

Abstract
The tumor stroma of most solid malignancies is characterized by a pathological accumulation of pro-angiogenic and pro-tumorigenic hyaluronan driving tumorigenesis and metastatic potential. Of all three hyaluronan synthase isoforms, HAS2 is the primary enzyme that promotes the build-up of tumorigenic HA in breast cancer. Previously, we discovered that endorepellin, the angiostatic C-terminal fragment of perlecan, evokes a catabolic mechanism targeting endothelial HAS2 and hyaluronan via autophagic induction. To explore the translational implications of endorepellin in breast cancer, we created a double transgenic, inducible Tie2CreERT2;endorepellin(ER)Ki mouse line that expresses recombinant endorepellin specifically from the endothelium. We investigated the therapeutic effects of recombinant endorepellin overexpression in an orthotopic, syngeneic breast cancer allograft mouse model. First, adenoviral delivery of Cre evoking intratumor expression of endorepellin in ERKi mice suppressed breast cancer growth, peritumor hyaluronan and angiogenesis. Moreover, tamoxifen-induced expression of recombinant endorepellin specifically from the endothelium in Tie2CreERT2;ERKi mice markedly suppressed breast cancer allograft growth, hyaluronan deposition in the tumor proper and perivascular tissues, and tumor angiogenesis. These results provide insight into the tumor suppressing activity of endorepellin at the molecular level and implicate endorepellin as a promising cancer protein therapy that targets hyaluronan in the tumor microenvironment.
Keywords: Proteoglycans, hyaluronan synthase, cancer growth, autophagy, ATG5
Introduction
The extracellular matrix (ECM) consists of a plethora of heterogeneous biological components that oversee an expanse of intricate and critical signaling pathways and functions. Acting as a deep reservoir of structural and signaling macromolecules including collagens, laminins, fibronectin, elastin, proteoglycans, glycoproteins and glycosaminoglycans (GAGs), the ECM coordinates seamlessly to influence a whole host of homeostatic and pathological functions, including proliferation, migration, tissue remodeling and regeneration, wound healing, inflammation and angiogenesis [1–3]. Just as its composition varies drastically among different organs and tissues, the ECM can dynamically evolve and diverge from homeostasis in disease pathologies including fibrosis and cancer, thereby positing the microenvironment itself as a major driver of pathogenicity [4–6]. A dominant component of the ECM, hyaluronan (HA) is a linear, non-sulfated glycosaminoglycan (GAG) that carries intrinsic properties integral to tissue homeostasis and stability. HA is synthesized ubiquitously in every tissue and body fluid by three members of the multi-pass transmembrane hyaluronan synthase family (HAS1–3) that simultaneously synthesize strands of HA at their cytosolic loop while directing the reducing end past the lipid bilayer into the extracellular space through a transmembrane pore. Newly-synthesized HA exists as viscoelastic, linear high molecular weight strands (~100–2000 kDa) of predictable glucuronic acid and N-acetyl-glucosamine disaccharide repetitions possessing anti-inflammatory and anti-angiogenic properties. Interestingly, breakdown fragments of HA—either through reactive oxygen species, hyaluronidases, UV rays, or mechanical forces—carry dynamic pro-inflammatory and pro-angiogenic capabilities. Thus, the broad, length-dependent effects of HA allow this simple glycosaminoglycan to regulate a whole host of functions including embryonic development, angiogenesis, migration, inflammation, and wound healing [7–11].
ECM remodeling in the context of cancer is often referred to as the tumor microenvironment and encompasses the dynamic crosstalk between cancer cells and their adjacent stroma [12]. In breast cancer, the tumor microenvironment is characterized by a dense ECM, and tumor cells proliferate in a background of increased linearized collagen deposition, reduced ECM remodeling and enhanced matrix cross-linking. This dense breast tumor ECM also confers greater resistance to breast cancer treatment [6]. Another defining characteristic of the breast tumor microenvironment is an increased deposition of extracellular HA. This pathological accumulation that is absent in normal breast epithelium is not only strongly correlated with poorer prognoses in cancer patients, but it also plays a key role in propelling matrix-driven breast tumor progression, angiogenesis and metastatic potential [12–14]. Furthermore, high levels of stromal HA in breast cancer cases are associated with large tumor size, lymph node infiltration, poor tumor differentiation, estrogen and progesterone receptor negativity, increased rate of relapse and decreased patient survival [12,15]. Indeed, HA content is a more accurate prognostic indicator of breast cancer recurrence above other common risk factors [16]. Conversely, murine mammary tumors deficient in stroma-derived HA show attenuated tumor angiogenesis [17].
Perlecan is a ubiquitous heparan sulfate proteoglycan known to be subjected to partial proteolysis [18], leading to release of bioactive fragments with signaling roles [19]. Perlecan harbors multiple biological functions [20–27] and is itself under complex transcriptional and post-transcriptional control [28–33]. Some of its most notable roles include regulating vasculogenesis and angiogenesis [34–36], ECM composition, adhesion, basement membrane physiology and growth factor/receptor activities [23,36–44] through a flurry of protein/protein interactions [40,45,46]. Endorepellin, the C-terminal domain V of perlecan [47,48], is a well-studied bioactive molecule that acts as an anti-angiogenic and pro-autophagic effector [49–56], similar in part to the role of decorin [57,58]. Through its three laminin-like globular (LG) domains, endorepellin signals as a dual receptor ligand via simultaneously binding vascular endothelial growth factor 2 (VEGFR2) and α2β1 integrin [59–64] and evoking the tyrosine phosphatase SHIP-1 [65]. As VEGFR2 and α2β1 receptors are nearly exclusively expressed on the surface of vascular endothelial cells, the angiostatic and pro-autophagic pathways induced by endorepellin are restricted to the vascular endothelium [62]. Through evoking angiostasis, endorepellin also exerts anti-oncogenic properties in vivo, specifically in human squamous carcinoma and murine Lewis lung carcinoma where it abrogates tumor angiogenesis and growth [66].
We recently discovered that endorepellin induces autophagic degradation of HAS2 in endothelial cells [67]. This novel bioactivity drastically suppresses stromal HA content in vitro and ex vivo, resulting in robust angiostasis [67]. This axis occurs through a signaling pathway beginning with endorepellin/VEGFR2 interaction and progressing to downstream AMPK activation and mTOR inhibition [64,67–69]. In this study, we investigated the efficacy of conditional expression of endorepellin in the angiogenic network of syngeneic breast carcinoma allografts. We report a profound oncosuppressive effect on breast cancer growth, angiogenesis and production of peritumor HA. Thus, endorepellin could represent a protein-based therapy in suppressing cancer growth and spread.
Results
HAS2 is an autophagic target in endothelial cells
We previously found that endorepellin evokes autophagic degradation of HAS2 in endothelial cells [67]. To establish the generality of this finding, we inhibited basal autophagy in human umbilical vein endothelial cells (HUVEC) via RNAi knockdown of autophagy protein 5 (ATG5). This critical autophagic effector, together with ATG12 and ATG16, functions as an E3-like enzyme conjugating LC3 to phosphatidylethanolamine (PE), a lipidation that facilitates the expanding autophagosomal membrane [70]. Transient knockdown of ATG5 significantly decreased ATG5 protein while concurrently upregulating levels of unconjugated LC3-I (Fig. 1, A and B), thereby validating successful attenuation of ATG5 function [71]. Notably, ATG5 suppression increased HAS2 protein levels by more than 2-fold (Fig. 1, A and B), establishing ATG5 as a critical player in maintaining basal autophagic degradation of HAS2. To further validate these in vitro observations, we performed in vivo studies. Specifically, wildtype C57BL/6J mice were fasted for 48 h and inhibited autophagy globally via chloroquine i.p. injections at 4 and 24 h prior to sacrifice. Immunofluorescence of cardiac frozen sections demonstrated a profound decrease of HAS2 in fasted mice that was even more pronounced with chloroquine treatment at both 4 and 24 h (Fig. 1C). Quantification of fluorescence intensity revealed a significant suppression of HAS2 (P<0.01, Fig. 1D) and an even greater accumulation evoked by chloroquine at 4 and 24 h (Fig. 1D). Furthermore, no structural changes in these cardiac tissues were seen on H&E staining (Fig. S1). Of note, we used an unbiased method for quantification: the images were first captured in the vehicle-treated samples and then the objective was moved to adjacent frozen sections maintaining constant exposure time, gain and intensity. Collectively, our results are paramount in demonstrating ATG5-mediated HAS2 degradation in an animal model and reinforces the generality of HAS2 regulation via autophagic catabolism in vivo.
Figure 1. Inhibiting autophagy via ATG5 knockdown in vitro or chloroquine (CQ) treatment in vivo blocks autophagic degradation of HAS2.
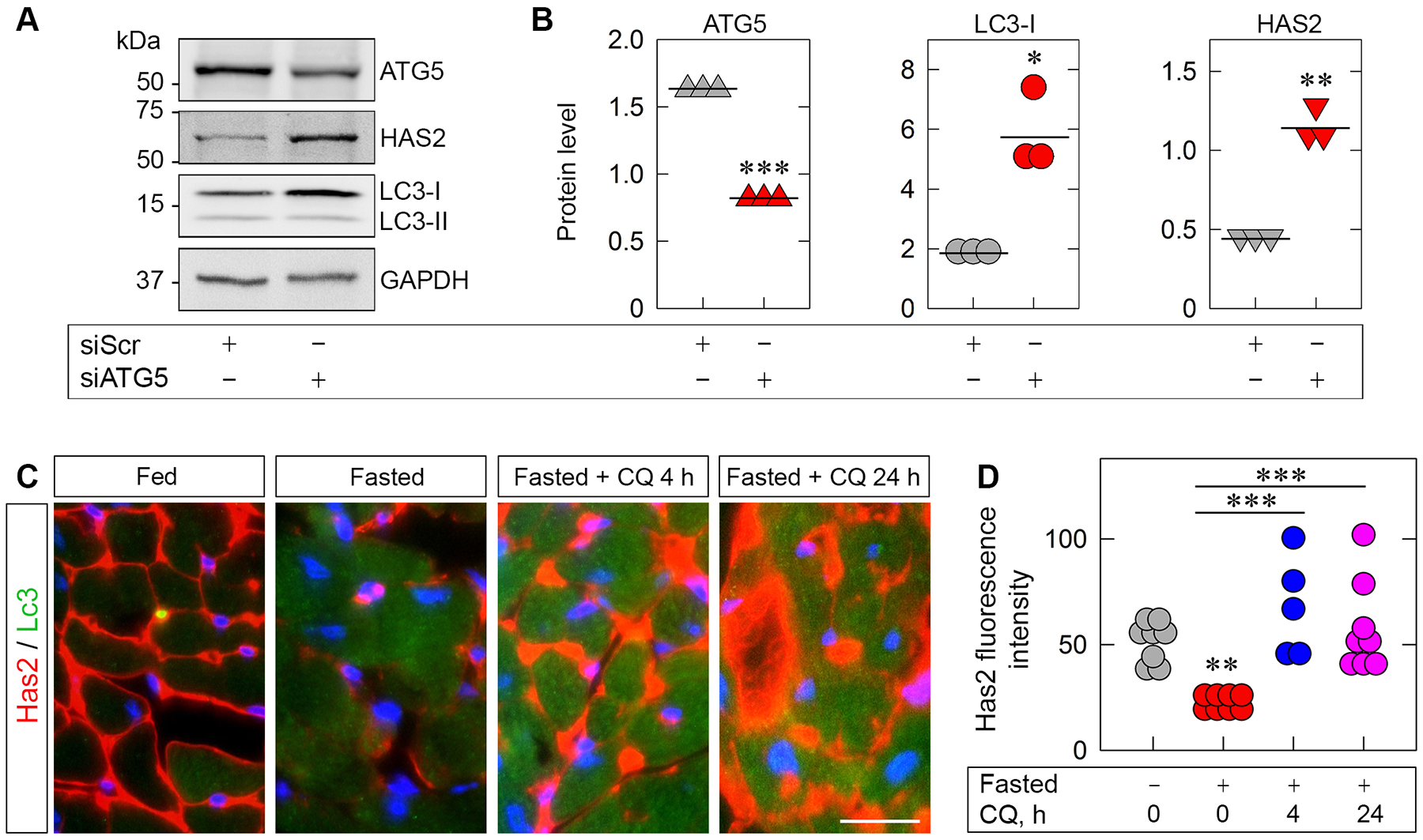
(A) Representative western blot (WB) of HUVEC whole-cell lysates pretreated with scrambled siRNA (siScr) or siRNA targeting ATG5 (siATG5). (B) Quantification of ATG5, LC3-I and HAS2 from A. Statistical analyses were calculated via two-tailed Student’s t test (*p < 0.05, **p <0.01, ***p<0.001). (C) Representative IHC of heart tissue from wild-type mice fasted for 48 h ±chloroquine (50 mg/kg) injected i.p. 4 and 24 h prior to sacrifice. Has2 is labeled in red and Lc3 in green; nuclei are blue. Scale bar, 20 μm. D, quantification of Has2 fluorescence intensity from C; n = 6–8 biological replicates. Statistical analyses were calculated via One-way ANOVA (**p < 0.01, ***p < 0.001).
Inducible Intratumor Overexpression of Endorepellin Decreases Breast Carcinoma Allograft Growth, Vasculature and HA Levels
To test the therapeutic efficacy of this angiostatic HAS2-HA regulatory axis in a disease driven by excessive deposition of HA [12–14], we investigated whether endorepellin could suppress growth, extracellular HA and angiogenesis in a breast cancer mouse model. First, we generated a novel, inducible endorepellin knock-in (ERKi) transgenic mouse by placing the ERKi construct within the Rosa26 allele on chromosome 6 (Fig. 2A), a common and effective site for the integration of transgene constructs [72]. The ERKi construct harbors a strong synthetic CAG (CMV enhancer, chicken β-actin promoter and rabbit β-globin splice acceptor site) promoter upstream of a neomycin-resistant STOP cassette flanked by LoxP sites followed by a His6-tagged recombinant endorepellin (ERHis6) sequence (Fig. 2A). The endorepellin transgene construct begins with a signal peptide sequence followed by amino acids 3687–4391 (region of Domain V) of the parent perlecan protein [47] and ending with a His6 tag. Domain V includes three laminin-type G modules (LG1–3) each separated by two EGF-like modules (Fig. S2). In the presence of Cre recombinase, the neomycin-resistant STOP cassette is excised, allowing for strong expression of the ERHis6 sequence (Fig. 2A). This advantageous Cre-inducible system not only prevents transgenic endorepellin overexpression from interfering with physiological development but also enables customization in the timing and location of endorepellin induction. We then validated ERHis6 expression in lung fibroblasts isolated from ERKi mice treated with either AdCre, an attenuated, type 5 human adenovirus expressing Cre driven by an upstream CMV promoter, or AdNull, the same adenoviral backbone carrying an empty CMV promoter as control. Immunoblotting of conditioned media of ERKi lung fibroblasts treated with AdCre for 24 or 48 h showed increasing levels of ERHis6 as compared to those treated with AdNull (Fig. S3).
Figure 2. Intratumor overexpression of His-tagged endorepellin (ERHis6) decreases syngeneic E0771 breast tumor allograft growth, angiogenesis and hyaluronan levels in Cre-inducible ERHis6 knock-in (ERKi) mice with validation of Tie2CreERT2 endothelial cell-specific Cre recombinase activity.
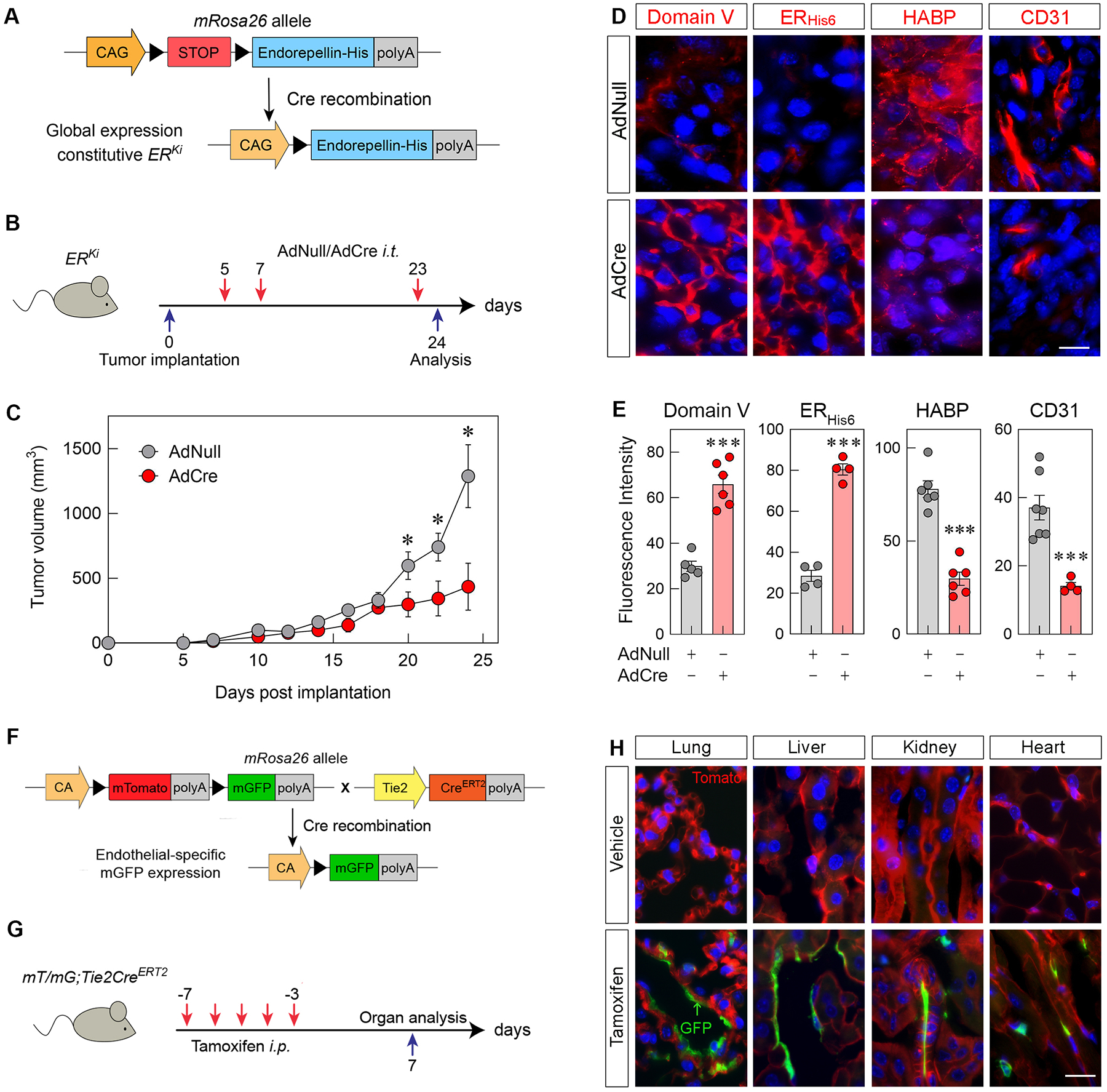
(A) Schematic diagram of the ERKi transgene construct before and after Cre recombination. (B) Schematic diagram of the time course treatment of AdNull or AdCre i.t. injections following tumor implantation. (C) Growth of E0771 murine orthotopic breast tumor allografts in ERKi mice treated with AdNull or AdCre; mean ±SEM, n=4 biological replicates. (D) Immunofluorescence of tumor allografts treated with i.t. injections of AdNull or AdCre and labeled for perlecan Domain V, ERHis6, HABP and CD31 (all red). Nuclei are blue; scale bar, 10 μm. (E) Quantification of fluorescence intensity of Domain V, ERHis6, HABP and CD31 in AdNull- or AdCre-treated allografts. Mean ±SEM, n = 3–7 biological replicates each representing 5–15 technical replicates. Statistics calculated via two-tailed Student’s t test (*p < 0.05, **p < 0.01, ***p<0.001). (F) Schematic of the mT/mG;Tie2CreERT2 transgene construct expressing membrane-targeted tdTomato (mT) globally before tamoxifen treatment and membrane-targeted GFP (mG) in endothelial cells following tamoxifen-induced intra-chromosomal recombination via Cre activation. (G) Schematic diagram of the time-course treatment of tamoxifen i.p. injections in mT/mG;Tie2CreERT2 mice. (H) immunofluorescence of lung, liver, kidney and heart tissue from mT/mG;Tie2CreERT2 mice injected with vehicle (corn oil) or tamoxifen. Scale bar, 15 μm.
To validate this mouse model, we designed a localized induction of transgenic ERHis6 following tumor implantation via multiple subcutaneous, intratumor (i.t.) injections of AdCre or AdNull (Fig. 2B). To avoid immune rejection of breast tumors, we utilized E0771, a syngeneic, malignant breast carcinoma cell line originally isolated from a metastatic lung nodule in C57BL/6 mice [73]. These cells have been since characterized as triple-negative, basal-like [74], and highly tumorigenic [75–77]. Following three injections of AdCre or AdNull at day 5, 7 and 23 post-tumor implantation, breast cancer allografts were significantly reduced by AdCre treatment vis-à-vis AdNull-treated counterparts (Fig. 2C). Next, we probed the allografts for Domain V of perlecan, His6 tag for transgenic endorepellin (ERHis6), HA binding protein (HABP), and the endothelial cell marker CD31. Of note, the custom antibody (anti-Domain V) for perlecan binds specifically to the entirety of domain V, which is the endorepellin region of the larger perlecan protein. For this reason, we will be referring to this antibody target as Domain V. Using an antibody against domain IV of perlecan, we have validated that both antibodies against Domains IV and V had identical distributions in the basement membrane structures in the glomeruli and renal tubules of murine kidneys and spaces of Disse lining the portal sinuses in murine liver tissue where perlecan and endorepellin reside (Fig. S4A). We found a stark upregulation of both Domain V and ERHis6 as well as a concurrent downregulation of HABP and CD31, markers of HA production and angiogenesis, respectively (Fig. 2, D and E). These results validate the successful intratumor introduction of Cre via AdCre and effective Cre recombinase activity in inducing transgenic endorepellin expression. Additionally, probing tumor allografts with anti-Domain IV antibody verified that tamoxifen treatment did not evoke significant changes in expression of endogenous, native perlecan (Fig. S4, B and C). This definitively proves that only transgenic endorepellin, measured specifically with anti-His6 antibody, is upregulated upon tamoxifen treatment and that endogenous perlecan levels remain unchanged (Fig. 2, D and E).
Importantly, induction of endorepellin throughout tumor progression resulted in a marked suppression of HA and downregulation of tumor vasculature as measured via HABP and CD31, respectively (Fig. 2E). Of note, through its specific binding to VEGFR2, endorepellin selectively evokes autophagic degradation of HAS2 in endothelial cells and not cancer cell lines including E0771 [67]. We conclude that localized expression of recombinant endorepellin during tumorigenesis abrogates extracellular HA and angiogenesis in the tumor microenvironment via downregulating HAS2 levels specifically in the tumor endothelium.
As perlecan proteoglycan is localized in the pericellular matrix and vascular basement membrane [78] and given the selectivity of endorepellin signaling in endothelia, we utilized Tie2CreERT2 (tamoxifen-inducible estrogen receptor fused to Cre recombinase driven by endothelium-specific Tie2 promoter) mice to allow for endothelial cell-specific expression of Cre recombinase [79–82]. To verify that these mice possessed endothelium-specific expression of Cre, we crossed Tie2CreERT2 with the mT/mG double fluorescent reporter mice that express membrane-targeted tdTomato (mT) in cells lacking Cre and membrane-targeted green fluorescent protein (GFP) (mG) in cells expressing Cre (Fig. 2F). We then induced robust activation of Cre recombinase in endothelia by daily intraperitoneal (i.p.) injections of tamoxifen over the course of 5 days and processed organs after 10 days (Fig. 2G). Analysis of the lungs, liver, kidneys, and heart in tamoxifen-treated mice demonstrated specific GFP signal in tissue endothelia vis-à-vis no GFP signal in organs of vehicle-treated mice (Fig. 2H). Collectively, these findings demonstrate successful endothelial cell-specific intrachromosomal recombination by Cre in tamoxifen-treated Tie2CreERT2 mice.
Conditional expression of endothelial-derived endorepellin in Tie2CreERT2;ERKi mice attenuates breast cancer growth, angiogenesis and peritumor HA
Having validated endothelial Cre activity in Tie2CreERT2 mice, we generated double transgenic Tie2CreERT2;ERKi mice allowing for the conditional expression of ERHis6 in the vasculature (Fig. 3A). The design of this mouse model ensures that recombinant endorepellin engages in autocrine and paracrine signaling as it binds its cognate receptors, VEGFR2 and α2β1 integrin, to promote a pro-autophagic and anti-angiogenic phenotype from the very cell type from which it was synthesized. As a proof-of-principle, we administered i.p. injections of tamoxifen to evoke endorepellin secretion from the vasculature. Notably, both treatments of i.p. tamoxifen and i.t. adenoviral vectors were well-tolerated in mice as we did not observe any noticeable side effects from either treatment vis-à-vis their control counterparts. In addition, treatments were administered in adult mice only, ensuring no aberrations in development. Three days following the last tamoxifen injection, we performed ex vivo aortic ring assays [83,84], probing the vascular sprouting from the aortic rings with anti-His6 mAb (ERHis6, green) and an endothelial cell marker isolectin B4 (IB4, red) (Fig. 3B). Aortic rings from tamoxifen-treated mice contained a significant upregulation in ERHis6 deposition vis-à-vis those from vehicle-treated mice (Fig. 3C).
Figure 3. Endothelial cell-specific expression of ERHis6 attenuates syngeneic E0771 breast tumor allograft volume, vasculature and hyaluronan levels in Tie2CreERT2;ERKi transgenic mice.
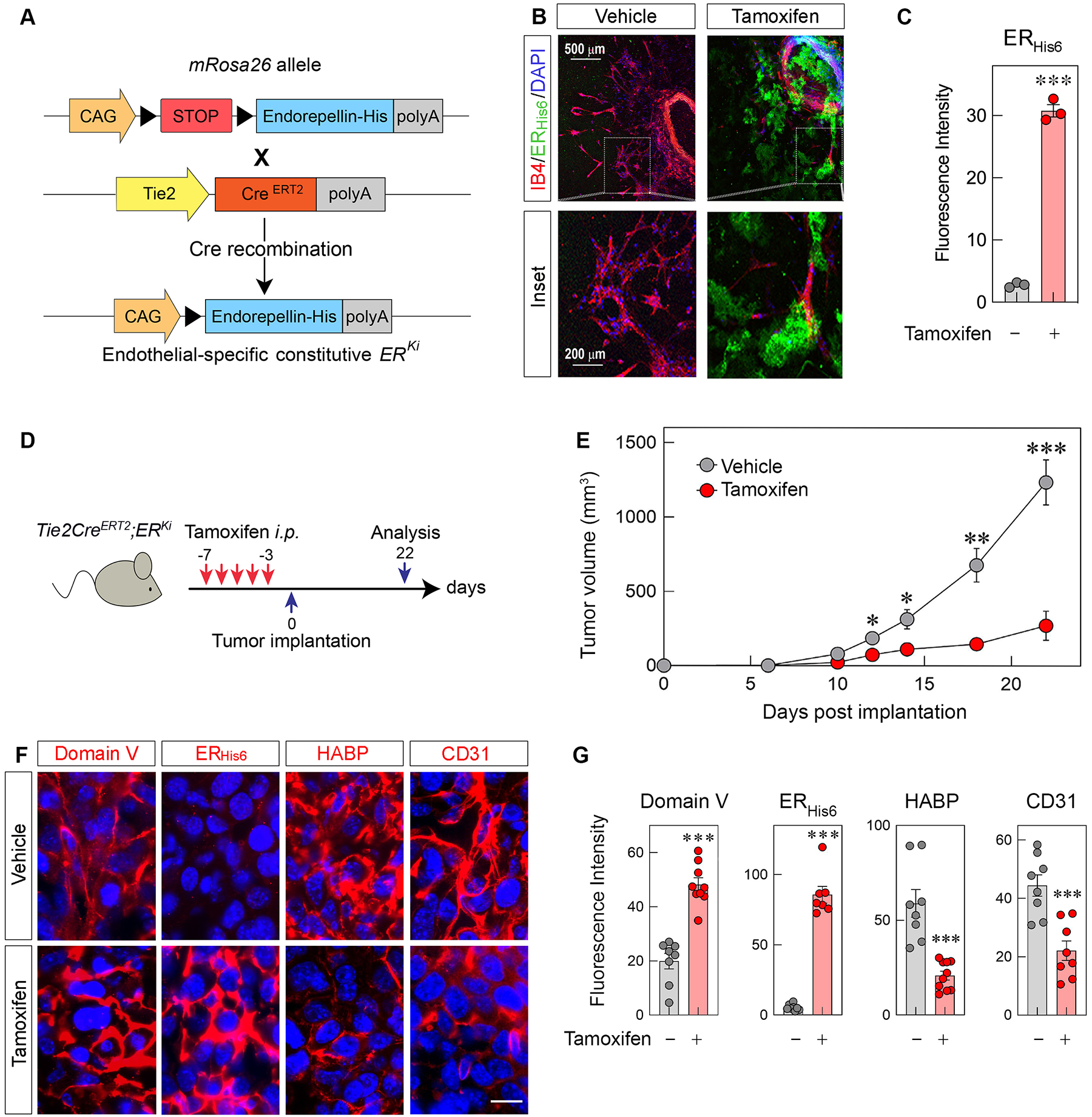
(A) Schematic diagram of the Tie2CreERT2;ERKi transgene construct before and after tamoxifen-induced Cre recombination. (B) Confocal images of ex vivo aortic rings of Tie2CreERT2;ERKi mice injected with i.p. vehicle or tamoxifen and labeled for the endothelial cell marker IB4 (red) and ERHis6, (green). Scale bar, 500 μm (top row) and 200 μm (bottom row, inset). (C) Quantification of mean fluorescence intensity of ERHis6 in the sprouted area of vehicle- or tamoxifen-treated rings; n = 3 mice and 3 biological replicates. (D) Schematic diagram of the time course treatment of i.p. tamoxifen (2 mg per injection) before E0771 implantation. (E) Growth of E0771 murine orthotopic breast tumor allografts in Tie2CreERT2;ERKi transgenic mice treated with vehicle (corn oil) or tamoxifen; mean ±SEM, n = 7 and 10 biological replicates for vehicle- and tamoxifen-treated allografts. (F) Immunofluorescence of tumor allografts treated with i.p. injections of vehicle or tamoxifen and labeled for Domain V, ERHis6, HABP and CD31 (all red). Nuclei are blue; scale bar, 10 μm. (G) Quantification of fluorescence intensity of Domain V, ERHis6, HABP and CD31 in vehicle- or tamoxifen-treated allografts. Mean ±SEM, n = 8–10 biological replicates each representing an average of 5–20 technical replicates. Statistical analyses were calculated via two-tailed Student’s t test (*p <0.05, **p <0.01, ***p<0.001).
We first administered daily i.p. injections of tamoxifen, then implanted syngeneic E0771 breast cancer cells into mammary fat pads and finally monitored the growth of the allografts (Fig. 3D). Tamoxifen-treated Tie2CreERT2;ERKi mice showed a marked, significant reduction in tumor volume (Fig. 3E). Furthermore, IHC analysis of allograft tumors revealed a marked increase in ERHis6, clearly shown by dual staining with antibodies against Domain V and anti-His6 mAb. In contrast, no ERHis6 positivity was observed in vehicle-treated carcinomas (Fig. 3F). As internal controls, we used HABP and CD31, two markers for HA production and angiogenesis, respectively. We found a marked suppression of both HABP and CD31 immunoreactivity (Fig. 3, F and G). Furthermore, regions of tumor expressing higher levels of Domain V do indeed correlate with lower levels of HABP and vice versa (Fig. S5). Collectively, these results indicate that conditional expression of endorepellin attenuates the HAS2-HA axis with a decrease in HA levels in the tumor stroma and concurrent suppression of tumor angiogenesis.
Differential transcriptomic landscape evoked by conditional endorepellin expression in the vasculature of breast carcinoma allografts
To further advance our knowledge of conditional ERHis6 and its transcriptomic effects on breast cancer allografts, we performed deep RNAseq analysis of Tie2CreERT2;ERKi allografts treated with tamoxifen vs vehicle. Among the differentially expressed genes, we found a global suppression in the gene expression in tamoxifen-treated allografts, shown both on heat map and volcano plot (Fig. 4, A and B). Using stringent parameters (>2-fold change and P<0.05) we found 58 upregulated and 294 downregulated genes (Supplementary Table 1 and 2, Fig. 4, A and B) and validated several of the target genes of interest via qPCR (Fig. 4C). The limited size of the upregulated gene dataset and the presence of pseudogenes and predicted genes lacking pathway or biological process annotations required for computational analyses limited the significance of these analyses. The computational tool Reactome [85] was nevertheless utilized to identify overrepresented biological processes and pathways mediated by upregulated genes. Reactome analysis identified YAP1- and WWTR1 (TAZ)-stimulated gene expression as the single biological pathway overrepresented among the upregulated genes (P=0.004, Supplementary Table 3). These transcriptional co-activators target genes that are critical to the regulation of cell proliferation and apoptosis.
Figure 4. Differential transcriptomic landscape of endorepellin expression in vasculature of E0771 breast allografts.
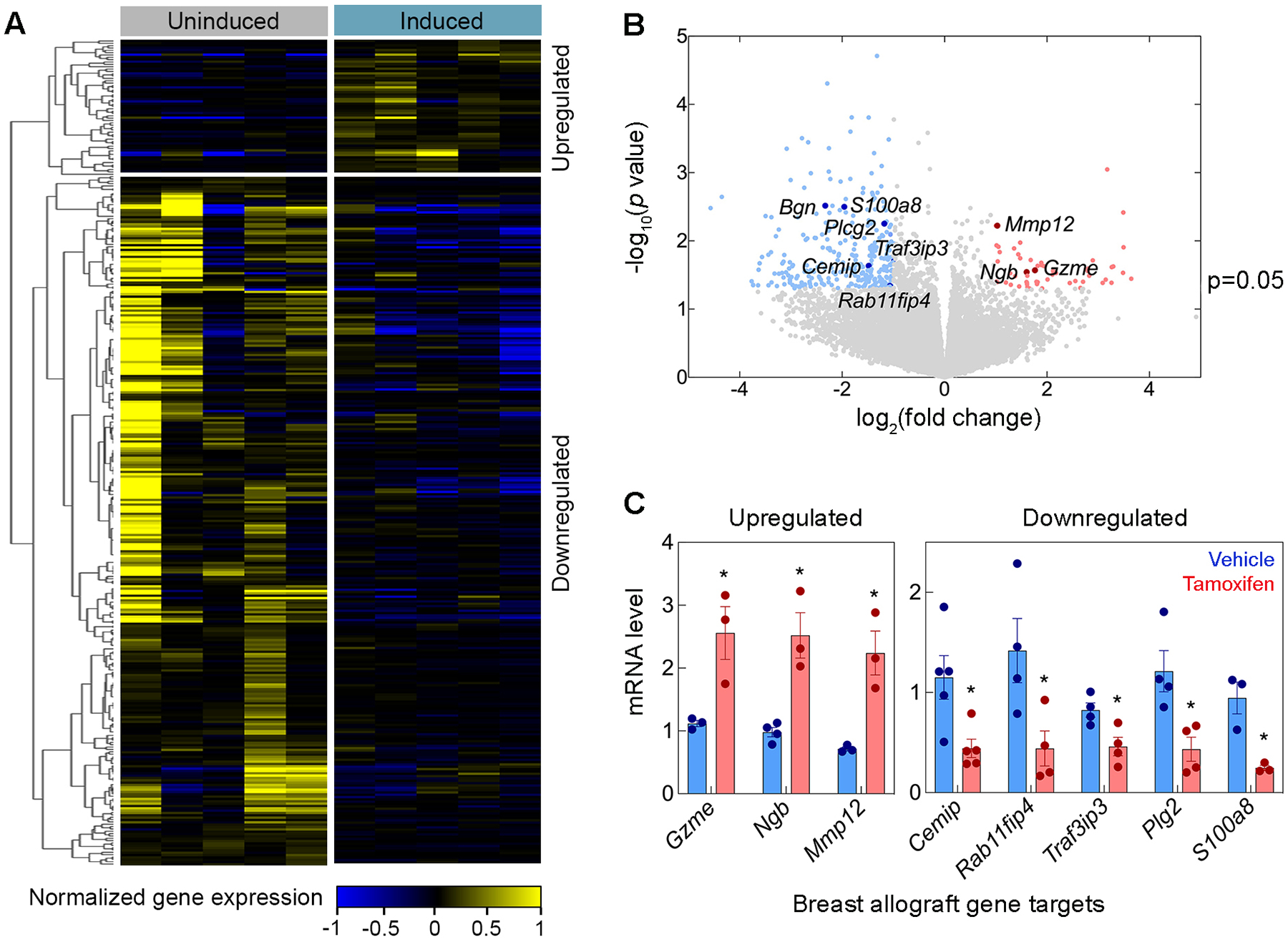
(A) Heat map and (B) volcano plot displays of differentially expressed genes from Tie2CreERT2;ERKi allografts treated with tamoxifen vs control using RNAseq analysis. Applying parameters of >2-fold change and a p<0.05 resulted in 58 upregulated genes and 294 downregulated genes. (C) Real time qPCR analysis of gene targets from both up- and down-regulated gene datasets confirmed RNAseq findings (blue for vehicle, red for tamoxifen). Statistical analyses were calculated via two-tailed Student’s t test (*p <0.05).
In the downregulated genes identified by RNAseq, more pathways were overrepresented. They were related to the immune system and gene expression as detailed below. Cytokine signaling in immune system (i.e., interleukin-10 signaling), the CLEC7A/inflammasome pathway and metal sequestration by antimicrobial proteins in the innate immune system were significantly overrepresented in downregulated genes (Supplementary Table 4). Other significantly enriched pathways associated with the immune system were the transcriptional regulation by RUNX3/RUNX3, regulators of immune response and cell migration, RUNX1/RUNX1 and FOXP3, regulators of regulatory T lymphocyte differentiation and the adaptive immune response (Supplementary Table 4).
The complete analysis of overrepresented Molecular Function and Biological Process Gene Ontology (GO) terms in downregulated genes was performed using Biological Networks Gene Ontology (BiNGO) [86] (Fig. S6 and S7, Supplementary Table 5 and 6). For each GO term category, BiNGO provides a layout of interconnected and colored circles corresponding to each annotation. The overrepresented molecular functions were associated with binding (i.e., receptor, protein, cytokine and chemokine binding) (Fig. 5). Another molecular function overrepresented among the downregulated genes was peptidase activity, including metalloprotease, exoprotease and endoprotease activity (Fig. 5). The most overrepresented biological processes were associated with the immune system (i.e., regulation of immune system process, regulation of immune response, immune response-regulating signaling pathway, and T cell activation) (Fig. 6). Other biological processes enriched in the downregulated genes were regulation of cytokine production, regulation of vascular endothelial growth and positive regulation of vascular endothelial growth factor production.
Figure 5. Analysis of downregulated genes with BiNGO for the GO term “Molecular Function”.
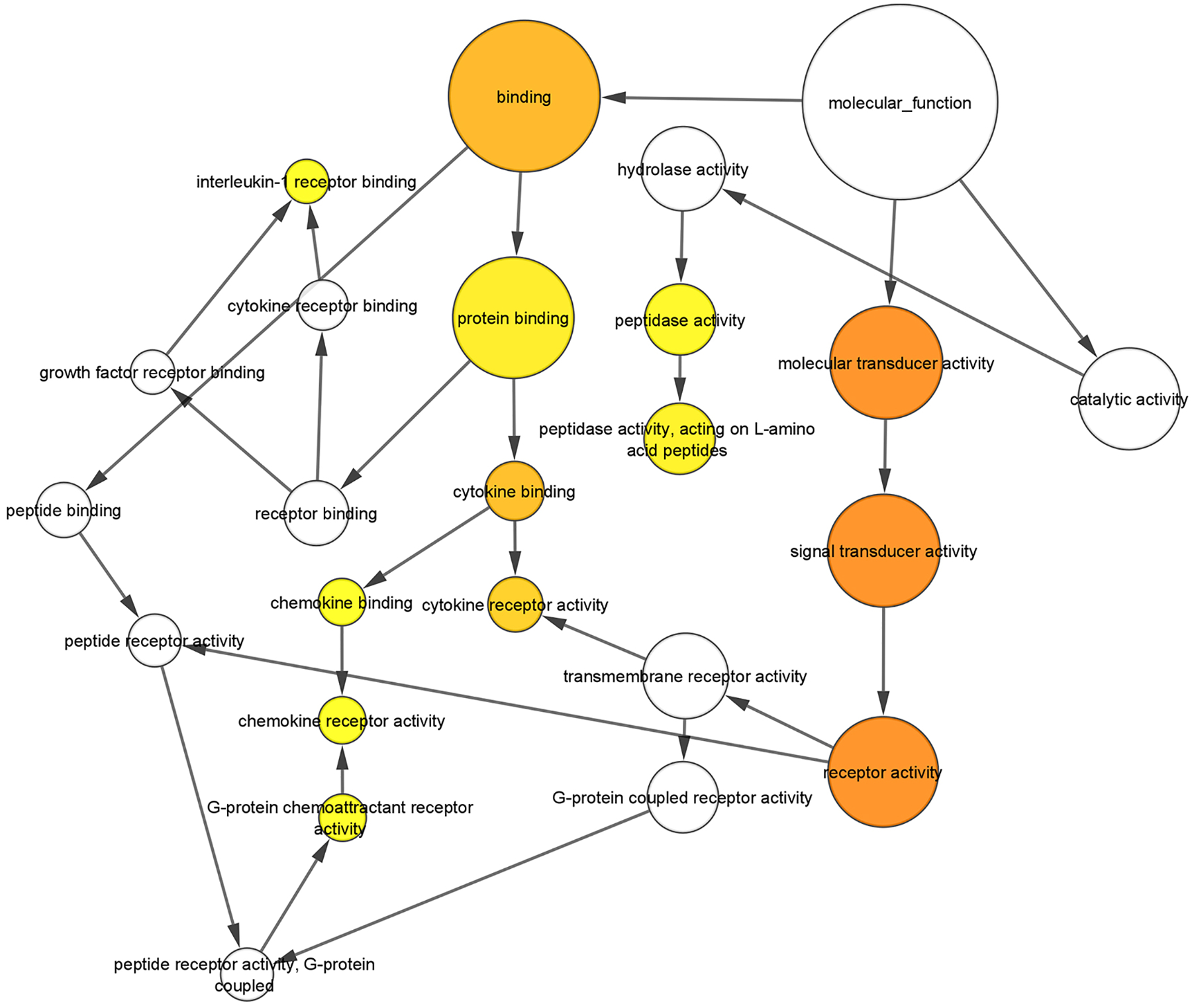
[86]. For each GO term category, BiNGO provides a layout of interconnected and colored circles corresponding to each annotation. The size of the nodes is proportional to the number of genes annotated to that node with a focus on the over-represented molecular functions associated with binding (i.e., receptor, protein, cytokine and chemokine binding) and peptidase activity (i.e., metalloprotease, exoprotease and endoprotease activity). The color of the node represents the corrected p-value. White nodes are not significantly overrepresented, whereas the other ones are, with a color scale ranging from yellow (p-value = 0.01) to dark orange (p-value = 10−7). The color saturates at dark orange for p-values that are more than 5 orders of magnitude smaller than the chosen significance level (0.01).
Figure 6. Analysis of downregulated genes with BiNGO for the GO term “Biological Process”.
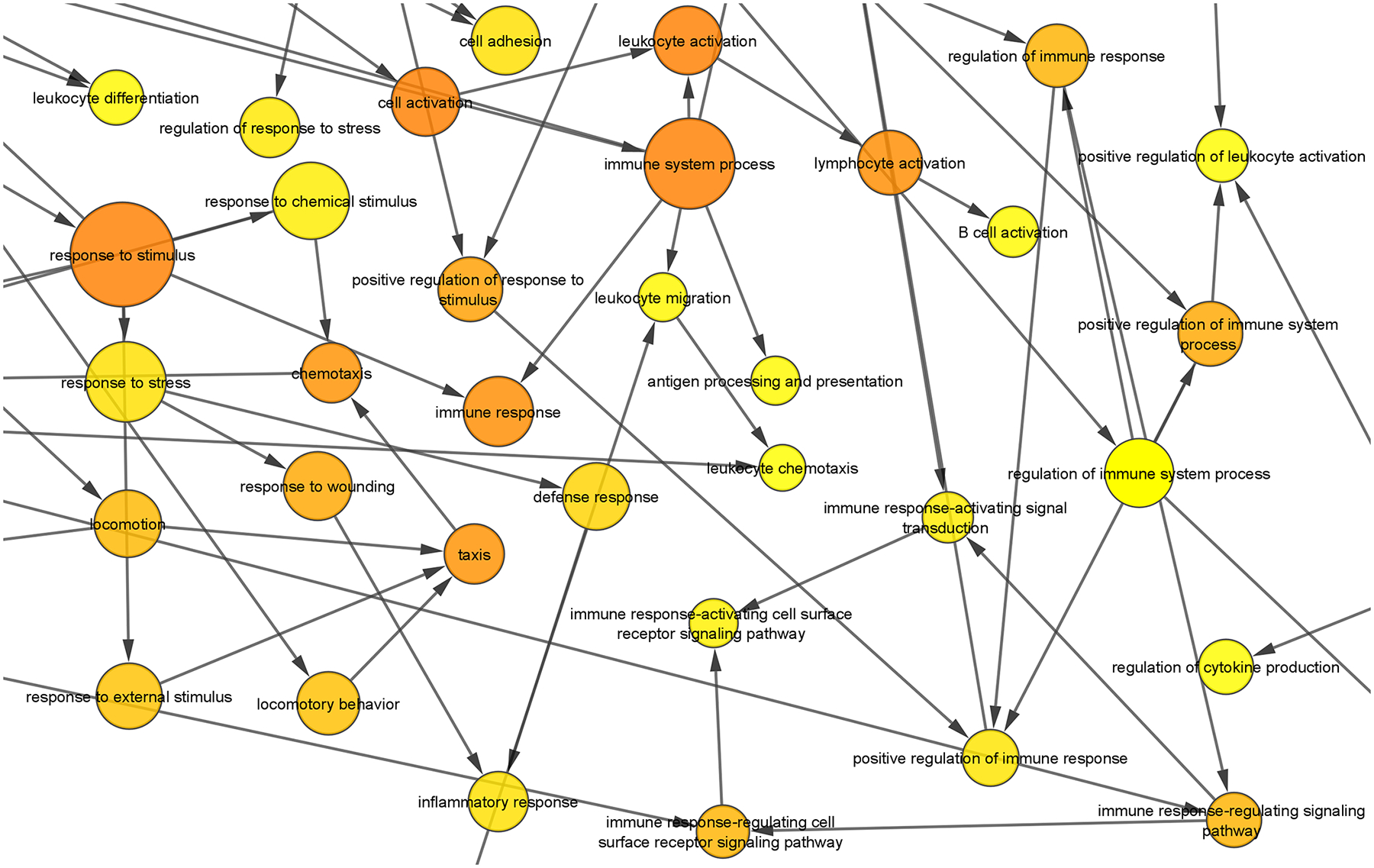
[86]. The size of the nodes is proportional to the number of genes annotated to that node with a focus on the overrepresented biological processes associated with the immune system. The color of the node represents the corrected p-value. White nodes are not significantly overrepresented, whereas the other ones are, with a color scale ranging from yellow (p-value = 0.0005) to dark orange (p-value = 5×10−9). The color saturates at dark orange for p-values which are more than 5 orders of magnitude smaller than the chosen significance level (0.0005).
Discussion
The tissue organization field theory that is gaining experimental support and widespread acceptance in cancer research proposes that tumorigenesis is critically driven by pathological interactions between a diseased stroma and its adjacent tissue [87]. Increasingly, experimental evidence substantiates the fundamental role of the stroma in tumorigenesis, thereby opening up the tumor stroma as a promising reservoir for therapeutic cancer targets and biomarkers [1,88–90]. Particularly in regards to breast cancer, there is a growing body of evidence revealing the deleterious clinical outcomes from breast cancer tumors containing an excessive level of stromal HA [8,11,64,91]. Thus, this exposes a great need for cancer therapeutics aimed at targeting HA in the breast cancer stroma. Furthermore, there is also the possibility of therapeutic clinical benefits as successfully depleting stromal HA in the breast tumor microenvironment would likely confer many onco-suppressive functions, including inhibiting tumor angiogenesis, invasiveness, growth, malignancy and aggressiveness [12–14,17].
Above HAS1 and HAS3, HAS2 is the definitive, major producer of oncogenic HA in the breast cancer stroma. HAS2 is overexpressed in breast cancer cells vis-à-vis normal breast tissue, and breast tumors expressing high levels of HAS2 are correlated with increased invasiveness and lymph node metastasis [91–93]. Increased HAS2 levels are also characteristic of breast cancer aggressiveness as HAS2 mRNA are significantly elevated in aggressive breast cancer cells as compared to non-aggressive cell lines [14]. HAS2 also plays a well-studied role in inducing epithelial-mesenchymal transition (EMT) evoked via transforming growth factor β (TGFβ). Not only does TGFβ upregulate HAS2 expression and HA synthesis, but RNAi-mediated knockdown of HAS2 in mammary epithelial cells inhibits expression of TGFβ-induced EMT markers by approximately 50% and abolishes TGFβ-mediated cell migration [94]. Moreover, genetic knockdown of HAS2 in malignant breast cancer attenuated breast tumor proliferation both in vivo and in vitro [92,95], significantly suppressed breast cancer-derived HA and abrogated cancer cell invasiveness and migration in vitro [95]. Separately, antisense inhibition of HAS2 mRNA prevented primary and secondary tumor formation of the highly invasive MDA-MB-231 breast cancer cell line in vivo, thereby increasing survival by 172% [96]. Overall, these results overwhelmingly posit HAS2 as not only a central player in breast cancer initiation, progression and invasion but also a prognostic marker in whose marked therapeutic suppression may yield clinically favorable outcomes.
Interestingly, a significant portion of disease-driving HA in the tumor microenvironment is produced by stromal cells, rather than the cancer cells. In a study of human breast cancer cases, both cancer cells and stromal cells (i.e. endothelial cells, fibroblasts and myofibroblasts) expressed HASes [97]. In breast cancer cells, the majority of extracellular HA from cancer cells were synthesized via HAS1, and HAS1 was the only HAS isoform that was associated with poor clinical outcomes such as high relapse rate and decreased survival. In contrast, HA synthesized from stromal cells in the tumor is synthesized by all HAS isoforms, and the expression of all three isoforms correlates with high relapse rate and short survival. However, whereas 99% of stromal cells express HAS2, 23% and 16% are negative for HAS1 and HAS3, respectively, again positing HAS2 as the indispensable, main producer of peritumor HA in breast cancer stroma [98]. Moreover, there is evidence of a paracrine interaction between breast cancer cells and endothelium in modulating endothelial expression of ECM molecules as breast cancer cells substantially upregulate the levels of HA and HAS2 in surrounding endothelium [99]. Similarly, stroma-derived HA secreted from fibroblasts in mammary cancer was critical in driving tumor angiogenesis and lymphangiogenesis via tumor-associated macrophage recruitment [17]. Indeed, breast cancer in particular presents with elevated levels of stromal HA especially in the invasive, leading edge of the tumor as opposed to the tumor center [100]. To add another layer of complexity, pro-angiogenic signals such a HA that are secreted from the tumor stroma may further upregulate HA synthesis in cancer cells, creating a deadly positive feedback loop. For example, global gene expression profiling in pancreatic cancer cells identified HAS2 as among the 0.3% genes that were upregulated as a result of co-culture with primary fibroblasts, further implicating HAS2 as being affected by tumor-stromal signaling and crosstalk [101]. Overall, these findings posit the tumor stroma as an essential source of HA in the tumor microenvironment and indicate that breast cancer-endothelium signaling is a critical proponent in driving the pro-angiogenic HA deposition in the tumor stroma. In agreement with this, our therapeutic usage of recombinant endorepellin in selectively diminishing HA secretion from the tumor endothelia directly targets this pro-angiogenic, pro-tumorigenic crosstalk at the breast cancer-endothelium interphase.
A few remaining factors represent limitations of this study and would be important to pursue as future studies. This includes the lack of in-depth studies demonstrating mechanisms of crosstalk between endothelial and tumor cells. Future studies focusing on these crosstalk pathways involving endorepellin is necessary and would build upon the substantial data of our study. Whereas the RNAseq data in this study were taken from tumor homogenates, further investigation of the transcriptomic signature driving this crosstalk could be performed using single cell RNAseq to individually extract endothelial, stromal, and tumor cell contributions. Notably, we did not expect HAS2 to appear among the differentially express genes as endorepellin does not affect HAS2 transcript levels in endothelial cells [67]. Interestingly, tumor lysates did not show changes in HAS2 protein levels (not shown). Since endorepellin specifically decreases HAS2 levels in endothelia and not tumor cells [67], it is most likely that decreases in HAS2 protein from the endothelia are not reflected in analysis of the whole tumor lysate. Future studies that separate endothelial cells from tumor cells would more accurately separate out effects of endorepellin on the endothelia vs tumor in the in vivo setting. Finally, there was no significant difference in tumor size between control- vs tamoxifen-treated allografts on day 10 post tumor implantation, which may implicate that early endorepellin expression does not affect the early stages of tumor implantation and survival. Further studies exploring the exact timing of endorepellin expression following tamoxifen induction would be beneficial in elucidating endorepellin’s role in tumor implantation, proliferation, and cell survival due to effects on angiogenesis.
Due to the intrinsic kinetics of HA turnover in the ECM, the therapeutic implications of targeting HA synthesis in the tumor microenvironment are striking. HA synthesis and metabolism are fast and dynamic processes. Stromal HA stands apart from the rest of its matrix constituents as it undergoes a rapid turnover of approximately 2 days from the time of synthesis to degradation and clearance. In fact, one-third of the total HA in the human body is turned over daily of which a substantial amount is cleared via the lymphatic system and removed by liver endothelial cells [14]. This dynamic kinetics speaks to the role of HA as a fast responder in the matrix as well as to the very likely expeditious therapeutic outcome of targeting HA synthesis in tumor endothelium. Thus, based on the favorable in vivo anti-tumorigenic effects of endorepellin in aggressive breast cancer (this work) as well as other carcinomas in previous work [66], we propose endorepellin as a promising protein therapy to suppress tumor angiogenesis and growth via suppressing endothelium-derived HA deposition in tumor microenvironments. As its mechanism of action is limited to targeting endothelium, we predict that co-treatment of endorepellin with other chemotherapeutics that directly target breast cancer cells would beget the best clinical outcome.
Collectively, analysis of the upregulated genes showed a general trend of suppressing endothelial cell-matrix adhesion as well as lymphatic vessel vasoconstriction, implicating the potential effects of endorepellin in angiogenic and lymphatic vascular remodeling and function. Interestingly, the upregulated gene dataset also suggested the effects of vascular endorepellin expression on cell growth and apoptosis through transcriptional co-activators YAP1 and TAZ, both of which regulate angiogenesis through VEGF signaling [102]. In fact, recent literature has revealed that YAP/TAZ are downstream effectors of VEGF binding to VEGFR2, such that endothelial-specific deletion of YAP/TAZ resulted in defective angiogenic sprouting in vessels exposed to VEGF both in vitro and in vivo [103]. Furthermore, AMPK, which is activated downstream of endorepellin-VEGFR2 signaling, inhibits YAP activity through phosphorylating at its S94 residue [104,105]. On the other hand, the YAP/TAZ axis is critical for autophagosomal degradation into autolysosomes [106], thereby implicating its activity to the pro-autophagic activity of endorepellin [68,107]. These results highlight YAP1/TAZ as potential downstream targets of endorepellin-VEGFR2 signaling, a biological link requiring future investigation that would further elucidate the complex outside-in signaling mechanisms of endorepellin.
Overwhelmingly, the downregulated dataset showed widespread suppression of multiple immunomodulatory pathways, including a variety of cytokine signaling, CLEC7A/inflammasome function, antimicrobial protein functions within the innate immune response, RUNX3-regulated immune cell migration, and regulatory T cell development via RUNX1 and FOXP3. This widens our understanding of endorepellin from the endothelial cell-matrix interface to regulating the larger immune response in the tumor microenvironment. Given the evolving focus on immunotherapies in cancer research, this presents an exciting avenue of further investigation in understanding how endorepellin itself could potentially play a role as a potent immune suppressor.
Experimental procedures
Cells and materials
Human umbilical vein endothelial cells (HUVEC) were grown and maintained in basal media supplemented with VascuLife EnGS LifeFactors Kit (Lifeline Cell Technology, Oceanside, CA). Cells were plated on 0.2% (w/v) gelatin-coated cell culture plates (Thermo Fisher Scientific, Waltham, MA) and utilized within the first five passages. Antibodies for Western Blotting (WB) were purchased from the following sources and utilized at the designated dilutions: GAPDH (14C10, 1:10,000 dilution; Cell Signaling, Danvers, MA), LC3B (L7543, 1:1,000 dilution; Sigma), and HAS2 (sc-514737 and sc-365263 at 1:250 and 1:150, respectively; Santa Cruz Biotechnology, Dallas, TX). The following siRNA was purchased from Santa Cruz Biotechnology and used at the designated amounts: ATG5 siRNA (sc-41445, 100 pM) and scramble control siRNA-A (sc-37007, 100 pM).
Transient siRNA-mediated knockdown
Transient knockdown of ATG5 in HUVEC was accomplished via transfection of validated siRNA directed against human ATG5 (sc-41445; Santa Cruz Biotechnology). Scrambled control siRNA-A (sc-37007; Santa Cruz Biotechnology) was used as control. Gelatinized six-well plates seeded with 2 × 105 HUVEC were grown at 37 °C and 5% CO2 until 70% confluence. ATG5 or scrambled siRNA duplex (100 pM) were added to Lipofectamine 2000 (Invitrogen) in serum-free OptiMEM (Gibco). Cells were washed with PBS, incubated with the OptiMEM solution for 6 h at 37 °C and 5% CO2, and incubated overnight with full HUVEC media in the same culture conditions. Verification of siRNA-mediated knockdown was accomplished via immunoblotting.
Mice generation and genotyping
Inducible ERKi mice expressing a LoxP-flanked stop cassette between the CAG promoter and the human endorepellin cDNA within the mRosa26 locus of C57/BL6J mice were generated by Cyagen. To generate an endothelial-specific overexpression of endorepellin, we crossed ERKi mice with Tie2CreERT2 mice generously provided by Dr. B. Eliceiri (UCSD) [79]. Tie2CreERT2 mice were also crossed with mT/mG reporter mice to create mT/mG;Tie2CreERT2. All animal experiments were conducted following the Guide for Care and Use of Laboratory Animals according to the policies of the Institutional Animal Care and Use Committee at Thomas Jefferson University. All mice utilized in tumor allograft experiments were females 2–4 months of age.
For mouse genotyping, genomic DNA from 0.25 cm mouse tail fragments were extracted via incubating in 200 ml DirectPCR Lysis Reagent for tails (Viagen Biotech, #101-T) and 60 mg Proteinase K (Sigma, P6556) at 55°C for 5–6 h, then 85°C for 45 min. The genotype was determined via a 25 ml PCR reaction—consisting of 100–500 ng genomic DNA, 1 μM each primer and ReadyMix REDTaq PCR Reaction Mix with MgCl2 (Sigma, R2648)-and Southern blotting in an 2% (w/v) agarose gel (Fisher, BP1356). PCR conditions were 95°C (30 s), 54–62°C (45 s) and 72°C (1 min) for 35 cycles. Primers to detect Cre (277 bp): 5’-AAACGTTGATGCCGGTGAAC-3’ and 5’-CAGGCTAAGTGCCTTCTCTACA-3’. Primers to genotype ERKi: 5’-GCTGGTTATTGTGCTGTCTCATCAT-3’ and 5’-GTCCATCTGCACGAGACTAGTGAG-3’ (360 bp for ERKi); 5’- GAGGGCTCAGTTGGGCTGTTTTGG −3’ and 5’- CACACCAGGTTAGCCTTTAAGC −3’ (300 bp for wild type).
Adenoviral treatment and western blot analysis of lung fibroblasts
Following dissection of lung tissue from ERKi heterozygous mice, lungs were finely chopped using scalpels and cultured in supplemented DMEM (Dulbecco’s Modified Eagle Medium, Thermofisher) with 10% (v/v) FBS (fetal bovine serum, Sigma) and Penicillin-Streptomycin (ThermoFisher) at 37°C and 5% (v/v) CO2 to isolate lung fibroblasts. Following 1–2 passages, 5×108 Pfu of either AdNull or AdCre (Vector Biolabs, 1300 and 1045N) were added to media for 24 or 48 h. After adenoviral treatment, conditioned fibroblast media were taken, vortexed in 5x sample buffer [84] and boiled for 5 min. Samples were then resolved on SDS-PAGE as described previously [108–110], transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA) and probed with primary monoclonal mouse anti-His6 antibodies (ab18184, 1:1000 dilution; Abcam) and secondary goat anti-mouse IgG antibody, horseradish peroxidase-conjugated (AP308P, 1:4000 dilution; EMD Millipore). Blots were then developed with SuperSignal West Pico Chemiluminescence substrate (Thermo Fisher Scientific), and signal detection was performed via an ImageQuant LAS-4000 (GE Healthcare) platform. The bottom half of the SDS-PAGE gel was removed, stained in Coomassie Brilliant Blue dye (20279, Thermo Scientific) for 1 h, destained in 45% (w/v) methanol and 10% (w/v) acetic acid overnight and rehydrated in water.
Mouse experiments, tumor RNA processing, and real time qPCR validation
Animal experiments were performed as per the Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University and in accordance with the German Animal Protection Law.
For in vivo autophagic flux experiments, wildtype C57BL/6 mice were obtained from The Jackson Laboratory and both male and female sex ranging from 5–6 months old were equally included in the study. Fasting protocol consisted of withholding food for 48 h with water accessible ad libitum. Autophagic flux experiments were performed with i.p. chloroquine injection of 50 mg/kg 4h or 24 h prior to sacrificing the animals. Following euthanasia, cardiac tissue was isolated and processed for IHC analysis.
In mT/mG;Tie2CreERT2 mice, tamoxifen (2 mg dissolved in corn oil; Sigma, H7904) or vehicle was administered i.p. for 5 consecutive days. Ten days after the last injection, mice were euthanized and organs (liver, lung, kidney, and heart) were removed and processed for IHC analysis. No primary or secondary antibodies were used given that all tissue were genetically engineered to fluoresce either red or green.
For targeted endorepellin overexpression in the tumor in ERKi mice, E0771 (1×106 cells; CH3 BioSystems) were implanted subcutaneously into the upper left mammary fat pad on day 0. Then, adenovirus vectors AdNull and AdCre (Vector Biolabs, 1300 and 1045N) were injected intratumorally (5×108 Pfu) on day 5, 7 and 23 following E0771 implantation. Mice were euthanized and tumors removed on day 24.
For endothelial-inducible endorepellin overexpression in Tie2CreERT2;ERKi mice, tamoxifen (2 mg dissolved in corn oil; Sigma, H7904) was administered i.p. for 5 consecutive days. After 3 days, murine E0771 breast adenocarcinoma cells (1×106 cells; CH3 BioSystems) were then implanted subcutaneously into the upper left mammary fat pad. Mice were euthanized and tumors removed on day 22. In all tumor allograft experiments, mice were euthanized before tumors reached 2000 mm3.
To process tumors for RNA analysis, organs were removed, frozen in liquid nitrogen and stored at −80°C. Small portions of tumor were crushed via mortar and pestle in liquid nitrogen for RNA extraction and resuspended in 500 μl TRIzol reagent (15596026; Invitrogen). RNA was then extracted from the resuspension using the Direct-zol RNA Miniprep Plus Kit (R2072; Zymo Research). The final RNA extracts were resuspended in nuclease-free water. A total of 2 μg RNA per tumor sample were sent to Genewiz for RNA sequencing. Another 1 μg of total RNA per tumor was used for real time quantitative PCR validation. The RNA was annealed with oligo (dT18–20) primers, and SuperScript Reverse Transcriptase III (Invitrogen) was utilized to synthesize cDNA. Amplicons representing target genes and the endogenous housekeeping gene, β-actin (Actb), were amplified in quadruple independent reactions using the Brilliant SYBR Green Master Mix II (Agilent Technologies). All samples were run on the Roche LightCycler 480-II Real Time PCR platform (Roche Applied Sciences), and cycle number (Ct) was recorded for each reaction. Fold changes were normalized to Actb and calculated using the ΔΔCt method (2−ΔΔCT). Data were derived from three independent biological replicates, each carried out in triplicate for every gene of interest. The primers used in qPCR analyses were as follows: Gzme forward, 5’- GATTCTCCTGACCCTACTTCTG −3’; Gzme reverse, 5’- GCCTCCACAGTATCTCCTATTAC −3’; Ngb forward, 5’- AGAATTCCTGGACCACATTAGG −3’; Ngb reverse, 5’- ACTGTCGAGAAGGAGCTGAG −3’; Mmp12 forward, 5’- CTTAACCCCAGCACATTTCG −3’; Mmp12 reverse, 5’- GTGACAGCATCAAAACTCAAGC −3’; Cemip forward, 5’- AATGGAGTCAAGACAACTGAGG −3’; Cemip reverse, 5’- TTGTAGGCAGTAAAGTGTCGG −3’; Rab11fip4 forward, 5’- CAGGAGTTAGACATGGACAGTC −3’; Rab11fip4 reverse, 5’- ACAGATGGAGTGAGGACGAG −3’; Traf3ip3 forward, 5’- GGCCAAGTGTCTATCCTCAAG −3’; Traf3ip3 reverse, 5’- CCTCAGAGCTTATCCCTGTTTC −3’; Plg2 forward, 5’- GGGTACAGTGAAGACATCGAG −3’; Plg2 reverse, 5’- TCGTAAAGGAAGAGCTGGTTG −3’; S100a8 forward, 5’- AGTGTCCTCAGTTTGTGCAG −3’; S100a8 reverse, 5’- ACTCCTTGTGGCTGTCTTTG −3’.
Immunoblotting
For WB of cell lysates, treated HUVEC were rinsed once in ice-cold phosphate-buffered saline (PBS) and lysed in radioimmune precipitation assay (RIPA) buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1% (v/v) Triton X-100, 0.1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 1 mM EDTA/EGTA/sodium orthovanadate, 10 mM β-glycerophosphate, and protease inhibitors (1 mM phenylmethylsulfonyl fluoride and 10 μg/ml leupeptin/tosylphenylalanyl chloromethyl ketone/aprotinin each)] for 15 min rocking on ice. Lysates were boiled for 3 min and resolved on SDS-PAGE as described before [108–110]. Proteins were then transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA), probed with primary and secondary antibodies, and developed with SuperSignal West Pico Chemiluminescence substrate (Thermo Fisher Scientific). Signal detection was performed via an ImageQuant LAS-4000 (GE Healthcare) platform.
Imaging studies
To process mouse tissue for IHC immediately following euthanasia, a portion of the organs were fixed in 4% (w/v) paraformaldehyde (T353–500, Fisher) for 4 h at RT (room temperature), then 15% sucrose (w/v in PBS) (S6–500, Fisher) for 4 h at RT and 30% (w/v in PBS) sucrose overnight at 4°C. Immunofluorescence was performed as described before [108,111]. Tissue were then embedded into OCT (4583, Tissue Tek), cut into 5 μm sections and mounted onto glass slides as previously described [83]. The following primary and secondary reagents were used to probe tissue sections for IHC: rat CD31 (553370, 1:100 dilution; BD Biosciences), mouse HAS2 (sc-365263, 1:50 dilution; Santa Cruz Biotechnology), rabbit perlecan domain V (1:150 dilution, custom manufactured) [63], rat perlecan domain IV (ab2501, 1:100 dilution; Abcam), mouse histidine (ab18184, 1:200 dilution; Abcam), biotinylated HA binding protein (385911, 5 μg/ml; Sigma), Alexa Fluor 594 goat anti-rat H+L IgG (A11007, 1:400; Invitrogen), Alexa Fluor 568 rabbit anti-mouse H+L IgG (A11061, 1:400; Invitrogen), Alexa Fluor 594 goat anti-rabbit H+L IgG (A11037, 1:400; Invitrogen), Alexa Fluor 594 Streptavidin (S32356, 1:400; Thermofisher), and Alexa Fluor 488 Streptavidin (S11223, 1:400; Thermofisher). Sections were mounted with Hardset antifade mounting medium with DAPI (H-1500, Vectashield) and visualized with an upright fluorescence microscope (DM5500B, Leica).
RNAseq data processing for differential expression
To investigate the effects of systemic endorepellin on the tumor transcriptome, we examined the gene expression signature of the allografts at day 22 post-treatment by performing high-throughput deep RNAseq (Genewiz, NJ), RNAseq data is publicly available as a gene expression omnibus (GEO) dataset (accession GSE209743). Following capillary electrophoresis to validate RNA integrity, mRNA was enriched and sequenced utilizing the Illumina® HiSeq® system configured at 2×150 bp. Sequence reads were trimmed to remove possible adapter sequences and poor-quality nucleotides using Trimmomatic v.0.3695 and mapped to the Mus musculus GRCm38 reference genome (ENSEMBL) using STAR aligner v.2.5.2b96.
A matrix of raw counts was obtained from Genewiz. Mouse ensemble IDs were converted to gene symbols using the biomaRt package in ‘R’. Initial filtering for genes with low abundance was performed by removing all genes with less than ten total counts across all samples, leaving 25,349 genes for downstream analysis. Differentially analysis was performed in DESeq2 [112] with the likelihood ratio test across all samples to identify genes that were dysregulated in the treatment group compared to control. The final list of differentially expressed genes were those with a p < 0.05 and log2FoldChange > 1 (greater than 2-fold change), yielding 294 genes upregulated and 58 genes downregulated in the treatment samples compared to control. A regularized log transformation was carried out using DESeq2 [112] for downstream visualization of differentially expressed genes. The transformed data was median centered for heatmap visualizations.
Gene ontology and pathway analyses of RNAseq data
Analyses of upregulated and downregulated genes listed (Supplementary Tables 1–6) were performed with bioinformatic tools as previously described [113]. The BiNGO tool [86] was used to determine the Gene Ontology (GO) terms that are statistically overrepresented in the upregulated and downregulated genes. Mus musculus was used as the reference organism, and the p-value was set at p<0.0005 and p<0.01 for enrichement analysis of the GO terms “Biological Process” and “Molecular Function” respectively. Pathway overrepresentation was studied using the Reactome knowledgebase (https://reactome.org) [85].
Aortic ring assays in 3D collagen
To induce endorepellin overexpression in the endothelium, tamoxifen (2 mg dissolved in corn oil; Sigma, H7904) was administered i.p. for 5 consecutive days in Tie2CreERT2;ERKi mice. After 3 days, mouse thoracic aortae were dissected and cross-sectioned into 1–2 mm rings. Rings were sandwiched between Collagen type I (1 mg/ml) and cultured in endothelial cell media at 37°C for 5–7 days as described previously [83,84,114]. Following fixation with 4% (w/v) paraformaldehyde and blocking in 2% (w/v) BSA (45 min), rings were incubated overnight at 4°C with anti-perlecan domain V antibody (1:50, custom rabbit antibody) then incubated with Alexa Fluor 594 donkey anti-rabbit H+L IgG (R37119, 1:400; ThermoFisher) and Alexa Fluor 488 Isolectin GS-IB4 (I21411, 1:200; ThermoFisher) for 3 h at RT. Confocal analysis via Zeiss LSM780 NLO multiphoton microscope and quantification of fluorescence intensity of angiogenic sprouts peripheral to the ring were performed as previously described [67,83,84].
Quantification and statistical analysis
Immunoblots were analyzed via scanning densitometry (ImageJ). Fluorescence was quantified and normalized using an unbiased method with the same exposure, gain, intensity, and objective. Images were first captured in the vehicle-treated samples then tamoxifen-treated samples. All initial fluorescent images were captured with the exact same gain, intensity, and brightness/contrast. For quantification, the images were processed through ImageJ by initially performing a standardized background subtraction. Then, images were further processed by splitting the color channels into greyscale images. This allowed the images to be assessed empirically by measuring the mean grey value of the sections which directly corresponded to fluorescent intensity. At least 10 images were taken per sample and subsequently averaged to generate the most representative value for each sample. The quantification presented is the arbitrary value for the average of mean grey values for all sections per sample.
Significance in experiments with two groups was determined by two-tailed unpaired Student’s t test, whereas statistical analyses for experiments with more than two groups were calculated via One-way ANOVA, followed by Tukey’s post hoc test. Mean differences were considered statistically significant at p < 0.05.
Supplementary Material
Acknowledgments
We thank Drs. B. Eliceiri and M. Risbud for generously providing the Tie2CreERT2 and mT/mG mice, respectively. We also thank Dr. M. Mongiat for providing the endorepellin sequence. We thank all the members of the Iozzo laboratory for helpful scientific input and M. Gubbiotti for her assistance in isolating ERKi lung fibroblasts. This work was supported in part by NIH grants CA039481 and CA245311 to RVI. Carolyn Chen was supported by a Dubbs Scholar Fellowship Award from Thomas Jefferson University and by the NIH training grant T32 AR052273.
Abbreviations
- ECM
extracellular matrix
- GAG
glycosaminoglycan
- HA
hyaluronan
- HAS
hyaluronan synthase
- TGFβ
transforming growth factor β
- VEGFR2
vascular endothelial growth factor 2
- LG
laminin-like globular
- HUVEC
human umbilical vein endothelial cells
- PE
phosphatidylethanolamine
- ATG
autophagy protein 5
- ERKi
endorepellin knock-in
- HABP
hyaluronan binding protein
- GO
gene ontology
- BiNGO
Biological Networks Gene Ontology
Footnotes
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.matbio.2023.03.005.
Data availability
All data are contained within the article and supplementary material. The RNAseq data can be accessed online as a GEO database under accession GSE209743.
References
- [1].Iozzo RV, Gubbiotti MA, Extracellular matrix: The driving force of mammalian diseases, Matrix Biol 71–72 (2018) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mongiat M, Buraschi S, Andreuzzi E, Neill T, Iozzo RV, Extracellular matrix: the gatekeeper of tumor angiogenesis, Biochem. Soc. Trans 47 (2019) 1543–1555. [DOI] [PubMed] [Google Scholar]
- [3].Karamanos NK, Theocharis AD, Neill T, Iozzo RV, Matrix modeling and remodeling: A biological interplay regulating tissue homeostasis and diseases, Matrix Biol. 75–76 (2019) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gialeli C, Theocharis AD, Karamanos NK, Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting, FEBS J. 278 (2011) 16–27. [DOI] [PubMed] [Google Scholar]
- [5].Sanderson RD, Elkin M, Rapraeger AC, Ilan N, Vlodavsky, Heparanase regulation of cancer, autophagy and inflammation: New mechanisms and targets for therapy, FEBS J. 284 (2016) 42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lu P, Takai K, Weaver VM, Werb Z, Extracellular matrix degradation and remodeling in development and disease, Cold Spring Harb. Perspect. Biol 3 (2011) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Simpson MA, Concurrent expression of hyaluronan biosynthetic and processing enzymes promotes growth and vascularization of prostate tumors in mice, Am. J. Pathol 169 (2006) 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Simpson MA, de la Motte C, Sherman LS, Weigel PH, Advances in hyaluronan biology: Signaling, regulation, and disease mechanisms, Int. J. Cell Biol 2015 (2015) 690572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tolg C, Yuan H, Flynn SM, Basu K, Ma J, Tse KCK, Kowalska B, Vulkanesku D, Cowman MK, McCarthy JB, Turley EA, Hyaluronan modulates growth factor induced mammary gland branching in a size dependent manner, Matrix Biol 63 (2017) 117–132. [DOI] [PubMed] [Google Scholar]
- [10].Slevin M, Kumar S, Gaffney J, Angiogenic oligosaccharides of hyaluronan induce multiple signaling pathways affecting vascular endothelial cell mitogenic and wound healing responses, J. Biol. Chem 277 (2002) 41046–41059. [DOI] [PubMed] [Google Scholar]
- [11].Garantziotis S, Savani RC, Hyaluronan biology: A complex balancing act of structure, function, location and context, Matrix Biol 78–79 (2019) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chanmee T, Ontong P, Itano N, Hyaluronan: A modulator of the tumor microenvironment, Cancer Lett. 375 (2016) 20–30. [DOI] [PubMed] [Google Scholar]
- [13].Slevin M, Krupinski J, Gaffney J, Matou S, West D, Delisser H, Savani RC, Kumar S, Hyaluronan-mediated angiogenesis in vascular disease: uncovering RHAMM and CD44 receptor signaling pathways, Matrix Biol 26 (2007) 58–68. [DOI] [PubMed] [Google Scholar]
- [14].Heldin P, Basu K, Olofsson B, Porsch H, Kozlova I, Kahata K, Deregulation of hyaluronan synthesis, degradation and binding promotes breast cancer, J. Biochem 154 (2013) 395–408. [DOI] [PubMed] [Google Scholar]
- [15].Auvinen P, Tammi R, Kosma VM, Sironen R, Soini Y, Mannermaa A, Tumelius R, Uljas E, Tammi M, Increased hyaluronan content and stromal cell CD44 associate with HER2 positivity and poor prognosis in human breast cancer, Int. J. Cancer 132 (2013) 531–539. [DOI] [PubMed] [Google Scholar]
- [16].Casalini P, Carcangiu ML, Tammi R, Auvinen P, Kosma VM, Valagussa P, Greco M, Balsari A, Ménard S, Tagliabue E, Two distinct local relapse subtypes in invasive breast cancer: effect on their prognostic impact, Clin. Cancer Res 14 (2008) 25–31. [DOI] [PubMed] [Google Scholar]
- [17].Kobayashi N, Miyoshi S, Mikami T, Koyama H, Kitazawa M, Takeoka M, Sano K, Amano J, Isogai Z, Niida S, Oguri K, Okayama M, McDonald JA, Kimata K, Taniguchi S, Itano N, Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization, Cancer Res. 70 (2010) 7073–7083. [DOI] [PubMed] [Google Scholar]
- [18].Whitelock JM, Murdoch AD, Iozzo RV, Underwood PA, The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin and heparanases, J. Biol. Chem 271 (1996) 10079–10086. [DOI] [PubMed] [Google Scholar]
- [19].Whitelock JM, Melrose J, Iozzo RV, Diverse cell signaling events modulated by perlecan, Biochemistry 47 (2008) 11174–11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Iozzo RV, Biosynthesis of heparan sulfate proteoglycan by human colon carcinoma cells and its localization at the cell surface, J. Cell Biol 99 (1984) 403–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hassell JR, Robey PG, Barrach HJ, Wilczek J, Rennard SI, Martin GR, Isolation of a heparan sulfate-containing proteoglycan from basement membrane, Proc. Natl. Acad. Sci. USA 77 (1980) 4494–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Murdoch AD, Dodge GR, Cohen I, Tuan RS, Iozzo RV, Primary structure of the human heparan sulfate proteoglycan from basement membrane (HSPG2/perlecan). A chimeric molecule with multiple domains homologous to the low density lipoprotein receptor, laminin, neural cell adhesion molecules, and epidermal growth factor, J. Biol. Chem 267 (1992) 8544–8557. [PubMed] [Google Scholar]
- [23].Cohen IR, Murdoch AD, Naso MF, Marchetti D, Berd D, Iozzo RV, Abnormal expression of perlecan proteoglycan in metastatic melanomas, Cancer Res. 54 (1994) 5771–5774. [PubMed] [Google Scholar]
- [24].Fuki I, Iozzo RV, Williams KJ, Perlecan heparan sulfate proteoglycan. A novel receptor that mediates a distinct pathway for ligand catabolism, J. Biol. Chem 275 (2000) 25742–25750. [DOI] [PubMed] [Google Scholar]
- [25].Whitelock JM, Iozzo RV, Heparan sulfate: a complex polymer charged with biological activity, Chem. Rev 105 (2005) 2745–2764. [DOI] [PubMed] [Google Scholar]
- [26].Sher I, Zisman-Rozen S, Eliahu L, Whitelock JM, Maas-Szabowski N, Yamada Y, Breitkreutz D, Fusenig NE, Arikawa-Hirasawa E, Iozzo RV, Bergman R, Ron D, Targeting perlecan in human keratinocytes reveals novel roles for perlecan in epidermal formation, J. Biol. Chem 281 (2006) 5178–5187. [DOI] [PubMed] [Google Scholar]
- [27].Templin AT, Mellati M, Soininen R, Hogan MF, Esser N, Castillo JJ, Zraika S, Kahn SE, Hull RL, Loss of perlecan heparan sulfate glycosaminoglycans lowers body weight and decreases islet amyloid deposition in human islet amyloid polypeptide transgenic mice, Protein Eng Des Sel 32 (2019) 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dodge GR, Kovalszky I, Hassell JR, Iozzo RV, Transforming growth factor β alters the expression of heparan sulfate proteoglycan in human colon carcinoma cells, J. Biol. Chem 265 (1990) 18023–18029. [PubMed] [Google Scholar]
- [29].Grässel S, Cohen IR, Murdoch AD, Eichstetter I, Iozzo RV, The proteoglycan perlecan is expressed in the erythroleukemia cell line K562 and is upregulated by sodium butyrate and phorbol ester, Mol. Cell. Biochem 145 (1995) 61–68. [DOI] [PubMed] [Google Scholar]
- [30].Cohen IR, Grässel S, Murdoch AD, Iozzo RV, Structural characterization of the complete human perlecan gene and its promoter, Proc. Natl. Acad. Sci. U. S. A 90 (1993) 10404–10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Iozzo RV, Pillarisetti J, Sharma B, Murdoch AD, Danielson KG, Uitto J, Mauviel A, Structural and functional characterization of the human perlecan gene promoter. Transcriptional activation by transforming factor-β via a nuclear factor 1-binding element, J. Biol. Chem 272 (1997) 5219–5228. [DOI] [PubMed] [Google Scholar]
- [32].Iozzo RV, Danielson KG, Transcriptional and post-transcriptional control of proteoglycan gene expression, Progr. Nucl. Acids Res. Mol. Biol 62 (1999) 19–53. [DOI] [PubMed] [Google Scholar]
- [33].Lord MS, Jung M, Cheng B, Whitelock JM, Transcriptional complexity of the HSPG2 gene in the human mast cell line, HMC-1, Matrix Biol. 35 (2014) 123–131. [DOI] [PubMed] [Google Scholar]
- [34].Zoeller JJ, McQuillan A, Whitelock J, Ho S-Y, Iozzo RV, A central function for perlecan in skeletal muscle and cardiovascular development, J. Cell Biol 181 (2008) 381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zoeller JJ, Whitelock J, Iozzo RV, Perlecan regulates developmental angiogenesis by modulating the VEGF-VEGFR2 axis, Matrix Biol. 28 (2009) 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aviezer D, Iozzo RV, Noonan DM, Yayon A, Suppression of autocrine and paracrine functions of basic fibroblast growth factor by stable expression of perlecan antisense cDNA, Mol. Cell. Biol 17 (1997) 1938–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Iozzo RV, Cohen I, Altered proteoglycan gene expression and the tumor stroma, Experientia 49 (1993) 447–455. [DOI] [PubMed] [Google Scholar]
- [38].Aviezer D, Hecht D, Safran M, Eisinger M, David G, Yayon A, Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis, Cell 79 (1994) 1005–1013. [DOI] [PubMed] [Google Scholar]
- [39].Whitelock JM, Graham LD, Melrose, Murdoch AD, Iozzo RV, Underwood PA, Human perlecan immunopurified from different endothelial cell sources has different adhesive properties for vascular cells, Matrix Biol. 18 (1999) 163–178. [DOI] [PubMed] [Google Scholar]
- [40].Gonzalez EM, Mongiat M, Slater SJ, Baffa R, Iozzo RV, A novel interaction between perlecan protein core and progranulin: Potential effects on tumor growth, J. Biol. Chem 278 (2003) 38113–38116. [DOI] [PubMed] [Google Scholar]
- [41].Chuang CY, Lord MS, Melrose J, Rees MD, Knox SM, Freeman C, Iozzo RV, Whitelock J, Heparan sulfate-dependent signaling of fibroblast growth growth factor 18 by chondrocyte-derived perlecan, Biochemistry 49 (2010) 5524–5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lord MS, Chuang CY, Melrose J, Davies MJ, Iozzo RV, Whitelock JM, The role of vascular-derived perlecan in modulating cell adhesion, proliferation and growth factor signaling, Matrix Biol 35 (2014) 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nakamura K, Ikeuchi T, Nara K, Rhodes CS, Zhang P, Chiba Y, Kazuno S, Miura Y, Ago T, Arikawa-Hirasawa E, Mukouyama YS, Yamada Y, Perlecan regulates pericyte dynamics in the maintenance and repair of the blood-brain barrier, J. Cell Biol 218 (2019) 3506–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ocken AR, Ku MM, Kinzer-Ursem TL, Calve S, Perlecan knockdown significantly alters extracellular matrix composition and organization during cartilage development, Mol. Cell Proteomics 19 (2020) 1220–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mongiat M, Otto J, Oldershaw R, Ferrer F, Sato JD, Iozzo RV, Fibroblast growth factor-binding protein is a novel partner for perlecan protein core, J. Biol. Chem 276 (2001) 10263–10271. [DOI] [PubMed] [Google Scholar]
- [46].Mongiat M, Fu J, Oldershaw R, Greenhalgh R, Gown A, Iozzo RV, Perlecan protein core interacts with extracellular matrix protein 1 (ECM1), a glycoprotein involved in bone formation and angiogenesis, J. Biol. Chem 278 (2003) 17491–17499. [DOI] [PubMed] [Google Scholar]
- [47].Mongiat M, Sweeney S, San Antonio JD, Fu J, Iozzo RV, Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan, J. Biol. Chem 278 (2003) 4238–4249. [DOI] [PubMed] [Google Scholar]
- [48].Gonzalez EM, Reed CC, Bix G, Fu J, Zhang Y, Gopalakrishnan B, Greenspan DS, Iozzo RV, BMP-1/Tolloid-like metalloproteases process endorepellin, the angiostatic C-terminal fragment of perlecan, J. Biol. Chem 280 (2005) 7080–7087. [DOI] [PubMed] [Google Scholar]
- [49].Iozzo RV, Schaefer L, Proteoglycan form and function: A comprehensive nomenclature of proteoglycans, Matrix Biol. 42 (2015) 11–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chen CG, Iozzo RV, Extracellular matrix guidance of autophagy: a mechanism regulating cancer growth, Open. Biol 12 (2022) 210304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gubbiotti MA, Neill T, Iozzo RV, A current view of perlecan in physiology and pathology: A mosaic of functions, Matrix Biol 57–58 (2017) 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Neill T, Andreuzzi E, Wang Z-X, Peiper SC, Mongiat M, Iozzo RV, Endorepellin remodels the endothelial transcriptome toward a pro-autophagic and pro-mitophagic gene signature, J. Biol. Chem 293 (2018) 12137–12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Karamanos NK, Piperigkou Z, Theocharis AD, Watanabe H, Franchi M, Baud S, Brezillon S, Gotte M, Passi A, Vigetti D, Ricard-Blum S, Sanderson RD, Neill T, Iozzo RV, Proteoglycan chemical diversity drives multifunctional cell regulation and therapeutics, Chem. Rev 118 (2018) 9152–9232. [DOI] [PubMed] [Google Scholar]
- [54].Schaefer L, Tredup C, Gubbiotti MA, Iozzo RV, Proteoglycan neofunctions: regulation of inflammation and autophagy in cancer biology, FEBS J. 284 (2017) 10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Curran CS, Keely PJ, Breast tumor and stromal cell response to TGF-β and hypoxia in matrix deposition, Matrix Biol. 32 (2013) 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Neill T, Buraschi S, Kapoor A, Iozzo RV, Proteoglycan-driven autophagy: A nutrient-independent mechanism to control intracellular catabolism, J. Histochem. Cytochem 68 (2020) 733–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Neill T, Schaefer L, Iozzo RV, Instructive roles of extracellular matrix on autophagy, Am. J. Pathol 184 (2014) 2146–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Neill T, Schaefer L, Iozzo RV, Decoding the matrix: Instructive roles of proteoglycan receptors, Biochemistry 54 (2015) 4583–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Woodall BP, Nyström A, Iozzo RA, Eble JA, Niland S, Krieg T, Eckes B, Pozzi A, Iozzo RV, Integrin α2β1 is the required receptor for endorepellin angiostatic activity, J. Biol. Chem 283 (2008) 2335–2343. [DOI] [PubMed] [Google Scholar]
- [60].San Antonio JD, Zoeller JJ, Habursky K, Turner K, Pimtong W, Burrows M, Choi S, Basra S, Bennett JS, DeGrado WF, Iozzo RV, A key role for the integrin α2β1 in experimental and developmental angiogenesis, Am. J. Pathol 175 (2009) 1338–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Simons M, Gordon E, Claesson-Welsh L, Mechanisms and regulation of endothelial VEGF receptor signalling, Nat. Rev. Mol. Cell Biol 17 (2016) 611–625. [DOI] [PubMed] [Google Scholar]
- [62].Goyal A, Pal N, Concannon M, Paulk M, Doran M, Poluzzi C, Sekiguchi K, Whitelock JM, Neill T, Iozzo RV, Endorepellin, the angiostatic module of perlecan, interacts with both the α2β1 integrin and vascular endothelial growth factor receptor 2 (VEGFR2), J. Biol. Chem 286 (2011) 25947–25962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bix G, Fu J, Gonzalez E, Macro L, Barker A, Campbell S, Zutter MM, Santoro SA, Kim JK, Höök M, Reed CC, Iozzo RV, Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through the α2β1 integrin, J. Cell Biol 166 (2004) 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chen CG, Iozzo RV, Angiostatic cues from the matrix: endothelial cell autophagy meets hyaluronan biology, J. Biol Chem 295 (2020) 16797–16812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Nyström A, Shaik ZP, Gullberg D, Krieg T, Eckes B, Zent R, Pozzi A, Iozzo RV, Role of tyrosine phosphatase SHP-1 in the mechanism of endorepellin angiostatic activity, Blood 114 (2009) 4897–4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bix G, Castello R, Burrows M, Zoeller JJ, Weech M, Iozzo RA, Cardi C, Thakur MT, Barker CA, Camphausen KC, Iozzo RV, Endorepellin in vivo: targeting the tumor vasculature and retarding cancer growth and metabolism, J. Natl. Cancer Inst 98 (2006) 1634–1646. [DOI] [PubMed] [Google Scholar]
- [67].Chen CG, Gubbiotti MA, Kapoor A, Han X, Yu Y, Linhardt RJ, Iozzo RV, Autophagic degradation of HAS2 in endothelial cells: A novel mechanism to regulate angiogenesis, Matrix Biol 90 (2020) 1–19. [DOI] [PubMed] [Google Scholar]
- [68].Poluzzi C, Casulli J, Goyal A, Mercer TJ, Neill T, Iozzo RV, Endorepellin evokes autophagy in endothelial cells, J. Biol. Chem 289 (2014) 16114–16128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Goyal A, Gubbiotti MA, Chery DR, Han L, Iozzo RV, Endorepellin-evoked autophagy contributes to angiostasis, J. Biol. Chem 291 (2016) 19245–19256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ye X, Zhou XJ, Zhang H, Exploring the role of autophagy-related gene 5 (ATG5) yields important insights into autophagy in autoimmune/autoinflammatory diseases, Front Immunol. 9 (2018) 2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N, Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice, Nature 441 (2006) 885–889. [DOI] [PubMed] [Google Scholar]
- [72].Nyabi O, Naessens M, Haigh K, Gembarska A, Goossens S, Maetens M, De Clercq S, Drogat B, Haenebalcke L, Bartunkova S, De Vos I, De Craene B, Karimi M, Berx G, Nagy A, Hilson P, Marine J-C, Haigh JJ, Efficient mouse transgenesis using Gateway-compatible ROSA26 locus targetting vectors and F1 hybrid ES cells, Nucleic Acids Res. 37 (2009) e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Casey A, Laster WR Jr., Ross GL, Sustained enhanced growth of carcinoma EO771 in C57 black mice, Proc. Soc. Exp. Biol Med 77 (1951) 358–362. [DOI] [PubMed] [Google Scholar]
- [74].Johnstone CN, Smith YE, Cao Y, Burrows AD, Cross RS, Ling X, Redvers RP, Doherty JP, Eckhardt BL, Natoli AL, Restall CM, Lucas E, Pearson HB, Deb S, Britt KL, Rizzitelli A, Li J, Harmey JH, Pouliot N, Anderson RL, Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer, Dis. Model. Mech 8 (2015) 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ewens A, Luo L, Berleth E, Alderfer J, Wollman R, Hafeez BB, Kanter P, Mihich E, Ehrke MJ, Doxorubicin plus interleukin-2 chemoimmunotherapy against breast cancer in mice, Cancer Res. 66 (2006) 5419–5426. [DOI] [PubMed] [Google Scholar]
- [76].Dong D, Stapleton C, Luo B, Xiong S, Ye W, Zhang Y, Jhaveri N, Zhu G, Ye R, Liu Z, Bruhn KW, Craft N, Groshen S, Hofman FM, Lee AS, A critical role for GRP78/BiP in the tumor microenvironment for neovascularization during tumor growth and metastasis, Cancer Res. 71 (2011) 2848–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zou Z, Bellenger S, Massey KA, Nicolaou A, Geissler A, Bidu C, Bonnotte B, Pierre AS, Minville-Walz M, Rialland M, Seubert J, Kang JX, Lagrost L, Narce M, Bellenger J, Inhibition of the HER2 pathway by n-3 polyunsaturated fatty acids prevents breast cancer in fat-1 transgenic mice, J. Lipid Res 54 (2013) 3453–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Iozzo RV, Basement membrane proteoglycans: from cellar to ceiling, Nat. Rev. Mol. Cell Biol 6 (2005) 646–656. [DOI] [PubMed] [Google Scholar]
- [79].Forde A, Constien R, Gröne H-J, Hämmerling G, Arnold B, Temporal Cre-mediated recombination exclusively in endothelial cells using Tie2 regulatory elements, Genesis 33 (2002) 191–197. [DOI] [PubMed] [Google Scholar]
- [80].Lee J, Borboa AK, Chun HB, Baird A, Eliceiri BP, Conditional deletion of the focal adhesion kinase FAK alters remodeling of the blood-brain barrier in glioma, Cancer Res. 70 (2010) 10131–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Monkley SJ, Kostourou V, Spence L, Petrich B, Coleman S, Ginsberg MH, Pritchard CA, Critchley DR, Endothelial cell talin 1 is essential for embryonic angiogenesis, Dev. Biol 349 (2011) 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Chislock EM, Ring C, Pendergast AM, Abl kinases are required for vascular function, Tie2 expression, and angiopoietin-1-mediated survival, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 12432–12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Chen C, Kapoor A, Iozzo RV, Methods for monitoring matrix-induced autophagy, Methods Mol. Biol 1952 (2019) 157–191. [DOI] [PubMed] [Google Scholar]
- [84].Kapoor A, Chen CG, Iozzo RV, A simplified aortic ring assay: A useful ex vivo method to assess biochemical and functional parameters of angiogenesis, Matrix Biol Plus 6–7 (2020) 100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R, Loney F, May B, Milacic M, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Weiser J, Wu G, Stein L, Hermjakob H, D’Eustachio P, The reactome pathway knowledgebase, Nucleic Acids Res. 48 (2020) D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Maere S, Heymans K, Kuiper M, BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks, Bioinformatics. 21 (2005) 3448–3449. [DOI] [PubMed] [Google Scholar]
- [87].Sonnenschein C, Soto AM, Carcinogenesis explained within the context of a theory of organisms, Prog. Biophys. Mol. Biol 122 (2016) 70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Maffini MV, Calabro JM, Soto AM, Sonnenschein C, Stromal regulation of neoplastic development: age-dependent normalization of neoplastic mammary cells by mammary stroma, Am. J. Pathol 167 (2005) 1405–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Barcellos-Hoff MH, Ravani SA, Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells, Cancer Res. 60 (2000) 1254–1260. [PubMed] [Google Scholar]
- [90].Guerra L, Odorisio T, Zambruno G, Castiglia D, Stromal microenvironment in type VII collagen-deficient skin: The ground for squamous cell carcinoma development, Matrix Biol 63 (2017) 1–10. [DOI] [PubMed] [Google Scholar]
- [91].Passi A, Vigetti D, Buraschi S, Iozzo RV, Dissecting the role of hyaluronan synthases in the tumor microenvironment, FEBS J. 286 (2019) 2937–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Li P, Xiang T, Li H, Li Q, Yang B, Huang J, Zhang X, Shi Y, Tan J, Ren G, Hyaluronan synthase 2 overexpression is correlated with the tumorigenesis and metastasis of human breast cancer, Int. J. Clin. Exp. Pathol 8 (2015) 12101–12114. [PMC free article] [PubMed] [Google Scholar]
- [93].Bernert B, Porsch H, Heldin P, Hyaluronan synthase 2 (HAS2) promotes breast cancer cell invasion by suppression of tissue metalloproteinase inhibitor 1 (TIMP-1), J. Biol Chem 286 (2011) 42349–42359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Porsch H, Bernert B, Mehic M, Theocharis AD, Heldin CH, Heldin P , Efficient TGFβ-induced epithelial-mesenchymal transition depends on hyaluronan synthase HAS2, Oncogene 32 (2013) 4355–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Li Y, Li L, Brown TJ, Heldin P, Silencing of hyaluronan synthase 2 suppresses the malignant phenotype of invasive breast cancer cells, Int. J. Cancer 120 (2007) 2557–2567. [DOI] [PubMed] [Google Scholar]
- [96].Udabage L, Brownlee GR, Waltham M, Blick T, Walker EC, Heldin P, Nilsson SK, Thompson EW, Brown TJ, Antisense-mediated suppression of hyaluronan synthase 2 inhibits the tumorigenesis and progression of breast cancer, Cancer Res. 65 (2005) 6139–6150. [DOI] [PubMed] [Google Scholar]
- [97].Passi A, Vigetti D, Hyaluronan as tunable drug delivery system, Adv. Drug Deliv. Rev 146 (2019) 83–96. [DOI] [PubMed] [Google Scholar]
- [98].Auvinen P, Rilla K, Tumelius R, Tammi M, Sironen R, Soini Y, Kosma VM, Mannermaa A, Viikari J, Tammi R, Hyaluronan synthases (HAS1–3) in stromal and malignant cells correlate with breast cancer grade and predict patient survival, Breast Cancer Res. Treat 143 (2014) 277–286. [DOI] [PubMed] [Google Scholar]
- [99].Gialeli C, Viola M, Barbouri D, Kletsas D, Passi A, Karamanos NK, Dynamic interplay between breast cancer cells and normal endothelium mediates the expression of matrix macromolecules, proteasome activity and functional properties of endothelial cells, Biochim. Biophys. Acta 1840 (2014) 2549–2559. [DOI] [PubMed] [Google Scholar]
- [100].Tammi RH, Kultti A, Kosma VM, Pirinen R, Auvinen P, Tammi MI, Hyaluronan in human tumors: pathobiological and prognostic messages from cell-associated and stromal hyaluronan, Semin. Cancer Biol 18 (2008) 288–295. [DOI] [PubMed] [Google Scholar]
- [101].Sato N, Maehara N, Goggins M, Gene expression profiling of tumor-stromal interactions between pancreatic cancer cells and stromal fibroblasts, Cancer Res. 64 (2004) 6950–6956. [DOI] [PubMed] [Google Scholar]
- [102].Elaimy AL, Amante JJ, Zhu LJ, Wang M, Walmsley CS, FitzGerald TJ, Goel HL, Mercurio AM, The VEGF receptor neuropilin 2 promotes homologous recombination by stimulating YAP/TAZ-mediated Rad51 expression, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 14174–14180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Kim J, Kim YH, Kim J, Park DY, Bae H, Lee DH, Kim KH, Hong SP, Jang SP, Kubota Y, Kwon YG, Lim DS, Koh GY, YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation, J. Clin. Invest 127 (2017) 3441–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Mo JS, Meng Z, Kim YC, Park HW, Hansen CG, Kim S, Lim DS, Guan KL, Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway, Nat. Cell Biol 17 (2015) 500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Wang W, Xiao ZD, Li X, Aziz KE, Gan B, Johnson RL, Chen J, AMPK modulates Hippo pathway activity to regulate energy homeostasis, Nat. Cell Biol 17 (2015) 490–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Black LM, Farrell ER, Barwinska D, Osis G, Zmijewska AA, Traylor AM, Esman SK, Bolisetty S, Whipple G, Kamocka MM, Winfree S, Spangler DR, Khan S, Zarjou A, El-Achkar TM, Agarwal A, VEGFR3 tyrosine kinase inhibition aggravates cisplatin nephrotoxicity, Am. J. Physiol Renal Physiol 321 (2021) F675–F688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Poluzzi C, Iozzo RV, Schaefer L, Endostatin and endorepellin: A common route of action for similar angiostatic cancer avengers, Adv. Drug Deliv. Rev 97 (2016) 156–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Rudnicka L, Varga J, Christiano AM, Iozzo RV, Jimenez SA, Uitto J, Elevated expression of type VII collagen in the skin of patients with systemic sclerosis, J. Clin. Invest 93 (1994) 1709–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Goldoni S, Owens RT, McQuillan DJ, Shriver Z, Sasisekharan R, Birk DE, Campbell S, Iozzo RV, Biologically active decorin is a monomer in solution, J. Biol. Chem 279 (2004) 6606–6612. [DOI] [PubMed] [Google Scholar]
- [110].Alvarez RJ, Sun MJ, Haverty TP, Iozzo RV, Myers JC, Neilson EG, Biosynthetic and proliferative characteristics of tubulointerstitial fibroblasts probed with paracrine cytokines, Kidney Int. 41 (1992) 14–23. [DOI] [PubMed] [Google Scholar]
- [111].Ryynänen M, Ryynänen J, Solberg S, Iozzo RV, Knowlton RG, Uitto J, Genetic linkage of Type VII collagen (COL7A1) to dominant dystrophic epidermolysis bullosa in families with abnormal anchoring fibrils, J. Clin. Invest 89 (1992) 974–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol 15 (2014) 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Vallet SD, Clerc O, Ricard-Blum S, Glycosaminoglycan-protein interactions: The first draft of the glycosaminoglycan interactome, J. Histochem. Cytochem 69 (2021) 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Baker M, Robinson SD, Lechertier T, Barber PR, Tavora B, D’Amico G, Jones DT, Vojnovic B, Hodivala-Dilke K, Use of the mouse aortic ring assay to study angiogenesis, Nat. Protoc 7 (2012) 89–104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the article and supplementary material. The RNAseq data can be accessed online as a GEO database under accession GSE209743.