

Summary
Effective therapeutic strategies are needed for non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations that acquire resistance to EGFR tyrosine kinase inhibitors (TKIs) mediated by epithelial to mesenchymal transition (EMT). We investigate cell surface proteins that could be targeted by antibody-based or adoptive cell therapy approaches and identify CD70 as being highly upregulated in EMT-associated resistance. Moreover, CD70 upregulation is an early event in the evolution of resistance and occurs in drug-tolerant persister cells (DTPCs). CD70 promotes cell survival and invasiveness, and stimulation of CD70 triggers signal transduction pathways known to be re-activated with acquired TKI resistance. Anti-CD70 antibody drug conjugates (ADCs) and CD70-targeting CAR T cell and CAR NK cells show potent activity against EGFR TKI resistant cells and DTPCs. These results identify CD70 as a therapeutic target for EGFR mutant tumors with acquired EGFR TKI resistance that merits clinical investigation.
eTOC Blurb
Nilsson et al. show that CD70 is highly upregulated in non-small cell lung cancer (NSCLC) tumors with acquired EGFR tyrosine kinase inhibitor (TKI) resistance that occurs independent of mesenchymal-epithelial transition (MET) or secondary EGFR mutations. Anti-CD70 antibody drug conjugates and CD70-targeting CAR T and CAR NK cells have activity against resistant cells as well as drug tolerant persister cells.
Graphical Abstract
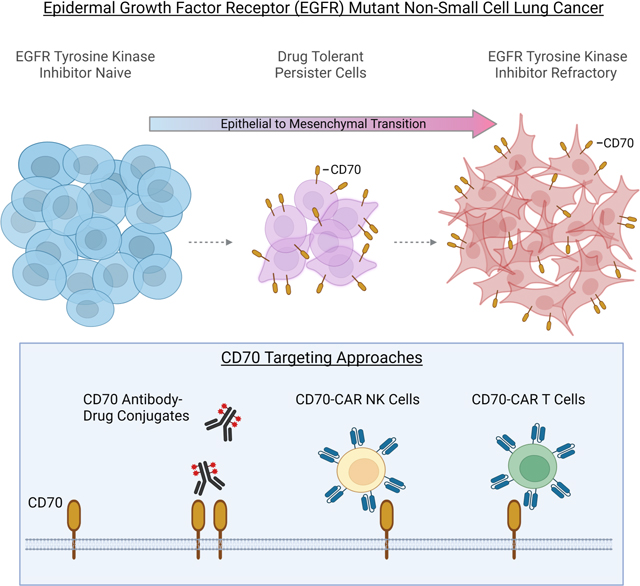
INTRODUCTION
Approximately 10–15% of patients with non-small cell lung cancer (NSCLC) harbor epidermal growth factor receptor (EGFR) activating mutations. The initial standard of care treatment for these patients are EGFR tyrosine kinase inhibitors (TKIs). Although these patients are initially highly responsive to TKIs, EGFR TKI-refractory disease inevitably emerges, with cancer progression after a median of 10–19 months 1–7. EGFR TKIs resistance can be broadly grouped into EGFR-dependent mechanisms such as the acquisition of additional EGFR mutations, and EGFR-independent mechanisms such as expression of alternate receptor tyrosine kinases such as MET 8; epithelial to mesenchymal transition (EMT)9–12; and small cell lung cancer (SCLC) transformation13. While secondary EGFR mutations or MET amplifications can, in some cases, can be treated with other EGFR inhibitors (e.g. the use of the third-generation TKI osimertinib for the T790M resistance mutations)5,14–16, or combinations of EGFR and MET inhibitors17,18, respectively, the vast majority of resistance to the most effective TKIs occurs through other, EGFR-independent mechanisms for which there is a paucity of targeted treatment options. The clinical need is heightened by the observation that these TKI-resistant tumors often adopt a broadly drug-resistant phenotype across most available drugs, particularly if they have undergone EMT19, and are not typically responsive to immune checkpoint inhibitors with an objective response rate of less than 10% 20–23. This highlights the critical need for new treatment approaches for NSCLC patients with EGFR TKI-refractory disease.
Here, we conducted an integrative analysis of putative cell surface targets on NSCLC cells with EGFR-independent resistance mechanisms such as EMT in effort to identify proteins that could be targeted by antibody-based or adoptive cell therapy approaches. We identified CD70 as being highly upregulated on EGFR TKI resistant cells where resistance occurred independent of EGFR or MET in cell lines and in clinical cases of TKI refractory EGFR mutant NSCLC. CD70 is normally expressed predominantly on lymphocytes; we found that EGFR TKI resistant cells markedly upregulated CD70 and stimulation of CD70 activated PI3K and AKT signal transduction pathways critical for cell survival and invasiveness. Moreover, we determined that overexpression of CD70 was an early event in the evolution of EGFR TKI resistance as it was highly expressed on EGFR TKI drug-tolerant persister cells (DTPCs). CD70-targeting approaches including anti-CD70 antibody drug conjugates (ADCs) and CD70-targeting chimeric antigen receptor (CAR) T cell and CAR NK cells had marked anti-tumor activity against EGFR TKI resistant, but not EGFR TKI-naïve, models in vitro and in vivo activity. Given that anti-CD70 based therapeutic strategies are in clinical development for other malignancies, these results support the future testing of CD70 targeting in NSCLC patients with acquired EGFR TKI resistance.
RESULTS
CD70 gene expression is upregulated in NSCLC cells with acquired EGFR TKI resistance
We generated EGFR TKI-resistant cells by culturing EGFR mutant NSCLC cell lines HCC827, HCC4006, H1975 and PC9 cells in the presence of EGFR inhibitors until resistant variants emerged. To identify potentially targetable cell surface proteins upregulated on cells with acquired resistance to EGFR TKIs, we interrogated RNA-seq data from EGFR mutant parental cells (HCC827 and HCC4006) and their associated erlotinib resistant (ER) variants previously shown to have developed resistance through EMT19 and filtered gene expression data to include only genes which transcribed proteins localized to the cell surface (Figure 1A). Cell surface genes which displayed a log2fold change of 3 or greater in expression in resistant cells as compared to parental cells are shown in Table S1 and Figure 1A. AXL was significantly upregulated in ER cells compared to parental lines which was consistent with previous reports 10,24. We identified EMP3 (p = 2.21 × 10−53) and CD70 (p=2.65 × 10−39) as the first and second most significantly upregulated genes in ER cells, respectively (Figure 1A; Figure S1A). Given that EMP3 is expressed in the normal lung, kidneys, and gastrointestinal track and that CD70 expression is highly restricted and only transiently expressed on activated B and T cells and mature dendritic cells and absent on normal epithelial tissue or hematopoietic cells 25–27, CD70 was selected as a top candidate cell surface gene for further study (Table S2). We next assessed CD70 mRNA expression in cell lines with acquired osimertinib resistance (OR) previously shown to be resistant to both erlotinib and osimertinib and to have undergone EMT19 (Figure S1B) and observed significant upregulation of CD70 mRNA in OR cells compared to parental cell lines (HCC4006 and H1975; p = 0.0075; Figure 1B). While CD70 RNA was elevated in EGFR TKI resistant cells as compared to parental cell lines, there was no significant difference in CD70 expression between EGFR wild-type and mutant cell lines (n = 95 EGFR wt, 16 EGFR mutant; Figure S1C) or in treatment-naïve lung adenocarcinoma tumor specimens from the PROSPECT28 or TCGA-LUAD clinical datasets (Figure S1D&E).
Figure 1. CD70 is elevated in NSCLC cells with acquired, EMT-associated EGFR TKI resistance.

(A) Differential expression of genes transcribing cell surface proteins in erlotinib resistant (ER) and parental cells. (B) Mean CD70 expression ± SD in parental (H1975, HCC4006) and osimertinib-resistant (OR) variants. *p = 0.008. (C) CD70 expression by Western blotting. (D-F) Mean CD70 positivity ± SD by flow cytometry (n = 3–6). (G) Mean CD70 positivity ± SD in HCC827 and YUL-0019 cells as compared to MDA-L-011 and MDA-L-004K (n = 2–5). *p = 0.003. (H) Expression of CD70 and EMT-related genes in patient-derived models of erlotinib resistance. (I) scRNAseq analysis of CD70 in T790M negative resistant cells that had undergone EMT31. (J) Change in expression of an EMT gene signature and CD70 in osimertinib-refractory and matched pre-treatment clinical samples. (K) Spearman correlation of differences in EMT-score and CD70 expression for 10 patients with matched pre- and post-osimertinib treatment. Student’s t test was used for B and G.
CD70 is elevated on the cell surface of EGFR TKI resistant cells that have undergone EMT
We next evaluated CD70 expression at the protein level and observed marked increases in CD70 expression by Western blotting in nearly all ER and OR variants compared to parental cells (Figure 1C). CD70 expression was not detected in HCC827 ER2 cells which acquired erlotinib resistance through MET amplification29 (Figure 1C) or PC9 ER2 and PC9 ER6 cells which acquired erlotinib resistance though T790M secondary EGFR mutations19 (Figure S1F). Likewise, flow cytometry analysis revealed elevated cell surface expression of CD70 in ER and OR cells that had acquired resistance through EMT (Figure 1D&E) but not in cells where resistance was mediated by MET amplification or T790M (Figure 1F). Because CD27 is the binding partner for CD70, we assessed CD27 expression in EGFR TKI resistant cells and observed no upregulation of CD27 at the RNA level (Figure S1G) or on the cell surface of EGFR TKI resistant cells (Figure S1H).
To determine whether CD70 was expressed in clinical cases of EGFR TKI resistance, we utilized patient-derived cell lines including MDA-L-011 which was established from a patient with an EGFRL858R-positive NSCLC who progressed on erlotinib and was previously shown to have undergone EMT19. CD70 was detected on the surface of MDA-L-011 cells by flow cytometry (Figure 1G). Likewise, CD70 was absent on YUL-0019 cells which were derived from a treatment-naïve EGFR exon 20 insertion mutant tumor but was highly expressed on MDA-L-004K cells which were derived from a NSCLC patient harboring an EGFR exon 20 insertion mutation who progressed on the EGFR TKI poziotinib (Figure 1G). Using publicly available transcriptomic data from cell lines derived from erlotinib-resistant patient biopsies30, we observed that tumor cells that developed EMT-associated EGFR TKI resistance had increased expression of CD70 (Figure 1H). Next, using single-cell RNA-seq (scRNA-seq), we analyzed specimens from two patients with EGFR mutant lung cancer that had acquired EGFR TKI resistance in which one developed resistance to afatinib and was T790M negative and one developed resistance to gefitinib and was T790M positive31. Previous analysis revealed that cluster 0 from the T790M-negative tumor expressed markers consistent with having undergone EMT31, and we observed elevated expression of CD70 in the EMT tumor cell population (Figure 1I, left). In contrast, CD70 was not detected in the T790M+ tumor (Figure 1I, right). Next, using a dataset of 10 pairs of matched baseline and osimertinib-refractory clinical samples32, we compared the change in EMT-score with the change in CD70 expression in samples collected at progression. Osimertinib-refractory tumors with a high degree of change in EMT score were significantly associated with the greatest increase in CD70 expression (p = 0.028; Figure 1J&K).
We next assessed protein levels of CD70 in a cohort of unmatched EGFR mutant NSCLC clinical specimens collected at baseline (n=16) or after progression on osimertinib (n=36) by immunohistochemistry. CD70 positivity was significantly increased in osimertinib refractory specimens as compared to the TKI naïve group (p = 0.028; Figure 2A&B). CD70 staining intensity was varied across osimertinib-refractory specimens, with 75% of specimens showing tumor cell expression of CD70 to be moderate or high (Figure 2C).
Figure 2. CD70 is increased in osimertinib-resistant NSCLC clinical samples.

(A) Violin plot of CD70 IHC score in specimens collected at baseline (n = 16) or after progression on osimertinib (n = 36). Dashed line = median; dotted lines = first and third quartile. Student’s t test. (B) Representative images of CD70 IHC. 400x magnification; scale bars = 20 μm. (C) CD70 staining intensity among osimertinib-refractory tumors. (D) Overall survival for EGFR TKI refractory NSCLC patients with high or low CD70. (E) Overall survival for NSCLC patients (TCGA-LUAD and GEO databases) with high or low CD70.
CD70 expression and its association with outcome in NSCLC patients
We next evaluated CD70 expression from NSCLC patients and its association with outcome by Kaplan-Meier analysis. Among a dataset of EGFR TKI refractory NSCLC patients (N=39)33 high CD70 expression was associated with strikingly 4.95 fold increase in the risk of death compared with the low CD70 group (p = 0.0017; Figure 2D). For comparison, we also evaluated the impact of CD70 in the overall lung adenocarcinoma population using the TCGA-LUAD and Gene Expression Omnibus (GEO) databases. High CD70 expression was also associated with a significantly worse overall survival (OS) than those with low CD70 expression with a more modest 1.95-fold increase in the risk of death observed (p = 1.6e-08; Figure 2E).
EMT induces CD70 upregulation
We next evaluated the potential role of EMT in modulating CD70 expression in tumor cells. Using TCGA data, we evaluated the correlation between a previously established EMT gene expression signature 24 and CD70 expression. In patients with NSCLC, CD70 expression was correlated with a mesenchymal phenotype (Figure 3A, left) and with expression of the EMT transcriptional regulator, ZEB134,35 (Figure 3A, right). Likewise, CD70 gene expression correlated with an EMT gene expression signature and ZEB1 in NSCLC cell lines (n = 118; Figure 3B). We previously reported that overexpression of ZEB1 in HCC827 cells induced a mesenchymal phenotype and EGFR inhibitor resistance19. Here, we determined that overexpression of ZEB1 resulted in increased CD70 mRNA and cell surface expression of CD70 as determined by real-time PCR and flow cytometry, respectively (Figure 3C&D).
Figure 3. CD70 expression is associated with EMT and epigenetic changes in EGFR mutant NSCLC cells.

(A and B) In NSCLC samples (TCGA-LUAD dataset; A) and across NSCLC cell lines (B), CD70 expression correlated with an EMT expression score (left) and ZEB1 (right). (C) Mean CD70 RNA expression ± SEM in HCC827 cells expressing ZEB1 (n = 3). *p = 0.0096. (D) Mean CD70 positivity ± SD in HCC827 cells with or without ZEB1 (n = 3). *p < 0.0001. (E) GSEA analysis of TGFB expression signature in ER cells that acquired resistance through EMT. (F) Correlation of CD70 and TGFB1 among NSCLC patients (TCGA). (G & H) TGFβ (10 ng/ml) increased ZEB1 (G) and CD70 (H) mRNA. Mean ± SD (n = 3–4). *p < 0.001. (I) Mean CD70 positivity ± SD following TGF-β treatment (n = 2). *p < 0.001. (J) CD70 expression is associated with reduced CD70 promoter methylation. (K) CD70 promoter methylation in epithelial (E) and mesenchymal (M) NSCLC cell lines. Box plots depict the median (line) as well as the 25th and 75th percentile, with whiskers showing 1.5x the interquartile range (IQR). (L) CD70 promoter methylation in parental (P) and osimertinib resistant (OR) and erlotinib resistant (ER) cells. (M) Mean CD70 ± SD expression following exposure to decitabine (DEC; 1 μM; n = 3). *p < 0.0001. Student’s t test was used for C, D, G, H, I, K, M.
See also Figure S2.
Activation of the transforming growth factor beta (TGF-β) signaling axis in EGFR mutant NSCLC cells is sufficient to induce EMT and EGFR TKI resistance36,37. Consistent with this earlier report, we observed that EGFR TKI resistant cells displayed an enriched TGF-β expression signature as determined by gene set enrichment analysis (GSEA; Figure 3E). Given that TGF-β has been shown to modulate the expression of CD70 in immune cells and lymphomas38, we assessed the relationship between TGF-β and CD70 expression in NSCLC. Our analysis of TCGA data revealed a significant positive correlation between TGFB1 expression and CD70 in NSCLC tumors (p < 0.001; Figure 3F). To evaluate the effect of TGF-β on CD70, we treated H1975, HCC827, and HCC4006 parental cells with 10 ng/ml TGF-β for 28 days and evaluated expression of ZEB1 and CD70. In all three cell lines, TGF-β induced expression of the EMT regulator ZEB1 (Figure 3G) as well as CD70 (Figure 3H). Cell surface expression of CD70 was also increased in all three cell lines following exposure to TGF-β (Figure 3I). Using a publicly available RNAseq dataset39, we observed that induction of EMT in HCC827 cells through knockdown of CDH1 similarly resulted in enhanced expression of CD70 (Figure S2A-E). Taken together, these results indicate that expression of CD70 in EGFR TKI resistant cells is linked to the acquisition of a mesenchymal phenotype.
Epigenetic regulation of CD70 in EGFR TKI resistant cells
Promoter methylation has been reported as a mechanism by which CD70 expression is regulated in immune and leukemia cells40–42. To assess the relationship between promoter methylation and CD70 expression in NSCLC cells, we compared CD70 mRNA expression as determined by RNA-seq and CD70 promoter methylation status in 68 NSCLC cell lines and observed a highly significant inverse correlation between CD70 promoter methylation and CD70 mRNA (p = 4.08e-5; Figure 3J). Moreover, we observed significantly lower CD70 promoter methylation in mesenchymal cell lines as compared to epithelial NSCLC cell lines (p = 0.034; Figure 3K). We next assessed CD70 promoter methylation in NSCLC cells with acquired EGFR TKI resistance and found that in ER and OR cells that developed resistance through EMT, CD70 promoter methylation was reduced, whereas in cells where resistance was mediated by MET amplification or T790M, CD70 promoter methylation was unchanged relative to parental cells (Figure 3L). We next treated HCC827 and H1975 cells with the hypomethylating agent, decitabine (1 μM), and observed a significant rise in CD70 mRNA expression (Figure 3M).
Activation of CD70 stimulates signal transduction and invasive pathways in EGFR TKI resistant cells
To determine whether CD70 promotes tumor cell proliferation in EGFR TKI resistant cells, we treated H1975 OR5 and H1975 OR16 cells with siRNA targeting CD70 and evaluated tumor cell growth rate by Cell Titer Glo assay. While siRNA targeting CD70 did not impact the growth rate of parental H1975 cells (Figure 4A), knockdown of CD70 significantly impaired the growth rate of H1975 OR5 and H1975 OR16 cells (Figure 4B&C; Figure S3A&B). Likewise, siRNA targeting CD70 significantly reduced the proliferation rate of HCC4006 OR2 and HCC827 ER6 cells (Figure S3C-E) but had a minimal effect on HCC827 parental cells (Figure S3F). Moreover, siRNA-mediated knockdown of CD70 significantly reduced tumor cell migration in EGFR TKI resistant cells as determined by Boyden chamber assay (Figure 4D&E; Figure S3G&H).
Figure 4. CD70 activates signal transduction in EGFR TKI resistant cells and regulates survival and invasive pathways.

(A-C) Growth rate of H1975, H1975 OR5, and H1975 OR16 following CD70 knockdown (n = 4). Data points are mean viability ± SD. p < 0.001. (D & E) Representative images and high-powered field (HPF) quantification of mean number of migrating H1975 OR16 cells ± SD after CD70 knockdown. *p < 0.0001. (n=3) Scale bars = 1000 μm. (F – I) p-AKT and p-ERK1/2 following stimulation with rhsCD27. (J) Mean number of migrating cells ± SEM following stimulation with rhsCD27 (n = 3). *p < 0.05; student’s t-test. (K) Mean CD70 positivity ± SD in HCC827 cells engineered to overexpress CD70. (L & M) Osimertinib (OSI) dose response curve for HCC827 cells with or without CD70 or rhsCD27. Mean ± SD (n = 3). One-way ANOVA was applied for A, B, C, E.
See also Figure S3.
Binding of CD27 to CD70 activates PI3K and MAPK pathways in lymphocytes 43,44. We stimulated EGFR TKI resistant cells with recombinant human soluble CD27 (rhsCD27) and assessed activation of these signal transduction pathways by Western blotting. rhsCD27 induced phosphorylation of Akt and ERK1/2 in all EGFR TKI resistant cell lines tested (Figure 4F-I) but did not trigger phosphorylation of Akt and ERK1/2 in parental cells (Figure S3I&J). Given that the PI3K/Akt and MAPK/ERK pathways regulate proliferation and migration, we next assessed the impact of rhsCD27 on these cellular programs. In HCC4006 OR2, HCC4006 OR7, and H1975 OR16, rhsCD70 treatment for 5 days did not enhance the number of viable cells alone or in the presence of osimertinib (Figure S3K-M). In H1975 OR5 cells, rhsCD27 induced a modest yet significant increase in viable cells (Figure S3N). Treatment with rhsCD27 did, however, induce a more striking increase in tumor cell migration in OR2 and OR5 EGFR TKI resistant cells (Figure 4J). These results suggest that the binding of CD27 to CD70 activates intracellular pathways and induces migration although there appeared to be less of an impact on cell survival in this setting.
CD70 expression alone is not sufficient to induce EGFR TKI resistance
To assessed whether expression of CD70 alone was sufficient to induce EGFR TKI resistance, HCC827 cells were transfected to overexpress CD70 (Figure 4K), and sensitivity to osimertinib was evaluated by Cell-Titer Glo assay. HCC827 CD70 cells were sensitive to EGFR inhibition and displayed IC50 values similar to HCC827 control cells (Figure 4L). Moreover, CD27 stimulation of HCC827 CD70 cells did not affect sensitivity to EGFR inhibition (Figure 4M). These results indicate that while CD70 is upregulated in cells with acquired EGFR TKI resistance, CD70 expression alone does not confer therapeutic resistance when expressed in the context of an epithelial phenotype.
Upregulation of CD70 is an early event in the evolution of EGFR TKI resistance
Re-activation of signal transduction pathways downstream of EGFR has been shown to be a critical step in the development of resistance to EGFR TKIs 45,46. Consistent with this, we observed that while treatment of EGFR mutant NSCLC cells with osimertinib resulted in inhibition of EGFR and diminished ERK phosphorylation, reactivation of ERK1/2 could be observed in the surviving population of cells after 72 hours (Figure 5A-C). Real-time PCR analysis revealed that within 72 hours of osimertinib treatment RNA levels of ZEB1 and CD70 were significantly upregulated (Figure 5D&E). Next, HCC4006 and HCC827 cells were treated with erlotinib for 10 days to generate drug-tolerant persister cells (DTPCs) and protein expression was evaluated by reverse phase protein array (RPPA). Erlotinib-treated DTPCs displayed an intermediate EMT phenotype with decreased expression of β-catenin and significantly increased expression of mesenchymal proteins including fibronectin, Axl, and ZEB119 (Figure 5F). Similar results were obtained with HCC4006 cells treated with osimertinib for 10 days (Figure 5G). HCC827, HCC4006 and H1975 osimertinib-treated DTPCs also displayed a significant increase in gene expression of ZEB1 and CD70 as compared to control cells (Figure 5H&I). Likewise, cell surface expression of CD70 was increased on DTPCs as determined by flow cytometry (Figure 5J-L) and Western blotting (Figure 5M). Taken together, these findings indicate that upregulation of CD70 occurs in DTPCs and hence is an early event in the acquisition of a drug resistant phenotype.
Figure 5. CD70 upregulation is an early event in the evolution of EGFR TKI resistance.

(A-C) Western blotting of HCC4006 (A), HCC827 (B), and H1975 (C) cells following treatment with osimertinib (OSI). (D & E) ZEB1 (D) and CD70 (E) RNA levels following OSI treatment (n = 3). *p < 0.05; **p < 0.001. (F & G) Protein expression by RPPA in control and DTPCs following erlotinib (ERL; F) and osimertinib treatment (OSI; G) (n = 3). (H & I) ZEB1 and CD70 RNA in osimertinib-treated DTPCs (n = 3). *p < 0.05; **p < 0.001. (J) CD70 positivity on osimertinib-derived DTPC (n = 3). *p = 0.016, HCC827; p = 0.0086, HCC4006. (K) Representative flow cytometry data for CD70 expression on H1975 DTPCs. (L) CD70 positivity on osimertinib-derived DTPC (n = 3). *p <0.0001. (M) CD70 expression in H1975 parental and DTPCs. All bars are mean ± SD. One-way ANOVA was applied for D and E; Student’s t test was applied for H, I, J, L.
CD70-Antibody drug conjugates (ADC) have activity against EGFR TKI resistant cells in vitro
We next assessed whether the CD70 antibody cusatuzumab conjugated to the toxin monomethyl auristatin E (MMAE) could effectively target CD70 expressing NSCLC cells in vitro. HCC827 cells with or without CD70 expression were treated with cusatuzumab-MMAE for 24 hours and cell viability was evaluated after 5 days. HCC827 CD70 cells but not HCC827 control cells were sensitive to CD70 ADC treatment (Figure 6A). Likewise, H1975 OR cells (Figure 6B&C) were more sensitive to cusatuzumab-MMAE than parental cells. Similar results were obtained using the CD70 ADC vorsetuzumab-MMAE (Figure S4A&B). We observed heterogeneity in sensitivity to cusatuzumab-MMAE among EGFR TKI resistant cells as HCC4006 OR and MDA-L-011 cells were resistant to CD70-ADC treatment despite being CD70 positive (Figure S4C). MDA-L-011 cells were found to be relatively resistant to the MMAE payload by Cell Titer Glo assay (Figure S4D).
Figure 6. CD70 expression can targeted in EGFR TKI resistant cells.

(A) HCC827 cells with or without CD70 expression treated with cusatuzumab (Cus)-MMAE. *p ≤ 0.01. (B) H1975 and OR cells treated with cusatuzumab-MMAE. *p ≤ 0.01. (C) Viability of H1975 and OR variants treated with cusatuzumab (Cus)-MMAE (3 μg/ml). *p < 0.5; **p < 0.0007. (D - G) Activity of CD70 CAR T cells against HCC827 cells with or without CD70 expression (D), HCC4006 OR cells (E), HCC4006 parental cells (F), or MDA-L-011 cells (G). *p < 0.01; *p < 0.001. (H) CD107a expression on CD70 CAR T cells following incubation with HCC827 cells, HCC827 cells expressing CD70, and DTPCs. *p < 0.01; *p < 0.001. (I) Activity of CD70 CAR T cells against HCC827 cells and HCC827 DTPCs. (J &K) Activity of CD70-targeting CAR NK cells against HCC827 cells with or without CD70 (J) or HCC4006 parental cells and HCC4006 OR variants (K). (L & M) Viability (L) and clonogenic growth (M) of HCC4006 (GFP+) and HCC4006 OR7 (GFP-) cells grown as a mixed culture and treated with CD70-CAR NK (trCD27) cells, and osimertinib (OSI; 200 nM). *p < 0.0001. For all graphs, data shown as mean ± SD (n = 3). Statistics were applied using multiple t tests (A, B, C), Student’s t test (H), or one-way ANOVA (M).
See also Figures S4 and S5.
CD70-directed CAR T cells effectively target EGFR TKI resistant cells and DTPCs
Adoptive cell therapies have shown promise in targeting therapeutic-resistant malignancies, although suitable targets for this approach have yet to be validated in the context of EGFR TKI resistance. To investigate this approach, we evaluated whether CAR T cells could be designed to target CD70 using three different approaches: i) by incorporating the scFv derived from cusatuzumab, ii) incorporating a CD70 monoclonal antibody (P91), or iii) using a truncated form of CD27 which can bind CD70 on target cells (Figure S5). We incubated HCC827 cells with or without CD70 expression with CD70-targeting CAR T cells for 4 hours and observed potent cell killing of HCC827 CD70 cells but not HCC827 control cells (Figure 6D) indicating that this CD70-targeting approach was specific and cytotoxic. Moreover, CD70-targeting CAR T cells effectively killed HCC4006 OR cells (Figure 6E) but not parental HCC4006 cells (Figure 6F). We next assessed whether CD70-targeting CAR T cells had activity against a patient-derived model of EGFR TKI resistance. MDA-L-011 cells were cultured with CAR T cells for 2 hours and cell lysis was evaluated by chromium release assay. CD70-targeting CAR T cells effectively killed MDA-L-011 cells with significantly greater activity than control T cells (Figure 6G).
Our analysis of DTPCs indicated that upregulation of CD70 is an early event in the development of resistance to EGFR TKIs. Therefore, we next assessed whether EGFR TKI DTPCs activate and can be targeted by CD70-targeting CAR T cells. HCC827 osimertinib-derived DTPCs were cultured with or without CD70targeting CAR T cells for 2 hours, and CD107a expression on T cells was evaluated by flow cytometry as a marker of T cell activation. HCC827 DTPCs triggered a significant upregulation of T cell-expressed CD107a as compared to HCC827 control cells (Figure 6H). Notably, HCC827 DTPCs induced CD107a on T cells at a level similar to that induced by HCC827 cells engineered to overexpress CD70, and CD70-targeting CAR T cells effectively killed HCC827 DTPCs but not parental cells (Figure 6I).
CD70-targeting CAR NK cells have activity against EGFR TKI resistant cells
We generated CD70-targeting CAR NK cells expressing the scFv derived from cusatuzumab, the CD70 antibody (P91), or a truncated form of CD27 (Figure S5). CD70-targeting CAR NK cells showed significantly greater activity against HCC827 cells expressing CD70 as compared to parental HCC827 cells (Figure 6J). Likewise, CD70-targeting CAR NK cells had greater killing activity against HCC4006 OR cells than HCC4006 parental cells (Figure 6K). In order to recapitulate the heterogeneous tumor microenvironment, we next co-cultured HCC4006 cells transfected to express GFP with non-GFP HCC4006 OR7 cells and treated the mixed cultures with wild-type NK cells or CD70-targeting CAR (trCD27) NK cells with or without osimertinib. After 7 days, the surviving fraction of each cell line was evaluated by flow cytometry to distinguish between HCC4006-GFP and HCC4006 OR7 cells (Figure 6L). In parallel, total cell viability was assessed by clonogenic assay (Figure 6M). Treatment with CD70 CAR NK cells preferentially killed HCC4006 OR7 cells whereas osimertinib preferentially killed HCC4006-GFP cells. EGFR inhibition in combination with CD70 targeting resulted in depletion of both parental and osimertinib resistant cells.
CD70 ADCs and CD70-targeting CAR NK cells have in vivo anti-tumor activity
We next assessed whether CD70-targeting strategies had activity against CD70 positive tumor cells in vivo. HCC827 CD70 cells were injected subcutaneously into mice. Once tumors reached approximately 80 mm3 animals were randomized to receive IgG control antibodies, the CD70 ADC cusatuzumab-MMAE, or the CD30 ADC brentuximab-MMAE which was utilized a negative control as it bears the same payload but targets CD30 which is not expressed on HCC827 cells. Tumor growth was inhibited by cusatuzumab-MMAE but not by IgG control antibodies or the control ADC (Figure 7A). In parallel, a subset of mice was randomized to receive control NK cells or NK cells expressing CARs targeting CD70. While treatment with control NK cells did not impact tumor growth, treatment with CD70-targeting CAR NK cells resulted in complete regression of HCC827 CD70 tumors (Figure 7B). Similarly, H1975 OR17 cells were injected subcutaneously into mice and once tumors reached approximately 90 mm3 animals were randomized to receive control antibodies, osimertinib, cusatuzumab-MMAE, or osimertinib in combination with cusatuzumab-MMAE. Cusatuzumab-MMAE treatment resulted in complete tumor regression of H1975 OR17 tumors (Figure 7C; p < 0.0001 vs control). Likewise, CD70-targeting CAR-NK cells demonstrated potent anti-tumor activity against H1975 OR17 tumors (Figure 7D; p < 0.0001).
Figure 7. CD70 targeting has potent anti-tumor cell activity in vivo.

(A) Growth rate of HCC827-CD70 xenografts treated with CD70-targeting cusatuzumab (Cus)-MMAE, control IgG antibodies, or an irrelevant ADC - brentuzimab (Bren)-MMAE) (n = 9 mice/group). *p < 0.0001 vs control. (B) Effect of wild-type (WT) NK cells or CD70-targeting CAR NK cells on HCC827-CD70 xenografts (n = 9 mice/group). *p < 0.0001. (C) Growth rate of H1975 OR17 xenografts treated with IgG antibodies, osimertinib (OSI), Cus-MMAE, or the combination (n = 7–8 mice/group). *p < 0.0001 vs control. (D) Growth H1975 OR17 xenografts treated with wild-type NK cells or CD70-targeting CAR NK cells (n = 7–8mice/group). *p < 0.0001 vs control; p<0.0001 vs control NK cells. For all graphs, data are mean ± SEM.
DISCUSSION
For NSCLC patients harboring EGFR activating mutations, EGFR TKI resistance commonly occurs via EGFR-dependent or RTK-bypass mechanisms- such as secondary EGFR mutations (e.g. T790M) or MET amplification- or through mechanisms associated with a “rewiring” of cell lineage, such as EMT. The latter is particularly problematic as there are no known targeted approaches approved in these cases. To address this critical unmet need, we have conducted an integrative analysis of cell surface proteins upregulated in EMT-associated TKI resistance and identified that CD70 is markedly upregulated on the cell surface of EGFR mutant NSCLC cells that have developed resistance independent of EGFR or MET. While CD70 expression alone was not sufficient to induce a resistant phenotype in cells that had not undergone EMT, in the context of EMT-associated TKI resistance CD70 regulated cell survival and invasiveness. Our preclinical findings were supported by clinical data indicating that CD70 is upregulated in EGFR TKI refractory patients. Moreover, we determined that the upregulation of CD70 is an early event in the evolution of EGFR TKI resistance occurring in DTPCs. Finally, we demonstrated that CD70 on resistant cells could be exploited using antibody-based and adoptive therapy approaches to effectively target EGFR TKI resistant cells.
CD70 has been considered an attractive therapeutic target for malignancies in which it is overexpressed as CD70 is highly restricted and nearly absent on normal tissue26,27. Indeed, high CD70 positivity have been observed in renal, melanoma, pancreatic, ovarian, and breast cancer cells47–50. A report by Jacobs et al. assessing the frequency of CD70 expression in lung cancer determined that approximately 16% of NSCLC patient biopsies were positive for CD70, and 40% of NSCLC samples with tumors staged at T4 were CD70 positive51. Among treatment-naïve EGFR mutant positive cases, CD70 expression was not observed51. Consistent with this, we found that CD70 expression was low or absent on EGFR mutant treatment-naïve NSCLC cells and that CD70 expression was elevated following TKI treatment. While factors within the tumor microenvironment may contribute to the development of TKI resistance and influence the resistant phenotype that emerges, our observation that CD70 is upregulated in cell line models of resistance as well as in clinical specimens suggests that this is a cell autonomous effect. Resistance to EGFR TKIs can occur through several mechanisms including secondary EGFR mutations, activation of bypass pathways, or EMT, and multiple mechanisms may be present in a single tumor or patient. Therefore, multiple targeting strategies may be needed to effectively target resistant disease. In our immunohistochemical analysis of osimertinib-refractory specimens, we observed moderate to high CD70 staining intensity in the majority of specimens, supporting a role for CD70 targeting in cases where EMT-associated resistance has occurred.
Our finding that CD70 can be detected in DTPCs indicates that CD70 upregulation is an early event in the evolution of EGFR TKI resistance. DTPCs are distinct from drug resistant cells in that DTPCs display a transient and flexible resistant state and these cells may remain quiescent or clinically invisible for prolonged periods of time, but eventually progress to become fully resistant cells which resume growth and metastatic spread. Given that DTPCs are not rapidly dividing it is expected that they may be refractory to ADC-based approaches that utilize a payload targeting dividing cells such as MMAE. However, we show that CD70 CAR T cells effectively target osimertinib DTPCs indicating that CD70-based adoptive therapy approaches may be highly effective early in the course of TKI treatment as a strategy to target DTPCs rather than waiting until the emergence of drug resistance. Upon binding of CD70 to CD27 metalloproteinases cleave the extracellular domain of membrane-bound CD27 to generate soluble CD27 (sCD27) which can be detected in the circulation52–54. In NSCLC patients, elevated sCD27 was associated with a worse prognosis51. Thus, it is feasible that circulating sCD27 could serve as a biomarker to identify EGFR mutant NSCLC patients likely to respond to CD70-based targeting approaches.
Here, we evaluated multiple targeting strategies to exploit the elevated CD70 expression on EGFR TKI resistant cells. While anti-CD70 ADCs were highly effective against the vast majority of resistant tumor cell lines tested, we did observe heterogeneity in sensitivity which could be explained by reduced sensitivity to the payload in one of the models. However, CAR T and CAR NK-based CD70 targeting approaches were highly effective against resistant cells including those that were resistant to anti-CD70 ADCs. NK cells are an attractive alternative CAR-engineered effector cells because while CAR T cells must be generated from the patient’s own lymphocytes, CAR NK cells do not require HLA matching and thus may represent an off-the-shelf approach for CD70-targeting adoptive therapy.
Acquired EGFR TKI resistance is associated with re-activation of downstream signaling pathways including the PI3K and MAPK pathways45,46. We observed that in EGFR TKI resistant cells, stimulation of CD70 with exogenous CD27 triggered activation of AKT and MAPK pathways. Therefore, it is feasible that within the tumor microenvironment interactions between CD70-positive DTPCs or EGFR TKI resistant cells and CD27-positive immune cells may facilitate tumor cell survival and invasiveness. Likewise, while this report focused on the cell autonomous role of CD70 in the context of EGFR TKI resistance, tumor cell expressed CD70 likely also contributes to immune cell evasion. Several studies have investigated the potential contribution of tumor-derived CD70 on immune escape and suggested that interactions between CD70-positive tumor cells and T cells can induce a shift towards a pro-tumor T regulatory phenotype51,55,56, induce apoptosis of lymphocytes57–59, or promote T cell exhaustion60. Future studies investigating the role of CD70 in facilitating anti-tumor immunity in EGFR mutant NSCLC are warranted as this patient population is refractory to immunotherapy20.
Our finding that CD70 is overexpressed in cells with acquired TKI resistance mediated by EMT has important implications beyond EGFR mutant NSCLC. In NSCLC preclinical models EMT has been shown to be a mechanism of resistance to other targeted agents including KRAS G12C inhibitors61 and ALK inhibitors62. Given our observation that CD70 expression is associated with a mesenchymal phenotype in cell lines and patient samples, CD70 may be a therapeutic target for NSCLC cells with acquired resistance to inhibitors beyond EGFR.
Effective therapeutic strategies are sorely needed for patients with EGFR mutant NSCLC with acquired EGFR TKI resistance. We report that CD70 is upregulated on EGFR TKI resistant cells that have undergone EMT and that CD70 expression on the surface of tumor cells can be exploited using ADC and CAR-based approaches. These findings support the future clinical testing of CD70-based targeting approaches in EGFR mutant NSCLC patients in conjunction with EGFR TKIs to eradicate DTPCs, and in patients with EMT-associated TKI resistance.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, John V. Heymach (jheymach@mdanderson.org).
Materials Availability
Cell lines generated in this study will be made available upon request and completion of a Material Transfer Agreement.
Data and Code Availability
The PROSPECT dataset has been detailed previously28, and PROSPECT gene expression data are available at GEO accession GSE42127. Gene expression data for EGFR TKI resistant cell lines are available at GSE121634 as described previously19. NSCLC cell line gene expression data are available at GEO accession GSE4824. Gene expression data from HCC827 cells with or without CDH1 knockdown are available at GSE12303139. Transcriptomic data from cell lines derived from erlotinib-resistant patient biopsies are available at GSE64322 (super-series GSE64766)30. scRNAseq data was reported previously31. The dataset of matched baseline and osimertinib-refractory clinical samples32 is associated with accession number dbGaP: phs002001. For the analysis of CD70 expression and outcome among EGFR TKI refractory patients, we utilized a publicly available clinical dataset for which sequencing data was deposited in the European Genome-phenome Archive (EGA, EGAS00001005389, http://www.ebi.ac.uk/ega/) and clinical records, mutations, gene expression data, were hosted in OncoSG (https://src.gisapps.org/OncoSG/) as reported previously33. Clinical outcome data was accessed using the portal: https://src.gisapps.org/OncoSG_public/study/summary?id=GIS023. The TCGA-LUAD clinical cohort has been previously reported63. Clinical information and gene expression data were obtained through the TCGA portal (https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm). TCGA-LUAD and publicly available datasets (GSE14814, GSE19188, GSE29013, GSE30219, GSE31210, GSE3141, GSE31908, GSE37745, GSE43580, GSE4573, GSE50081, GSE8894, CAARAY) were used to assess CD70 expression and overall survival through kmplotter.com64.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Cell lines were obtained from ATCC and Dr. John Minna (UT Southwestern). Cell lines were authenticated by DNA fingerprinting and tested for the presence of Mycoplasma. Under an Institutional Review Board-approved protocol at MD Anderson Cancer Center, MDA-L-011 cells were established from the pleural effusion of a patient with EGFRL858R-positive NSCLC with acquired resistance to erlotinib, and MDA-L-004K cells were established from an EGFR exon 20 insertion positive NSCLC patient with acquired resistance to poziotinib. HCC827 cells expressing ZEB1 were generated using Lipofectamine LTX (Thermofisher) with ZEB1 cloned into the pcDNA3.1(+) vector (79663, Addgene)19. YUL-0019 cells were obtained from Dr. Politi (Yale Medical School)65. EGFR TKI resistant HCC827, H1975, and HCC4006 were established as previously described19,66. rhCD27 Fc (382-CD-100, R&D systems) was used at a concentration of 500 ng/ml. NSCLC cell lines were cultured using RPMI media containing 10% FBS and 1% penicillin/streptomycin. PBMCs were cultured in RPMI supplemented with 10% FBS, 2 mM GlutaMAX, 100 U/mL penicillin, and 100 μg/mL streptomycin. CAR-T and CAR-NK cells were cultured in RPMI media containing 10% FBS and 1% penicillin/streptomycin media supplemented with 100 U/mL or 200 U/ml IL-2 (Peprotech), respectively. 293T cells were cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin. All cells were cultured at 37 °C.
Mice
All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Texas MD Anderson Cancer Center. Animals were housed under pathogen-free conditions in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the United States Department of Agriculture and NIH guidelines. Female NSG mice were purchased from Jackson Labs. At the time of the studies, the mice were 8 weeks old. Animals were randomly assigned to control or treatment groups. No statistical method was used to predetermine the sample size.
Human samples
Biospecimens including tissues, biopsies, and patient-derived cells were obtained after patients gave informed consent, under protocols approved by Institutional Review Boards at participating institutions. Studies were conducted in accordance with the Declaration of Helsinki. Osimertinib-refractory specimens were collected from EGFR mutant NSCLC patients at MD Anderson Cancer Center enrolled in a phase 1b trial (NCT04479306).
METHOD DETAILS
RT-PCR
Total RNA was isolated using TRIzol® Reagent (Invitrogen) according to the manufacturer’s protocol. RT-PCR was performed as previously described 66. For experiments involving decitabine treatment, cells were treated daily with decitabine (1 μM; Sigma) for 10 days; RNA was isolated as listed above.
Clinical cohorts
Clinical information and gene expression data for lung adenocarcinomas included in TCGA-LUAD dataset were obtained through the TCGA portal (https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm). We applied an EMT gene expression signature as previously described24. We analyzed CD70 expression and clinical outcome using kmplotter.com64 using the top and bottom tertile as the cutoff for high and low CD70 expression. For the analysis of CD70 expression and outcome among EGFR TKI refractory patients, we utilized a publicly available clinical dataset33 using the portal: https://src.gisapps.org/OncoSG_public/study/summary?id=GIS023. For the analysis of OS, we performed ROC analysis on the available DFS data and calculated Youden J to define the best threshold for defining the CD70 high cases. This cutoff was then applied to the OS data using log-rank test and cox-proportional hazard ratio. Single cell RNA sequencing analysis of clinical specimens was described previously31. Differences in change of EMT-Score and CD70 expression was calculated from a publicly available dataset with 10 pairs of matched baseline and post-osimertinib treated samples using RNAseq32. EMT-Score was calculated as highlighted previously67 and differences were calculated by subtracting the progression values from the baseline valued for each pair. Correlation was calculated using Spearman correlation. The Profiling of Resistance patterns and Oncogenic Signaling Pathways in Evaluation of Cancers of the Thorax (PROSPECT) dataset included 189 surgically resected tumors, collected between 2006 and 2010. Gene expression analysis has been reported previously68–70. Clinical specimens used for immunohistochemical analysis of CD70 were a mixture of biopsies and surgically resected tumors from NSCLC patients. Treatment naïve, EGFR mutant NSCLC tumor specimens (n = 16) were purchases from AMSBIO. The stages of these tumors ranged from stage I to III. The specific catalog/specimen ID numbers are as follows: NGT-NSCLC 2107243, NGT-NSCLC 2107245, NGT-NSCLC 2107249, NGT-NSCLC-1907106, NGT-NSCLC-1907128, NGT-NSCLC-1907132, NGT-NSCLC1907143, NGT-NSCLC-1907144, NGT-NSCLC-1907148, NGT-NSCLC-1907149, NGT-NSCLC-1907502, NGT-NSCLC-2106440, NGT-NSCLC-2106462, NGT-NSCLC-2106467, NGT-NSCLC-2106469, NGT-NSCLC-2107008. Osimertinib-refractory specimens were collected from EGFR mutant NSCLC patients at MD Anderson Cancer Center. These patients were treated with first line osimertinib and at the time of progression were enrolled in a phase 1b trial (NCT04479306). Biopsy was taken at the time of progression on osimertinib.
CD70 IHC
For IHC analysis of CD70, formalin-fixed paraffin embedded slides were incubated at 60C for 1 hour. Slides were deparaffinized and rehydrated using a series of washes (xylene 5 minutes, thrice; 100% ethanol for 2 minutes, twice; 95% ethanol, 3 minutes; 70% ethanol, 3 minutes; 50% ethanol, 3 minutes; water, 5 minutes, twice). Antigen retrieval was performed using Target retrieval solution (pH 9, Cat#S236783–2; Aligent) in a steamer for 35 minutes and then cooled for 30 minutes at room temperature. Endogenous peroxidases were blocked using 3% H202 for 12 minutes. Blocking was performed using a solution of 5% normal horse serum and 1% normal goat serum in PBS. Slides were then incubated in anti-CD70 antibodies (Cat#Ab300083, RRID: AB_2924231; Abcam) at a dilution of 1:500 in blocking buffer overnight at 4C. After washing in PBS, VECTASTAIN ABC kit (PK-6101; Vector Laboratories) was applied according the manufacturer’s instructions. After washing in PBS, slides were developed using ImmPACT DAB substrate kit (Cat#SK-4105, Vector Laboratories) for 5 minutes. Quantification of CD70 staining was performed by a pathologist who was blinded from the clinical information of the specimens. Overall scoring was calculated as positive staining intensity (0 = negative; 1 = weak; 2 = moderate; 3 = strong) multiplied by the percent positivity.
Generation of drug-tolerant persister cells
For RPPA analysis of DTPCs, HCC827 or HCC4006 cells were plated onto 15 cm tissue culture dishes and following a 24-hour incubation were treated with 500 nM erlotinib (Cat#S1023, Selleck Chemicals) or 200 nM osimertinib (Cat#S7297, Selleck Chemicals). Media was replenished every 2 days. Protein was isolated after 10 days and processed for analysis by RPPA as reported previously19. Similarly, HCC827 cells were treated with 100 nM osimertinib, HCC4006 cells were treated with 200 nM osimertinib, and H1975 cells were treated with 500 nM osimertinib. After 14 days RNA and protein were extracted for real-time PCR analysis and Western blotting, respectively, and cells were stained for flow cytometry analysis.
Reverse phase protein array
Whole cell lysates were collected and RPPA slides were printed from lysates as described previously 71. The SuperCurve method was used to quantify protein concentration 72. RPPA data were analyzed using R packages (version 2.10.0)73.
siRNA-mediated knockdown of CD70
CD70-targeting siRNA (siCD70–1, SI04277182; siCD70–3, SI00748713; siCD70–7, SI05078178) and control siRNA (1027281) were purchased from Qiagen. Cells were plated in antibiotic free media and after an overnight incubation siRNA was delivered using Lipofectamine RNAiMAX (Invitrogen). For assessing growth rate following CD70 knockdown, 72 hours after the addition of siRNA, cells were seeded into wells of a 364-well (200 cells/well). After 4 hours, baseline cell viability was evaluated by Cell Titer Glo (Promega). For 4 subsequent days, additional plates were read using Cell Titer Glo. Likewise, for evaluating the effect of CD70 knockdown on cellular migration, cells were seeded at a density of 30,000 cells/Boyden chamber (8.0-μm pore size; Fisher Scientific) 72 hours after introduction of siRNA and allowed to migrate for 24 hours. Migrating cells were counted by brightfield microscopy.
Western blotting
For analysis of protein abundance of CD70, were plated onto 10-cm dish and once cells reached 70% confluency, whole cell lysates were collected and analyzed by Western blotting. For experiments assessing the impact of rhsCD27 on signal transduction pathways, cells were plated into 10-cm dishes and after a 24-hour incubation period were serum starved for 24 hours. Cells were then stimulated with 500 ng/ml rhCD27 (382-CD-100, R&D systems) 74 for the indicated times. For experiments assessing the impact of osimertinib on EGFR and ERK1/2 activation, HCC4006, HCC827, and H1975 were treated with 200 nM osimertinib (S7297, Selleck Chemicals) for the indicated times and then washed protein lysates were collected and analyzed by Western blotting. Antibodies for detection of CD70 (Cat#72094; RRID: AB_2924230, Cell Signaling), p-ERK (Cat#9106; RRID: AB_331768, Cell Signaling), ERK (Cat#9102; RRID: AB_330744, Cell Signaling), EGFR (Cat#4267; RRID: AB_2246311, Cell Signaling), p-EGFR (Cat#3777; RRID:AB_2096270, Cell Signaling), p-Akt (Cat#9275; RRID: AB_329828, Cat#9271S; RRID: AB_329825, 13038, Cell Signaling), and Akt (Cat#9272; RRID: AB_329827, Cell Signaling) were used at a dilution of 1:1000. Antibodies for detection of vinculin (Cat#V9131; RRID:AB_477629, Sigma-Aldrich) and β-actin (Cat#A5441; RRID: AB_476744, Sigma-Aldrich) were used at a dilution of 1:10,000. HRP-conjugated secondary antibodies were obtained from BioRad at a dilution of 1:3000.
RNA sequencing and surfaceome analysis
As previously reported19, RNAseq expression profiling of ER cell lines was performed in triplicate using the Illumina Human 76nt PE format. For analysis of expression of surfaceome genes, RNAseq data was filtered to include only genes which transcribed proteins localized to the cell surface according to the subcellular location data of the Human Proteom Atlas (HPA, RRID:SCR_006710; https://www.proteinatlas.org/) and all genes encoding cluster of differentiation (CD) proteins.
DNA methylation analysis in NSCLC cell lines
Methylation status of 68 NSCLC cell lines was evaluated using the Illumina HumanMethylation27 beadchip as previously reported. This data was integrated along with gene expression profiling75. Additionally, primary cell lines as well as osimertinib and erlotinib resistant clones have been analyzed using Reduced Representation Bisulfite Sequencing (RRBS). Briefly, 100ng of RNA was processed using the Ovation RRBS Methyl-Seq kit (Tecan Group Ltd., Zurich, Switzerland). Sequencing was performed in a single Read 57 bp configuration on a Illumina HiSeq 3000 sequencer. Data processing was performed using Bismark v 0.22. Annotations of methylated regions was performed using the annotatr package and the Hg38 database.
Generation of CD70 CAR T and NK cells
CD70 CAR (trCD27) was generated using truncated CD27 (extracellular and transmembrane domains), 4–1BB intracellular domain, and CD3ζ intracellular domain. CD70 CAR (cusatuzumab) was generated using single-chain variable fragment derived from cusatuzumab, IgG2 CH2 and CH3 domains with N297Q mutation, CD28 transmembrane and intracellular domains, 4–1BB intracellular domain, and CD3ζ intracellular domain. Genes encoding these CARs were synthesized as gene fragments (Genewiz) and then cloned into an SFG retroviral expression vector (Cat#22493, Addgene). Retroviral supernatant was produced via co-transfection of 293T (Cat#CRL-3216, ATCC) cells with the plasmids encoding the CARs, the RD114 envelope protein, and packaging proteins (gag and pol). To generate CAR-T cells, PBMCs were activated with anti-CD3/CD28 Dynabeads (ThermoFischer Scientific) at a 1:1 ratio in RPMI-1640 complete media (10% FBS, 2 mM GlutaMAX, 100 U/mL penicillin, and 100 μg/mL streptomycin) supplemented with 100 U/mL IL-2 (Preprotech). T cells were transduced with retroviral supernatant on plates coated with Retronectin (Takara) on days 3 post activation. Anti-CD3/CD28 Dynabeads were removed 2 days later. CAR-T cells were then maintained at 0.5–1 × 106 cells/ml in the media supplemented with 100 U/mL IL-2 (Peprotech). To generate CAR-NK cells, a feeder cell line was generated by transducing K562 (Cat#CCL-243, ATCC) cells with retrovirus encoding membrane-bound IL-21, 4–1BBL, OX40L, and CD86, respectively. PBMCs were activated with 100 gray γ-ray irradiated feeder cells at a 1:2 PBMC:Feeder cell ratio in RPMI-1640 complete media supplemented with 200 U/mL IL-2 and 5 ng/mL IL-15 (ThermoFisher Scientific). NK cells were transduced with retroviral supernatant on plates coated with Retronectin (Takara) on days 4 or 5 post activation. CAR-NK cells were maintained at 0.5–1 × 106 cells/ml in the media supplemented with 200 U/mL IL-2. All CAR-T and CAR-NK cells were used after they reached a resting state, which is about 7 days after transduction and was confirmed by cell size on a flow cytometer.
CD70 CAR NK and T cell killing assays
To quantify killing activity of CD70 CAR-T and CAR-NK cells in vitro, luciferase (Luc) reporter assays and standard Chromium-51 (51Cr) release assays were performed as indicated. For Luc reporter assays, the tumor cell lines were transduced with pHIV-Luc-ZsGreen (Cat#39196, Addgene) lentivirus to generate LUC-stable-expression cell lines. Target cells (Luc expressing tumor cells) were pre-seeded into 96-well white cell culture plate (ThermoFischer Scientific) at 1 × 104 cells in 100 μl complete media overnight. Effector cells (CAR-T and CAR-NK cells) were added to each well basing on indicated effector:target (E:T) ratio. The plate was then incubated at 37 °C with 5% CO2 for 4 hours. Supernatant were gently discarded after the incubation, and 100 μl of D-luciferin (ThermoFischer Scientific) at 150 μg/ml were added. Luminescence was read using a FLUOstar OPTIMA multi-mode micro-plate reader (BMG Labtech). The percentage of specific lysis was calculated from luminescence as follows: ([Target only – experiment]/[Target only – no target] × 100). For standard 51Cr release assays, target cells were labeled with 51Cr at 37 °C for 2 hours and then co-cultured with effector cells for 4 hours. The supernatants were collected and the released 51Cr was measured with a gamma counter. The percentage of specific lysis was calculated from counts for 51Cr release as follows: ([NK cells – medium]/[Triton x-100 – medium] × 100).
CD107a assay on CAR T cells
HCC827 and persister cells were pre-seeded into 96-well plate at 1 × 104 cells in 100 μl complete media overnight. CAR-T cells were added to each well at a 1:1 ratio, as well as PE labeled anti-CD107a (Cat#555801; RRID:AB_396135, BD Biosciences) antibodies and GolgiStop (BD Biosciences). The reaction was incubated at 37 °C with 5% CO2 for 2 hours. The cells were collected and stained with anti-CD3 antibody (Cat#300325; RRID:AB_2616609, Biolegend) on ice for 30 minutes. Expression of CD107a was determined by flow cytometry.
In vivo targeting of CD70
HCC827 CD70 cells (5 × 106) or H1975 OR17 cells (3 × 106) were injected subcutaneously into 8-week-old female NSG (Cat#005557, Jackson Labs) mice. Animals were randomly assigned to control or treatment groups. For IgG and ADC treatments, animals were treated twice weekly (i.p.) with 3 mg/kg IgG control antibodies (Bio X Cell), brentuximab-MMAE (MD Anderson institutional pharmacy), or cusatuzumab-MMAE. For CAR-NK cell treatments, 1 × 107 of control NK cells (non-transduced), CD70 CAR-NK cells (trCD27), or CD70 CAR-NK cells (Cusatuzumab) were i.v. injected in 100 μL HBSS each week for 3 weeks. 10,000 units of IL-2 and 100 ng of IL-15 were i.p. injected in 200 μL HBSS (ThermoFischer Scientific) on the same day and the next day of NK cells treatment.
QUANTIFICATION AND STATISTICAL ANALYSIS
For in vitro studies, as specified in the figure legend we used Student’s t test (two-tailed) or one-way ANOVA to test significance. The association between CD70 expression and EMT score or other gene expression was calculated using the Pearson or Spearman correlation coefficients. For multiple testing, P values were adjusted using false discovery rate (FDR). The survival curve between CD70 Low and High groups was estimated using the Kaplan-Meier method. All P values were two-tailed and for all analyses, P ≤ 0.05 is considered statistically significant, unless otherwise specified.
Supplementary Material
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-ZEB1 | Cell Signaling | Cat#3396; RRID: AB_1904164 |
| Anti- E-cadherin | Cell Signaling | Cat#24E10; RRID: AB_2291471 |
| Anti Axl | Cell Signaling | Cat#8661; RRID: AB_11217435 |
| Anti-GAPDH | Cell Signaling | Cat#5174; RRID: AB_10622025 |
| Anti-CD70 (for Western Blotting) | Cell Signaling | Cat#72094S; RRID: AB_2924230 |
| Anti-CD70 (for immunohistochemistry) | Abcam | Cat#Ab300083; RRID: AB_2924231 |
| Anti-CD70 (for flow cytometry) | BD Biosciences | Cat#355104; RRID: AB_2561430 |
| Anti-CD27 | BD Biosciences | Cat#356410; AB_2561957 |
| Anti-vinculin | Sigma | Cat#V9131; RRID:AB_477629 |
| Anti-pAKT S473 | Cell Signaling | Cat#9271S; RRID: AB_329825 |
| Anti-pERK 1/2 | Cell Signaling | Cat#9106; RRID: AB_331768 |
| Anti-β-Tubulin | Cell Signaling | Cat#86298S; RRID: AB_2715541 |
| Anti-ERK1/2 | Cell Signaling | Cat#9102S; RRID: AB_330744 |
| Anti-AKT | Cell Signaling | Cat#9272; RRID: AB_329827 |
| Anti-pAKT T308 | Cell Signaling | Cat# 9275S; RRID: AB_329828 |
| Anti-pEGFR | Cell Signaling | Cat#3777; RRID:AB_2096270 |
| Anti-EGFR | Cell Signaling | Cat#4267; RRID: AB_2246311 |
| Anti-β-Actin | Sigma | Cat#A5441; RRID: AB_476744 |
| Anti-CD3 | Biolegend | Cat#300325; RRID:AB_2616609 |
| Anti-CD107a | BD Biosciences | Cat#555801; RRID:AB_396135 |
| Custuzumab-MMAE | MD Anderson Cancer Center | N/A |
| Vorsetuzumab-MMAE | MD Anderson Cancer Center | N/A |
| Brentuzimab-MMAE | MD Anderson Cancer Center Institutional Pharmacy | N/A |
| Bacterial and virus strains | ||
| Biological samples | ||
| Tumor biopsies (osimertinib refractory) | This paper | N/A |
| Tumor biopsies (baseline) | AMSBIO | |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant TGF-β | R&D systems | Cat#7754-BH-025 |
| MMAE | Selleck Chem | Cat#S7721 |
| Recombinant sCD27 | R&D systems | Cat#382-cd-100 |
| CellTiter-Glo® 2.0 Cell Viability Assay | Promega | Cat#G9243 |
| 4–15% Criterion TGX Precast Midi Protein Gel | BioRad | Cat#5671084 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | Cat#34580 |
| Erlotinib | Selleck Chem | Cat#S7788 |
| Osimertinib | Selleck Chem | Cat#S7297 |
| Decitabine | Selleck Chem | Cat#S1200 |
| Cell Lysis Buffer (10X) | Cell Signaling | Cat#9803 |
| Critical commercial assays | ||
| ImmPACT DAB peroxidase substrate | Vector laboratories | Cat#SK-4105 |
| VECTASTAIN Elite ABC kit Peroxidase HRP | Vector laboratories | Cat#PK-6101 |
| Deposited data | ||
| Gene expression data from PROSPECT clinical dataset | Ref: 68-70 | GSE42127 |
| Gene expression data for NSCLC cell lines | Ref: 19 | GSE121634 |
| Gene expression data for NSCLC cell lines | Ref: 9 | GSE4824 |
| Gene expression data for HCC827 with CDH1 knockdown | Ref: 39 | GSE123031 |
| Gene expression data from erlotinib-resistant biopsies | Ref: 30 | GSE64322 |
| Dataset of matched baseline and osimertinib-refractory samples | Ref: 32 | dbGaP: phs002001 |
| Dataset of EGFR TKI refractory patients | Ref: 33 | EGAS00001005389 |
| TCGA Dataset | Ref: 63 | TCGA-LUAD |
| LUAD dataset | Ref: 64 | GSE14814 |
| LUAD dataset | Ref: 64 | GSE19188 |
| LUAD dataset | Ref: 64 | GSE29013 |
| LUAD dataset | Ref: 64 | GSE30219 |
| LUAD dataset | Ref: 64 | GSE31210 |
| LUAD dataset | Ref: 64 | CAARAY |
| LUAD dataset | Ref: 64 | GSE8894 |
| LUAD dataset | Ref: 64 | GSE50081 |
| LUAD dataset | Ref: 64 | GSE4573 |
| LUAD dataset | Ref: 64 | GSE43580 |
| LUAD dataset | Ref: 64 | GSE37745 |
| LUAD dataset | Ref: 64 | GSE31908 |
| LUAD dataset | Ref: 64 | GSE3141 |
| Experimental models: Cell lines | ||
| Human: MDA-L-011 | MD Anderson Cancer Center | N/A |
| Human: MDA-L-004K | MD Anderson Cancer Center | N/A |
| Human: HCC827 | ATCC | Cat#CRL-2868 |
| Human: HCC4006 | ATCC | Cat#CRL-2871 |
| Human: H1975 | ATCC | Cat#CRL-5908 |
| Human: Raji | ATCC | Cat#CCL-86 |
| Human: YUL-0019 | Dr. Katerina Politi (Yale) | N/A |
| Human: 293T | ATCC | CRL-3216 |
| Human: K562 | ATCC | CCL-243 90071810 |
| Human: PC9 | Millipore Sigma | Cat#90071810 |
| Experimental models: Organisms/strains | ||
| NSG mice | Jackson Labs | Cat#005557 |
| Oligonucleotides | ||
| siRNA targeting CD70 #1 | Qiagen | Cat#SI04277182 |
| siRNA targeting CD70 #3 | Qiagen | Cat#SI00748713 |
| siRNA targeting CD70 #7 | Qiagen | Cat#SI05078178 |
| Control siRNA | Qiagen | Cat#1027281 |
| CD70 Real Time PCR primers | ThermoFisher | Cat#4331182; Hs00174297_m1 |
| ZEB1 Real Time PCR primers | ThermoFisher | Cat#4331182; Hs00232783_m1 |
| GAPDH Real Time PCR primers | ThermoFisher | Cat#4331182;Hs00266705_91 |
| Recombinant DNA | ||
| CD70 expression construct | Addgene | Cat#82003 |
| SFG retroviral expression vector | Addgene | Cat#22493 |
| pHIV-Luc-ZsGreen | Addgene | Cat#39196 |
| Software and algorithms | ||
| GraphPad Prism | Dotmatics | https://www.graphpad.com/ |
| FlowJo | Becton, Dickenson & Company | www.flowjo.com |
| Other | ||
Highlights.
CD70 is upregulated on EGFR mutant NSCLC cells that have undergone EMT
In EGFR inhibitor-resistant cells, CD70 regulates cell survival and invasiveness
CD70 upregulation occurs in drug tolerant persister cells (DTPCs)
Drug-resistant CD70+ tumors can be targeted with CD70-ADCs, CAR-Ts, and NKCARs
Acknowledgements
This work was supported by The Mugnaini Fund, the Emerson Collective, the David Bruton, Jr. Endowment, Rexanna’s Foundation for Fighting Lung Cancer, Lung SPORE grant 5 P50 CA070907, NIH CCSG (CA016672), NIH CCSG (CA016672)-Bioinformatics Shared Resource, 1R01CA247975, 1R01CA190628, 1R50CA265307, 1R01CA240257, 1R01CA234183–01A1, Lung Cancer Moon Shot Program, The Lerryn M. Carl Endowment, Stading Fund for EGFR inhibitor resistance, the Fox Lung EGFR Inhibitor Fund, the Hanlon Fund, the Richardson fund, the Kopelman Foundation, the Hallman fund, the Exon 20 group, and CPRIT Core Facility Support Grants (#RP120348 & #RP170002). The authors would like to thank the Science Park Next-Generation Sequencing Core as well as the MDACC Epigenetics core for supporting this project. Graphical Abstract made using BioRender.
Declaration of Interest
JVH serves on advisory committees for DAVA Oncology, Regeneron, BerGenBio, Jazz Pharmaceuticals, Curio Science, Immunocore, AstraZeneca, EMD Serono, Boehringer-Ingelheim, Catalyst, Genentech, GlaxoSmithKline, Guardant Health, Foundation medicine, Hengrui Therapeutics, Eli Lilly, Novartis, Spectrum, Sanofi, Takeda, Mirati Therapeutics, BMS, BrightPath Biotherapeutics, Janssen Global Services, Nexus Health Systems, Pneuma Respiratory, Kairos Venture Investments, Roche, Leads Biolabs, RefleXion, Chugai Pharmaceuticals, receives research support from Takeda, AstraZeneca, Boehringer-Ingelheim, and Spectrum, receives royalties and licensing fees from Spectrum Pharmaceuticals. DG serves as an advisor/consultant for Sanofi, GlaxoSmithKline, Janssen Research & Development, Ribon Therapeutics, Mitobridge, Eli Lilly, Menarini, Napa Therapeutics, and receives research funding from Janssen Research & Development, Takeda, AstraZeneca, Mitobridge, Ribon Therapeutics, NGM Biopharmaceuticals, Boehringer Ingelheim, Mirati Therapeutics. SSK. reports research support from Boehringer Ingelheim, Janssen, MiNA Therapeutics, MiRXES, and Taiho Therapeutics and honoraria from Boehringer Ingelheim, Bristol Meyers Squibb, AstraZeneca, Chugai Pharmaceutical, and Takeda Pharmaceuticals, all outside of the submitted work. XL receives consulting/advisory fees from EMD Serono (Merck KGaA), AstraZeneca, Spectrum Pharmaceutics, Novartis, Eli Lilly, Boehringer Ingelheim, Hengrui Therapeutics, Janssen, Blueprint Medicines, Sensei Biotherapeutics, and Abbvie, and Research Funding from Eli Lilly, EMD Serono, Regeneron, and Boehringer Ingelheim. MBN and JPR receive royalties and licensing fees from Spectrum Pharmaceuticals. MBN and JVH have filed a patent for CD70 targeting in EGFR TKI resistant NSCLC (17/611,019). JPR is currently a full-time employee and shareholder of AstraZeneca. YYE discloses research support from AstraZeneca, Takeda, Eli Lilly, Xcovery, Tuning Point Therapeutics, BluPrint, Elevation Oncology; advisory role for AstraZeneca, Eli Lilly, Takeda, Spectrum, Bristol Myers Squibb and Turning Point; Accommodation expenses Eli Lilly. HU receives research support from Takeda Pharmaceuticals and Boehringer Ingelheim. SH receives consulting fees from AstraZeneca and Boehringer Ingelheim and speaker fees from Qiagen.
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et al. (2009). Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. The New England journal of medicine 361, 947–957. 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 2.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, et al. (2012). Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. The Lancet. Oncology 13, 239–246. 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 3.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, et al. (2010). Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. The New England journal of medicine 362, 2380–2388. 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 4.Sequist LV., Martins RG., Spigel D., Grunberg SM., Spira A., Janne PA., Joshi VA., McCollum D., Evans TL., Muzikansky A., et al. (2008). First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 26, 2442–2449. 10.1200/JCO.2007.14.8494. [DOI] [PubMed] [Google Scholar]
- 5.Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WS, et al. (2017). Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med 376, 629–640. 10.1056/NEJMoa1612674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T, et al. (2018). Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. The New England journal of medicine 378, 113–125. 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 7.Ohashi K, Maruvka YE, Michor F, and Pao W. (2013). Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 31, 1070–1080. 10.1200/JCO.2012.43.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. (2007). MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043. 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 9.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, et al. (2013). An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 19, 279–290. 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al. (2012). Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nature genetics 44, 852–860. 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung JH, Rho JK, Xu X, Lee JS, Yoon HI, Lee CT, Choi YJ, Kim HR, Kim CH, and Lee JC (2011). Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer 73, 176–182. 10.1016/j.lungcan.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 12.Uramoto H, Iwata T, Onitsuka T, Shimokawa H, Hanagiri T, and Oyama T. (2010). Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res 30, 2513–2517. [PubMed] [Google Scholar]
- 13.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, et al. (2011). Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Science translational medicine 3, 75ra26. 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, and Halmos B. (2005). EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. The New England journal of medicine 352, 786–792. [DOI] [PubMed] [Google Scholar]
- 15.Pao W, and Miller VA (2005). Epidermal growth factor receptor mutations, small-molecule kinase inhibitors, and non-small-cell lung cancer: current knowledge and future directions. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 23, 2556–2568. [DOI] [PubMed] [Google Scholar]
- 16.Mok TS, Wu YL, and Papadimitrakopoulou VA (2017). Osimertinib in EGFR T790M-Positive Lung Cancer. The New England journal of medicine 376, 1993–1994. 10.1056/NEJMc1703339. [DOI] [PubMed] [Google Scholar]
- 17.Ou SI, Govindan R, Eaton KD, Otterson GA, Gutierrez ME, Mita AC, Argiris A, Brega NM, Usari T, Tan W, et al. (2017). Phase I Results from a Study of Crizotinib in Combination with Erlotinib in Patients with Advanced Nonsquamous Non-Small Cell Lung Cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 12, 145–151. 10.1016/j.jtho.2016.09.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu YL, Zhang L, Kim DW, Liu X, Lee DH, Yang JC, Ahn MJ, Vansteenkiste JF, Su WC, Felip E, et al. (2018). Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients With EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 36, 3101–3109. 10.1200/JCO.2018.77.7326. [DOI] [PubMed] [Google Scholar]
- 19.Nilsson MB, Sun H, Robichaux J, Pfeifer M, McDermott U, Travers J, Diao L, Xi Y, Tong P, Shen L, et al. (2020). A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components. Science translational medicine 12. 10.1126/scitranslmed.aaz4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, Huynh TG, Zhao L, Fulton L, Schultz KR, et al. (2016). EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clinical cancer research : an official journal of the American Association for Cancer Research 22, 4585–4593. 10.1158/1078-0432.CCR-15-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marcelo Vailati Negrao AR, Jacqulyne Ponville Robichaux, Le Xiuning, Nilsson Monique B., Wu Chang-jiun, Jianhua Zhang, Lara CA Landry, Emily Roarty, Waree Rinsurongkawong, Stephen Swisher, Simon George R., Andrew Futreal, Bonnie S. Glisson, Jianjun Zhang, John Heymach Association of EGFR and HER-2 exon 20 mutations with distinct patterns of response to immune checkpoint blockade in non-small cell lung cancer. J Clin Oncol 36, 2018. (suppl; abstr 9052). [Google Scholar]
- 22.Negrao MV, Skoulidis F, Montesion M, Schulze K, Bara I, Shen V, Xu H, Hu S, Sui D, Elamin YY, et al. (2021). Oncogene-specific differences in tumor mutational burden, PD-L1 expression, and outcomes from immunotherapy in non-small cell lung cancer. J Immunother Cancer 9. 10.1136/jitc-2021-002891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, Thai AA, Mascaux C, Couraud S, Veillon R, et al. (2019). Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol 30, 1321–1328. 10.1093/annonc/mdz167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, et al. (2013). An Epithelial-Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance. Clinical cancer research : an official journal of the American Association for Cancer Research 19, 279–290. 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nolte MA, van Olffen RW, van Gisbergen KP, and van Lier RA (2009). Timing and tuning of CD27-CD70 interactions: the impact of signal strength in setting the balance between adaptive responses and immunopathology. Immunol Rev 229, 216–231. 10.1111/j.1600-065X.2009.00774.x. [DOI] [PubMed] [Google Scholar]
- 26.Craddock C, Quek L, Goardon N, Freeman S, Siddique S, Raghavan M, Aztberger A, Schuh A, Grimwade D, Ivey A, et al. (2013). Azacitidine fails to eradicate leukemic stem/progenitor cell populations in patients with acute myeloid leukemia and myelodysplasia. Leukemia 27, 1028–1036. 10.1038/leu.2012.312. [DOI] [PubMed] [Google Scholar]
- 27.Bowman MR, Crimmins MA, Yetz-Aldape J, Kriz R, Kelleher K, and Herrmann S. (1994). The cloning of CD70 and its identification as the ligand for CD27. J Immunol 152, 1756–1761. [PubMed] [Google Scholar]
- 28.Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, et al. (2014). Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nature communications 5, 5241. 10.1038/ncomms6241 ncomms6241 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le X, Puri S, Negrao MV, Nilsson M, Robichaux JP, Boyle TA, Hicks JK, Lovinger K, Roarty EB, Rinsurongkawong W, et al. (2018). Landscape of EGFR -dependent and -independent resistance mechanisms to osimertinib and continuation therapy post-progression in EGFR-mutant NSCLC. Clinical cancer research : an official journal of the American Association for Cancer Research. 10.1158/1078-0432.CCR-18-1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, Moran T, et al. (2015). RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nature communications 6, 6377. 10.1038/ncomms7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kashima Y, Shibahara D, Suzuki A, Muto K, Kobayashi IS, Plotnick D, Udagawa H, Izumi H, Shibata Y, Tanaka K, et al. (2021). Single-Cell Analyses Reveal Diverse Mechanisms of Resistance to EGFR Tyrosine Kinase Inhibitors in Lung Cancer. Cancer research 81, 4835–4848. 10.1158/0008-5472.CAN-20-2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roper N, Brown AL, Wei JS, Pack S, Trindade C, Kim C, Restifo O, Gao S, Sindiri S, Mehrabadi F, et al. (2020). Clonal Evolution and Heterogeneity of Osimertinib Acquired Resistance Mechanisms in EGFR Mutant Lung Cancer. Cell Rep Med 1. 10.1016/j.xcrm.2020.100007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chua KP, Teng YHF, Tan AC, Takano A, Alvarez JJS, Nahar R, Rohatgi N, Lai GGY, Aung ZW, Yeong JPS, et al. (2021). Integrative Profiling of T790M-Negative EGFR-Mutated NSCLC Reveals Pervasive Lineage Transition and Therapeutic Opportunities. Clinical cancer research : an official journal of the American Association for Cancer Research 27, 5939–5950. 10.1158/1078-0432.CCR-20-4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist P, et al. (2007). The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26, 6979–6988. 10.1038/sj.onc.1210508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez-Tillo E, Lazaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, Engel P, and Postigo A. (2010). ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 29, 3490–3500. 10.1038/onc.2010.102. [DOI] [PubMed] [Google Scholar]
- 36.Yao Z., Fenoglio S., Gao DC., Camiolo M., Stiles B., Lindsted T., Schlederer M., Johns C., Altorki N., Mittal V., et al. (2010). TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proceedings of the National Academy of Sciences of the United States of America 107, 15535–15540. 10.1073/pnas.1009472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen H, Guan D, Shen J, Wang M, Chen X, Xu T, Liu L, and Shu Y. (2016). TGF-beta1 induces erlotinib resistance in non-small cell lung cancer by down-regulating PTEN. Biomed Pharmacother 77, 1–6. 10.1016/j.biopha.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 38.Yang ZZ, Grote DM, Xiu B, Ziesmer SC, Price-Troska TL, Hodge LS, Yates DM, Novak AJ, and Ansell SM (2014). TGF-beta upregulates CD70 expression and induces exhaustion of effector memory T cells in B-cell non-Hodgkin’s lymphoma. Leukemia 28, 1872–1884. 10.1038/leu.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Becker JH, Gao Y, Soucheray M, Pulido I, Kikuchi E, Rodriguez ML, Gandhi R, Lafuente-Sanchis A, Aupi M, Alcacer Fernandez-Coronado J, et al. (2019). CXCR7 Reactivates ERK Signaling to Promote Resistance to EGFR Kinase Inhibitors in NSCLC. Cancer research 79, 4439–4452. 10.1158/0008-5472.CAN-19-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riether C, Schurch CM, Buhrer ED, Hinterbrandner M, Huguenin AL, Hoepner S, Zlobec I, Pabst T, Radpour R, and Ochsenbein AF (2017). CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J Exp Med 214, 359–380. 10.1084/jem.20152008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu Q, Wu A, and Richardson BC (2005). Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J Immunol 174, 6212–6219. 10.4049/jimmunol.174.10.6212. [DOI] [PubMed] [Google Scholar]
- 42.Riether C, Pabst T, Hopner S, Bacher U, Hinterbrandner M, Banz Y, Muller R, Manz MG, Gharib WH, Francisco D, et al. (2020). Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat Med 26, 1459–1467. 10.1038/s41591-020-0910-8. [DOI] [PubMed] [Google Scholar]
- 43.Arens R, Nolte MA, Tesselaar K, Heemskerk B, Reedquist KA, van Lier RA, and van Oers MH (2004). Signaling through CD70 regulates B cell activation and IgG production. J Immunol 173, 3901–3908. 10.4049/jimmunol.173.6.3901. [DOI] [PubMed] [Google Scholar]
- 44.Garcia P, De Heredia AB, Bellon T, Carpio E, Llano M, Caparros E, Aparicio P, and Lopez-Botet M. (2004). Signalling via CD70, a member of the TNF family, regulates T cell functions. J Leukoc Biol 76, 263–270. 10.1189/jlb.1003508. [DOI] [PubMed] [Google Scholar]
- 45.Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, Pratilas CA, Rosen N, Gray NS, Wong KK, and Janne PA (2015). Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR-Mutant Lung Cancer. Cancer discovery 5, 960–971. 10.1158/2159-8290.CD-15-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ercan D, Xu C, Yanagita M, Monast CS, Pratilas CA, Montero J, Butaney M, Shimamura T, Sholl L, Ivanova EV, et al. (2012). Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer discovery 2, 934–947. 10.1158/2159-8290.CD-12-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Law CL, Gordon KA, Toki BE, Yamane AK, Hering MA, Cerveny CG, Petroziello JM, Ryan MC, Smith L, Simon R, et al. (2006). Lymphocyte activation antigen CD70 expressed by renal cell carcinoma is a potential therapeutic target for anti-CD70 antibody-drug conjugates. Cancer research 66, 2328–2337. 10.1158/0008-5472.CAN-05-2883. [DOI] [PubMed] [Google Scholar]
- 48.Pich C, Sarrabayrouse G, Teiti I, Mariame B, Rochaix P, Lamant L, Favre G, Maisongrosse V, and Tilkin-Mariame AF (2016). Melanoma-expressed CD70 is involved in invasion and metastasis. British journal of cancer 114, 63–70. 10.1038/bjc.2015.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryan MC, Kostner H, Gordon KA, Duniho S, Sutherland MK, Yu C, Kim KM, Nesterova A, Anderson M, McEarchern JA, et al. (2010). Targeting pancreatic and ovarian carcinomas using the auristatin-based anti-CD70 antibody-drug conjugate SGN-75. British journal of cancer 103, 676–684. 10.1038/sj.bjc.6605816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu L, Yin B, Yi Z, Liu X, Hu Z, Gao W, Yu H, and Li Q. (2018). Breast cancer stem cells characterized by CD70 expression preferentially metastasize to the lungs. Breast Cancer 25, 706–716. 10.1007/s12282-018-0880-6. [DOI] [PubMed] [Google Scholar]
- 51.Jacobs J, Zwaenepoel K, Rolfo C, Van den Bossche J, Deben C, Silence K, Hermans C, Smits E, Van Schil P, Lardon F, et al. (2015). Unlocking the potential of CD70 as a novel immunotherapeutic target for non-small cell lung cancer. Oncotarget 6, 13462–13475. 10.18632/oncotarget.3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loenen WA, De Vries E, Gravestein LA, Hintzen RQ, Van Lier RA, and Borst J. (1992). The CD27 membrane receptor, a lymphocyte-specific member of the nerve growth factor receptor family, gives rise to a soluble form by protein processing that does not involve receptor endocytosis. Eur J Immunol 22, 447–455. 10.1002/eji.1830220224. [DOI] [PubMed] [Google Scholar]
- 53.Kato K, Chu P, Takahashi S, Hamada H, and Kipps TJ (2007). Metalloprotease inhibitors block release of soluble CD27 and enhance the immune stimulatory activity of chronic lymphocytic leukemia cells. Exp Hematol 35, 434–442. 10.1016/j.exphem.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 54.Hintzen RQ, de Jong R, Hack CE, Chamuleau M, de Vries EF, ten Berge IJ, Borst J, and van Lier RA (1991). A soluble form of the human T cell differentiation antigen CD27 is released after triggering of the TCR/CD3 complex. J Immunol 147, 29–35. [PubMed] [Google Scholar]
- 55.Jacobs J, Deschoolmeester V, Zwaenepoel K, Rolfo C, Silence K, Rottey S, Lardon F, Smits E, and Pauwels P. (2015). CD70: An emerging target in cancer immunotherapy. Pharmacol Ther 155, 1–10. 10.1016/j.pharmthera.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 56.Silence K, Dreier T, Moshir M, Ulrichts P, Gabriels SM, Saunders M, Wajant H, Brouckaert P, Huyghe L, Van Hauwermeiren T, et al. (2014). ARGX-110, a highly potent antibody targeting CD70, eliminates tumors via both enhanced ADCC and immune checkpoint blockade. MAbs 6, 523–532. 10.4161/mabs.27398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chahlavi A., Rayman P., Richmond AL., Biswas K., Zhang R., Vogelbaum M., Tannenbaum C., Barnett G., and Finke JH. (2005). Glioblastomas induce T-lymphocyte death by two distinct pathways involving gangliosides and CD70. Cancer research 65, 5428–5438. 10.1158/0008-5472.CAN-04-4395. [DOI] [PubMed] [Google Scholar]
- 58.Diegmann J, Junker K, Loncarevic IF, Michel S, Schimmel B, and von Eggeling F. (2006). Immune escape for renal cell carcinoma: CD70 mediates apoptosis in lymphocytes. Neoplasia 8, 933–938. 10.1593/neo.06451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wischhusen J, Jung G, Radovanovic I, Beier C, Steinbach JP, Rimner A, Huang H, Schulz JB, Ohgaki H, Aguzzi A, et al. (2002). Identification of CD70-mediated apoptosis of immune effector cells as a novel immune escape pathway of human glioblastoma. Cancer research 62, 2592–2599. [PubMed] [Google Scholar]
- 60.Wang QJ, Hanada K, Robbins PF, Li YF, and Yang JC (2012). Distinctive features of the differentiated phenotype and infiltration of tumor-reactive lymphocytes in clear cell renal cell carcinoma. Cancer research 72, 6119–6129. 10.1158/0008-5472.CAN-12-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adachi Y, Ito K, Hayashi Y, Kimura R, Tan TZ, Yamaguchi R, and Ebi H. (2020). Epithelial-to-Mesenchymal Transition is a Cause of Both Intrinsic and Acquired Resistance to KRAS G12C Inhibitor in KRAS G12C-Mutant NonSmall Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 26, 5962–5973. 10.1158/1078-0432.CCR-20-2077. [DOI] [PubMed] [Google Scholar]
- 62.Fukuda K, Takeuchi S, Arai S, Katayama R, Nanjo S, Tanimoto A, Nishiyama A, Nakagawa T, Taniguchi H, Suzuki T, et al. (2019). Epithelial-to-Mesenchymal Transition Is a Mechanism of ALK Inhibitor Resistance in Lung Cancer Independent of ALK Mutation Status. Cancer research 79, 1658–1670. 10.1158/0008-5472.CAN-18-2052. [DOI] [PubMed] [Google Scholar]
- 63.Comprehensive molecular profiling of lung adenocarcinoma. (2014). Nature 511, 543–550. 10.1038/nature13385 nature13385 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gyorffy B, Surowiak P, Budczies J, and Lanczky A. (2013). Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 8, e82241. 10.1371/journal.pone.0082241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robichaux JP, Elamin YY, Tan Z, Carter BW, Zhang S, Liu S, Li S, Chen T, Poteete A, Estrada-Bernal A, et al. (2018). Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat Med 24, 638–646. 10.1038/s41591-018-0007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nilsson MB, Sun H, Diao L, Tong P, Liu D, Li L, Fan Y, Poteete A, Lim SO, Howells K, et al. (2017). Stress hormones promote EGFR inhibitor resistance in NSCLC: Implications for combinations with beta-blockers. Science translational medicine 9. 10.1126/scitranslmed.aao4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mak MP, Tong P, Diao L, Cardnell RJ, Gibbons DL, William WN, Skoulidis F, Parra ER, RodriguezCanales J, Wistuba II, et al. (2016). A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clinical cancer research : an official journal of the American Association for Cancer Research 22, 609–620. 10.1158/10780432.CCR-15-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang H, Xiao G, Behrens C, Schiller J, Allen J, Chow CW, Suraokar M, Corvalan A, Mao J, White MA, et al. (2013). A 12-gene set predicts survival benefits from adjuvant chemotherapy in non-small cell lung cancer patients. Clin Cancer Res 19, 1577–1586. 10.1158/1078-0432.CCR-12-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, et al. (2005). Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 97, 339–346. 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 71.Byers LA, Sen B, Saigal B, Diao L, Wang J, Nanjundan M, Cascone T, Mills GB, Heymach JV, and Johnson FM (2009). Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 15, 6852–6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nanjundan M., Byers LA., Carey MS., Siwak DR., Raso MG., Diao L., Wang J., Coombes KR., Roth JA., Mills GB., et al. (2010). Proteomic profiling identifies pathways dysregulated in non-small cell lung cancer and an inverse association of AMPK and adhesion pathways with recurrence. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 5, 1894–1904. 10.1097/JTO.0b013e3181f2a266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Arbiser JL, Moses MA, Fernandez CA, Ghiso N, Cao Y, Klauber N, Frank D, Brownlee M, Flynn E, Parangi S, et al. (1997). Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proceedings of the National Academy of Sciences of the United States of America 94, 861–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dang LV, Nilsson A, Ingelman-Sundberg H, Cagigi A, Gelinck LB, Titanji K, De Milito A, Grutzmeier S, Hedlund J, Kroon FP, and Chiodi F. (2012). Soluble CD27 induces IgG production through activation of antigenprimed B cells. J Intern Med 271, 282–293. 10.1111/j.1365-2796.2011.02444.x. [DOI] [PubMed] [Google Scholar]
- 75.Lin SH, Wang J, Saintigny P, Wu CC, Giri U, Zhang J, Menju T, Diao L, Byers L, Weinstein JN, et al. (2014). Genes suppressed by DNA methylation in non-small cell lung cancer reveal the epigenetics of epithelialmesenchymal transition. BMC Genomics 15, 1079. 10.1186/1471-2164-15-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The PROSPECT dataset has been detailed previously28, and PROSPECT gene expression data are available at GEO accession GSE42127. Gene expression data for EGFR TKI resistant cell lines are available at GSE121634 as described previously19. NSCLC cell line gene expression data are available at GEO accession GSE4824. Gene expression data from HCC827 cells with or without CDH1 knockdown are available at GSE12303139. Transcriptomic data from cell lines derived from erlotinib-resistant patient biopsies are available at GSE64322 (super-series GSE64766)30. scRNAseq data was reported previously31. The dataset of matched baseline and osimertinib-refractory clinical samples32 is associated with accession number dbGaP: phs002001. For the analysis of CD70 expression and outcome among EGFR TKI refractory patients, we utilized a publicly available clinical dataset for which sequencing data was deposited in the European Genome-phenome Archive (EGA, EGAS00001005389, http://www.ebi.ac.uk/ega/) and clinical records, mutations, gene expression data, were hosted in OncoSG (https://src.gisapps.org/OncoSG/) as reported previously33. Clinical outcome data was accessed using the portal: https://src.gisapps.org/OncoSG_public/study/summary?id=GIS023. The TCGA-LUAD clinical cohort has been previously reported63. Clinical information and gene expression data were obtained through the TCGA portal (https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm). TCGA-LUAD and publicly available datasets (GSE14814, GSE19188, GSE29013, GSE30219, GSE31210, GSE3141, GSE31908, GSE37745, GSE43580, GSE4573, GSE50081, GSE8894, CAARAY) were used to assess CD70 expression and overall survival through kmplotter.com64.