


Abstract
A robust Tn7-based in vitro transposition system is described that displays little target site selectivity, allowing the efficient recovery of many different transposon insertions in target DNAs ranging from small plasmids to cosmids to whole genomes. Two miniTn7 derivatives are described that are useful for the analysis of genes: one a derivative for making translational and transcriptional target gene fusions and the other a derivative that can generate 15 bp (5 amino acid) insertions in target DNAs (proteins).
INTRODUCTION
Transposable elements have been exploited by both nature and humans to modify genomes and genes. Transposable elements are conveniently and widely used as insertional mutagens that can provide physical markers, and transcriptional and translational fusions to a gene of interest (1–4). The ease of generating transposon derivatives that are useful tools is facilitated by detailed understanding of the proteins and DNAs that are necessary for and participate in transposition. To be most useful, the transposition reaction must provide a large number of intermolecular simple insertions of a single element into a target DNA. Moreover, there should be a low degree of target site selection by the element so that insertions can readily be obtained at many different positions. Also, the recombination sequences at the ends of the element should be simple so that mini-transposon derivatives that will provide transcriptional and translation fusions can be readily constructed. It is also useful to be able to carry out the mutagenizing transposition reactions in vitro (5) as this strategy can avoid features of the target DNA that may impose biases on insertions that are not observed on naked DNA in vitro (6). The ability to carry out transposition in vitro can also facilitate the use of transposable elements for the analysis of genes and genomes from organisms that do not have highly developed genetic systems. Several transposition systems have been used for in vitro mutagenesis (5–11).
In this work, we describe a simplified in vitro transposition system based on the bacterial transposon Tn7 (12,13), which is highly efficient in generating intermolecular simple insertions with only little target site selectivity. Thus, insertions at many different target sites in gene-sized intervals are readily obtained. Furthermore, predominately, one insertion is made into a given target molecule and little intramolecular recombination occurs, avoiding the complexity of dealing with DNA rearrangements other than simple insertions. Although wild-type Tn7 is most notable for its considerable target site preferences (13), in this simplified system the protein that regulates the transposase to execute breakage and joining in response to particular target signals has been modified so as to effectively abolish target site selectivity (14,15).
We have also generated several miniTn7 derivatives whose terminal recombination sequences are altered so that both transcriptional and translational fusions can be readily obtained and another derivative that allows simple recovery of genes (or other DNA sequences) that contain ‘linker insertions’ to facilitate identification of critical amino acids (base pairs).
MATERIALS AND METHODS
Media, chemicals and enzymes
Luria broth (LB) and agar were prepared as described by Miller (16). Carbenicillin was used at 100 µg/ml, chloramphenicol was used at 100 µg/ml and kanamycin was used at either 100 or 20 µg/ml as indicated. DNA modifying and restriction enzymes were purchased from commercial sources and used according to manufacturers’ instructions. Pfu polymerase was purchased from Stratagene and used according to manufacturer’s instructions. Taq polymerase was purchased from Boehringer Mannheim Biochemicals and used as described below.
Bacterial strains and plasmids
Subcloning efficiency DH5-α competent cells were purchased from Gibco BRL and used according to the manufacturer’s instructions for all experiments except those described in Figure 4; for the experiments of Figure 4, competent cells were prepared using a rubidium chloride procedure (New England Biolabs #401-Q).
Figure 4.

(A) Distribution of 63 insertions generated by TnsABCA225V in vitro transposition of miniTn7 from pEMΔ into target plasmid pER183, recovered by transformation and analyzed by DNA sequencing; junction sequences were determined utilizing Tn7 end-specific sequencing primers for both ends. Left→right insertions are indicated on the top, and right→left insertions are on the bottom. Essential regions of the target excluded by selection are indicated in gray. (B) Base composition of pER183, presented as percentage A+T for each 20 bp along the length of the target sequence. (C) Diagram of the nucleotide sequences flanking the 63 positions of Tn7 insertion in pER183, centered on the 5 bp target duplication. The vertical axis denotes the percentage by which the A+T content for a given position deviates from the average A+T content of the entire plasmid. The horizontal axis represents nucleotide position relative to the 5 bp target sequence duplication, with positions to the left of the insertion site indicated as negative numbers and positions to the right as positive numbers.
The target plasmid pRM2 contains bases –342 to +165 of attTn7 cloned into pUC18 (17). Target plasmid pET15b is commercially available (Novagen). The target cosmid pES185 (ES3) contains ∼40 kb of genomic DNA from Edwardsiella tarda and was kindly provided by Evelyn Strauss (18).
The donor plasmid pEMΔ carries a miniTn7 element comprised of the 166 terminal bases of the left end of Tn7 and 199 bases of the right end flanking a gene conferring resistance to kanamycin, miniTn7 L1–166–KmR–R1–199, whose flanking sequences are a chromosomal TnsE site (19).
The donor plasmid pMCB31 (pUC::miniTn7 L1–166–SalI–SpeI–KmR–NotI–SalI–R1–199 or miniTn7-SpeI–KmR–NotI) is a pUC19-based donor derived from pEMΔ. A segment from pEMΔ containing the miniTn7 element but lacking a flanking SalI site was cloned into a pUC vector whose polylinker was modified to remove its SalI site; thus, the only Sal sites in this construct are those flanking the KmR segment. The internal SalI–KmR–SalI cassette was then replaced by a SalI–SpeI–KmR–NotI–SalI cassette generated by PCR.
The donor plasmid pMCB40 (p-oriγR6K:: miniTn7 SpeI–KmR–NotI) carries a miniTn7 element in a plasmid backbone that will replicate only in cells providing the replicator protein Π. pBW30, a gift from Barry Wanner (Purdue University, West Lafayette, IN), is a conditional replicon carrying an R6Kγ origin (20). This conditional replicon requires the Π protein for replication and can be maintained only in cells with a resident pir gene, which encodes the Π protein (e.g. BW23474); the pir+ strain used was BW23474 [Escherichia coli Δlac-169 robA1 creC510 hsdR514 ΔuidA (MluI):: pir-116 endA (BT333) recA1], generously provided by Barry Wanner. The donor plasmid pMCB40 was generated by transposing the miniTn7 element from pMCB31 into pBW30 in vitro, transforming the products into BW23474 cells, selecting for kanamycin resistance and screening for carbenicillin sensitivity, resulting in a conditional replicon donor plasmid with a single antibiotic marker located within the element.
The donors pMCB62 and pMCB64 were generated using the Original TA Cloning Kit (Invitrogen) according to the manufacturer’s instructions.
The donor plasmid pMCB62 (pTA::R1–41 OPEN–KmR–R1–41 OPEN), in which there are open reading frames through the ends of the Tn7 element, was generated by amplifying the miniTn7 R1–R41–KmR–R1–41 from pLA5 (21) with a mutagenic oligonucleotide NLC685, which contained R1–41 OPEN sequences in it, and the flanking 10 bp sequences of pEMΔ, and ligating it into the TA vector pCR2.1 (Invitrogen).
The donor plasmid pMCB64 (pTA::miniTn7 R1–70 OPEN–KmR–R1–70*) was generated by a three part PCR reaction. The first part entailed annealing the two oligonucleotides NLC685 and NLC673 and extending with Taq polymerase. The second entailed using the resultant PCR fragment as a primer to amplify the miniTn7 R1–70–KmR–R1–70 element from pLA55 (21). In the third reaction, the low concentration product that resulted from the second amplification was further amplified with the Tn7-specific primer NLC685, and was ligated into pCR2.1.
The miniTn7 PmeI element was constructed by amplifying the miniTn7 element in pMIM (19) with oligonucleotide NLC 573, which contains a PmeI site near the tips of the transposon, and inserting the resulting product into the TA vector pCR2.1.
The marker plasmid pMCB20 was isolated from a TnsABC+D transposition reaction in which miniTn7L166–KmR–R199 from pEMΔ was transposed into the attTn7 site on pRM2.
Primers
Oligonucleotides used for cloning and the various PCR amplifications to analyze the products of transposition are:
NLC94, 5′-AAAGTCCAGTATGCTTTTTCACAGCATAAC-3′;
NLC95, 5′-ATAATCCTTAAAAACTCCATTTCCACCCCT-3′;
NLC501, 5′-AGTTGGCAGGATGTTTGATTAAAAACAT-3′;
NLC573, 5′-GGATCCACGCGTGTTTAAACACAATAAAGT-3′;
NLC641, 5′-ATGACAGAGAGAGATAAGTCTTCAGT-3′;
NLC642, 5′-ATGTGACAAACCGTCATCTTCGGC-3′;
NLC646, 5′-ACTTTATTGTCATAGTTTAGATCTATTTTG-3′;
NLC673, 5′-GTCGACTCTAGAGGATCCCATAGTTTGAGA-CTTTTTTGCCATAGTTTCGATCTT-3′;
NLC685, 5′-ATTGCTCTAAATGTGGGCGGACAAAAAAGT-CTCAAACTGGACAAAAAAGAT-3′.
Tns proteins
TnsA was purified as a fusion protein to a chitin binding domain through affinity chromatography to a chitin column, followed by release by intein-mediated cleavage (New England Biolabs). TnsA at ∼100 µg/ml was stored in 25 mM HEPES pH 7.7, 150 mM NaCl, 1 mM DTT, 1 mM EDTA, 10% glycerol (v/v), 0.25 mM PMSF at –80°C. TnsB was TnsB-His (22), a derivative containing a C-terminal polyhistidine tag, and was stored in 25 mM HEPES pH 8.0, 500 mM KCl, 2 mM DTT, 1 mg/ml BSA, 25% glycerol at –80°C. The purification of TnsC and TnsCA225V is a modified procedure of Gamas and Craig (23), which is described by Stellwagen and Craig (15). Both proteins were stored in 25 mM HEPES pH 8.0, 1 M NaCl, 2.5 mM DTT, 1 mM ATP, 10 mM MgCl2, 0.1 mM EDTA, 10 mM CHAPS, 10% glycerol at –80°C. TnsD was TnsD-His (24), a derivative containing a C-terminal polyhistidine tag, and was purified by Ni+2 chromatography before being stored in 50 mM Tris pH 7.5, 2 mM DTT, 500 mM KCl, 1 mM EDTA, 25% glycerol at –80° C.
Transposition reactions in vitro
The in vitro transposition reactions were carried out as described by Bainton et al. (19) with indicated modifications. Reaction mixtures, 100 µl in volume, contained (final concentration) 26 mM HEPES, 4.2 mM Tris pH 7.6, 50 µg/ml BSA, 2 mM ATP pH 7.0, 2.1 mM DTT, 0.05 mM EDTA, 0.2 mM MgCl2, 0.2 mM CHAPS, 28 mM NaCl, 21 mM KCl, 1.35% glycerol, 40 ng TnsA (12.8 nM), 25 ng TnsB (3 nM), TnsC as indicated and 22 ng TnsD (3.7 nM) as indicated; reactions in which TnsD was omitted were supplemented with a compensatory volume of TnsD storage buffer. TnsC (50 ng) was used in the reactions of Figures 2, 3, 6, 7 and 8, and 100 ng TnsC was used in the reactions of Figures 4 and 5. The reactions in Figure 4 also contained 100 µg/ml yeast tRNA. The reactions contained 100 ng miniTn7-containing donor DNA (0.25 nM for pEMΔ; the target DNAs were used at 400 ng (1.9 nM for pRM2).
Figure 2.
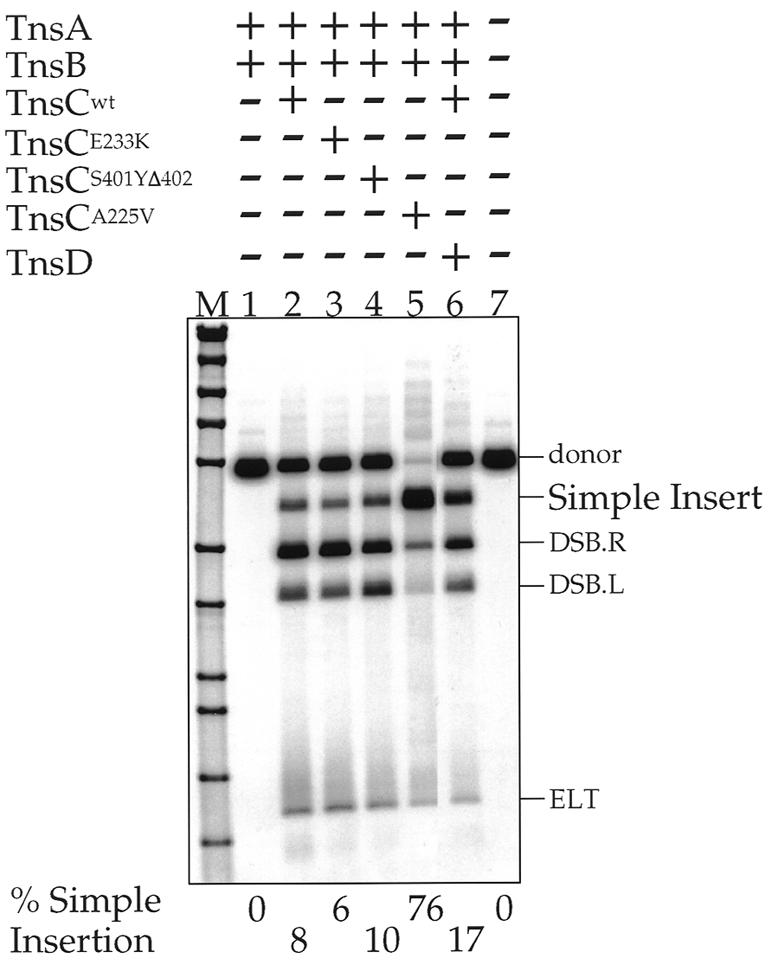
A Southern blot of NdeI-digested in vitro transposition products resolved on an agarose gel, transferred to a membrane and detected by using a transposon-specific probe. TnsABCmut reactions are compared to the TnsABCwt and TnsABC+D reactions; the percentage of donor substrate converted to simple insertion is shown beneath each lane. The various DNAs are labeled as described in Figure 1.
Figure 3.

Histograms showing the recovery of transposition products via transformation of simple insertion products from in vitro TnsABCmut and TnsABCwt transposition reactions. The donor plasmid used was pMCB40 (p-oriγR6K:: miniTn7 SpeI–KmR–NotI) which cannot replicate in the transformed strain. Three differently sized target plasmids, pRM2, pET15b and ES3 were used.
Figure 6.

(A) Schematic illustrating the positions of 12 different target-specific PCR primers used to survey the positions of TnsABC225V-mediated insertions into into pRM2. (B) Southern blots of PCR products derived from amplification of transposition reactions using the indicated target primers and hybridized to end-labeled oligonucleotides, NLC646 and NLC685.
Figure 7.


(A) Sequence of the Tn7 ends in two miniTn7 elements: nucleotide substitutions made to wild-type Tn7R end sequences are indicated in bold. Diagrams of the termini of the donor plasmids containing Tn7 termini that have open reading frames through them; the amino acid translation is shown beneath each end sequence, with ‘*’ indicating stop codons. B, BamHI site; X, XbaI site; S, SalI site; P, PstI site. These read-through elements are R1–41 OPEN–KmR–R1–41 OPEN (pMCB62) and R1–70 OPEN–KmR–R1–70* (pMCB64). R70* is a partially mutated right end that is mutated as in the R1–41 OPEN element and wild-type from R42–R70. (B) Southern blots of ScaI-digested transposition products of in vitro TnsABCA225V reactions for the two donor plasmids in (A), as detected by a hybridization with a transposon specific probe; the target plasmid for these experiments was pRM2. The reactions were incubated for various times as indicated. About 10% of R1–41 OPEN–KmR–R1–41 OPEN was converted to simple insertions; ∼58% of R1–70 OPEN–KmR–R1–70* was converted to simple insertion. The DNA species are labeled as in Figure 1.
Figure 8.

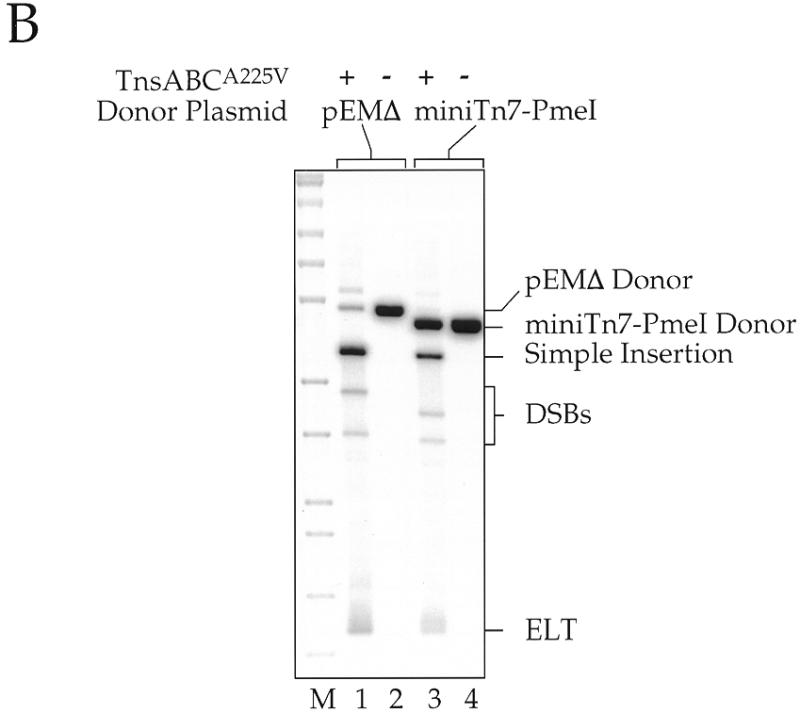
(A) Schematic diagram for making a 5 amino acid (15 bp) insertion in a target gene. A miniTn7 element containing PmeI sites close to the transposon termini is inserted into the target DNA at many different sites by transposition. The target plasmid with its inserted miniTn7 element DNA is then isolated, digested with PmeI and religated, resulting in a 15 bp insertion in the target DNA, 5 bp from each end of Tn7 and 5 bp from the target site duplication. Some of the inserted amino acids are dictated by the Tn7 end sequences whereas others will vary due to variations in the target sequence. (B) Southern blot using a transposon-specific probe of restriction digested products of in vitro transposition reactions using miniTn7 PmeI as a donor; pRM2 was the target DNA. Reactions containing pEMΔ were digested with NdeI and those using miniTn7 PmeI were digested with ScaI. DNA species are labeled as in Figure 1.
Figure 5.
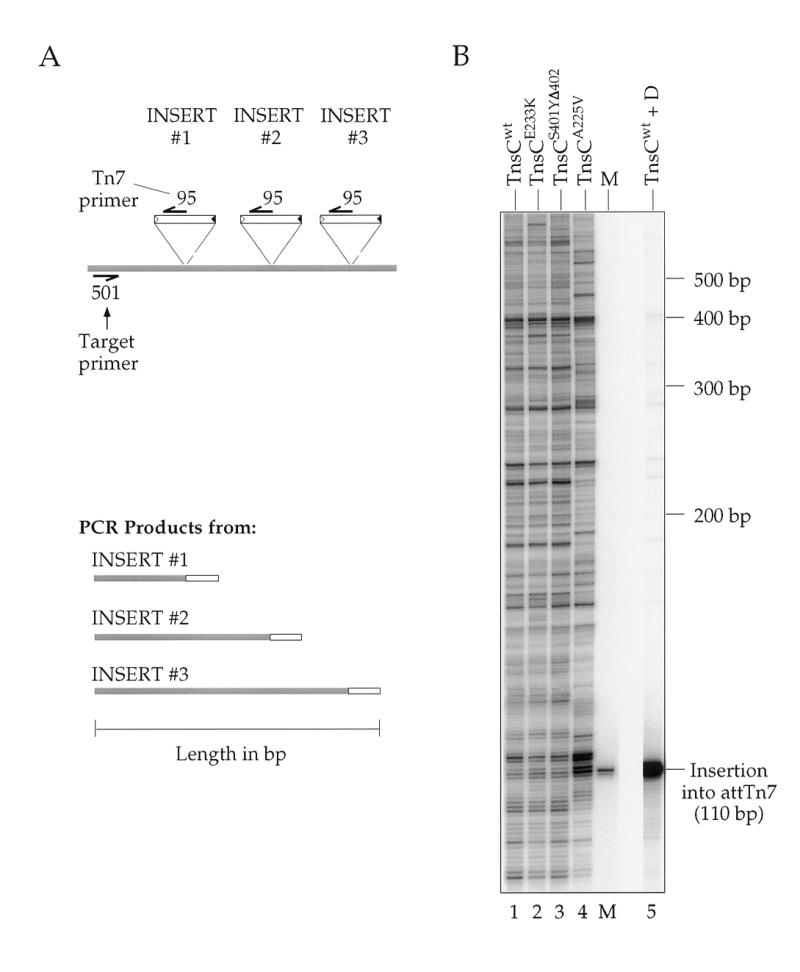
Analysis by a Southern blot of PCR products amplified from in vitro reactions using different TnsCs resolved on a denaturing gel and hybridized to a transposon-specific probe. (A) Diagram depicting three independent insertions which might occur in a given transposition reaction, and the corresponding products that would arise from PCR amplification with target-specific primer NLC501 and Tn7 end-specific primer NLC95. The shaded regions indicate target DNA or the portion of DNA in a PCR product that is derived from target template. (B) The profiles for three gain-of-function mutant TnsC reactions are compared to those for TnsABCwt and TnsABC+D in target plasmid pRM2. The amplicon chosen encompasses the attTn7 insertion site, allowing observation of the strong preference the TnsABC+D reaction (lane 5) displays for this position. The marker (M) is a PCR product amplified from an isolated insertion into attTn7 via a TnsABC+D reaction.
In all the experiments except those in Figure 4, the target proteins (TnsC ± TnsD), the target DNA and small molecules were incubated for 30 min at 30°C as a ‘pre-incubation’ step; TnsA, TnsC, MgOAc2 at a final concentration of 15 mM and the donor DNA were then added and the reactions were allowed to proceed for an additional 45 min at 30°C. In the reactions in Figure 4, there was no pre-incubation step and the reactions were incubated for 30 min at 30°C. Products were phenol extracted, ethanol precipitated and resuspended in water in preparation for subsequent analyses.
Analysis of transposition products by gel electrophoresis and Southern blotting
For Figures 2, 7B and 8B, transposition products were digested with restriction enzymes as indicated, electrophoresed overnight at 50 V on a 0.6% agarose gel, transferred to a membrane by Southern blotting and analyzed by hybridization to a Tn7-specific probe.
Probe labeling
Oligonucleotide probes were 5′ end-labeled with [γ-32P]ATP substrate and bacteriophage T4 polynucleotide kinase (New England Biolabs) for 45 min at 37°C. Labeled probes were separated from unincorporated label by size exclusion through a G50 Nick Spin Column (Pharmacia).
Transformation of transposition products
The recovery efficiencies of simple insertions from TnsABCmut and TnsABCwt reactions via transformation as shown in Figure 3 were compared by transforming subcloning efficiency DH5-α competent cells (Gibco Life Sciences) with one-twentieth of the phenol-extracted, ethanol-precipitated transposition reactions (Fig. 3). Transformations were plated on LB plates containing 100 µg/ml kanamycin to select for the transposon. Unreacted donor molecules were not detected because replication of pMCB40 is not supported in DH5α cells which are pir–.
In the experiment of Figure 4, unreacted pEMΔ donor molecules were destroyed by digestion with BstEII and simple insertions were selected by transformation of ER1821, using plates containing 15 µg/ml chloroamphenicol and 20 µg/ml kanamycin.
Sequencing
Sequencing of recovered transposition products was done using primers NLC 94, NLC 95 and NLC 646 with an ABI 377 sequencer and dye termination chemistry. Sequences were processed using the programs Seq.ed, Factura, Autoassembler and GCK2 (Textco).
Analysis of transposition by PCR
The equivalent of 2 µl of the 100 µl transposition reaction was used as the template in a given PCR amplification of a product pool. An aliquot of 100 pg of plasmid pMCB20 was used when amplifying a marker product for size comparison on the high resolution denaturing gels. Thirty temperature cycles of 94°C for 1 min, 55°C for 1.5 min and 72°C for 1.5 min were run for all amplifications, followed by a single 5 min incubation at 72°C. PCR products were ethanol precipitated and resuspended in a reduced volume of water in preparation for loading on a high resolution denaturing gel.
High resolution denaturing gels
The resuspended PCR products were electrophoresed on either a 5, 6 or 7% polyacrylamide denaturing gel, electrotransferred to Gene Screen Plus membrane (Du Pont) and visualized by hybridization at 50°C with a Tn7-specific end-labeled oligonucleotide probe (NLC95 for left end and NLC94, NLC646 and/or NLC685 for right end). Blots were exposed overnight to phosphorimager screens (Molecular Dynamics), which were scanned the following day.
RESULTS
We have previously reconstituted several in vitro Tn7 transposition systems using purified Tn7-encoded transposition (Tns) proteins (15,19). Insertion into attTn7, a specific bacterial site used as an ‘attachment’ site by Tn7, requires four of the five Tns proteins: the Tn7 transposase, which is comprised of TnsA and TnsB (TnsAB), an ATP-utilizing protein TnsC, which mediates communication between TnsAB, and TnsD, a site-specific attTn7 binding protein. The cofactors Mg2+ and ATP are also required (13). Our working model of Tn7 transposition is that TnsD bound to attTn7 ‘activates’ TnsC by facilitating the formation of TnsC–ATP bound to a target DNA. This target can then interact with and activate TnsAB to promote insertion adjacent to the TnsD binding site in attTn7 (19,25). Little Tn7 transposition is observed in vivo or in vitro with just TnsABCwt alone. We have recently isolated several gain-of-function TnsC mutants which have lost their dependence on TnsD such that considerable intermolecular recombination is promoted in vivo (14) and in vitro (26) with these mutant TnsCs and wild-type TnsAB. One such mutant of particular interest is TnsCA225V, which we have previously observed efficiently promotes insertion into a variety of target sites (15). In this work, we analyze TnsCA225V target site selection in vitro in detail.
Tn7 recombination can be readily detected in vitro (19). Recombination proceeds through a series of double-strand breaks (DSBs; Double Strand Break Right and Double Strand Break Left), generating Tn7 elements disconnected from the donor backbone at either Tn7 end (DSBs) and excised linear transposons (ELTs) with DSBs at both ends (Fig. 1); the 3′OH ends of Tn7 then join to staggered positions 5 bp apart on the target DNA to form a simple insertion flanked by small gaps that are intracellularly repaired by the host repair system. Figure 2 displays the products of Tn7 recombination as evaluated by restriction enzyme digestion, gel electrophoresis, Southern blotting and hybridization to a transposon-specific probe; recombination products can also be readily detected by ethidium bromide staining after gel electrophoresis (not shown). To increase the amount of TnsC-dependent recombination, we omitted tRNA (A.Stellwagen and N.L.Craig, unpublished observation), increased TnsC in the reaction and extended reaction incubation time as compared to our standard reaction conditions for TnsABC+D transposition; high levels of recombination intermediates can be observed under these conditions with certain TnsC mutants and with TnsC+D. No recombination is detected with just the transposase TnsAB (Fig. 2, lane 1); in the presence of TnsABCwt, some recombination products, mostly DSBs and ELTs and a small amount of simple insertion, are seen. Little increase in recombination is seen in vitro with the gain-of-function mutants TnsCE233K and TnsCS401YΔ402. In contrast, highly efficient recombination is seen with TnsCA225V (15); indeed, under these conditions more recombination is seen with TnsABCA225V than with TnsABCwt+D. Thus, in our standard reaction containing 100 ng of donor DNA, ∼75 ng of donor is converted to product, corresponding to ∼60 ng of simple insertion using a 3.2 kb target plasmid (pRM2).
Figure 1.

Substrates, reaction intermediates and products of Tn7 transposition. The donor molecule is disconnected from the donor backbone by DSBs at the ends of Tn7, generating Double-Strand Break Left (DSB.L) and Double-Strand Break Right (DSB.R) intermediates and an excised linear transposon (ELT); the ELT is joined to the target DNA to generate a simple insertion. Target DNA is represented by broken lines, donor DNA by solid lines. The filled triangle represents the right end of Tn7 and the open triangle, the left end. NdeI and ScaI sites are indicated in the donor backbone; NdeI was used to linearize pEMΔ and ScaI linearizes the other donors used.
Recovery of transposition products in vivo
The in vitro simple insertion product of Tn7 transposition is a target molecule containing the Tn7 element covalently linked to target DNA at its 3′ ends and flanked at its 5′ ends by short gaps that result from the staggered positions of target joining (27). These gaps are repaired by host systems that likely remove the Tns proteins from the ends of the transposon following transposition (C.Johnson and N.L.Craig, unpublished observation) and execute repair. The efficient recovery in vivo of transposition products from in vitro recombination reactions requires that the Tns proteins be removed from the ends prior to transformation; failure to do so results in an at least 100-fold decrease in the number of transformants recovered (data not shown). Recombination products are efficiently recovered when treated by phenol extraction followed by ethanol precipitation, or by heat treatment at 75°C.
Another issue that arises when attempting to uniquely recover transposition products is the fate of unreacted donor DNAs that will, for example, carry the same drug resistances as the simple insertion product of transposition. We have successfully used several strategies to eliminate donor recovery including selective restriction of the donor backbone and use of a transposon donor that has a conditional origin of replication in its backbone. Such unreacted donor molecules cannot replicate in strains lacking the appropriate replication initiation protein. For example, in the transformation experiments shown in Figure 3, we used a transposon donor containing an R6Kγ origin of replication and transformed into a strain lacking the Π protein which is required the the initiation of replication of this plasmid.
As shown in Figure 3, many thousands of simple insertion transposition products resulting from transposon insertion into small (3–9 kb) plasmids can be readily recovered, even when the host strain exhibits relatively low transformability for supercoiled DNA (~2 × 107 transformants/µg DNA in these experiments). When the recipient cells are of higher transformability or when electroporation is used, the number of Tn7 recombinants recovered increases roughly in proportion to the increase in efficiency of transformation for supercoiled control DNA (data not shown).
It is also notable that large numbers of insertions can be readily obtained when the target DNA is a cosmid, or even when sheared chromosomal DNA is used as a target (8).
Transposition promoted by TnsABCA225V displays low target site selectivity
To evaluate the target site specificity of the TnsABCA225V insertions, we determined the point of element insertion by DNA sequencing of 66 transformants recovered from an in vitro transposition reaction using an 8.9 kb target plasmid, pER183 (28), a pACYC-based plasmid containing the chromosomal mcrB and mcrC genes (Fig. 4). We used primer sites inside the Tn7 ends; although related, the ends of Tn7 are distinct so that primers specific to the Tn7-left end and Tn7-right end are possible (see Materials and Methods).
In one case, there were two insertions in one target plasmid; this plasmid was not analyzed further. Also, we failed to recover sequencing data from two plasmids, probably because of low DNA concentrations. In the remaining 63 plasmids used for specificity analysis, two plasmids contained insertions at the same position in opposite orientation, the remaining 61 insertions were dispersed all over the target plasmid, avoiding the selected for CmR and ori regions. There was no obvious transposon orientation bias in insertion although the ends of Tn7 are functionally and structurally distinct (21). In 61 plasmids, Tn7’s characteristic target site duplication was observed but in two plasmids, we found that there were sequence changes in the region of target site duplication; the original sequence was used in the specificity analysis.
Examination of the insertion sites of the 63 miniTn7 insertions revealed little sequence similarity in the 5 bp duplicated at the point of insertion, i.e. between the positions of Tn7 end joining. A Poisson distribution analysis of these insertion sites revealed that the observed insertion sites are not significantly different from random; a χ2 analysis of the target site duplication revealed only the weak consensus NYTRN, with only 10 of the characterized 63 insertions actually matching this consensus (data not shown).
To probe the role(s) of the target sequences flanking the target site duplication, we also compared the sequences of the regions flanking each insertion, 50 bp to each side of each insertion (Fig. 4). We find that there is no obvious primary sequence similarity between these insertion regions. Most notable is a preference for AT-rich sequences, especially in one region ∼15–30 bp rightwards of the point of insertion. However, no obvious preference for insertion into A+T-rich regions is observed.
These TnsABCA225V insertion products were obtained from transposition reactions by transformation; we have also directly analyzed the distribution of insertions obtained in TnsABCwt reactions and have similarly observed relatively little target site selectivity (see below).
Using PCR to analyze target site selectivity in vitro
A very useful method for analyzing the distribution of transposon insertions in a particular target region is to use PCR in which one primer is complementary to one end of the transposon and the other is complementary to a chosen target position (29). PCR products of differing sizes will be obtained when the transposon inserts at various distances from the target primer site (Fig. 5A). The distribution of insertions can readily be analyzed at high resolution by displaying them on a denaturing gel, followed by Southern blotting and hybridization with a transposon-specific probe. Such an analysis of TnsABCwt, TnsABCA225V and TnsAB with other TnsC mutant insertions is shown in Figure 5B. Most notable is that the PCR fragments are of very many different lengths; indeed, a band corresponding to transposon insertion at virtually every internucleotide position can be observed. However, the observed PCR products do vary in intensity, revealing that the frequency of insertion is not uniform at every internucleotide junction. It should be noted that while many of the positions of the insertions using TnsCA225V are similar to those obtained with the other TnsCs, the frequency of insertions at particular positions using TnsCA225V does vary. The basis of this variation is not yet understood.
Thus, the picture that emerges from this analysis is that TnsCA225V promotes insertion at many sites. Although the distribution of insertions is not completely ‘random’, selectivity appears to be low enough that multiple insertions can be readily recovered in small target windows of 50–100 bp.
We also examined insertion throughout the target plasmid pRM2 (17) using multiple primers (Fig. 6). Again, this analysis emphasizes that insertions occur at many different junctions although some junctions are preferred more than others. There are some regions, usually at long distances from the target primer, that appear to show some regional specificity. However, an expanded view of these regions using a target primer closer to the apparent ‘hot region’ often shows that, as elsewhere, insertions occur at many different positions. Gel compressions, particularly of longer PCR products, can also contribute to the impression of regional specificity.
Manipulation of Tn7 end sequences to generate target gene fusions upon insertion
The sequences at the two ends of the transposon itself are key recombination substrates, being sites of action of recombination proteins. A particularly useful application of transposons is to make translational fusions to various reporter genes. This often requires mutation of the ends, which spares their recombination ability but allows at least one open translational frame across the end of the element.
In most transposable elements, the sequences at the ends are arranged as inverted repeats. Detailed studies of several elements have shown that this inverted repeat consists of an interior transposase binding site and, at the extreme termini, several base pairs that are critical for the breakage and joining steps that follow transposase binding (30). Some elements, including Tn7, also have multiple transposase binding sites interior to the terminal inverted repeat. A miniTn7 element containing three TnsB binding sites (one in the inverted repeat and two more distal ones) in a 166 bp left-end segment and four TnsB sites in a right-end segment is functional for transposition in vivo. An element containing a 70 bp Tn7 R segment is also highly functional (21,31). However, no transposition has been detected in vivo with elements in which either end is composed only of the terminal 41 bp right-end fragment that includes the terminal inverted 28 bp and most of another TnsB binding site (21,32).
We have now found that in in vitro reactions, in contrast to the in vivo situation, the shorter Tn7 ends are also active, allowing more flexibility in end modification for generating miniTn7 elements useful in making gene fusions and other derivatives. Some recombination is observed in vitro with a miniTn7 element R1–41–KmR–R1–41 (Fig. 7), albeit at reduced frequency from that observed with elements containing larger Tn7 end segments. Furthermore, we have modified this R1–41 such that translation is open through all three frames of the R1–41 end, leading to construction of the miniTn7 element R1–41 OPEN–KmR–R1–41 OPEN, allowing for translational fusions. We have also generated a miniTn7 element in which one R1–70 end is translationally open in all three frames, R1–70 OPEN; the other end, R1–70*, has also been modified from the wild-type sequence (see Materials and Methods). The miniTn7 element R1–70 OPEN–KmR–R1–70* transposes efficiently in vitro (Fig. 7B), allowing easy recovery of insertions that will result in translation fusions to the target DNA.
Using Tn7 to make 5 amino acid insertions in proteins
A particularly interesting form of mutagenesis is a ‘linker’ scanning approach whereby manipulation of the target gene using transposon insertion in vivo generates a target protein that has a number of extra amino acids inserted at various particular positions (33). This approach has proved useful in identifying critical regions missed by other types of mutagenesis. We have engineered a miniTn7 element that will insert 5 amino acids into a target protein at any position of Tn7 insertion in the corresponding gene by in vitro transposition (Fig. 8A).
We have previously described a miniTn7 element in which the terminal inverted repeats at the ends of Tn7 have been rendered symmetric by site-directed mutagenesis to give L = R1–19, L20–199–KmR–R1–R199 (19). As shown in Figure 8, we constructed a derivative of this element that contains a PmeI site closely juxtaposed to the ends of Tn7. The introduction of these PmeI sites involved changing R4–R8, a region that we have shown in other work can be substantially altered and still retain transposition activity (P.Gary and N.L.Craig, unpublished observation), and changing R9, a position in the first TnsB binding site. This element, miniTn7 PmeI, transposes efficiently in vitro (Fig. 8B).
Following transposition, the interior KmR segment of the miniTn7 PmeI element is excised by PmeI digestion and the target DNA is then religated, resulting in a 15 bp insertion in the target gene. The 15 bp derive from 5 bp from the duplicated target sequence and 5 bp from each end of modified miniTn7 PmeI–KmR–PmeI. These extra 15 bp will introduce 5 amino acids into the target protein, depending on the frame of the insertion. Because the PmeI recognition site includes a stop codon, only four of the six possible frames across each 15 bp insertion are open. Therefore, only two out of every three possible ‘random’ miniTn7 PmeI insertions in a target gene will actually result in the insertion of new in-frame amino acids into the corresponding protein. The introduction of a ‘stop’ codon via a miniTn7 PmeI insertion can also be exploited for making deletion derivatives of target genes.
DISCUSSION
We have described here a simplified in vitro transposition system based on the bacterial transposon Tn7 that is highly useful for the analysis of genes and genomes. Perhaps the most notable feature of this system is that many insertions at many different sites can be readily obtained. The use of an in vitro transposition system for mutagenesis (5,6) avoids execution of the transposition in vivo where intracellular DNA structure could influence transposition. Moreover, since the recombination reactions are performed in vitro, genetic manipulation of a heterologous system which may be technically difficult or unfamiliar to many workers is avoided. There are many uses of such transposon insertions: they can provide primer binding sites for DNA sequencing and they can be insertional mutagens that may be very efficiently used on relatively small DNAs such as plasmids or cosmids. Reintroduction of these derivatized molecules into an appropriate host can then be followed by screening for mutant phenotypes, allowing efficient recovery of the insertionally mutated genes. Indeed, the efficiency of the Tn7 recombination system in vitro is sufficiently robust that whole genome mutagenesis can be executed in vitro and mutations of interest recovered by transformation when homologous recombination is efficient in the organism of interest (8). Several other systems have also been used for in vitro insertional mutagenesis (6,7,9–11).
We have also described several miniTn7 elements whose terminal recombination sequences have been altered in ways that allow for both transposition and recovery of usefully derivatized target genes. One of these derivatives has a truncated end that is ‘open’ in all three reading frames and is bounded by a polylinker segment allowing the convenient construction of elements that will make translational and transcriptional fusions into target genes upon insertion.
We have also constructed an element that allows for Tn7-mediated insertion of 5 amino acids into target proteins via element insertion by transposition followed by excision of an internal segment by restriction digestion and subsequent religation of the target backbone. Other studies have shown that this type of ‘linker insertion’ mutagenesis can be particularly useful as it allows direct identification of the regions of proteins that can be substantially altered without loss of function which then leads to identification of critical areas of the protein.
Target site selection in the TnsABC system
These studies have also provided interesting information about target site selection by Tn7. For wild-type Tn7 transposition, insertion into attTn7 requires the core machinery consisting of the transposase TnsA+B and TnsC, an ATP-dependent non-sequence-specific DNA binding protein that activates the transposase when it interacts with the specific attTn7 binding protein TnsD (19,26). We hypothesize that TnsE, Tn7’s other target selecting protein, also interacts with and activates TnsC. When TnsABC are wild-type, little transposition is observed. In this work, we have exploited a TnsC ‘gain-of-function’ mutant that can activate TnsA+B in the absence of TnsD or TnsE to give very robust levels of recombination in vivo (14) and in vitro (15). Other studies of this TnsCA225V mutant suggest that this TnsC’s gain-of-function phenotype results from a reduced level of ATP hydrolysis which results in a larger proportion of TnsC existing in an active ATP-bound state that binds more efficiently to target DNA (A.Stellwagen and N.L.Craig, unpublished observation).
We have described several different types of experiments that have demonstrated that TnsABCA225V recombination occurs with relatively low target site selectivity, inserting at many different sites but favoring AT-rich regions. Other experiments have shown that the recognition of distorted DNA by TnsC can play a key role in target site selection; thus, TnsABCA225V recombination may favor AT-rich regions because of their potential for distortion (J.Rao, P.Kuduvalli and N.L.Craig, unpublished observations).
Several other in vitro systems have been described for use in genome analysis (6,9–11). The Tn7 system is notable for its high efficiency and for the high number of different target sites that are used.
CONCLUSIONS
We have described a robust Tn7-based in vitro transposition system that displays little target site selectivity and have also described several miniTn7 elements that allow either the easy recovery of fusions or of target genes that have been insertionally mutated such that 5 amino acids ‘linker’ insertions are made in targeted proteins. This system should be a valuable and useful tool for the analysis of genes and genomes.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Barton Slatko for his assistance with DNA sequencing and sequence analysis. We also thank Patti Eckhoff for her assistance with preparation. This work was supported by NIH grant NIGM RO1 GM53824 to N.L.C.; M.C.B. and N.L.C. are employees of the Howard Hughes Medical Institute. Under a licensing agreement between the Johns Hopkins University and New England Biolabs, N.L.C. is entitled to a share of royalty received by the University from the sales of the licensed technology. The terms of this agreement are being managed by the University in accordance with its conflict of interest policies.
REFERENCES
- 1.Kleckner N., Bender,J. and Gottesman,S. (1991) Methods Enzymol., 204, 139–180. [DOI] [PubMed] [Google Scholar]
- 2.Kleckner N., Roth,J. and Botstein,D. (1977) J. Mol. Biol., 116, 125–159. [DOI] [PubMed] [Google Scholar]
- 3.Kaiser K., Sentry,J.W. and Finnegan,D.J. (1995) In Sherratt,D.J. (ed.), Eukaryotic Transposable Elements as Tools to Study Gene Structure and Function. IRL Press, Oxford, pp. 69–100.
- 4.Berg C.M. and Berg,E. (1995) In Sherratt,D.J. (ed.), Transposable Elements as Tools for Molecular Analyses in Bacteria. IRL Press, Oxford, pp. 38–68.
- 5.Waddell C.S. and Craig,N.L. (1988) Genes Dev., 2, 137–149. [DOI] [PubMed] [Google Scholar]
- 6.Devine S.E. and Boeke,J.D. (1994) Nucleic Acids Res., 22, 3765–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akerley B.J., Rubin,E.J., Camilli,A., Lampe,D.J., Robertson,H.M. and Mekalanos,J.J. (1998) Proc. Natl Acad. Sci. USA, 95, 8927–8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gwinn M.L., Stellwagen,A.E., Craig,N.L., Tomb,J.-F. and Smith,H.O. (1997) J. Bacteriol., 179, 7315–7320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffin T.J.IV, Parsons,L., Leschziner,A.E., DeVost,J., Derbyshire,K.M. and Grindley,N.D. (1999) Nucleic Acids Res., 27, 3859–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haapa S., Taira,S., Heikkinen,E. and Savilahti,H. (1999) Nucleic Acids Res., 27, 2777–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goryshin I.Y., Miller,J.A., Kil,Y.V., Lanzov,V.A. and Reznikoff,W.S. (1998) Proc. Natl Acad. Sci. USA, 95, 10716–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barth P.T., Datta,N., Hedges,R.W. and Grinter,N.J. (1976) J. Bacteriol., 125, 800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Craig N.L. (1995) Curr. Top. Microbiol. Immunol., 204, 27–48. [DOI] [PubMed] [Google Scholar]
- 14.Stellwagen A. and Craig,N.L. (1997) Genetics, 145, 573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stellwagen A. and Craig,N.L. (1997) EMBO J., 16, 6823–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, NY.
- 17.McKown R.L., Orle,K.A., Chen,T. and Craig,N.L. (1988) J. Bacteriol., 170, 352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strauss E.J., Ghori,N. and Falkow,S. (1997) Infect. Immunol., 65, 3924–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bainton R.J., Kubo,K.M., Feng,J.-N. and Craig,N.L. (1993) Cell, 72, 931–943. [DOI] [PubMed] [Google Scholar]
- 20.Wilmes-Riesenberg M.R. and Wanner,B.L. (1992) J. Bacteriol., 174, 4558–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arciszewska L.K., Drake,D. and Craig,N.L. (1989) J. Mol. Biol., 207, 35–52. [DOI] [PubMed] [Google Scholar]
- 22.Gary P.A., Biery,M.C., Bainton,R.J. and Craig,N.L. (1996) J. Mol. Biol., 257, 301–316. [DOI] [PubMed] [Google Scholar]
- 23.Gamas P. and Craig,N.L. (1992) Nucleic Acids Res., 20, 2525–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharpe P. and Craig,N.L. (1998) EMBO J., 17, 5822–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stellwagen A. and Craig,N.L. (1998) Trends Biochem. Sci., 23, 486–490. [DOI] [PubMed] [Google Scholar]
- 26.Stellwagen A.E. (1997) Thesis dissertation. TnsC: An ATP-dependent Regulator of Tn7 Transposition. University of California at San Francisco, San Francisco, CA, p. 192.
- 27.Bainton R., Gamas,P. and Craig,N.L. (1991) Cell, 65, 805–816. [DOI] [PubMed] [Google Scholar]
- 28.Dila D., Sutherland,E., Moran,L., Slatko,B. and Raleigh,E.A. (1990) J. Bacteriol., 172, 4888–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pryciak P.M., Sil,A. and Varmus,H.E. (1992) EMBO J. 11, 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahillon J. and Chandler,M. (1998) Microbiol. Rev., 62, 725–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeBoy R. and Craig,N.L. (1996) J. Bacteriol., 178, 6184–6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arciszewska L.K. and Craig,N.L. (1991) Nucleic Acids Res., 19, 5021–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hallet B., Sherratt,D.J. and Hayes,F. (1997) Nucleic Acids Res., 25, 1866–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]