 0.076) was found in patients with low expression of KLF10 [14]. Despite a non-significant correlation with local tumor control and tumor size, partly due to small sample size, KLF10 was a significant prognostic factor for overall survival (p = 0.013) of this cohort of patients with resected PDAC [14]. These results suggest that KLF10 may modulate tumor growth of PDAC.
Fig. 1.

KLF10 deficiency correlated with accelerated pancreatic tumor growth. A Representative H&E (upper panel) and KLF10 immunostaining (middle and lower panel) of human pancreatic normal tissues (left panel), intraepithelial pancreatic neoplasm (PanIN, middle panel), and invasive pancreatic adenocarcinoma (PDAC, right panel). Original magnification: ×100 (mid-panel), ×400 (lower panel). B Violin plots of KLF10 transcript levels of pancreatic normal (yellow) versus tumor (blue) tissues from two representative databases of Gene Expression Omnibus 16515 (n = 52) and Oncomine Logsdon n = 20). *p < 0.05 and **p < 0.01, respectively. C Local recurrence-free survival curves of 105 patients of resected PDAC with low (n = 66) versus high (n = 39) expression of KLF10 immunostaining as described in “Materials and methods” (HR: 1.70, p = 0.092). D Tumor stage (T1/2 versus T3/4) of 105 patients of resected PDAC with high and low expression of KLF10 immunostaining as described in “Materials and methods”. For T3/4, p = 0.076. E Malignant progression to PanIN or PDAC of pancreas tissue in 18 to 24 week-old transgenic mice including Pdx-1-Cre/LSL-K-RasG12D (KC), Pdx-1-Cre/LSL-KLF10 (KLF10 L/L) and Pdx-1-Cre/LSL-KrasG12D/LSL-KLF10 (KKC) mice. *p < 0.05. F Representative H&E (upper panel) and KLF10 staining (lower panel) of pancreas tissue from KC, KLF10 L/L and KKC mice of 18 to 24 week-old. G Representative immunoblots (left panel) and colony formation (middle panel) of Panc-1 cells with vector control (pLKO) or KLF10mRNA silencing (shKLF10). Quantitative bar graphs (right panel) represent cumulated data from three independent experiments of colony formation assay. *p < 0.05. H Representative images of IVIS during 2–6 weeks after tumor implantation (left panel) and resected tumors at 6th week (middle panel) from orthotopic murine model of Panc-1 pLKO (yellow) and Panc-1-pLKO-shKLF10 (blue) as described in “Materials and methods”. Cumulated IVIS signal (right panel) of at least six mice in each experimental group that were injected with Panc-1-pLKO and Panc-1-pLKO-shKLF10 as indicated. Each point represents mean ± standard error (SE). *p < 0.05
In the murine model of spontaneous pancreatic cancer, we found that all LSL: KrasG12D; Pdx1-Cre (KC) mice developed PanIN lesions without invasive carcinoma when they were 18–24 week-old (Fig. 1E). KLF10-knockout (KLF10 L/L) mice of similar age did not have pancreatic malignancy. However, invasive PDAC developed in over 50% of 18–24 week-old KC mice with additional KLF10 depletion (KKC mice) at the frontiers of PanIN lesions (Fig. 1E). In addition, IHC revealed prominent ductal dysplasia, tumor invasion, and fibrotic stroma with down-regulated KLF10 expression in the majority of the pancreatic tumors of KKC mice (Fig. 1F). KLF10 L/L mice, by contrast, exhibited no malignant transformation, and KC mice had various levels of PanIN lesions with moderate KLF10 immunostaining (Fig. 1F).
An in vitro clonogenic assay using Panc-1 cells with KLF10 mRNA knockdown (Panc-1-pLKO-shKLF10) revealed an increased colony number and size when compared with Panc-1 cells of the vector control (Panc-1-pLKO; Fig. 1G). Similar findings were observed in murine cell lines established from KPC (Pdx1-Cre; KrasG12D; Trp53LoxP/LoxP) mice versus those from KKPC (Pdx1-Cre; KrasG12D; Trp53LoxP/LoxP; KLF10L/L) mice (Additional file 1: Fig. S1A). We used a murine orthotopic tumor model by implanting Panc-1-Luc cells in the pancreas, as described in “Materials and methods”. The tumor growth rate was significantly higher in mice that received implantation of Panc-1-pLKO-shKLF10 compared with those that received Panc-1-pLKO (Fig. 1H). These findings suggest that KLF10 plays a pivotal role in pancreatic tumorigenesis.
KLF10 modulated stem cell phenotypes of PDAC
We evaluated whether KLF10 expression modulates stem cell phenotypes of PDAC. Sphere formation ability was measured and compared between Panc-1 cells with and without KLF10 mRNA depletion. The number of spheres formed was significantly higher for Panc-1-pLKO-shKLF10 cells compared with Panc-1-pLKO cells when using the criteria described in “Materials and methods” (Fig. 2A). Induction of KLF10 overexpression significantly suppressed the activity of sphere formation in Panc-1 cells (Fig. 2C and Additional file 1: Fig. S1C).
Fig. 2.

KLF10 modulated stem cell phenotypes of PDAC. A Representative pictures of sphere formation (left panel) of Panc-1-pLKO and Pan-1-pLKO-shKLF10 cells. Original magnification: (left upper panels) 1 × 100, (left lower panels) 1 × 400. Quantitative bar plots (right panel) represent mean ± SE of cumulated data from three independent experiments. **p < 0.01. B Representative flow cytometry (left panel) of stem cell markers including CD44, CD24, c-MET and CD326 of Panc-1-pLKO and Panc-1-pLKO-shKLF10. Quantitative bar plots (right panel) represent mean ± SE of cumulated data from three independent experiments of flow cytometry measuring each stem cell markers. *p < 0.05 and **p < 0.01, respectively. C (Upper panel) immunoblots of KLF10 expression in MiaPaCa cells with conditional overexpression of KLF10 (pLVX-KLF10) as described in “Materials and methods”. Dox represents doxycylin. β-Actin was used as internal control. (Lower panel) Quantitative bar plots of sphere formation from three independent data of MiaPaCa cells with pLVX vector control or MiaPaCa-pLVX-KLF10 treated without or with Dox. **p < 0.01. D (Left upper panels) immunofluorescence staining of CD47 (green) in MiaPaCa-pLVX or MiaPaCa-pLVX-KLF10 without or with Dox. Nuclei were counterstained with DAPI. Quantitative bar plots (right upper panel) represent mean ± SE from three independent experiments. *p < 0.05 and **p < 0.01, respectively. (Lower left panels) representative flow cytometry of CD24 expression of MiaPaCa-pLVX, and MiaPaCa-pLVX-KLF10 without or with Dox treatment. (Lower right panel) quantitative bar plots represent mean ± SE from three independent experiments. **p < 0.01. E Representative immunoblots of KLF10 and phospho-AMPK in Panc-1 cells treated without or with 2 mM metformin and 0–30 µM Compound C as described in “Materials and methods”. F Representative images, ×100 (left upper panels) and ×400 (left lower panel), of sphere formation from Panc-1-pLKO and Panc-1-pLKO-shKLF10 treated without or with 2 mM metformin as described in “Materials and methods”. Quantitative bar plots (right panel) of mean ± SE from cumulated data of three independent experiments. *p < 0.05. G In vivo limiting dilution assay. Number of mice with tumor growth after implanted subcutaneously with Panc-1-pLKO and Panc-1-LKO-shKLF10 cells of various cell number indicated as described in “Materials and methods”. H Tumor growth curves of mice after 1 × 104 cells of Panc-1-pLKO and Panc-1-pLKO-shKLF10 implanted. Each point represents mean ± SE of cumulated data from at least six mice. *p < 0.05
We measured the co-expression of CD24, CD44, c-Met, and CD326 to detect pancreatic cancer stem cells. Panc-1-pLKO-shKLF10 cells had a three- to five fold increase in stem cell marker expression compared with Panc-1-pLKO cells (Fig. 2B). Similar observations were made for murine cell lines derived from KPC versus those from KKPC mice (Additional file 1: Fig. S1B). In addition, conditionally overexpressing KLF10 in MiaPaCa cells revealed that the levels of CD47 [15], CD24, and CD44 expression on tumor cells were reduced by more than 50% (Fig. 2D and Additional file 1: Fig. S1D).
Our previous study revealed that metformin, an oral anti-diabetic agent, increased the radiation sensitivity of PDAC cells by elevating KLF10 expression [8]. AMP-activated protein kinase (AMPK) was demonstrated to activate and stabilize KLF10 via phosphorylation at Thr189 [16]. In this study, we found that KLF10 expression was modulated by metformin via phosphorylating AMPK. Compound C, an AMPK inhibitor, suppressed AMPK phosphorylation and KLF10 expression (Fig. 2E). Sphere formation in Panc-1-pLKO-shKLF10 cells could be reduced by metformin, which elevated KLF10 expression (Fig. 2F).
We used in vivo limiting dilution assay to evaluate tumorigenic ability. When a 1 × 104 or lower cell amount was implanted, the number of mice with tumor formation differed significantly between Panc-1-pLKO and Panc-1-pLKO-shKLF10 cells (Fig. 2G). Tumor size was significantly larger in the mice implanted with 1 × 104 of Panc-1-pLKO-shKLF10 cells than in those implanted with Panc-1-pLKO cells (Fig. 2H).
These results suggest that KLF10 is significantly associated with stem cell phenotypes in PDAC tumorigenesis and progression, which might be reversed by metformin.
KLF10 deficiency facilitated the Notch signaling pathways
A microarray analysis was used to identify differentially expressed genes in Panc-1 with or without KLF10 depletion as described in “Materials and methods”. After KLF10 depletion, an ingenuity pathway analysis (IPA) revealed that pathways, including the Wnt and Notch signaling pathways, were highly upregulated in the “cancer signal pathway” profile (Fig. 3A). A gene set enrichment analysis (GSEA) revealed that the Notch signaling pathway, which may modulate stem cell phenotypes, was one of the most differentially upregulated by Panc-1-pLKO-shKLF10 cells (Fig. 3B, D). We measured representative transcripts of candidate molecules from 10 signaling pathways essential to cancer stem cell survival to understand the signals governing stem cell homeostasis in Panc-1 cells with or without KLF10 deficiency (Fig. 3C and Additional file 6: Table S1A). Notch signaling was one of the most upregulated in Panc-1-pLKO-shKLF10 compared with Panc-1-pLKO cells. Since Notch signal transcript was the most upregulated one in the 10-candidate signaling pathway of cancer stem cell and in both IPA and GSEA studies, we chose Notch pathway rather than WNT/b-catenin or EMT pathway for further investigation.
Fig. 3.

KLF10 deficiency facilitated the Notch signal pathway. A The most differentially expressed “cancer signaling pathways” associated with KLF10 depletion in Panc-1 cells by IPA. Graphs in green or blue show category scores as − log [p-value]. Ratio (black dot) indicates the molecules from the data set that map to the pathway listed divided by the total number of molecules that map to the pathway from within the IPA database. B The most enriched pathways associated with Panc-1-pLKO-shKLF10 cells by GSEA analysis. Red indicates positive and yellow indicates negative normalized enrichment score. C Quantitative RT-PCR of candidate signal pathways molecules related to stem cell phenotypes were measured in Panc-1-pLKO (yellow) and Panc-1-pLKO-shKLF10 (blue) cells. Data were presented in mean ± SE from cumulated results of three independent experiments. ***p < 0.005. D GSEA analysis of Panc-1-pLKO-shKLF10 versus Panc-1-pLKO showing upregulated HALLMARK_NOTCH_SIGNALING (left panel). Heat map of Notch signal pathway molecules expression of Panc-1-pLKO and Panc-1-pLKO-shKLF10 cells in GSEA analysis (right panel). E Quantitative RT-PCR of candidate molecules of Notch signal pathway in Panc-1-pLKO (yellow) and Panc-1-pLKO-shKLF10 (blue) cells. Data were presented in mean ± SE from cumulated results of three independent experiments. **p < 0.01 and ***0.005, respectively. F Representative immunoblots of Notch signal pathway molecules in Panc-1-pLKO and Panc-1-pLKO-shKLF10 cells. β-Actin was used as internal control. G Representative databases from Oncomine showing mild to moderate inverse correlation between the transcript levels of KLF10 versus Notch-3 (left panel); and KLF10 versus Notch-4 (right panel). H (Left panel) representative IHC staining of KLF10, NICD-3 and -4 in tumor tissues from two patients of PDAC showing low (upper panel, EI score = 4) and high (lower panel, EI score = 9) KLF10 expression. (Right panel) correlation of KLF10 versus NICD-3 (upper panel, n = 31) and KLF10 versus NICD-4 (lower panel, n = 29) expression on IHC in pancreatic tumors tissues from the cohort mentioned in “Materials and methods”. The correlation coefficients R were − 0.16 and − 0.3 with p = 0.05 and 0.04, respectively
The Notch signaling molecules were validated in RNA and protein expression levels in Panc-1 cells with or without KLF10 depletion (Fig. 3E, F, Additional file 2: Fig. S2A and Additional file 6: Table S1B) as well as in MiaPaCa cells with or without ectopic KLF10 overexpression (Additional file 2: Fig. S2B, C). In Panc-1-pLKO-shKLF10 cells, we found increased expression of the Notch ligands, receptors, activated intracellular domain, activating enzymes and down-stream effectors, which correlates with the heatmap of the Notch signaling pathway from GSEA (Fig. 3D, right panel). Among all, Notch-3 and Notch-4 in both full length and activated forms (i.e., Notch-3 and -4 intracellular domains, NICD-3/4) were all significantly upregulated in Panc-1-pLKO-shKLF10 compared with Panc-1-pLKO cells (Fig. 3E, 3F and Additional file 2: Fig. S2A).
Transcriptomic data from Oncomine revealed a low-to-moderate inverse correlation between expression of KLF10 and Notch-3/4 in several databases (Fig. 3G). IHC of 31 and 29 tumor specimens, respectively, from the cohort of PDAC patients described in “Materials and methods”, also showed a trend of inverse correlation between KLF10 and NICD-3/4 (Fig. 3H).
We concluded that the Notch signaling pathway is activated by KLF10 deficiency in PDAC.
KLF10 suppressed Notch-3 and Notch-4 gene transcription
To delineate the molecular mechanisms of KLF10 in regulating Notch-3 and Notch-4 transcription, we evaluated candidate binding sites in the promoter region of the human Notch-3 and Notch-4 genes by using the Eukaryotic Promoter Database (Swiss Institute of Bioinformatics, Lausanne, Switzerland). Since KLF10 belongs to the specificity protein-1 (SP-1)-like transcription factor family, over 20 putative SP-1 binding elements were recognized from the gene promoters of human Notch-3 and Notch-4 with predicted p < 0.01 (Fig. 4A, left panel, predicted p < 0.001). To validate if KLF10 indeed acted through these elements, the luciferase reporter genes were constructed to be driven by the human Notch-3/4 gene promoter region containing the wild-type and deleted SP-1 binding motifs, respectively (Fig. 4A, right panel; Additional file 3: Fig. S3B, C). Luciferase activities of wild-type Notch-3/4 gene promoters were significantly decreased by HA-KLF10 in Panc-1 cells (Fig. 4B). However, reduced luciferase activity of Notch-3/4 gene promoters with deletion of − 986 to − 726 and − 938 to − 797 was not observed (Fig. 4C).
Fig. 4.

KLF10 competed with ELF3 and transcriptionally suppressed Notch-3 and -4. A Primer maps with predicted SP-1/KLF10 binding sites (left panel, predicted p < 0.001) and various designed deletion (right panel) of promoter regions of Notch-3 and -4 for luciferase reporter assay. B Luciferase activity of Panc-1 cells transfected with vector control (yellow), Notch-3(blue), and Notch-4(green) gene promoter plasmids without (filled) or with (diagonal stripe) hemagglutinin (HA) tagged-KLF10 transfection (OE). The bar graphs were mean ± SE from cumulated data of three independent experiments. *p < 0.05 and **p < 0.01, respectively. C Luciferase activity of Panc-1 cells, transfected with Notch-3 (yellow) or Notch-4 (blue) gene promoter plasmids with full length (filled) or with various deletion (diagonal stripe, diagonal check) indicated. The bar graphs were mean ± SE from cumulated data of three independent experiments. *p < 0.05 and **p < 0.01, respectively. D (Upper panel) primer maps of Notch-3/4 gene promoters for CHIP-PCR assay. (Lower panel) CHIP assay. DNA fragments of Panc-1 cells were immune-precipitated with KLF10, IgG or positive control (P) followed by PCR amplification of the Notch-3/4 gene promoter region that contains KLF10 binding sites (input), as described in “Materials and methods” section. E Quantitative PCR of the Notch-3/4 gene promoter region using samples from MiaPaCa (yellow) or Panc-1 (blue) cells prepared as described in D. The bar graphs were mean ± SE from cumulated data of three independent experiments. F MiaPaCa cells with conditional over-expression of KLF10 under Dox treatment were immuno-precipitated with KLF10 antibody and quantitative PCR of the Notch-3 (yellow) and Notch-4 (blue) gene promoter regions. The bar graphs were mean ± SE from cumulated data of three independent experiments. *p < 0.05. G Primer maps of predicted ELF3 binding sites on the promoter regions of Notch-3 (upper panel) and Notch-4 (left panel) genes. H Luciferase reporter assays of Notch-3 (left panel) and Notch-4 (right panel) under conditions of various combination of HA-KLF10 and ELF3 (yellow) and ELF3-mutant (ELF3-S68A, blue) as indicated. The bar graphs were mean ± SE from cumulated data of three independent experiments. **p < 0.01
To demonstrate that KLF10 directly bound to Notch-3 and Notch-4 gene promoters, we conducted the Chromatin Immunoprecipitation (ChIP) assay in Panc-1 cells with an anti-KLF10 antibody and polymerase chain reaction (PCR) primers spanning each SP-1-binding site of the Notch-3/4 gene promoter regions (Fig. 4D, upper panel; Additional file 3: Fig. S3A). We observed physical binding of KLF10 with the promoters of Notch-3 and Notch-4 genes (Fig. 4D, lower panel). CHIP-PCR demonstrated physical binding of KLF10 with promoters of Notch-3 and -4 in Panc-1 and MiaPaCa cells (Fig. 4E). Overexpressing KLF10 in MiaPaCa cells exhibited enhanced binding of KLF10 with promoters of Notch-3 and Notch-4 genes (Fig. 4F).
Because E74-like ETS transcription factor 3 (ELF3) was reported to bind to Notch-3 gene promoter with transcription regulation in K-RAS-mediated lung adenocarcinoma [17], we evaluated the interaction of KLF10 and ELF3 in Panc-1 cells. The predicted ELF3-binding sites of Notch-3/4 gene promoters highly overlapped with those of KLF10 (Fig. 4G). We constructed plasmids expressing wild-type and mutated ELF3, as described in “Materials and methods” (Additional file 3: Fig. S3D). Luciferase expression in the Notch-3/4 gene promoter-driven reporter assay increased in proportion with increasing amount of ELF3 over KLF10. The phenomenon was not as significant in competition assay using mutated ELF3 with KLF10 (Fig. 4H). Phosphorylation of serine 68 of ELF3 was demonstrated to participate in nuclear import, accumulation and retention of ELF3 in Kras mutated lung adenocarcinoma [17] The partial competition between S68A ELF3 mutant and KLF10, especially in Notch-3 gene promoter activity, may be attributed to multiple binding sites of ELF3 on Notch-3 promoter with various affinity and inherent levels of KLF10/ELF3 in Panc-1 cells. Several GEO databases of pancreatic adenocarcinoma revealed a positive correlation of ELF3 with Notch-3 and -4 transcripts (Additional file 3: Fig. S3E, left panel). An inverse correlation of ELF3 and KLF10 expression was also found (Additional file 3: Fig. S3E, right panel).
The findings suggest that ELF3 and KLF10 competed for activation and suppression, respectively, of the Notch-3 and -4 promoter activity (Fig. 4H).
Inhibition of Notch signal reversed stem cell phenotypes induced by KLF10 deficiency
To demonstrate that the Notch signaling pathway contributed to KLF10 in modulating stem cell phenotypes of PDAC, we silenced mRNA of Notch-3 or -4 genetically in Panc-1-pLKO-shKLF10 cells. The immunoblots revealed that Notch-3, Notch-4, NICD-3, NICD-4, Hes 7, and c-Myc were elevated by KLF10 depletion in Panc-1 cells. The phenomenon was reversed by genetically depleting Notch-3 and -4, respectively (Fig. 5A). Sphere formation and expression levels of CD24 and c-Met in Panc-1-pLKO-shKLF10 were also reversed by Notch-3/4 knockdown (Fig. 5B, C). Despite small difference in elevating Notch-3 and -4 levels by KLF10 knock-down, there was no evident disparity of biologic outcomes of Notch-3/4 depletion in Panc-1 shKLF10 cells. Pharmacologically, N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT, a γ-secretase inhibitor) was used to suppress Notch signal transduction. NICD-3 and -4 as well as Hes 7, and c-Myc were down-regulated in Panc-1-pLKO-shKLF10 after DAPT treatment (Fig. 5D). Since the level of presenilin enhancer, gamma-secretase subunit (PSENEN, PEN-2), was elevated after KLF10 mRNA silencing (Fig. 3F), the therapeutic effect of DAPT was more prominent on Panc-1-pLKO-shKLF10 compared with that on Pan-1pLKO cells (Fig. 5D). The enhanced sphere formation and stem cell markers of CD24 and c-Met in Panc-1-pLKO-shKLF10 were suppressed by DAPT (Fig. 5E, F, Additional file 4: Fig. S4A, B).
Fig. 5.

Inhibition of Notch signal reversed stem cell phenotypes induced by KLF10 depletion. A (Upper panels) representative immunoblots of KLF10 and Notch signal pathway molecules as indicated in Panc-1-pLKO or Panc-1-pLKO-shKLF10 cells without or with Notch-3 (left panel) or Notch-4 (right panel) genes silencing. β-Actin was used as internal control. (Lower panels) the bar graphs were mean ± SE from cumulated data of three independent experiments. Levels of signal molecule expression in Panc-1-pLKO (yellow) or Panc-1-pLKO-shKLF10 (blue) cells without (filled) or with Notch 3/4 (shN3/4, diagonal stripe) receptor mRNA silencing were measured. *p < 0.05. B Sphere formation of Panc-1-pLKO (yellow) or Panc-1-pLKO-shKLF10 (blue) cells without (filled) or with Notch 3 (shN3, diagonal stripe) or Notch 4 (shN4, diagonal check) receptor mRNA silencing. The bar graphs were mean ± SE from cumulated data of three independent experiments. *p < 0.05 and **p < 0.01, respectively. C (Left panels) representative flow cytometry of CD24 and c-Met on Panc-1-pLKO (upper panels) and Panc-1-pLKO-shKLF10 (lower panels) cells without or with Notch-3/4 mRNA silencing as indicated. (right panels) Expression of CD24 (left panel) and c-Met (right panel) on Panc-1-pLKO (yellow) or Panc-1-pLKO-shKLF10 (blue) cells without (filled) or with Notch-3 (sN3, diagonal stripe) or Notch-4 (sN4, diagonal check) mRNA silencing. The bar graphs were mean ± SE from cumulated data of three independent experiments. **p < 0.01 and ***p < 0.005, respectively. D (Left panel) representative immunoblots of KLF10 and Notch signal molecules in Panc-1-pLKO or Panc-1-pLKO-shKLF10 cells without or with 20 µM DAPT treatment. β-Actin was used as internal control. (Right panel) the bar graphs were mean ± SE from cumulated data of three independent experiments. Levels of signal molecule expression in Panc-1-pLKO (yellow) or Panc-1-pLKO-shKLF10 (blue) cells without (filled) or with DAPT (diagonal stripe) treatment were measured. *p < 0.05. E Sphere formation of Panc-1-pLKO (yellow) or Panc-1-pLKO- shKLF10 (blue) cells without (filled) or with (diagonal stripe) 20 µM DAPT treatment. The bar graphs were mean ± SE from cumulated data of three independent experiments. *p < 0.05. F (Upper panels) representative flow cytometry of CD24 and c-Met on Panc-1-pLKO (left panels) and Panc-1-pLKO-shKLF10 (right panels) cells without or with DAPT treatment as indicated. (Lower panels) expression of CD24 (left panel) and c-Met (right panel) on Panc-1-pLKO (yellow) or Panc-1-pLKO-shKLF10 (blue) cells without (filled) or with (diagonal stripe) 20 µM DAPT treatment. The bar graphs were mean ± SE from cumulated data of three independent experiments. *p < 0.05. G Representative IVIS images of mice at 2–6 weeks after implanting orthotopically with Panc-1-pLKO or Panc-1-pLKO-shKLF10 cells without or with DAPT 15 mg/kg intra-peritoneally for 3 weeks as described in “Materials and methods” (left panel). Representative photos of tumors resected from mice experiments mentioned above (right panel). H Cumulated IVIS signal of at least six mice in each experimental group that were implanted with Panc-1-pLKO (yellow) and Panc-1-pLKO-shKLF10 (blue) without (solid) or with (dashed line) DAPT treatment for 3 weeks as indicated. Each point represents mean ± SE. *p < 0.05. I Representative H&E (upper panel) and IHC staining, as described in “Materials and methods”, of KLF10, NICD-3 and -4, as indicated, in tumors resected from mice experiments of G
In the orthotopic murine model, the tumor growth of Panc-1-pLKO-shKLF10 cells was ameliorated by DAPT treatment (Fig. 5G, H). The histology of tumors from mice implanted with Panc-1-pLKO-shKLF10 after DAPT treatment revealed significant loss of immune-labeling of NICD-3 and -4 but no effect on KLF10 expression (Fig. 5I).
We concluded that the suppression of the Notch signaling pathway genetically or pharmacologically might reverse the stem cell phenotypes of PDAC with KLF10 deficiency.
Metformin and evodiamine cooperatively reduced stem cell phenotypes and tumor growth of PDAC
Because of the notorious toxicity and lack of survival benefit of γ-secretase inhibitors in clinical application [18, 19], we evaluated evodiamine, a specific Notch-3 inhibitor that was reported to reverse tumor growth in non-small cell lung cancer with minimal side effects [12]. In Panc-1 cells, evodiamine exhibited relative suppression of NICD-3 but not of NICD-1 and -4 (Fig. 6A, left panel). Because of the low-to-moderate inverse correlation between KLF10 and Notch-3, we used metformin and evodiamine combination treatment in Panc-1 cells to increase KLF10 while suppressing Notch-3 signaling for better therapeutic outcomes. The combination of metformin and evodiamine in Panc-1-pLKO-shKLF10 suppressed NICD-3 and -4 and elevated KLF10 expression (Fig. 6A, right panel). The sphere-forming activity and the stem cell markers expression decreased by concomitant metformin and evodiamine compared to either reagent alone (Fig. 6B, C). The murine orthotopic model treated with the combination of metformin and evodiamine showed reduced tumor formation of Panc-1-pLKO-shKLF10 without evident body weight loss or toxicity (Fig. 6D–F and Additional file 5: Fig. S5). IHC of tumors in mice implanted orthotopically with Panc-1-pLKO-shKLF10 and treated with the combination of metformin and evodiamine revealed increased KLF10 and reduced NICD-3 and -4 expression (Fig. 6G).
Fig. 6.

Metformin and evodiamine cooperatively reduced stem cell phenotypes and tumor growth of PDAC. A Representative immunoblots of KLF10 and NICD-1, -3, and -4 in Panc-1 cells treated with various dosage of evodiamine (EVO) as indicated. β-Actin was used as internal control (left panel). Representative immunoblots of KLF10 and NICD-3 and -4 in Panc-1-shKLF10 treated with combination of 2 mM metformin (M) and/or 80 µM evodiamine (E) (right panel). B Sphere formation of Panc-1-pLKO (yellow) and Panc-1-pLKO-shKLF10 (blue) without treatment (filled) or treated with metformin alone (diagonal stripe) evodiamine alone (diagonal check) or with combination treatment (unfilled). The bar graphs were mean ± SE from cumulated data of three independent experiments. *p < 0.05. C Expression of CD24 (left panel) and c-MET (right panel) on Panc-1-pLKO (yellow) and Panc-1-pLKO-shKLF10 (blue) without treatment (filled), treated with metformin alone (diagonal stripe), with evodiamine alone (diagonal check), or with combination treatment (unfilled). The bar graphs were mean ± SE from cumulated data of three independent experiments. *p < 0.05 and **p < 0.001 respectively. D Schema of orthotopic murine model treated with metformin (120 mg/kg) and/or evodiamine (15 or 30 mg/kg). E (Left panel) representative IVIS images of mice bearing Panc-1-shKLF10 and treated with metformin (Met) and/or evodiamine (EVO). (Right panel) representative photos of tumors resected from murine experiments mentioned above. F Cumulated IVIS signal of at least six mice in each experimental group that were implanted with Panc-1-shKLF10 and received PBS (dashed yellow), metformin alone (solid yellow), evodiamine alone (dashed blue), or combination (solid blue) for 3 weeks, as indicated. Each point represents mean ± SE. *p < 0.05 and **p < 0.01. G Representative H&E (upper panel) and IHC staining, as described in “Materials and methods”, of KLF10, NICD-3 and NICD-4, as indicated, in tumors resected from mice experiments of E
The results suggest that the combination of metformin and evodiamine suppressed tumor growth of KLF10-deficient PDAC cells by elevating KLF10 and suppressing Notch-3/4 signals without any substantial toxicity (Fig. 7).
Fig. 7.
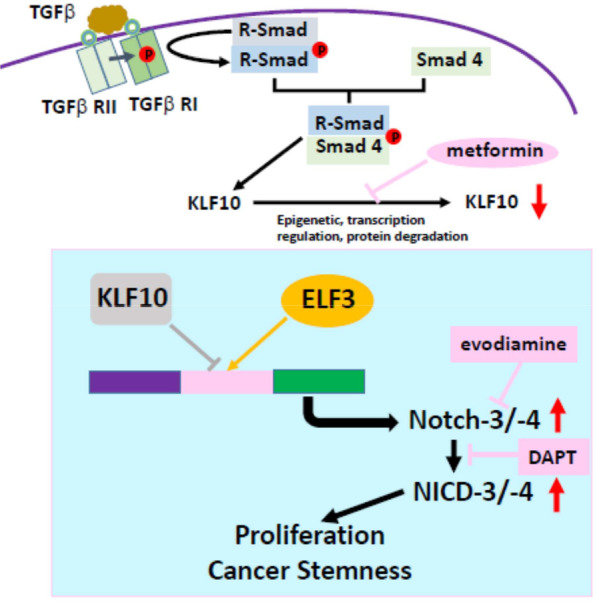
KLF10/Notch signal pathway in modulating proliferation and stemness of PDAC. Loss of KLF10, due to epigenetic, transcriptional regulation or protein degradation in PDAC, interferes the balance of ELF3 and KLF10 competing for activation and suppression, respectively, of the Notch-3 and -4 promoter activity which modulates cancer cell proliferation and stemness phenotypes. Elevating KLF10 expression level by metformin, upregulating Notch 3 transcription by evodiamine or NICD by DAPT, ameliorates PDAC progression induced by KLF10 deficiency
Discussion
PDAC is one of the most complex and aggressive malignancies, exhibiting rapid progression, and is resistant to most cancer therapies. A recent study revealed that cancer cell stemness in PDAC is a major factor that contributes to tumorigenesis, progression, and therapy resistance and are associated with poor clinical outcomes [20]. Our previous studies have shown that KLF10 is not only a prognostic indicator for survival but also a predictor for therapeutic outcomes in PDAC [3, 7, 8]. This study demonstrated that KLF10 regulated sphere formation, expression of stem cell markers and tumorigenesis in PDAC cells. The IPA and GSEA of the microarray study revealed that the Notch signaling pathway was significantly upregulated in Panc-1-pLKO-shKLF10. KLF10 bound to the promoter regions of Notch-3 and -4 genes, and transcriptionally suppressed Notch signal transduction. Genetically depleting Notch-3/4, or pharmacologically suppressing Notch signal by γ-secretase inhibitor ameliorated the stem cell phenotypes and tumor growth induced by KLF10 deficiency in PDAC cells. The combination of metformin, which upregulated KLF10 expression, and evodiamine, a non-toxic Notch-3 inhibitor, did not only reduced sphere formation activity and stem cell markers expression in Panc-1-pLKO-shKLF10 cells, but also exhibited its therapeutic efficacy in suppressing the tumor growth of the Panc-1shKLF10 cells in orthotopic implant mice.
Unlike many other signaling pathways that can be transduced via kinase cascades, the Notch signaling pathway lacks a mediator to amplify signal. Several studies have found that the Notch signal is controlled by the spatiotemporal production and maintenance of Notch receptors and ligands on the cell surface, type of ligand binding to the receptor, glycosylation of the EGF domain that influences ligand-receptor binding, and the amount and stability of NICD [21, 22]. In addition, Notch gene mutations reflecting context-specific selective pressure altered Notch function in various cancers, although mutational activation of Notch is rare-to-absent in PADC [23, 24].
The KLF family transcription factors were demonstrated to regulate Notch signals. Notch-1, DLL4 and Hes 1 were direct transcriptional targets of KLF4 in promoting angiogenesis [25]. KLF9 suppressed Notch-1 transcription in glioblastoma multiforme derived from neurospheres [26]. In this study, we found that KLF10 transcriptionally suppressed Notch-3 and -4 and other Notch signaling molecules. Whether KLF family members can share common promoter binding sites on Notch signal targets and regulate the balance between stemness and differentiation in a coordinated manner warrants further exploration.
Given KLF10 is an early response gene of TGFβ, there is also an interplay between Notch and TGFβ signals. In the presence of CSL and NICD, Smad 3 could be recruited to CSL (CBF-1/RBP-JK, Su(H), Lag-1)-binding sites on DNA [27]. KLF10 might also contribute to Smad 3 recruitment in regulating Notch signaling because the levels of phospho-Smad 3 were significantly higher in the tissue of KLF10 L/L mice [28]. Previous studies, including our own, have revealed that KLF10 transcriptionally suppressed Sirtuin 6 and Slug to modulate EMT in PDAC [6, 7]. KLF10 might also modulate stem cell phenotypes of PDAC through EMT and metabolic reprogramming [29, 30].
The Notch signaling pathway exerts both oncogenic and tumor suppressive functions, depending on the cellular context [31]. Recent studies have revealed that real-time Notch signaling dynamics represent another critical component of the pathway function [32]. The conserved Notch target genes responded differently to the level changes of NICD. Sustained or pulsatile Notch activation, NICD dimerization and chromatin opening initiate different gene expression [33, 34]. We found that ELF3 and KLF10 exerted positive and negative effects on Notch-3 and Notch-4 gene transcription, respectively. ELF3, playing an important oncogenic role in several tumors including PDAC, was reported to positively regulate Notch-3 gene transcription and modulate tumor-initiating cells in K-Ras-mediated lung adenocarcinoma [17]. Furthermore, ELF3 showed an upregulated expression in PDAC and was associated with EMT and poor prognosis [35]. Levels of Notch-3 and -4, which affect the dynamics of the Notch signal, may be adjusted by modulating the ELF3/KLF10 level. Transcription factors including E26 transformation specific (ETS) family for binding serine 68 resides within the pointed domain of ELF3 [17] or epigenetic regulation of KLF10 by interacting with corepressor such as histone deacetylases may also contribute to the Notch signal regulated by ELF3/KLF10.
In contrast to Notch-1 and -2, which were expressed widely in many tissues, Notch-3 was found most abundantly in vascular smooth muscle and pericytes, while Notch-4 was highly expressed on endothelial cells [36]. The levels of Notch-1 and -3 were increased in PDAC tissues [37]. Different Notch receptors have discrete functions in PDAC due to the unique downstream targets and expression patterns of the receptors. In mice with oncogenic K-Ras, the deletion of both Notch-1 alleles accelerated PanIN progression; whereas the deletion of Notch-2 prolonged survival and delayed PDAC development [31, 38, 39]. The expression of Notch-3 and -4 in PDAC has been correlated with malignant phenotypes and drug resistance [40–43]. Notch-3 was reported to inhibit signaling from the Notch-1 by competing for access to RBP-JK and common coactivator [44]. When expressed in cis, the extracellular domain of Notch-4 also inhibited Notch-1 signaling [45]. The opposite effect of the Notch receptors’ signal warrants caution when using a global Notch inhibition approach for treatment purposes.
In addition to γ-secretase inhibitor, we used evodiamine to suppress Notch-3 expression. Evodiamine, an alkaloid derived from Euodiae Fructus, has low toxicity and suppresses Notch-3 by activating DNA methyltransferase 3A induced Notch-3 methylation in non-small-cell lung cancer [12]. Despite its diverse biological effects, we found that evodiamine minimally changed the expression levels of Notch-1 and -4 in PDAC cells. In contrast to some reports of evodiamine’s hepatotoxicity and cardiotoxicity [46], the significant inhibitory effect of evodiamine on tumor size of Panc-1-pLKO-shKLF10 did not result in substantial body weight loss of the mice. Recent studies have also demonstrated the therapeutic efficacy of Notch-3-inhibiting antibodies and Notch-3-targeted antibody–drug conjugate in multiple murine cancer models [47, 48]. Because KLF10 might modulate EMT and metabolic reprogramming to regulate cancer stemness, we combined evodiamine with metformin, which elevated KLF10 expression by modulating AMPK phosphorylation [8], in the orthotopic murine model of PDAC. The combination of evodiamine and metformin therapy achieved significant tumor growth delay without obvious toxicity in mice implanted with Panc-1-pLKO-shKLF10 cells.
In conclusion, we demonstrated a novel signaling pathway of stem cell regulation in PDAC with KLF10 deficiency. We showed that KLF10 competes with ELF3 to bind to Notch-3 and Notch-4 genes promoters for transcription regulation. Loss of KLF10, observed in two-thirds of PDAC patients, favored Notch-3 and-4 expression and the further Notch signal transduction, resulting in the development of stem cell phenotype and tumorigenesis in PDAC. Elevating KLF10 expression and suppressing Notch signaling, either genetically or pharmacologically, ameliorated the malignant progression of PDAC with KLF10 deficiency.
Limitations
To fully exploit the therapeutic potential of our findings, additional work is needed to decipher the KLF10-modulated Notch signaling pathway molecules beyond Notch-3 and -4. The interactions between KLF10 family members and the Notch signaling pathways should also be explored to find potential counterbalances and dynamics in modulating stem cell phenotypes. We used metformin, DAPT, and evodiamine in the murine tumor model to demonstrate that KLF10, the Notch signaling pathway and Notch-3 are potential therapeutic targets in PDAC with KLF10 deficiency. The development of a KLF10 inducer and Notch signal inhibitor with high specificity and low toxicity is warranted to validate the importance of the KLF10/Notch signaling pathways in PDAC.
Conclusions
A spontaneous murine model of PDAC with additional KLF10 depletion reveals accelerated malignant progression and identifies a mechanism of cancer stem cell phenotype involving Notch signaling activation, suggesting a potential therapeutic target.
Supplementary Information
Additional file 1: Figure S1. Representative colony formation of primary murine PDAC cells from KPCmice as indicated.quantitative bar graphs of mean ± SE from cumulated data of three independent experiments. *p < 0.05,representative flow cytometry of CD133 and CD326 on murine PDAC cell lines from KPC and KKPC mice as indicated.quantitative bar graphs of mean ± SE from cumulated data of three independent experiments from each cell lines developed from KPCor KKPCmice as indicated. *p < 0.05,representative sphere formation of MiaPaCa cells of vector controlor with conditional KLF10 overexpressionwithout or with doxycyclinetreatment as indicated. Original magnification 100× and 400× of upper and lower panel, respectively.immunofluorescence stain of CD44on MiaPaCa-pLVX or MiaPaCa-pLVX-KLF10 without or with Dox treatment as indicated. DAPI was used for nuclei stain.quantitative bar graphs of mean ± SE from cumulated data of three independent experiments from MiaPaCa-pLVXor MiaPaCa-pLVX-KLF10 cells withoutor withDox treatment as indicated. *p < 0.05.
Additional file 2: Figure S2. Representative immunofluorescence stain of fluorescence conjugated signal molecules as indicated on spheres of Panc-1-pLKO and Panc-1-pLKP-shKLF10. DAPIwas used for nuclei staining. Original magnification 400×.Representative immunofluorescence stain of KLF10, Notch-3 and -4 as indicated on spheres of MiaPaCa-pLVX or MiaPaCa-pLVX-KLF10 without or with Dox treatment. DAPIwas used for nuclei staining. Original magnification 400×.Representative immunoblots of Notch signal molecules expression of MiaPaCa-pLVX or MiaPaCa-pLVX-KLF10 without or with Dox treatment. β-actin was used as internal control.
Additional file 3: Figure S3. Primer design ofNotch3/4 promoters for ChIP assay. Primer design for cloning ofNotch-3 andNotch-4 promoters of various deletions.Primer design for ELF3 and mutant S68ELF3.Representative GEO databases of correlation between ELF 3 versus Notch-3, Notch-4, and KLF10transcripts levels in pancreatic adenocarcinoma.
Additional file 4: Figure S4. Representative sphere formation of Panc-1-pLKO and Panc-1-pLKO-shKLF10 without or with Notch-3 or Notch-4 depletion as indicated. Original magnification 100×Representative sphere formation of Panc-1-pLKO-shKLF10 without or with 5 µM DAPT treatment. Original magnification 100×.
Additional file 5: Figure S5. Body weight of mice implanted orthotopically with Panc-1-pLKO-shKLF10 and treated with PBS, metformin, evodiamineor concomitant metformin and evodiamine. Each point represents mean ± SE from cumulated data of at least 5 mice.
Additional file 6: Table S1. 10 pathway QPCR primer sequence.Notch pathway QPCR primer sequence.
Additional file 7. Additional materials and methods.
Acknowledgements
We thank all the patients and physicians participating the clinical trials [ClinicalTrials.gov. identifier: NCT 00994721; NCT01666184]; the research nurses and statisticians of Taiwan Cooperative Oncology Group, NHRI, Taiwan; Dr. Shu-ling Peng and Dr. Chen Chang, from National Cheng Kung University Hospital, Tainan, for interpreting tumor specimens of PDAC patients. We thank Ms. Fang-Yu Tsai and the core Instrument Center of NHRI for providing service for microarray analysis; and the laboratory animal center of NHRI for animal study. This work was supported by grants MOST 109-2314-B-400-020-MY3 and NHRI CA-110~111-PP-14.
Abbreviations
- KLF10
Krüppel-like factor 10
- PDAC
Pancreatic adenocarcinoma
- KC mice
LSL: KrasG12D; Pdx1-Cre mice
- KLF10L/L
KLF10Loxp/Loxp mice
- KPC cells
Primary cancer cell lines from Pdx-1Cre, LSL-KrasG12D and p53Loxp/Loxp mice
- KKPC cells
Primary cancer cell lines from Pdx-1Cre, LSL-KrasG12D p53Loxp/Loxp and KLF10Loxp/Loxp mice
- NOD/SCIDmice
Nonobese diabetic /severe combined immunodeficient mice
- Panc-1-pLKO-shKLF10
Stably depleting KLF10 in Panc-1 cells
- MiaPaCa-pLVX-KLF10
Stable doxycycline inducible clones of KLF10 insert under the TRE3G promoter in MiaPaCa cells
- Panc-1-Luc
Panc-1 cells labeled with firefly luciferase plasmid vector
- ELF3
E74-like ETS transcription factor 3
- AMPK
Adenosine monophosphate-activated protein kinase
- NICD
Notch intracellular domains
- TIEG1
TGFβ-inducible early gene-1
- EMT
Epithelial–mesenchymal transition
- SP-1
Specific protein-1
- CHIP
Chromatin immunoprecipitation
- qRT-PCR
Quantitative real-time polymerase chain reaction
- IPA
Ingenuity pathway analysis
- GSEA
Gene set enrichment analysis
- PBS
Phosphate-buffered saline
- IVIS
In vivo imaging system
- EI score
Extent and intensity score of immunohistochemical stain
- Compound C
Dorsomorphin
- DAPT
N-[N-(3,5-Difluorophenacetyl-l-alanyl)]-(S)-phenylglycine t-butyl ester
Author contributions
HJC, KHC conceived and designed research. YCT, SSJ, SEC, SLC contributed to the development of methodology and analytic tools. HJC, YCT contributed to the pathological evaluation of murine and human tumor tissues. YCT, HJC performed research and collected data. TSH provided valuable conception and support. JRH provided transgenic murine model. HJC wrote the manuscript with help from TSH and other co-authors.
Funding
This work was supported by the grants MOST 109-2314-B-400-020-MY3 and NHRI CA-110~111-PP-14.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. The microarray data have been deposited in GEO under accession number GSE218172. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Declarations
Ethics approval and consent to participate
Tissue specimens were collected from pancreatic cancer patients recruited for clinical trials (ClinicalTrails.gov. identifier: NCT 00994721; NCT01666184), and the study was approved by the institutional review board of the National Health Research Institutes in Taiwan (approval number: EC1010502). The study was performed in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ercan G, Karlitepe A, Ozpolat B. Pancreatic cancer stem cells and therapeutic approaches. Anticancer Res. 2017;37(6):2761–2775. doi: 10.21873/anticanres.11628. [DOI] [PubMed] [Google Scholar]
- 2.Hwang YC, Yang CH, Lin CH, Ch'ang HJ, Chang VHS, Yu WCY. Destabilization of KLF10, a tumor suppressor, relies on thr93 phosphorylation and isomerase association. Biochim Biophys Acta. 2013;1833(12):3035–3045. doi: 10.1016/j.bbamcr.2013.08.010. [DOI] [PubMed] [Google Scholar]
- 3.Chang VH, Chu PY, Peng SL, Mao TL, Shan YS, Hsu CF, Lin CY, Tsai KK, Yu WC, Ch'ang HJ. Kruppel-like factor 10 expression as a prognostic indicator for pancreatic adenocarcinoma. Am J Pathol. 2012;181(2):423–430. doi: 10.1016/j.ajpath.2012.04.025. [DOI] [PubMed] [Google Scholar]
- 4.Tachibana I, Imoto M, Adjei PN, Gores GJ, Subramaniam M, Spelsberg TC, Urrutia R. Overexpression of the TGFbeta-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J Clin Invest. 1997;99(10):2365–2374. doi: 10.1172/JCI119418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weng CC, Hawse JR, Subramaniam M, Chang VHS, Yu WCY, Hung WC, Chen LT, Cheng KH. KLF10 loss in the pancreas provokes activation of SDF-1 and induces distant metastases of pancreatic ductal adenocarcinoma in the Kras(G12D) p53(flox/flox) model. Oncogene. 2017;36(39):5532–5543. doi: 10.1038/onc.2017.155. [DOI] [PubMed] [Google Scholar]
- 6.Mishra VK, Subramaniam M, Kari V, Pitel KS, Baumgart SJ, Naylor RM, Nagarajan S, Wegwitz F, Ellenrieder V, Hawse JR, Johnsen SA. Kruppel-like transcription factor KLF10 suppresses TGFbeta-induced epithelial-to-mesenchymal transition via a negative feedback mechanism. Cancer Res. 2017;77(9):2387–2400. doi: 10.1158/0008-5472.CAN-16-2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai YC, Chen SL, Peng SL, Tsai YL, Chang ZM, Chang VH, Ch'ang HJ. Upregulating sirtuin 6 ameliorates glycolysis, EMT and distant metastasis of pancreatic adenocarcinoma with kruppel-like factor 10 deficiency. Exp Mol Med. 2021;53(10):1623–1635. doi: 10.1038/s12276-021-00687-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang VH, Tsai YC, Tsai YL, Peng SL, Chen SL, Chang TM, Yu WC, Ch'ang HJ. Krupple-like factor 10 regulates radio-sensitivity of pancreatic cancer via UV radiation resistance-associated gene. Radiother Oncol. 2017;122(3):476–484. doi: 10.1016/j.radonc.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 10.Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24(3):148–154. doi: 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]
- 11.Paris D, Quadros A, Patel N, DelleDonne A, Humphrey J, Mullan M. Inhibition of angiogenesis and tumor growth by beta and gamma-secretase inhibitors. Eur J Pharmacol. 2005;514(1):1–15. doi: 10.1016/j.ejphar.2005.02.050. [DOI] [PubMed] [Google Scholar]
- 12.Su T, Yang X, Deng JH, Huang QJ, Huang SC, Zhang YM, Zheng HM, Wang Y, Lu LL, Liu ZQ. Evodiamine, a novel NOTCH3 methylation stimulator, significantly suppresses lung carcinogenesis in vitro and in vivo. Front Pharmacol. 2018;9:434. doi: 10.3389/fphar.2018.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang DH, Hsu CF, Lin CY, Guo JY, Yu WC, Chang VH. Kruppel-like factor 10 upregulates the expression of cyclooxygenase 1 and further modulates angiogenesis in endothelial cell and platelet aggregation in gene-deficient mice. Int J Biochem Cell Biol. 2013;45(2):419–428. doi: 10.1016/j.biocel.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 14.Pen SL, Shan YS, Hsiao CF, Liu TW, Chen JS, Ho CL, Chou WC, Hsieh RK, Chen LT, Ch'ang HJ. High expression of kruppel-like factor 10 or Smad4 predicts clinical benefit of adjuvant chemoradiotherapy in curatively resected pancreatic adenocarcinoma: from a randomized phase III trial. Radiother Oncol. 2021;158:146–154. doi: 10.1016/j.radonc.2021.02.037. [DOI] [PubMed] [Google Scholar]
- 15.Cioffi M, Trabulo S, Hidalgo M, Costello E, Greenhalf W, Erkan M, Kleeff J, Sainz B, Jr, Heeschen C. Inhibition of CD47 effectively targets pancreatic cancer stem cells via dual mechanisms. Clin Cancer Res. 2015;21(10):2325–2337. doi: 10.1158/1078-0432.CCR-14-1399. [DOI] [PubMed] [Google Scholar]
- 16.Chen YC, Chen RJ, Peng SY, Yu WCY, Chang VH. Therapeutic targeting of nonalcoholic fatty liver disease by downregulating SREBP-1C expression via AMPK-KLF10 axis. Front Mol Biosci. 2021;8:751938. doi: 10.3389/fmolb.2021.751938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ali SA, Justilien V, Jamieson L, Murray NR, Fields AP. Protein kinase Ciota drives a NOTCH3-dependent stem-like phenotype in mutant KRAS lung adenocarcinoma. Cancer Cell. 2016;29(3):367–378. doi: 10.1016/j.ccell.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imbimbo BP, Giardina GA. Gamma-secretase inhibitors and modulators for the treatment of Alzheimer’s disease: disappointments and hopes. Curr Top Med Chem. 2011;11(12):1555–1570. doi: 10.2174/156802611795860942. [DOI] [PubMed] [Google Scholar]
- 19.McCaw TR, Inga E, Chen H, Jaskula-Sztul R, Dudeja V, Bibb JA, Ren B, Rose JB. Gamma secretase inhibitors in cancer: a current perspective on clinical performance. Oncologist. 2021;26(4):e608–e621. doi: 10.1002/onco.13627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu H, Fang X, Luo Q, Ouyang G. Cancer stem cells: a potential target for cancer therapy. Cell Mol Life Sci. 2015;72(18):3411–3424. doi: 10.1007/s00018-015-1920-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moretti J, Brou C. Ubiquitinations in the notch signaling pathway. Int J Mol Sci. 2013;14(3):6359–6381. doi: 10.3390/ijms14036359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen W, Huang J, Wang Y. Biological significance of NOTCH signaling strength. Front Cell Dev Biol. 2021;9:652273. doi: 10.3389/fcell.2021.652273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aster JC, Pear WS, Blacklow SC. The varied roles of Notch in cancer. Annu Rev Pathol. 2017;12:245–275. doi: 10.1146/annurev-pathol-052016-100127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mullendore ME, Koorstra JB, Li YM, Offerhaus GJ, Fan X, Henderson CM, Matsui W, Eberhart CG, Maitra A, Feldmann G. Ligand-dependent Notch signaling is involved in tumor initiation and tumor maintenance in pancreatic cancer. Clin Cancer Res. 2009;15(7):2291–2301. doi: 10.1158/1078-0432.CCR-08-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hale AT, Tian H, Anih E, Recio FO, 3rd, Shatat MA, Johnson T, Liao X, Ramirez-Bergeron DL, Proweller A, Ishikawa M, Hamik A. Endothelial Kruppel-like factor 4 regulates angiogenesis and the Notch signaling pathway. J Biol Chem. 2014;289(17):12016–12028. doi: 10.1074/jbc.M113.530956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ying M, Sang Y, Li Y, Guerrero-Cazares H, Quinones-Hinojosa A, Vescovi AL, Eberhart CG, Xia S, Laterra J. Kruppel-like family of transcription factor 9, a differentiation-associated transcription factor, suppresses Notch1 signaling and inhibits glioblastoma-initiating stem cells. Stem Cells. 2011;29(1):20–31. doi: 10.1002/stem.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blokzijl A, Dahlqvist C, Reissmann E, Falk A, Moliner A, Lendahl U, Ibanez CF. Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J Cell Biol. 2003;163(4):723–728. doi: 10.1083/jcb.200305112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heo SH, Jeong ES, Lee KS, Seo JH, Lee WK, Choi YK. Knockout of kruppel-like factor 10 suppresses hepatic cell proliferation in a partially hepatectomized mouse model. Oncol Lett. 2017;13(6):4843–4848. doi: 10.3892/ol.2017.6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu M, Liang K, Zhen J, Zhou M, Wang X, Wang Z, Wei X, Zhang Y, Sun Y, Zhou Z, Su H, Zhang C, Li N, Gao C, Peng J, Yi F. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling. Nat Commun. 2017;8(1):413. doi: 10.1038/s41467-017-00498-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Avila JL, Kissil JL. Notch signaling in pancreatic cancer: oncogene or tumor suppressor? Trends Mol Med. 2013;19(5):320–327. doi: 10.1016/j.molmed.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seymour PA, Collin CA, Egeskov-Madsen AR, Jorgensen MC, Shimojo H, Imayoshi I, de Lichtenberg KH, Kopan R, Kageyama R, Serup P. Jag1 modulates an oscillatory Dll1-Notch-Hes1 signaling module to coordinate growth and fate of pancreatic progenitors. Dev Cell. 2020;52(6):731–747.e8. doi: 10.1016/j.devcel.2020.01.015. [DOI] [PubMed] [Google Scholar]
- 33.Nandagopal N, Santat LA, LeBon L, Sprinzak D, Bronner ME, Elowitz MB. Dynamic ligand discrimination in the notch signaling pathway. Cell. 2018;172(4):869–880.e19. doi: 10.1016/j.cell.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobia FM, Preusse K, Dai Q, Weaver N, Hass MR, Chaturvedi P, Stein SJ, Pear WS, Yuan Z, Kovall RA, Kuang Y, Eafergen N, Sprinzak D, Gebelein B, Brunskill EW, Kopan R. Notch dimerization and gene dosage are important for normal heart development, intestinal stem cell maintenance, and splenic marginal zone B-cell homeostasis during mite infestation. PLoS Biol. 2020;18(10):e3000850. doi: 10.1371/journal.pbio.3000850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kong L, Liu P, Zheng M, Wang Z, Gao Y, Liang K, Wang H, Tan X. The miR-1224-5p/ELF3 axis regulates malignant behaviors of pancreatic cancer via PI3K/AKT/notch signaling pathways. Onco Targets Ther. 2020;13:3449–3466. doi: 10.2147/OTT.S248507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferreira A, Aster JC. Seminars in cancer biology. London: Academic Press; 2021. Notch signaling in cancer: complexity and challenges on the path to clinical translation. [DOI] [PubMed] [Google Scholar]
- 37.Song HY, Wang Y, Lan H, Zhang YX. Expression of Notch receptors and their ligands in pancreatic ductal adenocarcinoma. Exp Ther Med. 2018;16(1):53–60. doi: 10.3892/etm.2018.6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanlon L, Avila JL, Demarest RM, Troutman S, Allen M, Ratti F, Rustgi AK, Stanger BZ, Radtke F, Adsay V, Long F, Capobianco AJ, Kissil JL. Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res. 2010;70(11):4280–4286. doi: 10.1158/0008-5472.CAN-09-4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mazur PK, Einwachter H, Lee M, Sipos B, Nakhai H, Rad R, Zimber-Strobl U, Strobl LJ, Radtke F, Kloppel G, Schmid RM, Siveke JT. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc Natl Acad Sci USA. 2010;107(30):13438–13443. doi: 10.1073/pnas.1002423107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mann CD, Bastianpillai C, Neal CP, Masood MM, Jones DJ, Teichert F, Singh R, Karpova E, Berry DP, Manson MM. Notch3 and HEY-1 as prognostic biomarkers in pancreatic adenocarcinoma. PLoS ONE. 2012;7(12):e51119. doi: 10.1371/journal.pone.0051119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eto K, Kawakami H, Kuwatani M, Kudo T, Abe Y, Kawahata S, Takasawa A, Fukuoka M, Matsuno Y, Asaka M, Sakamoto N. Human equilibrative nucleoside transporter 1 and Notch3 can predict gemcitabine effects in patients with unresectable pancreatic cancer. Br J Cancer. 2013;108(7):1488–1494. doi: 10.1038/bjc.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qian CJ, Chen YY, Zhang X, Liu FQ, Yue TT, Ye B, Yao J. Notch4 inhibition reduces migration and invasion and enhances sensitivity to docetaxel by inhibiting Akt/fascin in pancreatic cancer cells. Oncol Lett. 2016;12(5):3499–3505. doi: 10.3892/ol.2016.5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao J, Qian C. Inhibition of Notch3 enhances sensitivity to gemcitabine in pancreatic cancer through an inactivation of PI3K/Akt-dependent pathway. Med Oncol. 2010;27(3):1017–1022. doi: 10.1007/s12032-009-9326-5. [DOI] [PubMed] [Google Scholar]
- 44.Beatus P, Lundkvist J, Oberg C, Lendahl U. The notch 3 intracellular domain represses notch 1-mediated activation through Hairy/Enhancer of split (HES) promoters. Development. 1999;126(17):3925–3935. doi: 10.1242/dev.126.17.3925. [DOI] [PubMed] [Google Scholar]
- 45.James AC, Szot JO, Iyer K, Major JA, Pursglove SE, Chapman G, Dunwoodie SL. Notch4 reveals a novel mechanism regulating Notch signal transduction. Biochim Biophys Acta. 2014;1843(7):1272–1284. doi: 10.1016/j.bbamcr.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 46.Sun Q, Xie L, Song J, Li X. Evodiamine: a review of its pharmacology, toxicity, pharmacokinetics and preparation researches. J Ethnopharmacol. 2020;262:113164. doi: 10.1016/j.jep.2020.113164. [DOI] [PubMed] [Google Scholar]
- 47.Varga J, Nicolas A, Petrocelli V, Pesic M, Mahmoud A, Michels BE, Etlioglu E, Yepes D, Haupl B, Ziegler PK, Bankov K, Wild PJ, Wanninger S, Medyouf H, Farin HF, Tejpar S, Oellerich T, Ruland J, Siebel CW, Greten FR. AKT-dependent NOTCH3 activation drives tumor progression in a model of mesenchymal colorectal cancer. J Exp Med. 2020;217(10):e20191515. doi: 10.1084/jem.20191515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geles KG, Gao Y, Giannakou A, Sridharan L, Yamin TT, Zhang J, Karim R, Bard J, Piche-Nicholas N, Charati M, Maderna A, Lucas J, Golas J, Guffroy M, Pirie-Shepherd S, Roy M, Qian J, Franks T, Zhong W, O'Donnell CJ, Tchistiakova L, Gerber HP, Sapra P. NOTCH3-targeted antibody drug conjugates regress tumors by inducing apoptosis in receptor cells and through transendocytosis into ligand cells. Cell Rep Med. 2021;2(5):100279. doi: 10.1016/j.xcrm.2021.100279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Representative colony formation of primary murine PDAC cells from KPCmice as indicated.quantitative bar graphs of mean ± SE from cumulated data of three independent experiments. *p < 0.05,representative flow cytometry of CD133 and CD326 on murine PDAC cell lines from KPC and KKPC mice as indicated.quantitative bar graphs of mean ± SE from cumulated data of three independent experiments from each cell lines developed from KPCor KKPCmice as indicated. *p < 0.05,representative sphere formation of MiaPaCa cells of vector controlor with conditional KLF10 overexpressionwithout or with doxycyclinetreatment as indicated. Original magnification 100× and 400× of upper and lower panel, respectively.immunofluorescence stain of CD44on MiaPaCa-pLVX or MiaPaCa-pLVX-KLF10 without or with Dox treatment as indicated. DAPI was used for nuclei stain.quantitative bar graphs of mean ± SE from cumulated data of three independent experiments from MiaPaCa-pLVXor MiaPaCa-pLVX-KLF10 cells withoutor withDox treatment as indicated. *p < 0.05.
Additional file 2: Figure S2. Representative immunofluorescence stain of fluorescence conjugated signal molecules as indicated on spheres of Panc-1-pLKO and Panc-1-pLKP-shKLF10. DAPIwas used for nuclei staining. Original magnification 400×.Representative immunofluorescence stain of KLF10, Notch-3 and -4 as indicated on spheres of MiaPaCa-pLVX or MiaPaCa-pLVX-KLF10 without or with Dox treatment. DAPIwas used for nuclei staining. Original magnification 400×.Representative immunoblots of Notch signal molecules expression of MiaPaCa-pLVX or MiaPaCa-pLVX-KLF10 without or with Dox treatment. β-actin was used as internal control.
Additional file 3: Figure S3. Primer design ofNotch3/4 promoters for ChIP assay. Primer design for cloning ofNotch-3 andNotch-4 promoters of various deletions.Primer design for ELF3 and mutant S68ELF3.Representative GEO databases of correlation between ELF 3 versus Notch-3, Notch-4, and KLF10transcripts levels in pancreatic adenocarcinoma.
Additional file 4: Figure S4. Representative sphere formation of Panc-1-pLKO and Panc-1-pLKO-shKLF10 without or with Notch-3 or Notch-4 depletion as indicated. Original magnification 100×Representative sphere formation of Panc-1-pLKO-shKLF10 without or with 5 µM DAPT treatment. Original magnification 100×.
Additional file 5: Figure S5. Body weight of mice implanted orthotopically with Panc-1-pLKO-shKLF10 and treated with PBS, metformin, evodiamineor concomitant metformin and evodiamine. Each point represents mean ± SE from cumulated data of at least 5 mice.
Additional file 6: Table S1. 10 pathway QPCR primer sequence.Notch pathway QPCR primer sequence.
Additional file 7. Additional materials and methods.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. The microarray data have been deposited in GEO under accession number GSE218172. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.