

Abstract
Objectives:
Lupus T cells demonstrate aberrant DNA methylation patterns dominated by hypomethylation of interferon-regulated genes. The objective of this study was to identify additional lupus-associated DNA methylation changes and determine the genetic contribution to epigenetic changes characteristic of lupus.
Methods:
Genome-wide DNA methylation was assessed in naïve CD4+ T cells from 74 lupus patients and 74 age-, sex-, and race-matched healthy controls. We applied a trend deviation analysis approach, comparing methylation data in our cohort to over 16,500 samples. Methylation quantitative trait loci (meQTL) analysis was performed by integrating methylation profiles with genome-wide genotyping data.
Results:
In addition to the previously reported epigenetic signature in interferon-regulated genes, we observed hypomethylation of the promoter region of microRNA genes in the miR-17-92 cluster in lupus patients. Members of this microRNA cluster play an important role in regulating T cell proliferation and differentiation. Expression of two microRNAs in this cluster, miR-19b1 and miR-18a, showed a significant positive correlation with lupus disease activity. Among miR-18a target genes, TNFAIP3, which encodes a negative regulator of NFkB, was downregulated in lupus CD4+ T cells. Patient meQTL show overlap with genetic risk loci for lupus, including CFB and IRF7. The lupus risk allele in IRF7 (rs1131665) was associated with significant IRF7 hypomethylation. However, less than 1% of differentially methylated CpG sites in lupus patients were associated with an meQTL, suggesting minimal genetic contribution to lupus-associated epigenotypes.
Conclusion:
The lupus defining epigenetic signature, characterized by robust hypomethylation of interferon-regulated genes, does not appear to be determined by genetic factors. Hypomethylation of the miR-17-92 cluster that plays an important role in T cell activation is a novel epigenetic locus for lupus.
Keywords: lupus, epigenetics, methylation, microRNA, meQTL
Introduction
Systemic lupus erythematosus (lupus or SLE) is a heterogeneous autoimmune disease of incompletely understood etiology. The disease is characterized by a loss of immunotolerance and the development of autoantibodies against nuclear antigens. Severe manifestations of lupus have significant impact on quality of life and can lead to organ damage and mortality in affected patients, particularly among patients of non-European genetic ancestry [1, 2]. Genetic risk contributes to the development of lupus, but the estimated heritability of lupus is ~30% [3–5]. Indeed, monozygotic twin studies in lupus suggest a substantial non-genetic contribution to the etiology of lupus [6]. Environmental exposures across the lifespan that can directly impact epigenetic regulation and cellular function are suggested to be involved in the pathogenesis of lupus [7, 8].
DNA methylation is an epigenetic mechanism that regulates gene expression through the enzyme-mediated addition of a methyl group to the cytosine bases in the genome. DNA methylation is heritable across cell generations and can promote gene silencing, making it an important component in regulating the plasticity of immune cell identity and function [9]. Early work demonstrated that adoptive transfer of CD4+ T cells treated ex vivo with DNA methyltransferase (DNMT) inhibitors was sufficient to cause lupus-like disease in mice [10] mimicking the DNA methylation inhibition in patients with drug-induced lupus [11]. Since then, other studies have observed that CD4+ T cells of lupus patients show a distinct shift in global DNA methylation compared to healthy individuals, potentially in part due to defective MEK/ERK signaling, suppressing DNMT1 activity in CD4+ T cells, and leading to hypomethylation and overexpression of costimulatory genes [12–16].
We have previously observed a robust hypomethylation signature in interferon-regulated genes defining lupus patients [17, 18]. Our initial findings in CD4+ T cells were subsequently confirmed and expanded to other cell types by our group and others [19–21]. In CD4+ T cells, we observed hypomethylation in interferon-regulated genes at the naïve CD4+ T cell stage, preceding transcriptional activity. This epigenetic “poising” or “priming” of interferon-regulated genes was independent of disease activity [18]. The genetic contribution to this lupus-associated epigenotype is currently unknown.
Methylation quantitative trait loci (meQTL) are genetic polymorphisms that are associated with the methylation state of CpG sites either through direct nucleotide change within the CpG dinucleotide or intermediary mechanisms. Prior studies of lupus patients show an enrichment of meQTL associated with type I interferon genes, genetic risk loci, and specific clinical manifestations in whole blood and neutrophils [22–24]. Furthermore, our previous work suggests that meQTL might at least in part explain differences in DNA methylation between African-American and European-American lupus patients [22].
Herein, we evaluated genome-wide DNA methylation data in naïve CD4+ T cells from a large cohort of lupus patients compared to matched healthy controls. We integrated DNA methylation and genotyping data to better understand the influence of genetic factors upon the DNA methylation changes observed in lupus.
Methods
Study participants and demographics.
74 female lupus patients and 74 female healthy age (± 5 years), race, and sex-matched controls were recruited as previously described [25, 26] (Supplementary Table 1). All patients fulfilled the American College of Rheumatology (ACR) classification criteria for SLE [27]. Institutional review boards at our participating institutions approved this study. All participants signed a written informed consent prior to participation.
Sample collection, DNA isolation, and data generation.
Genomic DNA samples for this study were collected from naïve CD4+ T cells as previously described [18]. Briefly, magnetic beads and negative selection was used to isolate naïve CD4+ T cells from whole blood samples collected from lupus patients and controls. Genomic DNA was directly isolated from collected cells and bisulfite converted using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA, USA). The Illumina HumanMethylation450 BeadChip (Illumina, San Diego, CA, USA) was used to measure DNA methylation levels at over 485,000 methylation sites across the genome.
Epigenome-wide association study.
Epigenome-wide association study (EWAS) for identifying associations between specific CpG sites and disease status was performed using GLINT [28, 29]. Covariates for age, race, and technical batch were included for the analysis prior to other preprocessing. No outliers beyond four standard deviations were detected in the first two components of the principal component analysis (PCA) space, all 148 samples were included in the analysis. Reference-less cell type composition correction was performed using ReFACTor, with six components used in the downstream analysis to account for any cell-type heterogeneity in the samples. An additional covariate was included to account for effects of genetic admixture using the EPISTRUCTURE algorithm included in GLINT. Cell-type composition covariate components generated by ReFACTor were included at this step to reduce bias from potential cell-type heterogeneity, and polymorphic CpG sites were excluded from this step and the EWAS. Using the initial age, race, and technical batch covariates, along with six ReFACTor components and one EPISTRUCTURE component, logistic regression for disease status was performed across all CpG sites, excluding the polymorphic and unreliable cross-reactive probes previously described in the literature, as well as CpG sites with low variance (standard deviation <0.01) [30, 31].
Differential DNA methylation analysis of gene promoters.
Raw .idat files were used to generate methylation beta value profiles across all samples using GenomeStudio (Illumina, San Diego, CA, USA) after background subtraction and normalizing to internal control probes. Missing probe values were imputed using sklearn.impute.KNNImputer, and ComBat was used to correct for batch effects associated with sample preparation [32–34]. Ensembl gene loci for hg19 were downloaded using PyEnsembl [35]. For each gene, loci for 1500 base pairs upstream of the transcription start site [36] to the TSS were mapped to the overlapping CpG probes using PyBedtools, and the mean of the associated probes for each gene was used as the representative methylation value for the resulting 20,437 mapped genes [37]. Differential methylation analysis comparing patients and controls was performed on the mean TSS1500 methylation using limma, and false discovery rate adjustment using the Benjamini-Hochberg method was used to correct P-values for multiple testing. Gene Ontology Enrichment for Biological Process terms was performed on the differentially methylated gene list using Enrichr with the mapped promoter gene list used as the background [38, 39].
Trend deviation analysis.
DNA methylation data derived using the Illumina 450k methylation array from 23,415 samples were downloaded from Gene Expression Omnibus (GEO) [40]. To reduce batch effects, samples from experiments with less than 50 samples were omitted, and the resulting samples were quantile normalized [41]. A matrix of pairwise Pearson’s correlation values for DNA methylation levels was computed across TSS1500 gene promoters in 16,541 samples across 37 tissues to create a multi-tissue correlation network (Supplementary Figure 1). The differentially methylated genes in lupus naïve CD4+ T cells were clustered by their correlation in global signature created from the GEO data. Hierarchical clustering was performed using Scipy’s hierarchical linkage. KEGG enrichment analysis was performed using Enrichr [42], and P-values were reported after false-discovery rate adjustment.
The goal of a trend deviation analysis is to detect correlation patterns among differentially methylated genes in large DNA methylation datasets. A correlation in methylation among a set of differentially methylated genes between patients and controls suggests a trend is being observed, reinforcing the significance and robustness of the differential DNA methylation detected between patients and controls.
Genotyping.
Genomic DNA isolated from naïve CD4+ T cells was used as input for the Infinium Global Screening Array-24 v2.0 (Illumina, San Diego, CA, USA). Single nucleotide polymorphisms (SNPs) with a genotyping call rate < 98%, minor allele frequencies (MAF) < 5%, and deviating from Hardy-Weinberg equilibrium (HWE; P-value < 1E-3) were filtered out. Samples were removed if they had a genotyping call rate < 95%. Sex chromosomes were not analyzed. About 100,000 independent SNPs were pruned and used to perform PCA with Eigensoft (v.6.1.4) software [43]. Genotyping data were analyzed using PLINK (v.1.9) [44]. Genotype profiles were generated for n = 63 patients and n = 68 controls.
Methylation quantitative trait loci (meQTL) analysis.
Raw .idat files were used to generate methylation profiles using minfi (v.1.32.0) [45, 46] and to check median intensity values and reported sex in the R statistical computing environment (v.3.6.3) [47]. Probes with less than three beads and zero intensity values across all samples were removed using the DNAmArray package (v.0.1.1) [48]. Background signal and dye bias were corrected, followed by normalization of signal intensities using functional normalization in the preprocessFunnorm.DNAmArray function [48, 49] using the first three principal component values calculated from signal intensities of control probes present on all array spots to correct for technical variation. Any probe with a detection P-value < 0.01 or returned signal intensities in fewer than 98% of samples was removed. This resulted in a total of 476,767 probes used for further analysis. Signal intensities were then converted to M-values with a maximum bound of ±16. M-values were used for meQTL analysis and converted to beta values (0–100% methylation scale) using minfi for reporting.
We removed any probe for meeting any of the following technical criteria: A unique probe sequence of less than 30bp, mapping to multiple sites in the genome, polymorphisms that cause a color channel switching in type I probes, inconsistencies in specified reporter color channel and extension base, mapping to the Y chromosome, and/or having a polymorphism within 5bp of the 3’ end of the probe with a minor allele frequency > 1% with the exception of CpG-SNPs with C>T polymorphisms which were retained [50]. Batch correction for chip ID was performed using the ComBat function in the sva (v.3.34.0) package [51]. After technical filtering, there were a total of 421,214 probes used for meQTL analysis.
We implemented a mixed correspondence analysis with the PCAmixdata package (v.3.1) [52] to calculate eigenvalues using patient medication data for prednisone, hydroxychloroquine, azathioprine, mycophenolate mofetil, and cyclophosphamide. The top four components accounted for a cumulative 89.3% of variability in the medication data. Each component value was used as an independent variable in regression analysis to adjust for medication usage across individuals. MeQTL association analysis was performed in patients and controls separately using methylation M-value profiles and corresponding sample genotypes. Age, the top four medication components, and top ten genotype principal components were included as covariates to build a linear model for detecting meQTL using MatrixEQTL (v.2.3) [53]. Cis-meQTL were defined as CpG sites with methylation values associated with a SNP within a conservative 1000bp of the CpG dinucleotide. We used a Benjamini-Hochberg False Discovery Rate (FDR)-adjusted P-value cutoff of < 0.05 as a threshold for significant associations. The above EWAS results were compared with the meQTL results to determine overlap of lupus-associated differentially methylated CpG sites and those CpG sites in an meQTL.
Functional Enrichment Analysis.
ToppGene Suite was used for functional enrichment analysis [54] of Molecular Function and Biological Process Gene Ontologies and KEGG Pathways in meQTL. P-values were derived using a hypergeometric probability mass function, and a Benjamini-Hochberg FDR–adjusted P-value cutoff of < 0.05 was used as a threshold of significance. A minimum membership of 3 genes and maximum of 2,000 genes in each term was used as a threshold for inclusion. IFN-regulated genes were identified using the set of genes associated with the CpG site in each meQTL as input using Interferome (v.2.01) [55]. The type I interferon response genes were defined as genes with an expression fold change of 1.5 or greater between type I interferon-treated and untreated samples using gene expression datasets from all available CD4+ T cell experiments in the Interferome database.
For the analysis of miR-18a-regulated genes, literature-based network association analysis was performed using IRIDESCENT to create a weighted network of published relationships as previously described [56].
MicroRNA expression microarray.
MicroRNA expression was measured in naïve CD4+ T cells from a subset of lupus patients and healthy matched controls (n = 16). Cells were immediately lysed with TRIzol Reagent (ThermoFisher Scientific, NY, USA) followed by storage at −80C. Total RNA was isolated using the Direct-zol RNA MiniPrep Kit (Zymo Research, CA, USA) following the manufacturer’s directions. The Affymetrix miRNA 4.1 Array Strip (Affymetrix, CA, USA) was used to measure expression of over 2,000 premature and 2,500 mature human microRNA sequences. RNA sequences were polyadenylated and ligated to a biotin-labeled oligomer using the FlashTag Biotin HSR RNA Labeling Kit (Affymetrix, CA, USA). Biotin-labeled sequences were hybridized to array probes and washed then stained with streptavidin-PE. The Affymetrix Expression Console & Transcriptome Analysis Console 2.0 software (Affymetrix, CA, USA) was used to analyze biotin/streptavidin-PE fluorescence measurements. All samples passed signal intensity, polyadenylation, and ligation quality controls. Signal intensities were background adjusted and normalized. Log2-transformed expression values for each probeset was calculated using a robust multi-array average model (23). The Pearson r correlation coefficient for median expression values of probes for miR-17, miR-18a, miR-19a, miR-19b1, and miR-20a and patient Systemic Lupus Erythematosus Disease Activity (SLEDAI) score were calculated using GraphPad Prism (v9.3.0) (GraphPad Software, CA, USA).
Results
Differential methylation of gene promoters in naïve CD4+ T cells isolated from lupus patients.
A comparison of DNA methylation profiles from circulating naïve CD4+ T cells isolated from 74 lupus patients and 74 age, sex and race matched healthy controls revealed a total of 2,627 CpGs out of 334,337 total CpG sites included in the EWAS with a significant difference in average methylation. Significant hypomethylation in interferon-regulated genes was observed, consistent with previous reports (Supplementary Table 2). Average promoter methylation for each gene was calculated by including all CpG sites on the array within 1500bp of the associated gene’s transcription start site (TSS). A total of 51 genes showed a significant difference in average promoter methylation between lupus patients and controls (17 hypomethylated and 34 hypermethylated in patients compared to controls) (Table 1) (Figure 1). Biological Process Gene Ontology enrichment analysis of differentially methylated promoter regions did not show significant enrichment compared to the background of all gene promoters after adjusting for multiple testing (Supplementary Table 3).
Table 1.
Genes with differentially methylated promoter regions in naive CD4+ T cells of lupus patients compared to healthy controls.
| Gene | Δβ | −log10 (FDR-adjusted P-value) | t-statistic |
|---|---|---|---|
|
| |||
| IFI44L | −0.177 | infinity | −10.757 |
| DTX3L | −0.130 | infinity | −11.566 |
| BST2 | −0.089 | 11.323 | −9.285 |
| RABGAP1L | −0.088 | 9.165 | −8.421 |
| BCL2L14 | −0.086 | 5.520 | −6.908 |
| MIR19B1 | −0.059 | 3.169 | −5.846 |
| IFI44 | −0.059 | 2.057 | −5.304 |
| MIR20A | −0.055 | 3.088 | −5.807 |
| MIR17 | −0.054 | 6.882 | −7.487 |
| MIR18A | −0.051 | 6.537 | −7.342 |
| MIR19A | −0.049 | 4.771 | −6.579 |
| IKZF4 | −0.048 | 3.289 | −5.902 |
| MX1 | −0.046 | 10.624 | −9.004 |
| TRIM34 | −0.045 | 2.184 | −5.367 |
| ODF3B | −0.034 | 1.712 | −5.128 |
| GNG2 | −0.033 | 2.138 | −5.344 |
| FAM177B | −0.025 | 1.897 | −5.223 |
| MZF1 | 0.008 | 1.493 | 5.014 |
| SSBP4 | 0.015 | 1.344 | 4.934 |
| ATP6V0D1 | 0.018 | 2.594 | 5.569 |
| DCUN1D1 | 0.025 | 2.068 | 5.309 |
| C14orf93 | 0.025 | 1.922 | 5.236 |
| TIPARP | 0.026 | 2.069 | 5.310 |
| LMBRD1 | 0.027 | 2.211 | 5.381 |
| HAVCR2 | 0.027 | 2.574 | 5.560 |
| KIAA1949 | 0.030 | 3.158 | 5.841 |
| GPD2 | 0.032 | 1.953 | 5.251 |
| CNTF | 0.033 | 1.705 | 5.124 |
| CD47 | 0.034 | 4.259 | 6.350 |
| ARHGAP9 | 0.036 | 3.339 | 5.926 |
| IL27RA | 0.036 | 1.367 | 4.946 |
| RAP1A | 0.036 | 2.573 | 5.559 |
| LAMA3 | 0.037 | 1.445 | 4.988 |
| ABI3 | 0.037 | 1.436 | 4.983 |
| FAM102A | 0.038 | 3.161 | 5.842 |
| CXCR5 | 0.039 | 1.439 | 4.985 |
| DPEP2 | 0.040 | 1.889 | 5.219 |
| DYRK2 | 0.041 | 3.924 | 6.197 |
| TMEM71 | 0.044 | 2.757 | 5.649 |
| ADORA2A | 0.046 | 2.234 | 5.392 |
| SEPT9 | 0.047 | 2.036 | 5.293 |
| PSMB4 | 0.052 | 2.935 | 5.734 |
| TOM1 | 0.055 | 5.415 | 6.862 |
| PRIC285 | 0.057 | 9.934 | 8.729 |
| LTB | 0.062 | 2.036 | 5.293 |
| MIR1205 | 0.067 | 1.698 | 5.121 |
| ACER3 | 0.073 | 2.612 | 5.578 |
| BCL9L | 0.079 | 4.034 | 6.248 |
| MDS2 | 0.080 | 3.149 | 5.836 |
| SNORA5B | 0.083 | 1.712 | 5.128 |
| PTPRCAP | 0.091 | 3.620 | 6.057 |
FDR correction was performed using the Benjamini-Hochberg method with an FDR-adjusted P value threshold of < 0.05. Δβ methylation difference in median methylation value of CpG sites within 1500bp upstream of the associated gene’s transcription start site (TSS1500) between lupus patients and healthy controls.
Figure 1.
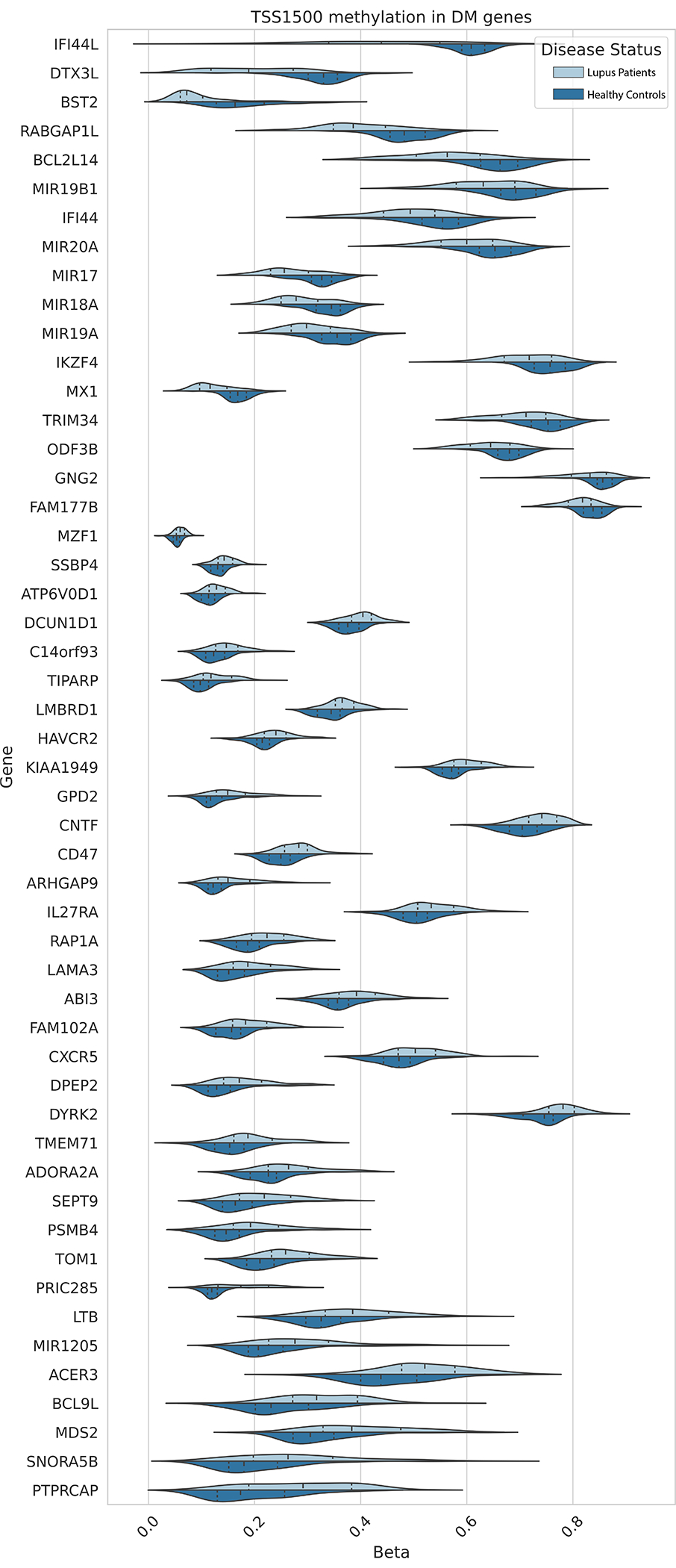
Distribution of average CpG methylation levels within 1500bp of the TSS for the respective genes differentially methylated in naïve CD4+ T cells of lupus patients compared to healthy controls.
The pairwise correlation of the 51 gene promoters identified above was calculated across a collection of 16,541 samples from 37 tissues available in GEO. Hierarchical clustering of correlations showed that 21 out of the 51 gene promoters were highly correlated. KEGG Pathway enrichment analysis showed a significant enrichment for three pathways among the 21 correlated gene promoters: “microRNAs in cancer” (P-value = 3.86E-04), “cytokine-cytokine receptor interaction” (P-value = 4.34E-02), and “rheumatoid arthritis” (P-value = 4.34E-02) (Table 2) (Figure 2). The microRNAs in cancer” pathway included genes encoding miR-17, miR-18a, miR-19a, miR-19b1, and miR-20a. Four of seven CpG sites used to calculate the average promoter methylation (TS1500) in this locus showed a significant reduction in median methylation in lupus patients compared to healthy controls (Figure 3A). These sites: cg17799287 (Δβ= 5.5%; P-value = 2.05E-03), cg07641807 (Δβ = −4.4%; P-value = 1.71E-02), cg23665802 (Δβ= −5.8%; P-value = 1.19E-02), and cg02297838 (Δβ= −4.9%; P-value = 3.48E-02) were all hypomethylated in lupus patients compared to healthy controls and overlapped with enhancers and regions flanking TSSs in peripheral naïve CD4+ T cells using data collected from the Epigenome Roadmap [57] and visualization using the WashU Epigenome Browser [58]. We examined expression levels of the microRNAs included in the “microRNAs in cancer” pathway (miR-17, miR-18a, miR-19a, miR-19b1, and miR-20a) in naïve CD4+ T cells of a subset of our lupus patients (n = 16) and healthy matched controls (n = 16). We did not observe a difference in expression between patients and controls. However, two microRNAs, miR-18a-5p and miR-19b1–5p, showed a significant positive correlation (hsa-miR-18a-5a P-value = 0.038 & hsa-miR-19b1-5p P-value = 0.042) between median expression levels and SLEDAI scores in lupus patients (Figure 3B) (Supplementary Table 4).
Table 2.
KEGG Pathway gene enrichment of 21 gene promoters highly correlated with each other in multi-tissue DNA methylation data constructed from 16,541 samples available through Gene Expression Omnibus.
| Pathway (KEGG_2019_Human) | P-value | FDR-adjusted P-value | Odds Ratio | Genes |
|---|---|---|---|---|
|
| ||||
| MicroRNAs in cancer | 1.21E-05 | 0.00039 | 20.92 | MIR19B1;MIR20A;MIR17;MIR18A;MIR19A |
| Cytokine-cytokine receptor interaction | 0.0034 | 0.043 | 11.28 | CNTF;CXCR5;LTB |
| Rheumatoid arthritis | 0.0041 | 0.043 | 23.52 | LTB;ATP6V0D1 |
Figure 2.
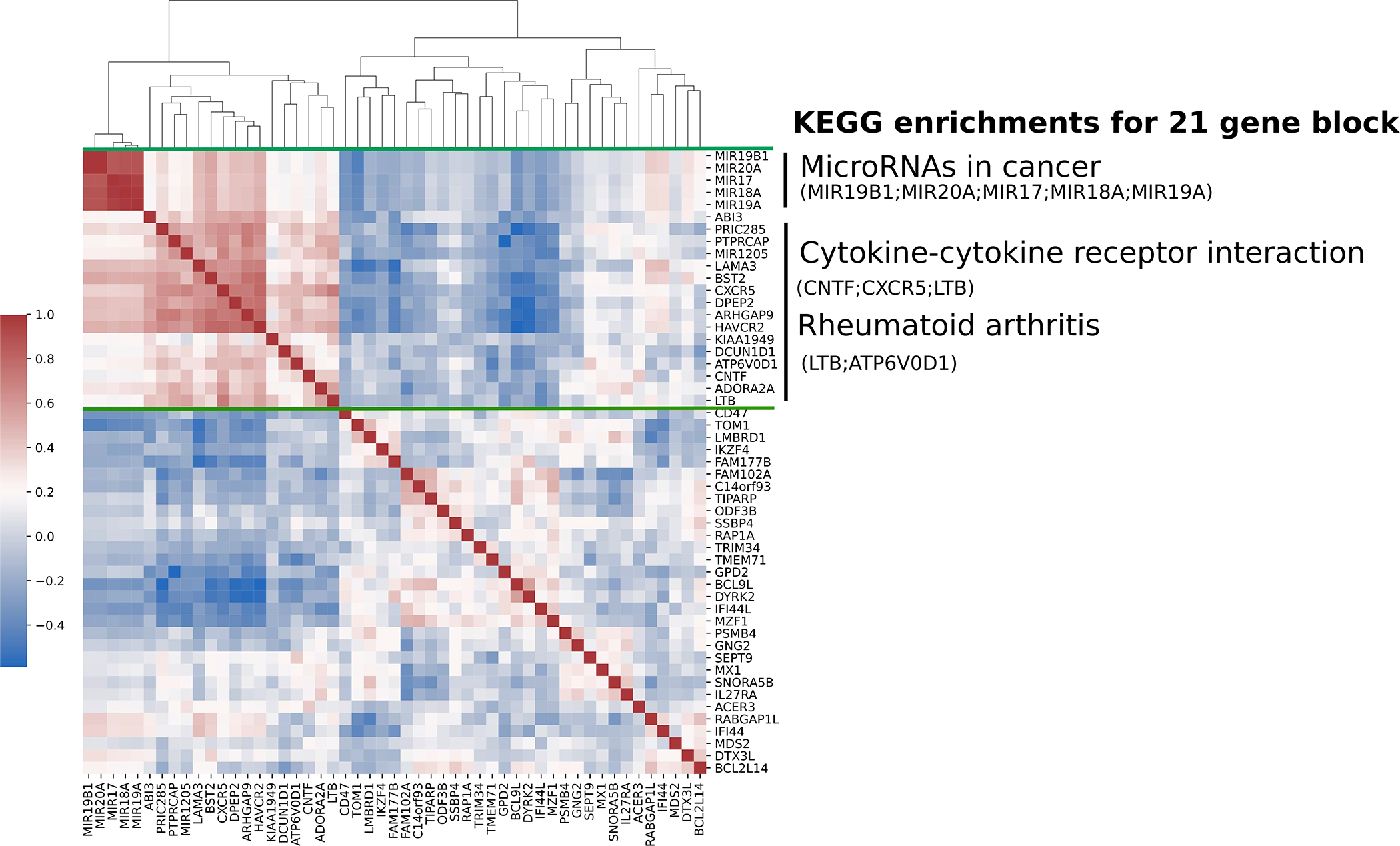
Heatmap of hierarchical clustering of pairwise Pearson correlation coefficient values of 51 differentially methylated gene promoters (TSS1500) in global tissue signature derived from 16,541 samples. Range from +1 (red) to −1 (blue), represent a greater to lower correlation in global tissue, respectively. KEGG pathways are significantly enriched (FDR-adjusted P-value < 0.05) in a block of 21 genes (green bars).
Figure 3.
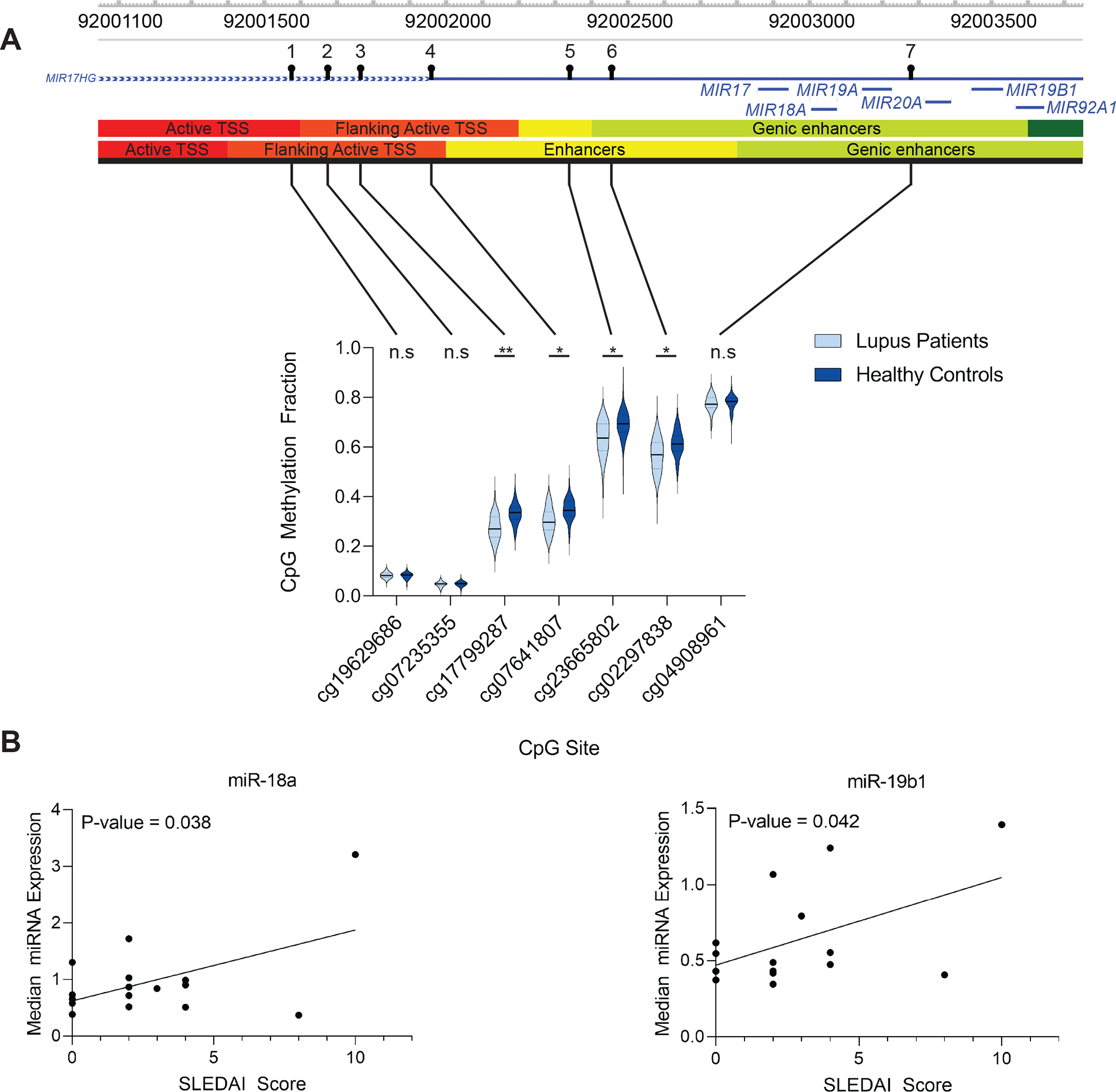
(A) Violin plots of the seven CG probes in lupus patients and healthy controls used to calculate the average promoter methylation (TSS1500) for the miR-17-92 cluster. The solid black bar represents the median value and the dashed lines the first and third quartiles. Genomic visualization and annotation are from WashU Epigenome Browser using AuxillaryHMM tracks from peripheral naïve CD4+ T cell tissues (E038 and E039, top and bottom tracks, respectively). For P-values: n.s. = not significant, * = P < 0.05, ** = P < 0.01. (B) Correlation of median microRNA expression in naïve CD4+ T cells of a subset (n = 16) of lupus patients with SLEDAI score. Hsa-miR-18a-5p and hsa-miR-19b1-5p had a Pearson correlation (r) of 0.52 (P-value = 0.038) and 0.51 (P-value = 0.042), respectively.
Examining publicly available microRNA expression data from total CD4+ T cells revealed overexpression of miR-18a in lupus patients compared to healthy control individuals [59]. In these same samples, a total of 74 miR-18a-target genes were downregulated in lupus patients compared to controls. Using a literature-based network association analysis, we identified 15 of these 74 genes with relatedness to lupus (Supplementary Figure 2). TNFAIP3, which encodes a negative regulator of NFkB targeted by miR-18a, was downregulated in lupus CD4+ T cells compared to controls. We examined the expression of MIR17HG, which encodes the miR-17-92 cluster, in single cell RNA-sequencing data from lupus nephritis tissue samples generated by the Accelerating Medicines Partnership (AMP) project [60]. We show evidence for MIR17HG mRNA expression in multiple immune cells infiltrating the kidneys of patients with lupus nephritis, including multiple T cell subsets, albeit in a small percentage of kidney infiltrating cells. While over 8% of tissue-resident macrophages in lupus nephritis tissues express MIR17HG mRNA, the highest levels of expression observed appears to be in T cell subsets (Supplementarty Figure 3).
Naïve CD4+ T cell methylation quantitative trait loci (meQTL) in lupus patients.
Global genotype profiles were generated in a subset of patients and controls and compared with global DNA methylation profiles to identify CpG sites with allele-specific methylation associations. There was no significant difference in the average age (years) between the patient (n = 63) and control (n = 68) subsets (patient average age = 41.6; patient age SD = 12.8; control average age = 40.8; control age SD = 12.5; t-test statistic = 0.381; two-tailed P-value = 0.704). Allele-specific DNA methylation associations were measured as meQTL where the CpG site was within 1000bp of the measured SNP separately in patients and controls. After adjusting for age, genetic background, and medication use in patients, we identified 5,785 meQTL present in the naïve CD4+ T cells of lupus patients with an FDR-adjusted P-value < 0.05 (Supplementary Table 5). These meQTL include 4,649 unique CpG sites and 4,120 unique polymorphisms. A linear model adjusting for age and genetic background was fit to controls separately. We identified a total of 7,331 meQTL with an FDR-adjusted P-value < 0.05 in controls (Supplementary Table 6). These meQTL include 5,885 unique CpG sites and 5,138 unique polymorphisms.
Of 2,627 CpG sites differentially methylated between patients and controls, we identified 17 (0.65%) and 34 (1.29%) CpG sites that overlapped with CpG sites included in meQTL in patients and controls, respectively (Figure 4A and B). We examined the overlap of meQTL in lupus patients and healthy controls and identified a total of 3,957 meQTL (68.4% of lupus patient meQTL and 54.0% of healthy control meQTL) shared between both patients and controls (Supplementary Table 7). This shared set of meQTL contained 8 (0.3%) CpG sites that we identified as differentially methylated between lupus patients and controls (Figure 4C).
Figure 4.
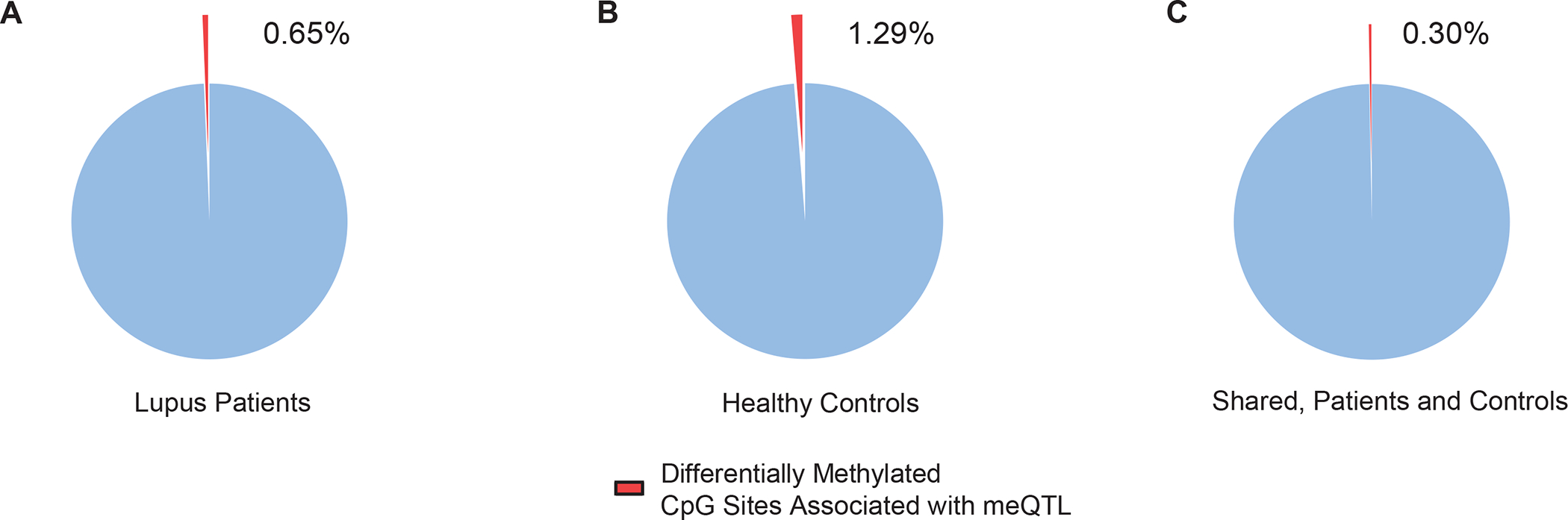
Proportion of differentially methylated CpG sites in naïve CD4+ T cells of lupus patients compared to healthy controls associated with an meQTL in (A) lupus patients, (B) healthy controls, and (C) the subset of meQTL shared between lupus patients and healthy controls.
Functional enrichment analysis was performed using genes associated with CpG sites in our meQTL shared between patients and controls. Functional enrichment analysis revealed multiple ontologies and pathways for cell adhesion (“cell-cell adhesion”; P-value = 1.04E-12, “biological adhesion”; P-value = 6.80E-12, “cell adhesion”; P-value = 8.25E-12, “Cell adhesion molecules (CAMs)”; P-value = 2.25E-06), transporter associated with antigen processing (TAP) proteins and antigen presentation (“TAP binding”; P-values = 1.59E-7, “peptide antigen binding”; P-value = 4.40E-5), and immune disorder pathways (“Type I diabetes mellitus”; P-value = 1.92E-8, “Graft-versus-host disease”; P-value = 4.38E-7) (Supplementary Table 8).
There were 1,828 meQTL detected only in lupus patients but not in controls. These were enriched in gene ontologies and pathways related to tissue growth and development (“animal organ morphogenesis”; P-value = 8.44E-10, “urogenital system development”; P-value = 1.05E-07) and gene silencing (“negative regulation of gene silencing by miRNA”; P-value = 2.54E-6, “negative regulation of posttranscriptional gene silencing”; P-value = 5.41E-6) (Supplementary Table 9).
We compared our list of meQTL in lupus patients to previously identified lupus susceptibility loci from genome-wide association studies [4, 61–64]. We found 41 meQTL with CpG site-associated genes that overlapped with 20 unique lupus risk loci genes (Supplementary Table 10). This included interferon regulatory factor genes IRF5 and IRF7. We found three meQTL in naïve CD4+ T cells that included, or were in high LD (r2 ≥ 0.80) with, a known lupus genetic risk variant (Table 3) [65]. We also performed a similar analysis using data previously collected from the neutrophils of lupus patients to determine if these effects were present across tissues [22]. We found meQTL associated with lupus risk variants in CFB (rs170942) and IRF7 (rs1131665) in both naïve CD4+ T cells and granulocytes isolated from lupus patients. In addition, an meQTL associated with the TMEM86B-PTPRH locus was observed in naïve CD4+ T cells. When we compared the lupus risk allele with DNA methylation levels, we found that the presence of the risk allele at rs1270942 (CFB) is associated with increased DNA methylation of cg16505946. The presence of the risk allele at rs1131665 (IRF7) (Figure 5) and rs56154925 (TMEM86B-PTPRH) was associated with decreased DNA methylation of cg16486109 and cg01414877, respectively. The direction of the risk allele-DNA methylation association in the CFB and IRF7 meQTL was the same in both naïve CD4+ T cells and granulocytes.
Table 3.
MeQTL in naive CD4+ T cells and granulocytes of lupus patients that include a known lupus risk variant.
| Lupus Naïve CD4+ T cell meQTL | |||||
|---|---|---|---|---|---|
|
| |||||
| CpG Site | meQTL SNP | Lupus Risk SNP# | Risk SNP-associated Gene | Lupus Risk Allele | Direction of CpG methylation associated with risk allele |
|
| |||||
| cg16505946 | rs558702 | rs1270942 | CFB | C | ↑ |
| cg16486109 | rs1131665 | rs1131665 | IRF7 | A | ↓ |
| cg01414877 | rs56154925 | rs56154925 | TMEM86B-PTPRH | C | ↓ |
|
| |||||
| #rs558702 and rs1270942 have an LD r2 ≥ 0.80. | |||||
| Lupus Granulocyte meQTL | |||||
|
| |||||
| CpG Site | meQTL SNP | Lupus Risk SNP# | Risk SNP-associated Gene | Lupus Risk Allele | Direction of CpG methylation associated with risk allele |
|
| |||||
| cg16505946 | rs558702 | rs1270942 | CFB | C | ↑ |
| cg16486109 | rs1131665 | rs1131665 | IRF7 | A | ↓ |
Figure 5.
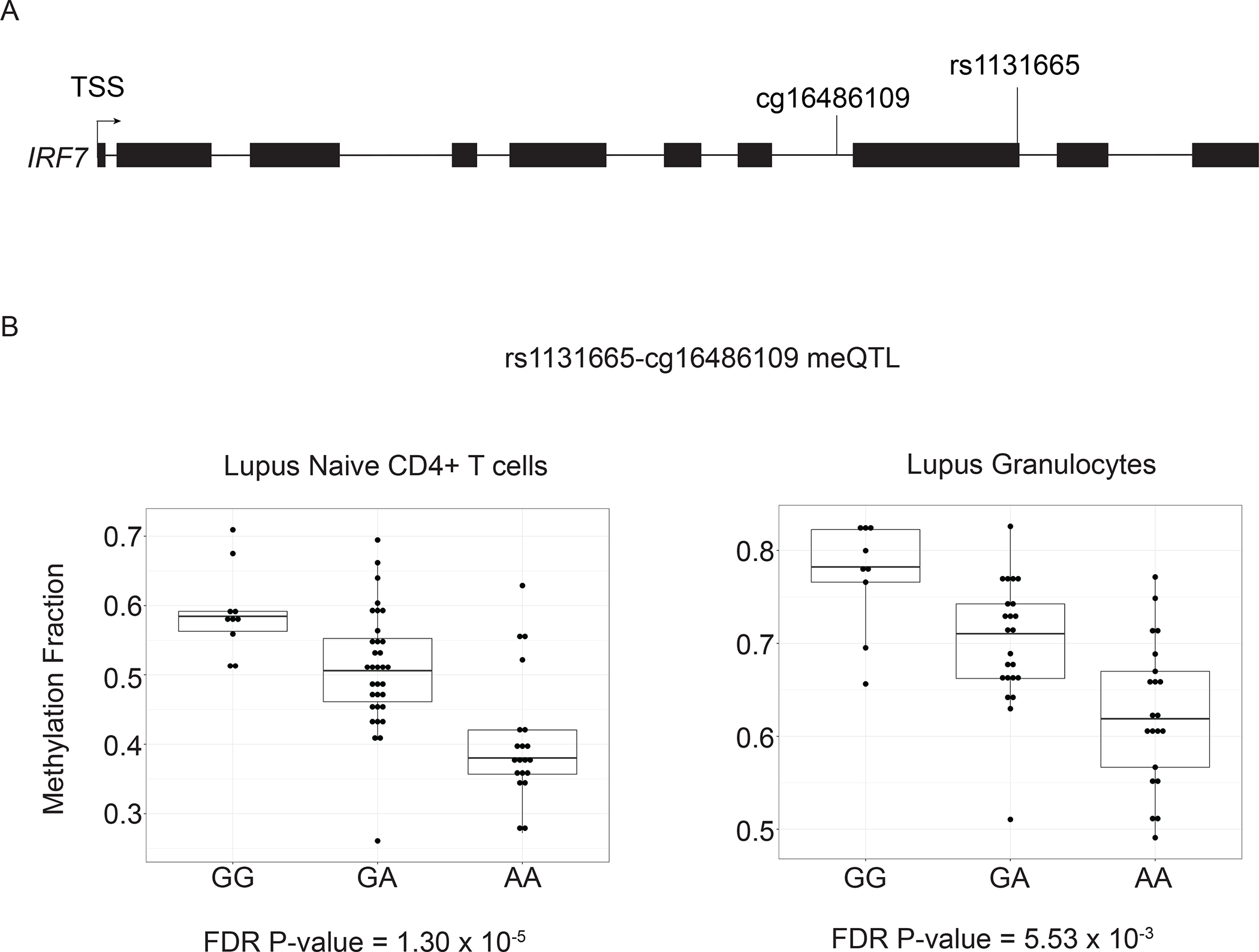
(A) Gene structure diagram of IRF7 depicting the location of rs1131665 and cg16486109. (B) The presence of the lupus risk allele at rs1131665 shows a significant negative correlation with DNA methylation of cg16486109 located in IRF7.
We examined the overlap between genes associated with CpG sites in meQTL we identified in lupus patient naïve CD4+ T cells and genes that respond to type I interferon treatment in CD4+ T cells, to better understand the association between patient genetics and type I interferon-response gene methylation differences in lupus. A total of 101 unique type I interferon-response genes were identified as meQTL in our data (Supplementary Table 11).
Because IRF7 is a master regulator of type I interferon response [66], and the lupus-associated epigenotype is dominated by hypomethylation in interferon-regulated genes, we examined if rs1131665 (IRF7) had an effect on the methylation levels of the 2,627 CpGs differentially methylated in lupus naïve CD4+ T cells identified in this study. This trans-meQTL analysis revealed no significant difference in methylation levels across these CpG sites based on rs1131665 genotypes, among lupus patients (ANOVA analysis, data not shown).
Discussion
We generated genome-wide DNA methylation data in naïve CD4+ T cells from a large cohort of lupus patients and matched healthy controls. Implementing an innovative trend deviation analysis, we identified a cluster of microRNAs (miR-17, miR-18a, miR-19a, miR-19b1, miR-20a) among differentially methylated loci in lupus patients. Promoter methylation analysis revealed significant hypomethylation in this microRNA cluster in lupus patients compared to controls. Trend deviation analysis suggested a coordinated, disease-associated change in promoter methylation for these microRNAs. Indeed, the expression of miR-18a and miR-19b1 included within this cluster positively correlated with disease activity, as measured using SLEDAI score, in our lupus patients. MicroRNAs play an important role in post-transcriptional gene regulation by targeting specific complementary gene transcripts for degradation [67]. Peripheral blood cells in lupus patients show altered expression of microRNAs [68]. Some dysregulated microRNAs in lupus target DNA methyltransferase 1 (DNMT1), and as a result, contribute to altered DNA methylation patterns in lupus CD4+ T cells [69–71]. MiR-17, miR-18a, and miR-20a form the “miR-17 family” while miR-19a and miR-19b1 form the “miR-19 family”, which are grouped by sequence homology and encoded in a single polycistronic microRNA gene called the “miR-17-92 cluster”. This cluster has been well-studied as an oncogene and an immune regulator [72]. Average promoter methylation of miR-17, miR-18a, miR-19a, miR-19b1, and miR-20a was reduced by ~5% in lupus patients compared to controls, which has not been previously described in immune cells of lupus patients. Enterovirus 71 infection has been observed to suppress miR-17-92 cluster expression by increasing DNMT-mediated promoter methylation [73], and chemical inhibition of DNMT1 activity in bleomycin-induced lung fibrosis mouse model increases miR-17-92 cluster expression in lung fibroblasts [74]. This suggests that miR-17-92 cluster promoter methylation plays an important role in regulating the expression of its members.
miR-17-92 cluster genes play a vital role in regulating T cell activities including proliferation and differentiation. Overexpression of miR-17-92 cluster genes promotes lymphoproliferative disease and autoimmunity in mice by targeting critical immunotolerance regulators Bim and PTEN [75]. Conditional knock out of miR-17-92 cluster in a murine model of chronic graft-versus-host disease (cGVHD) reduced disease-associated T cell infiltration and IgG deposition in the skin [76]. In cGVHD mice, miR-17-92 cluster expression in CD4+ T cells supports Th1, Th17, and Tfh cell differentiation. Loss of miR-17-92 cluster expression leads to a corresponding reduction in Tfh-dependent germinal center formation and plasma cell differentiation [76]. MiR-17, miR-18a, miR-19a, and miR-20a are overexpressed in splenic T cells of MRL/lpr mice [77]. Similarly, miR-17, miR-17a, miR-18a, miR-19a, miR-19b1, and miR-20a are overexpressed in circulating CD4+ T cells of lupus patients [78]. MiR-19b1 expression, specifically, has a significant positive correlation with disease activity as measured by SLEDAI score [78]. MiR-17 and miR-20 are downregulated in circulating PBMCs [79], B cells [80], and as circulating free microRNAs [81] in lupus patients compared to healthy controls, suggesting tissue-specific and microRNA-specific expression patterns. Of the miR-17-92 cluster microRNAs identified as differentially methylated in our analysis, only miR-18a and miR-19b1 showed a significant positive correlation between median expression and disease activity in naïve CD4+ T cells of lupus patients, consistent with these prior observations. MiR-19b1 promotes proliferation of mature CD4+ T cells, Th1 differentiation and IFN-γ production, and suppresses inducible Treg differentiation [82]. MiR-18a expression increases rapidly early on in CD4+ T cell activation [83, 84], and suppresses Th17 cell differentiation through direct targeting of critical Th17 transcription factor transcripts including SMAD4, HIF1A, and RORA in human CD4+ T cells in vitro and in vivo murine airway inflammation models [83]. We did not observe a difference in the expression of members in the miR-17-92 cluster between lupus patients and controls in naïve CD4+ T cells, likely because these microRNAs are upregulated upon T cell activation. Evidence for hypomethylation in lupus in naïve CD4+ T cells suggests epigenetic priming of this locus, similar to what we previously observed in interferon-regulated gene loci in lupus [18].
Consistent with our DNA methylation data and the epigenetic priming concept in naïve CD4+ T cells discussed above, gene expression data in total CD4+ T cells isolated from lupus patients compared to normal healthy controls revealed upregulation of miR-18a in lupus and concomitant downregulation of several genes known to be targeted by miR-18a [59]. Of 74 miR-18a target genes downregulated in lupus CD4+ T cells, our literature-based analysis highlighted 15 genes, including HIF1A which is involved in T cell differentiation as discussed above. The most robustly lupus-related gene was TNFAIP3, which encodes the NFkB negative regulator A20. Indeed, the genetic association between TNFAIP3 loss of function polymorphisms and lupus has been repeatedly confirmed [85].
Single cell RNA sequencing data from lupus nephritis kidney tissues revealed evidence for expression of MIR17HG, which encodes the miR-17-92 cluster, in kidney-infiltrating immune cells, including multiple T cell subsets. Further studies are needed to determine if altered DNA methylation at the miR-17-92 cluster promoter is associated with expression changes with a causal role in the development of lupus, and to determine if methylation levels at this locus can be used as biomarker for monitoring disease activity.
We used analysis of meQTL to identify allele-specific DNA methylation associations across the genome of naïve CD4+ T cells from lupus patients and healthy controls. Our primary objective was to understand to what extent are DNA methylation changes associated with lupus (the lupus-defining epigenetic profile), explained by genetic factors. We found that < 1% of differentially methylated sites in lupus patients compared to healthy controls were associated with a cis-meQTL. This suggests that almost all of the DNA methylation alterations observed in lupus are not associated with local allelic differences in the genome, suggesting a greater contribution from non-genetic and possibly environmental factors to epigenetic dysregulation in lupus. A previous study of meQTL in whole blood of lupus patients found that a majority of meQTLs were shared between patients and controls [24]. We observed that about 68% of meQTL in lupus patients and 54% of meQTL in healthy controls were shared by both groups, supporting this observation.
Our prior analysis of granulocytes from a cohort of lupus patients identified overlap in meQTL genes and lupus genetic risk loci [22]. MeQTL pairs including ARID5B (cg13344587-rs10821936), HLA-DQB1 (cg13047157-rs9274477), and IRF7 (cg16486109-rs1131665) were found in both neutrophils and naïve CD4+ T cells from lupus patients. Risk loci genes unique to naïve CD4+ T cell meQTLs included CD80 (cg06300880-rs3915166), TYK2 (cg06622468-rs280501), IKBKE (cg22577136-rs17020312), and CTLA4 (cg05092371-rs16840252, cg05092371-rs4553808). Naïve CD4+ T cell-specific meQTL risk loci genes are related to signal response and activation in CD4+ T cells compared to the more general DNA repair and type I interferon signaling seen in the shared meQTL risk loci genes. Disease-relevant meQTL show tissue-specific patterns which should be considered when teasing apart their potential impact.
We identified three meQTL that include SNPs previously identified as lupus genetic risk variants. One meQTL is in the complement factor B gene CFB (cg16505946-rs558702) where the risk allele is associated with increased DNA methylation of the nearby CpG site. Complement factor B (CFB) combines with C3 to form the C3 convertase after cleavage by complement factor D as part of the alternative complement pathway. Complement pathway defects have long been studied as a model of monogenic lupus and contribute to increased risk of polygenic lupus [65]. We identified an additional meQTL that included a known lupus risk variant in IRF7 (cg16486109-rs1131665). Rs1131665 is a missense variant in the inhibitory domain of IRF7 (Q412R). This lupus-associated amino acid change was demonstrated to enhance IRF7-induced expression response in a luciferase reporter assay [86]. This same risk allele is also associated with decreased DNA methylation of cg16486109. Though the relative DNA methylation fractions are different between naïve CD4+ T cells and granulocytes of lupus patients, the direction of the allele-specific DNA methylation is the same. This suggests that the observed meQTL effect may be present in other lymphoid and myeloid tissues, potentially including plasmacytoid dendritic cells, which are major producers of type I interferons. We describe a direct association between a lupus risk allele and local hypomethylation of a CpG site in IRF7 in lupus. This observation provides new insights regarding possible biological mechanisms underlying pathogenic consequences of lupus-associated genetic polymorphisms.
In summary, we investigated genome-wide DNA methylation changes in naïve CD4+ T cells from an extended cohort of lupus patients and controls, and using a methylation trend deviation analysis method, we showed promoter hypomethylation of the miR-17-92 cluster that has a significant regulatory role in T cells growth, function, and differentiation. Combining genome-wide DNA methylation and genotyping data, we were able to determine genetic contribution to the lupus-defining epigenotype. Our data indicate that epigenetic changes characteristic of lupus are not under direct genetic influence. This suggests a more important role for non-genetic factors in the epigenetic dysregulation observed in lupus patients, including the robust demethylation of interferon-regulated genes.
Supplementary Material
Key messages.
What is already known on this topic:
Lupus is characterized by robust DNA hypomethylation in interferon-regulated genes. However, the genetic contribution to the lupus-associated epigenotype is unknown.
What this study adds –
Our results suggest that genetic factors do not significantly contribute to the lupus-associated DNA methylation profiles. We also report a novel epigenetic locus for lupus in a microRNA cluster involved in T cell function. Further, we provide a prototype example showing how a lupus risk genetic variant might mediate functional pathogenic effects through altering DNA methylation levels.
How this study might affect research, practice or policy –
This study highlights the importance for non-genetic factors in determining epigenetic changes characteristic of lupus.
Funding Information
This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) grant number R01 AI097134 to Dr. Sawalha. Dr. Wren is supported by NIH grants number P30 AG050911 and P20 GM103636.
Footnotes
Competing interests: None of the authors has any financial conflict of interest to disclose
Conflict of interest: The authors have declared that no conflict of interest exists
Ethical approval information: The study was approved by the institutional review boards and the ethics committees at all participating institutions and all study participants signed an informed written contest.
Patient and public involvement: It was not appropriate or possible to involve patients or the public in the design, or conduct, or reporting, or dissemination plans of our research.
Data sharing statement:
All data are included in the manuscript or supplementary material.
References
- 1.Olesinska M, Saletra A. Quality of life in systemic lupus erythematosus and its measurement. Reumatologia. 2018; 56(1):45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lewis MJ, Jawad AS. The effect of ethnicity and genetic ancestry on the epidemiology, clinical features and outcome of systemic lupus erythematosus. Rheumatology (Oxford). 2017. Apr 1; 56(suppl_1):i67–i77. [DOI] [PubMed] [Google Scholar]
- 3.Kwon YC, Chun S, Kim K, Mak A. Update on the Genetics of Systemic Lupus Erythematosus: Genome-Wide Association Studies and Beyond. Cells. 2019. Sep 30; 8(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris DL, Sheng Y, Zhang Y, Wang YF, Zhu Z, Tombleson P, et al. Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat Genet. 2016. Aug; 48(8):940–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun C, Molineros JE, Looger LL, Zhou XJ, Kim K, Okada Y, et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat Genet. 2016. Mar; 48(3):323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Generali E, Ceribelli A, Stazi MA, Selmi C. Lessons learned from twins in autoimmune and chronic inflammatory diseases. J Autoimmun. 2017. Sep; 83:51–61. [DOI] [PubMed] [Google Scholar]
- 7.Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010. Feb; 20(2):170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005. Jul 26; 102(30):10604–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, et al. A Critical Role for Dnmt1 and DNA Methylation in T Cell Development, Function, and Survival. Immunity. 2001; 15(5):763–774. [DOI] [PubMed] [Google Scholar]
- 10.Quddus J, Johnson KJ, Gavalchin J, Amento EP, Chrisp CE, Yung RL, et al. Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J Clin Invest. 1993. Jul; 92(1):38–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J Immunol. 1988. Apr 1; 140(7):2197–2200. [PubMed] [Google Scholar]
- 12.Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990. Nov; 33(11):1665–1673. [DOI] [PubMed] [Google Scholar]
- 13.Lu Q, Wu A, Tesmer L, Ray D, Yousif N, Richardson B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J Immunol. 2007. Nov 1; 179(9):6352–6358. [DOI] [PubMed] [Google Scholar]
- 14.Lu Q, Wu A, Richardson BC. Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J Immunol. 2005. May 15; 174(10):6212–6219. [DOI] [PubMed] [Google Scholar]
- 15.Lu Q, Kaplan M, Ray D, Ray D, Zacharek S, Gutsch D, et al. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum. 2002. May; 46(5):1282–1291. [DOI] [PubMed] [Google Scholar]
- 16.Sawalha AH, Jeffries M, Webb R, Lu Q, Gorelik G, Ray D, et al. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008. Jun; 9(4):368–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeffries MA, Dozmorov M, Tang Y, Merrill JT, Wren JD, Sawalha AH. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics. 2011. May; 6(5):593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coit P, Jeffries M, Altorok N, Dozmorov MG, Koelsch KA, Wren JD, et al. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J Autoimmun. 2013. Jun; 43:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coit P, Yalavarthi S, Ognenovski M, Zhao W, Hasni S, Wren JD, et al. Epigenome profiling reveals significant DNA demethylation of interferon signature genes in lupus neutrophils. J Autoimmun. 2015. Apr; 58:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hedrich CM, Mabert K, Rauen T, Tsokos GC. DNA methylation in systemic lupus erythematosus. Epigenomics. 2017. Apr; 9(4):505–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ballestar E, Sawalha AH, Lu Q. Clinical value of DNA methylation markers in autoimmune rheumatic diseases. Nat Rev Rheumatol. 2020. Sep; 16(9):514–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coit P, Ortiz-Fernandez L, Lewis EE, McCune WJ, Maksimowicz-McKinnon K, Sawalha AH. A longitudinal and transancestral analysis of DNA methylation patterns and disease activity in lupus patients. JCI Insight. 2020. Nov 19; 5(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lanata CM, Paranjpe I, Nititham J, Taylor KE, Gianfrancesco M, Paranjpe M, et al. A phenotypic and genomics approach in a multi-ethnic cohort to subtype systemic lupus erythematosus. Nat Commun. 2019. Aug 29; 10(1):3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imgenberg-Kreuz J, Carlsson Almlof J, Leonard D, Alexsson A, Nordmark G, Eloranta ML, et al. DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann Rheum Dis. 2018. May; 77(5):736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coit P, Ognenovski M, Gensterblum E, Maksimowicz-McKinnon K, Wren JD, Sawalha AH. Ethnicity-specific epigenetic variation in naive CD4+ T cells and the susceptibility to autoimmunity. Epigenetics Chromatin. 2015; 8:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mok A, Solomon O, Nayak RR, Coit P, Quach HL, Nititham J, et al. Genome-wide profiling identifies associations between lupus nephritis and differential methylation of genes regulating tissue hypoxia and type 1 interferon responses. Lupus Sci Med. 2016; 3(1):e000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997. Sep; 40(9):1725. [DOI] [PubMed] [Google Scholar]
- 28.Flanagan JM. Epigenome-wide association studies (EWAS): past, present, and future. Methods Mol Biol. 2015; 1238:51–63. [DOI] [PubMed] [Google Scholar]
- 29.Rahmani E, Yedidim R, Shenhav L, Schweiger R, Weissbrod O, Zaitlen N, et al. GLINT: a user-friendly toolset for the analysis of high-throughput DNA-methylation array data. Bioinformatics. 2017. Jun 15; 33(12):1870–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013. Feb; 8(2):203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rahmani E, Shenhav L, Schweiger R, Yousefi P, Huen K, Eskenazi B, et al. Genome-wide methylation data mirror ancestry information. Epigenetics Chromatin. 2017; 10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007. Sep; 3(9):1724–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: Machine learning in Python. the Journal of machine Learning research. 2011; 12:2825–2830. [Google Scholar]
- 34.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007. Jan; 8(1):118–127. [DOI] [PubMed] [Google Scholar]
- 35.Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, et al. The Ensembl genome database project. Nucleic Acids Res. 2002. Jan 1; 30(1):38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Consortium F, the RP, Clst, Forrest AR, Kawaji H, Rehli M, et al. A promoter-level mammalian expression atlas. Nature. 2014. Mar 27; 507(7493):462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dale RK, Pedersen BS, Quinlan AR. Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics. 2011. Dec 15; 27(24):3423–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016. Jul 8; 44(W1):W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015. Apr 20; 43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013. Jan; 41(Database issue):D991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang T, Guan W, Lin J, Boutaoui N, Canino G, Luo J, et al. A systematic study of normalization methods for Infinium 450K methylation data using whole-genome bisulfite sequencing data. Epigenetics. 2015; 10(7):662–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. 2020. Mar; 17(3):261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006. Aug; 38(8):904–909. [DOI] [PubMed] [Google Scholar]
- 44.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015; 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014. May 15; 30(10):1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fortin JP, Triche TJ, Jr., Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017. Feb 15; 33(4):558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Team RC. R: A Language and Environment for Statistical Computing. 2020. [Google Scholar]
- 48.Sinke L, van Iterson, Maarten, Davy Cats, Roderick Slieker& Bas Heijmans. DNAmArray: Streamlined workflow for the quality control, normalization, and analysis of Illumina methylation array data. Zenodoa. 2019. [Google Scholar]
- 49.Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014. Dec 3; 15(12):503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou W, Laird PW, Shen H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2017. Feb 28; 45(4):e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012. Mar 15; 28(6):882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chavent M, Kuentz-Simonet V, Labenne A, Saracco J. Multivariate analysis of mixed data: The R Package PCAmixdata. arXiv preprint arXiv:14114911. 2014. [Google Scholar]
- 53.Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012. May 15; 28(10):1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009. Jul; 37(Web Server issue):W305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013. Jan; 41(Database issue):D1040–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Knight JS, Meng H, Coit P, Yalavarthi S, Sule G, Gandhi AA, et al. Activated signature of antiphospholipid syndrome neutrophils reveals potential therapeutic target. JCI Insight. 2017. Sep 21; 2(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015. Feb 19; 518(7539):317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li D, Hsu S, Purushotham D, Sears RL, Wang T. WashU Epigenome Browser update 2019. Nucleic Acids Res. 2019. Jul 2; 47(W1):W158–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao M, Liu S, Luo S, Wu H, Tang M, Cheng W, et al. DNA methylation and mRNA and microRNA expression of SLE CD4+ T cells correlate with disease phenotype. J Autoimmun. 2014. Nov; 54:127–136. [DOI] [PubMed] [Google Scholar]
- 60.Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol. 2019. Jul; 20(7):902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. 2015. Dec; 47(12):1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009. Nov; 41(11):1234–1237. [DOI] [PubMed] [Google Scholar]
- 63.Alarcon-Riquelme ME, Ziegler JT, Molineros J, Howard TD, Moreno-Estrada A, Sanchez-Rodriguez E, et al. Genome-Wide Association Study in an Amerindian Ancestry Population Reveals Novel Systemic Lupus Erythematosus Risk Loci and the Role of European Admixture. Arthritis & rheumatology (Hoboken, NJ). 2016. Apr; 68(4):932–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee YH, Bae SC, Choi SJ, Ji JD, Song GG. Genome-wide pathway analysis of genome-wide association studies on systemic lupus erythematosus and rheumatoid arthritis. Mol Biol Rep. 2012. Dec; 39(12):10627–10635. [DOI] [PubMed] [Google Scholar]
- 65.Harley ITW, Sawalha AH. Systemic lupus erythematosus as a genetic disease. Clin Immunol. 2022. Mar; 236:108953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005. Apr 7; 434(7034):772–777. [DOI] [PubMed] [Google Scholar]
- 67.Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019. Jan; 20(1):21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen N, Liang D, Tang Y, de Vries N, Tak PP. MicroRNAs--novel regulators of systemic lupus erythematosus pathogenesis. Nat Rev Rheumatol. 2012. Dec; 8(12):701–709. [DOI] [PubMed] [Google Scholar]
- 69.Pan W, Zhu S, Yuan M, Cui H, Wang L, Luo X, et al. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol. 2010. Jun 15; 184(12):6773–6781. [DOI] [PubMed] [Google Scholar]
- 70.Zhao S, Wang Y, Liang Y, Zhao M, Long H, Ding S, et al. MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum. 2011. May; 63(5):1376–1386. [DOI] [PubMed] [Google Scholar]
- 71.Qin H, Zhu X, Liang J, Wu J, Yang Y, Wang S, et al. MicroRNA-29b contributes to DNA hypomethylation of CD4+ T cells in systemic lupus erythematosus by indirectly targeting DNA methyltransferase 1. J Dermatol Sci. 2013. Jan; 69(1):61–67. [DOI] [PubMed] [Google Scholar]
- 72.Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013. Dec; 20(12):1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fu Y, Zhang L, Zhang R, Xu S, Wang H, Jin Y, et al. Enterovirus 71 Suppresses miR-17-92 Cluster Through Up-Regulating Methylation of the miRNA Promoter. Front Microbiol. 2019; 10:625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dakhlallah D, Batte K, Wang Y, Cantemir-Stone CZ, Yan P, Nuovo G, et al. Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013. Feb 15; 187(4):397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008. Apr; 9(4):405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu YX, Schutt S, Paz K, Zhang MM, Flynn RP, Bastian D, et al. MicroRNA-17-92 is required for T-cell and B-cell pathogenicity in chronic graft-versus-host disease in mice. Blood. 2018. Apr 26; 131(17):1974–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dai R, Zhang Y, Khan D, Heid B, Caudell D, Crasta O, et al. Identification of a common lupus disease-associated microRNA expression pattern in three different murine models of lupus. PLoS One. 2010. Dec 10; 5(12):e14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qin HH, Zhu XH, Liang J, Wu JF, Yang YS, Xu JH. The expression and significance of miR-17-92 cluster miRs in CD4+ T cells from patients with systemic lupus erythematosus. Clin Exp Rheumatol. 2013. May-Jun; 31(3):472–473. [PubMed] [Google Scholar]
- 79.Dai Y, Huang YS, Tang M, Lv TY, Hu CX, Tan YH, et al. Microarray analysis of microRNA expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus. 2007; 16(12):939–946. [DOI] [PubMed] [Google Scholar]
- 80.Te JL, Dozmorov IM, Guthridge JM, Nguyen KL, Cavett JW, Kelly JA, et al. Identification of unique microRNA signature associated with lupus nephritis. PLoS One. 2010. May 11; 5(5):e10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carlsen AL, Schetter AJ, Nielsen CT, Lood C, Knudsen S, Voss A, et al. Circulating microRNA expression profiles associated with systemic lupus erythematosus. Arthritis Rheum. 2013. May; 65(5):1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang S, Li C, Olive V, Lykken E, Feng F, Sevilla J, et al. Molecular dissection of the miR-17-92 cluster’s critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood. 2011. Nov 17; 118(20):5487–5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Montoya MM, Maul J, Singh PB, Pua HH, Dahlstrom F, Wu N, et al. A Distinct Inhibitory Function for miR-18a in Th17 Cell Differentiation. J Immunol. 2017. Jul 15; 199(2):559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Teteloshvili N, Smigielska-Czepiel K, Kroesen BJ, Brouwer E, Kluiver J, Boots AM, et al. T-cell Activation Induces Dynamic Changes in miRNA Expression Patterns in CD4 and CD8 T-cell Subsets. Microrna. 2015; 4(2):117–122. [DOI] [PubMed] [Google Scholar]
- 85.Adrianto I, Wen F, Templeton A, Wiley G, King JB, Lessard CJ, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet. 2011. Mar; 43(3):253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fu Q, Zhao J, Qian X, Wong JL, Kaufman KM, Yu CY, et al. Association of a functional IRF7 variant with systemic lupus erythematosus. Arthritis Rheum. 2011. Mar; 63(3):749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are included in the manuscript or supplementary material.