

Abstract
Hydroxychloroquine (HCQ) is an anti-malarial drug but also widely used to treat autoimmune diseases like arthritis and lupus. Although there have been multiple reports of the adverse effect of prolonged HCQ usage on the outer retina, leading to bull’s-eye maculopathy, the effect of HCQ toxicity on the inner retina as well as on overall visual functions has not been explored in detail. Furthermore, lack of an established animal model of HCQ toxicity hinders our understanding of the underlying molecular mechanisms. Here, using a small clinical study, we confirmed the effect of HCQ toxicity on the inner retina, in particular the reduction in central inner retinal thickness, and established a mouse model of chronic HCQ toxicity that recapitulates the effects observed in human retina. Using the mouse model, we demonstrated that chronic HCQ toxicity results in loss of inner retinal neurons and retinal ganglion cells (RGC) and compromises visual functions. We further established that HCQ treatment prevents autophagosome–lysosome fusion and alters the sphingolipid homeostasis in mouse retina. Our results affirm the notion that HCQ treatment causes early damage to the inner retina and affects visual functions before leading to characteristic toxicity in the macular region of the outer retina, ‘bull’s-eye maculopathy.’ We also provide insights into the underlying molecular mechanisms of HCQ retinal toxicity that may involve autophagy–lysosomal defects and alterations in sphingolipid metabolism.
Keywords: HCQ (Hydroxychloroquine), Retina, Autophagy, Lysosome, Sphingolipids
Introduction
Hydroxychloroquine (HCQ) is an anti-malarial drug well known for its anti-inflammatory and analgesic properties and widely used in various autoimmune diseases, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and also in dermatologic conditions [1]. HCQ received substantial attention recently with regard to its possible beneficial effect on COVID19 infection, which ultimately resulted in unresolved controversies rather than in any clear indications [2, 3]. HCQ is derived from chloroquine (CQ), which replaced the use of CQ in most of the world due to its lesser systemic toxicity and better tolerability [4, 5]. CQ and HCQ are known to cause retinal toxicity [6–8]. Although HCQ retinal toxicity is an infrequent event, HCQ use is still a concern because the resulting vision loss can be potentially severe and irreversible [8, 9]. Classical CQ and HCQ retinal toxicities are clinically described as pigment mottling within the macular area and present primarily in the outer retina, creating a characteristic ‘bull’s-eye maculopathy’ (BEM) [10, 11]. In later stages of toxicity, atrophy of the retinal pigment epithelium (RPE) and neurosensory retina spreads outward over the entire fundus. CQ and HCQ retinal atrophy is generally irreversible and may progress even after discontinuation of the medication [10, 12–14]. Recent studies suggest that the outer retinal toxicity represents end-stage, irreversible pathology, and that these drugs affect inner retina long before developing outer retinal pathology [15]. With the aid of modern retinal imaging technology such as multifocal electroretinography (mf-ERG) and spectral-domain optical coherence tomography (SD-OCT) and advanced methods of analysis, significant alteration has been reported in the macular region, especially affecting the retinal ganglion cells (RGC) and inner plexiform layers (IPL) [11, 16]. A clearer understanding of the mechanisms of actions of HCQ in retina could facilitate early detection of initial stages of pathology and provide an indication of patients who should discontinue the drug to avoid permanent vision loss.
The mechanism of CQ and HCQ retinal toxicity is not well understood. Although there have been studies on animal models for determining general pathology of CQ toxicity in 1960s to 1980s (mouse, rats, cat, monkey), there is no reported study on toxicity, pathology and mechanism of HCQ retinal toxicity in animal models [17–20]. Chronic exposure to CQ has been shown to result in marked histopathological changes in inner retinal neurons, specifically affecting retinal ganglion cells [17, 20]. The lack of animal models has hampered attempts to understand the mechanisms of retina-specific toxicity of HCQ at cellular and molecular levels in the human retina.
In this current report, we used retinal SD-OCT imaging in patients with long-term HCQ usage to confirm that inner retinal changes occur prior to the appearance of outer retinal toxicity. Importantly, we were able to develop a mouse model of chronic HCQ exposure that expressed outer and inner retinal structural and functional changes that recapitulate the human retinal HCQ pathology. By biochemical and molecular analyses, we observed lysosomal dysfunction and lysosomal metabolism of certain lipids, specifically sphingolipids, which are associated with retinal dysfunction and cell death.
Materials and Methods
Study patients and retinal image analysis
In this study, we included data of nineteen patients (n = 19; 18 females, 1 male) from prospective recruitment (n = 12) and retrospective chart review (n = 7) with history of chronic use of HCQ to treat autoimmune diseases (both RA and SLE) at the Dean McGee Eye Institute, University of Oklahoma Health Sciences Centre (Table 1). We also included data from thirty-seven (n = 37) age-matched healthy control subjects (28 females, 9 males) (Table 1). All patients were examined by two authors (HKP and VS). This study followed the Declaration of Helsinki Principles and was approved by an institutional review board at the University of Oklahoma Health Sciences Center. Informed consent was obtained from all participants. Exclusion criteria included known retinal diseases, glaucoma, intraocular pressure higher than 20 mmHg or a history of ocular hypertension, uveitis, optic nerve diseases, refractive error of more than ± 6D sphere or ± 3D cylinder, previous intraocular or refractive surgery and image quality that precluded a high-quality OCT examination.
Table 1.
Description of human subjects recruited and analyzed to determine the effect of long-term HCQ treatment in central and inner retina. P-values are generated by comparing means by unpaired t test with Welch’s correction and by comparing variances by F test. A p- value < 0.05 is considered significant. (M: Median)
| Healthy Control | HCQ | P-values (t-test*; F-test#) | |
|---|---|---|---|
|
| |||
| Number | 37 | 19 | |
| Age | 57.66 ± 11.39 (M: 59.45) | 59.4 ± 11.92 (M: 61.96) | 0.60*; 0.78# |
| HCQ (mg/kg/day) | 5.04 ± 1.36 (M: 4.65) | ||
| HCQ Duration (year) | 8.36 ± 7.9 (M: 4.0) | ||
| HCQ Cumulative dose (gram) | 16.04 ± 16.72 (M: 5.76) | ||
| RNFL thickness (average of 4 quadrants from both eyes) (qM) | 90.16 ± 11.76 (M: 89.88) | 86.57 ± 12.31 (M: 84.75) | 0.23*; 0.79# |
| GC + IPL thickness (qM) | 78.12 ± 5.52 (M: 78.0) | 73.03 ± 14.44 (M: 78.5) | 0.043*; 0.0001# |
| Central (macular) cube volume (mm3) | 9.93 ± 0.38 (M: 9.95) | 9.51 ± 0.69 (M: 9.7) | 0.019*; 0.0023# |
| Central (macular) cube thickness (qM) | 276.24 ± 10.66 (M: 276.0) | 264.08 ± 18.98 (M: 269.5) | 0.016*; 0.0031# |
Spectral-domain optical coherence tomography (SD-OCT) imaging was performed using Zeiss Cirrus 4000 (Cirrus HD-OCT 4000, version 5.0; Carl Zeiss Meditec, Dublin, CA, USA). Macular scans were performed using the Radial Lines protocol, which provided twelve, 6-mm scans centered at the fovea. Scan acquisition time required for each of the Radial Lines scans was 0.27 s. The scans had a depth resolution of 3 mm per pixel and spatial resolution of 6 mm per pixel. Horizontal and vertical SD-OCT images from the Radial Lines scans were exported in TIF format. Automated image segmentation and analysis were performed using a dedicated software program developed in Matlab (The Mathworks Inc., Natick, MA, USA) and previously described [9, 21]. The program enabled the measurement of the thickness profiles for the entire retina and retinal layers. Retinal nerve fiber layer (RNFL) and ganglion cells + inner plexiform layer (GC + IPL) thickness data were collected at 200 μm intervals along 6 mm lengths of the radial line scans, along with measurements of central (macular) cube volume (mm3) and average central (macular) cube thickness (μM). Using GraphPad Prism software, the means were compared using unpaired t test with Welch’s correction and the variance were compared using F test. Linear correlation between HCQ duration and retinal measurements as well as cumulative dosage and retinal measurements was determined by Pearson correlation coefficient (r) from the treated group. Statistical significance was accepted at P < 0.05.
Animal care and HCQ treatment
All the animals utilized in this study were born and raised in the University of Tennessee Health Science Center (UTHSC) vivarium following its guidelines of animal housing. The mice were maintained in dim (5–10 lx) cyclic light (12 h. ON/OFF) from birth. For chronic HCQ treatment, 4–6-month-old C57/BL6 mice received intra-peritoneal (IP) injection of HCQ at a dose of 10 mg/kg body weight twice a week for three months, which is equivalent to 2.86 mg/kg/day. Human dosage ranged from 3.5 to 9.3 mg/kg/day with a median value of 4.65 mg/kg/day (Table 1). Thus, the mouse dose of HCQ was comparatively lower than human dosage. Littermate mice that were injected with saline served as controls. Visual functions were measured at one-month intervals starting prior to receiving HCQ injections and continuing through the end of treatment period. After each time point, mice were euthanized, and retinas were harvested for biochemical and histopathological analyses. All procedures were reviewed and approved by the UTHSC Institutional Animal Care and Use Committee (IACUC).
SD-OCT analysis of retinal layers in mice
The thickness of different retinal layers was measured by spectral-domain optical coherence tomography (SD-OCT) using Bioptigen Envisu Image Guided SD-OCT system (Lecia Microsystems, Buffalo Grove, IL). Briefly, after three months of HCQ treatment, mice from both treated and control groups were anesthetized and placed in the imaging platform. The posterior segment of the eye was imaged, and the thickness of different retinal layers was measured and analyzed using the manufacturer supplied software for small animal models.
Electroretinogram (ERG)
Both scotopic and photopic flash ERGs were recorded with Diagnosys Espion E2 ERG system (Diagnosys LLC, Lowell, MA) as previously described [22]. Detailed procedure is described in SI Methods.
Optokinetic reflex measurements
The visual acuity and contrast sensitivity were measured by optokinetic tracking (OKT) using OptoMotry system of Cerebral Mechanics (Lethbridge, AB, Canada). Both visual acuity and contrast sensitivity were measured for the control and HCQ-treated mice immediately before starting HCQ treatment and at monthly intervals until three months of treatment. The visual acuity was assessed at 100% contrast by varying spatial frequency threshold while the contrast sensitivity was measured at a 0.042 cycles per degree (c/d) spatial frequency.
Cell culture
Rat Retinal Muller cell line (rMC1) was obtained from Kerafast Inc., Boston, MA (Cat#ENW001) and cultured following the manufacturer guidelines. The cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2. For HCQ treatment, cells were plated in 12-well plates with 2 × 105 cells/well (passage 17–19) and allowed to grow for 24 h. They were treated with 20 μM of HCQ for 24 h. and subsequently harvested for biochemical and sphingolipid analyses.
Western Blot Analysis
Total retinal proteins were isolated from control and HCQ-treated mice retina as well as rMC1 cells using T-PER tissue protein extraction reagent (Thermo Scientific, Rockford, IL) containing protease inhibitor cocktail (Roche, Indianapolis, IN). Proteins were separated by denaturing gel electrophoresis and transferred to PVDF membrane using Bio-Rad (Hercules, CA) transfer module. The membrane was probed for different autophagosomal–lysosomal pathway proteins as mentioned in the Results section. The procedural details were described in SI Methods.
Immunofluorescence
For immunohistochemical analysis of mouse retina, whole eyes were harvested from control and treated groups after three months of HCQ treatment. The eyes were fixed in PREFER fixative for 20 min and embedded in paraffin and sectioned at 7 μm thickness. Retinal sections were probed for different autophagosomal–lysosomal pathway proteins. For immunocytochemical analysis, cells were seeded on glass coverslips and cultured overnight. Next day, cells were treated with 20 μM of HCQ for 24 h. After treatment period, cells were fixed with 4% paraformaldehyde and probed for different autophagosomal–lysosomal pathway proteins. All the images were obtained using Zeiss 710 confocal microscope. Detailed procedure can be found in SI Methods.
Sphingolipid Analysis
After three months of HCQ treatment, retinas from both HCQ-treated and untreated controls were harvested, snap-frozen in liquid nitrogen and stored in −80°C freezer until further analysis. For the rat Muller cells, after 24 h. of HCQ treatment, treated and untreated cells were snap-frozen in liquid nitrogen and stored in −80°C freezer until further analysis. The sphingolipid analysis was performed using LC–MS following previously published protocol [23, 24]. Detailed procedures are described in SI Methods.
Gene Expression Analysis
Total RNA was extracted from control and HCQ-treated mice after one- and three-month treatment using PureLink RNA Easy kit using manufacturer’s protocol (Life Technologies, CA). Quantitative RT-PCR was performed according to previously published procedure [24]. The primer sequences are listed in Supplementary Table 1.
Statistical Analysis
Statistical analyses was performed using GraphPad Prism 8 (GraphPad Software, San Diego, CA) and SPSS 22.0 (SPSS, Inc., Chicago, IL) software. Nonparametric unpaired t-test with Welch’s correction was used for comparing means and F test for comparing variances between the comparing groups. Multiple linear regression analyses were carried out to assess the impact of age on the outcome variables. Univariate linear correlation between HCQ duration and retinal measurements as well as cumulative dosage and retinal measurements was determined by Pearson correlation coefficient (r) from the treated group. Statistical significance was accepted at P < 0.05.
RESULTS
Long-term HCQ treatment causes inner retinal neuronal loss in human subjects
HCQ causes irreversible toxicity to the retina, called BEM, which is progressive despite discontinuation of the medication [25]. Recent evidence suggests HCQ toxicity is not limited to the RPE and outer retina but also affects inner retinal neurons [15, 16, 26]. To confirm the effect of HCQ in the inner retina, we analyzed retinal SD-OCT images of 19 human subjects who had received HCQ treatment for a range of 1–25 years. Five of them developed toxicity and discontinued the treatment. In this group, the median duration of HCQ was 4.0 years with a median daily dose of 4.65 mg/kg, resulting in a median cumulative dose of 5.76 g over the period of treatment (Table 1). We compared their data with the analysis of SD-OCT images from 37 age-matched healthy controls (Table 1). We found significant reductions in the central macular thickness (6 mm radial scan of the macula), central cube volume and in the thickness of the combined ganglion cell and inner plexiform layer (GC + IPL) in the HCQ-treated group (Table 1).
Interestingly, our correlation analysis revealed that all the three parameters of the central retinal analysis, namely central cube volume and GC + IPL thickness, were inversely correlated, significantly, with the duration of HCQ exposure (Fig. 1a and b) as well as with the cumulative dose (Fig. 1c and d). The effect of ‘age’ was not significant on the duration of HCQ exposure as well as the cumulative dose by multivariate regression analysis (Supplementary Table 2). Our results confirm that long-term HCQ treatment in humans causes inner retinal neuronal loss before developing the characteristic HCQ toxicity, i.e., bull’s-eye maculopathy and RPE atrophy [10, 11].
Fig. 1.
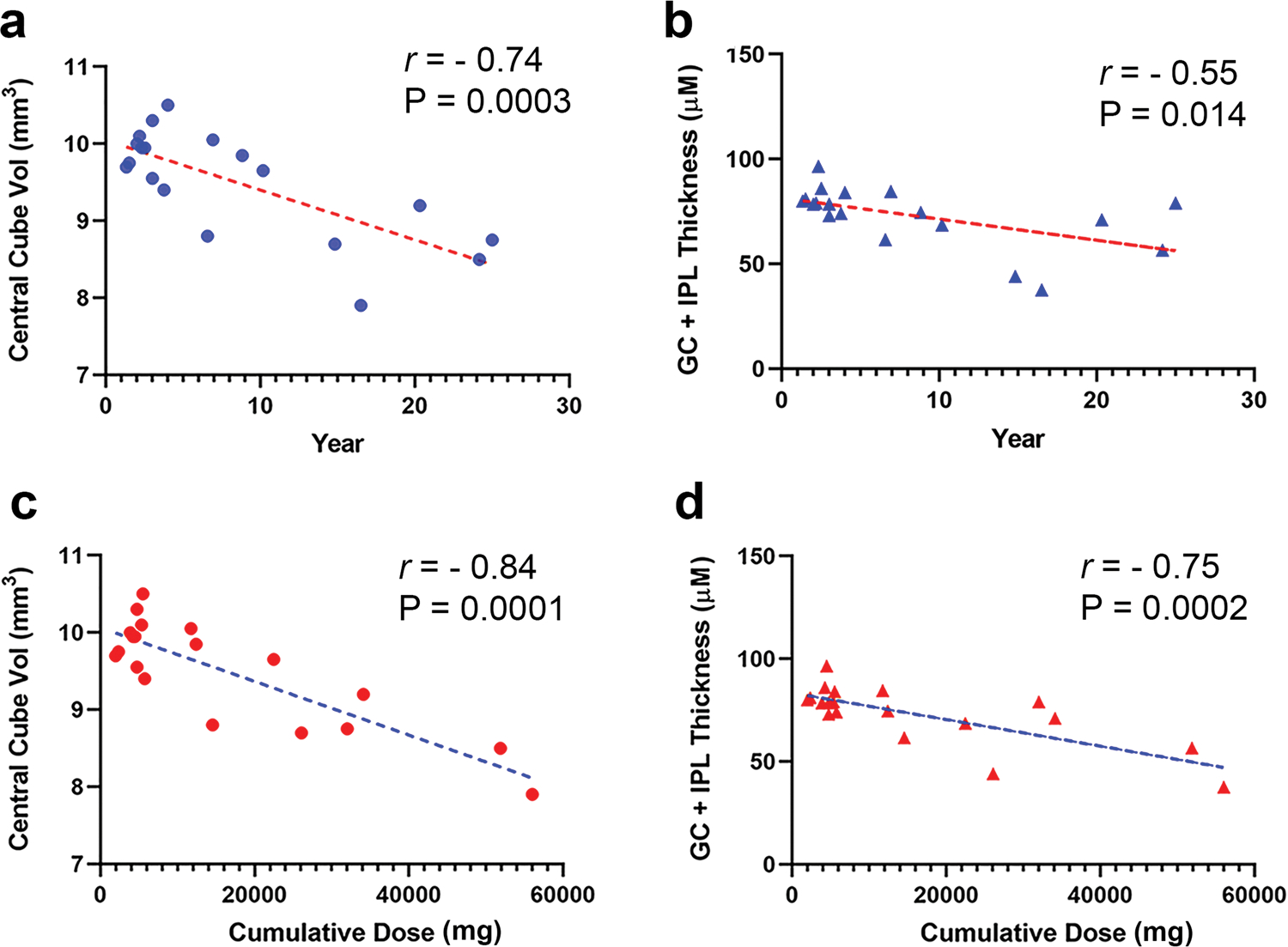
Correlation analysis of HCQ-treated subjects for retinal parameters with duration of the treatment and the cumulative doses. The different retinal parameters including central cube volume and IPL-GC layer thickness of the treated subjects were correlated with duration of treatment (a and b) and cumulative dose (c and d) using Pearson’s linear correlation. All the parameters were inversely correlated with both duration and cumulative dose of the treatment with high significance
Chronic HCQ exposure leads to loss of inner retinal cells and compromises visual function in mice
To study the mechanism of HCQ-mediated retinal toxicity, we developed a mouse model of chronic HCQ exposure. The mice were exposed to HCQ for a period of 3 months at a dose of 10 mg/kg body weight twice a week as described in the Methods section. As in human patients, we observed a significant reduction in the IPL-GC layer thickness in the HCQ-treated mice (Fig. 2a and Supplementary Fig. S1). In addition to the changes in the thickness of GC + IPL, we also observed a reduction in retinal ganglion cells numbers in the treated mice as compared to untreated controls (Fig. 2b, c). The RGC loss was more significant in the central retina as compared to medial and distal retina, resembling the pattern of retinal toxicity in human, which is more concentrated to the central retina [27, 28]. Similar to the RPE atrophy reported in chronic HCQ toxicity in human [13, 29], we observed increased death signals in the retinal RPE layer as well as the RGC layer in treated mice by labeling with single-stranded DNA (ssDNA) (Fig. 2d, e). These data indicate that the HCQ-treated mice suffer a loss of inner retinal neurons and this model of chronic HCQ exposure shows early retinal pathology similar to that observed in human patients with HCQ treatment.
Fig. 2. Retinal structure and functions were altered in mouse model of chronic HCQ exposure.
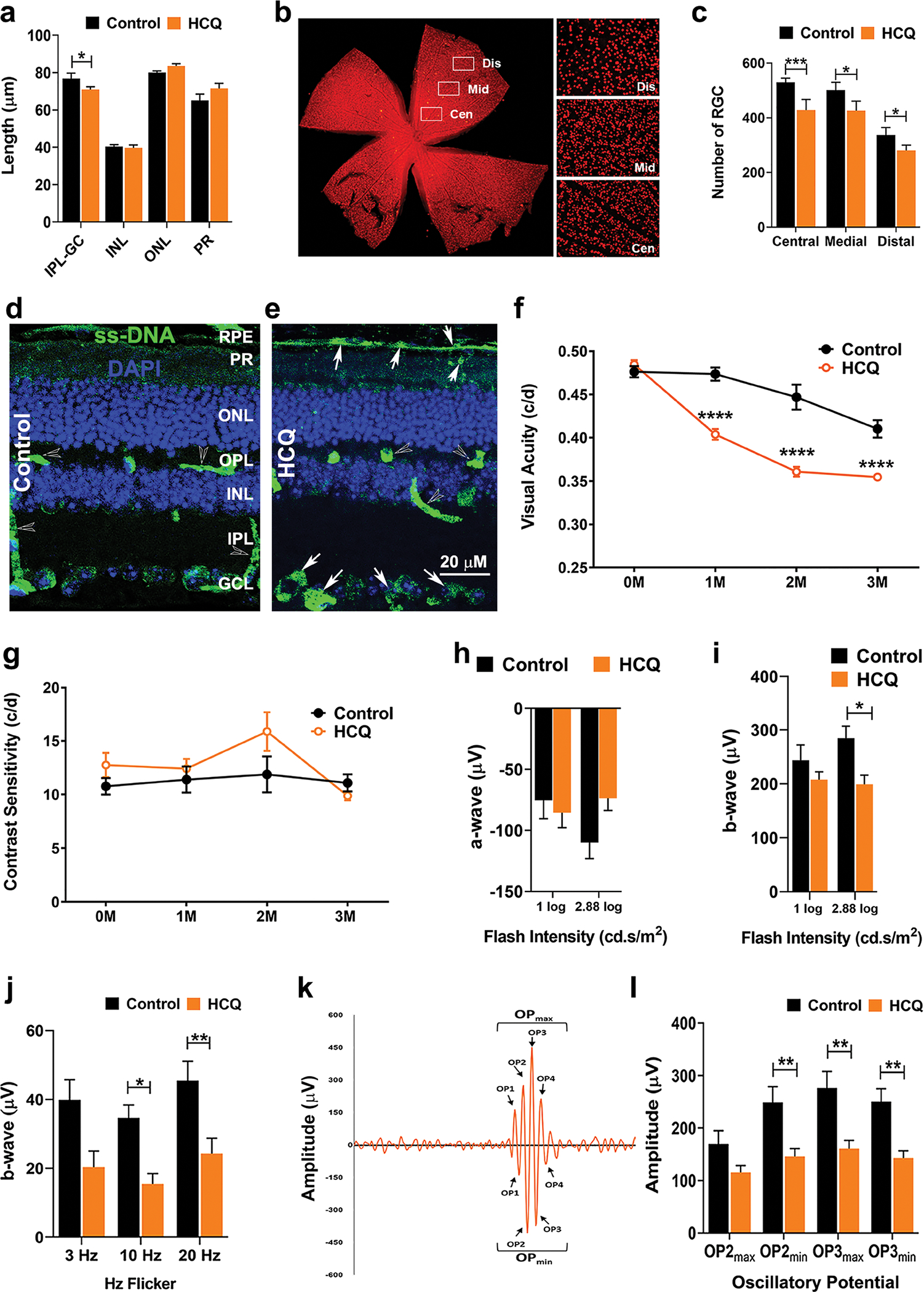
(a) Thickness of different retinal layers of mice after 3 months of HCQ treatment along with untreated controls as measured by SD-OCT. (b) Retinal flat mount of untreated mice immunohistochemically stained for Brn3a showing the retinal ganglion cells (RGC) and the three arbitrary regions of the retina used for measurement of RGC numbers. (c) RGC numbers from the three regions marked in b in control and HCQ-treated mice. (d and e) Representative image of immunohistochemistry of control (d) and 3 months of HCQ-treated (e) mice retina showing the level of single-stranded DNA (ssDNA). Arrows indicate higher level of ssDNA at retinal pigment epithelial (RPE) and RGC cell layers in the treated retina. Visual acuity (f) and contrast sensitivity (g) of control and HCQ-treated mice (n = 10 and 12, respectively) from pre-treatment to 3 months post-treatment. (h–l) Electroretinogram (ERG) of control and HCQ-treated mice (n = 12 and 20, respectively) after 3 months of treatment showing scotopic a-wave (h), b-wave (i) and photopic b-wave showing Hz flickers (j). (k) Representative diagram of oscillatory potential (OP) of b-wave in scotopic ERG showing minimum and maximum OPs measured and (l) graphical representation of OP2 and OP3. (All values are mean ± SEM; t-test; *p < 0.05, **p < 0.01, ***p < 0.001)
We then tested for visual acuity (VA) and contrast sensitivity (CS) of the treated mice using Optokinetic Tracking (OKT). One month of HCQ exposure caused a significant reduction in VA that consistently declined over the treatment period (three months); however, no significant change in CS was observed within the same time frame (Fig. 2f, g). Significant reduction in retinal rod function, as measured by electroretinogram (ERG) scotopic b-waves, at higher flash intensity (2.88 log) was observed with no significant changes in scotopic a-wave (Fig. 2h, i). The cone function, on the other hand, was significantly reduced in HCQ-treated mice (Fig. 2j). The oscillatory potentials (OP) generated via scotopic ERGs were also significantly reduced in the HCQ-treated mice (Fig. 2k, l). Together these data suggest that chronic HCQ treatment affects both inner and outer retinal neuronal cells leading to compromised visual and retinal functions.
We measured the expression of some relevant marker genes of retinal cells and found significant decrease in the levels of cone gene—Opsin1 medium wave sensitive (Opn1mw), Cone arrestin (Arr3), rod gene—Rod arrestin (Sag1) and bipolar cell marker, Protein kinase C-α (Pkcα) and RPE 65 in the retina of the mice treated with HCQ (Fig. 3). These data indicate that both types of photoreceptors as well as bipolar cells were affected by chronic HCQ treatment. We did not observe any significant difference in the expression of Rhodopsin (Rod), Retinal dystrophy (Mertk), Glial fibrillary acidic protein (Gfap), Chemokine C–C motif ligand 2 (Ccl2) and Early growth response gene (Egr1) (Fig. 3).
Fig. 3. HCQ exposure affects expression of retinal cellular markers.
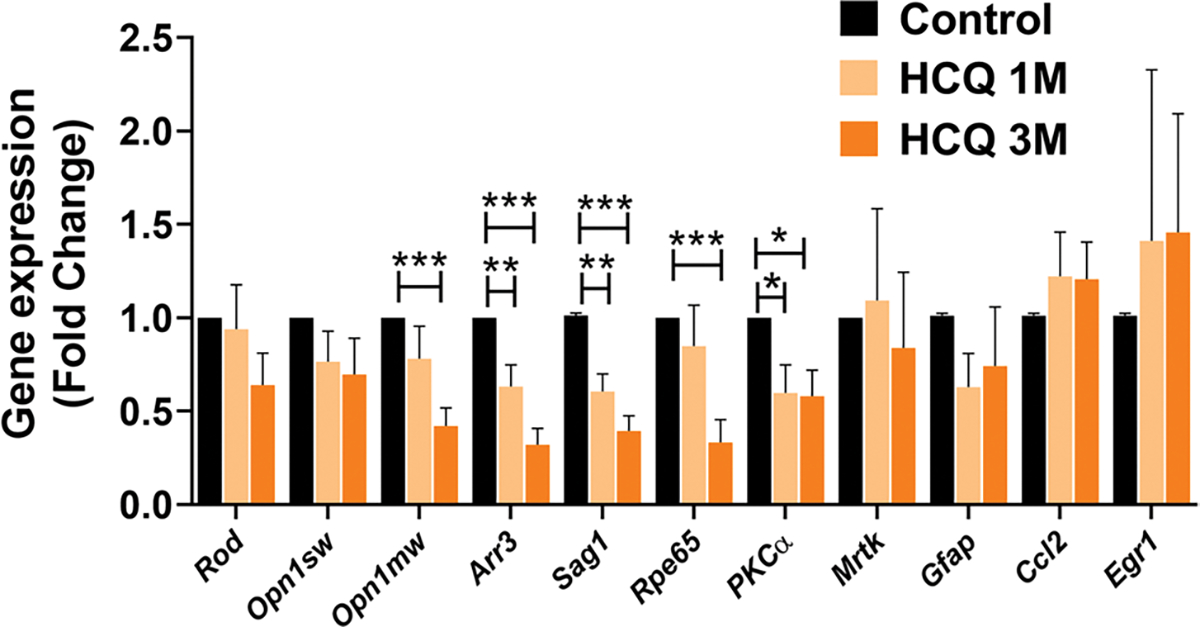
Expression analysis of relevant marker genes of retinal cells by qRT-PCR in control along with 1 and 3 months of HCQ-treated mice (n = 6). The result shows significant difference in markers of rods (Sag1), cones (Opn1mw, Arr3) and bipolar cells (Pkcα) of the retina along with retinal pigment epithelium (RPE) marker Rpe65 in the treated retina. (Values are mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001)
In conclusion, our mouse model of chronic HCQ exposure produces clinical and structural features of retinal toxicity resembling that seen in humans. Furthermore, this model also emphasizes the fact that chronic HCQ exposure affects inner retinal neurons as well as photoreceptors and RPE cells, and thus compromises visual functions at early stages of exposure. We propose that our model could potentially provide the basis for further understanding the mechanism of action of HCQ-mediated human retinal toxicity.
HCQ exposure affects the autophagosome–lysosome pathway
HCQ is a well-known lysosomotropic agent and is known to effect lysosomal homeostasis and function by increasing its pH [1, 30]. By gene expression analysis, we found a significant increase in Autophagy-related 4b (Atg4b) and lysosomal/autophagy transcription factor (TcFEB) genes, and a significant decrease in Lysosomal associated membrane protein 2 (Lamp-2) gene in the treated mouse retina (Fig. 4a). We observed an increase in the level of LC3B proteins in the treated retina over the course of treatment (Fig. 4b) and a significant decrease in the ratio of LC3B I/II (Fig. 4c). The autophagy marker protein, p62, was also increased in the treated mice retina along with the lysosome-associated membrane protein, LAMP1 and the early and late-endosomal markers Rab5 and Rab7, respectively [31], in the treated retinas (Fig. 4d). By immunohistochemical analysis of the retina, LC3B levels was appeared to be increased throughout the retinal layers in treated mice (Fig. 4e i–vi, arrows). We also observed increased co-localization of LC3B and Rab7, particularly in the ganglion cell layer (Fig. 4e vi, arrows). Together these data indicate a possible alteration of the autophagosomal–lysosomal pathway in HCQ-treated retina.
Fig. 4. HCQ exposure affects autophagosome–lysosomal pathway.
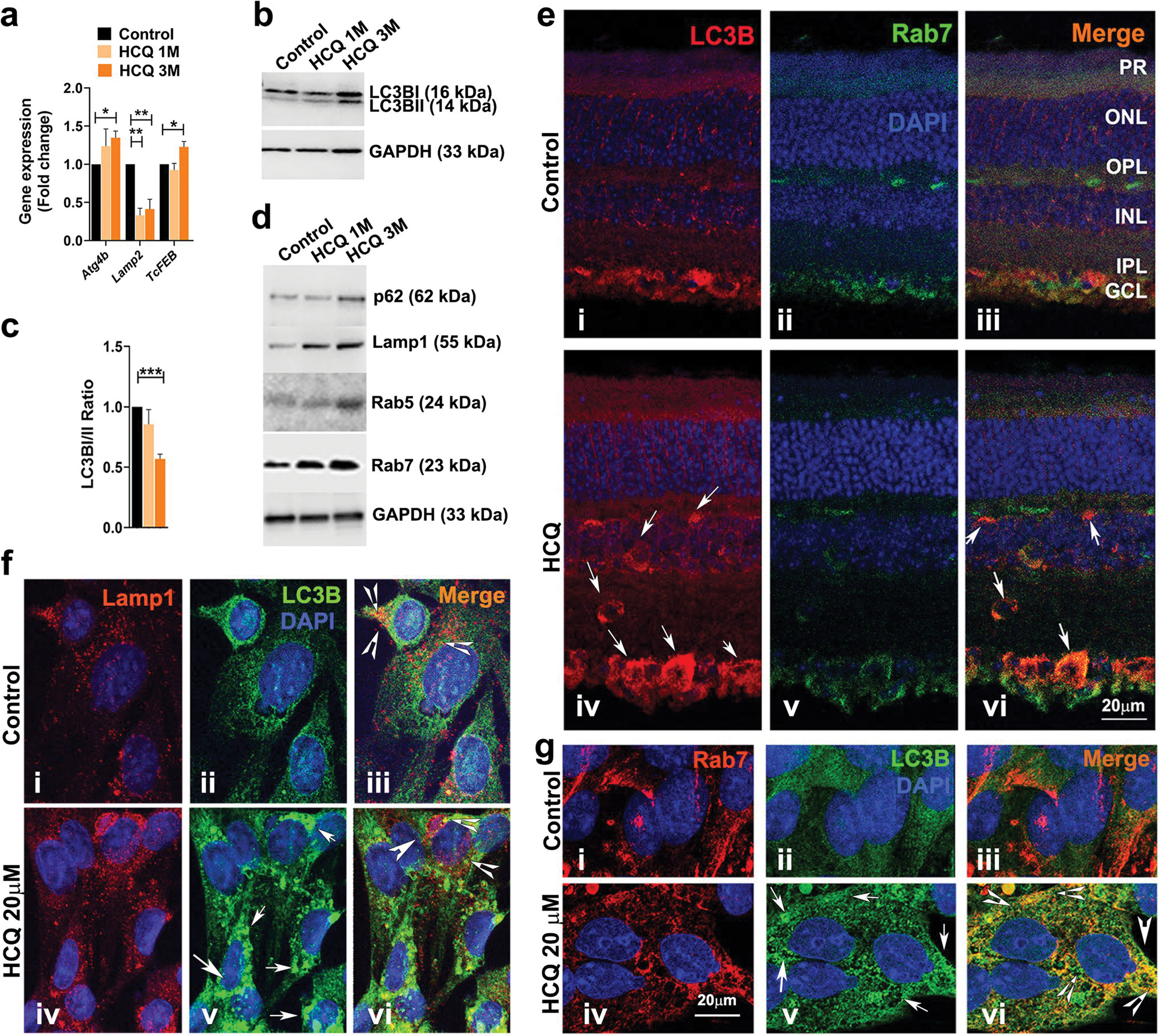
(a) Expression analysis of relevant autophagosome–lysosomal pathway genes by qRT-PCR from control and HCQ-treated mice retina (n = 6). (b and d) Western blot analysis of autophagosome–lysosomal pathway proteins from retinas described in a (n = 3 per group). (c) Quantification analysis of the protein expression in (b) showing ratio of LC3B I and II. (e) Representative image of immunohistochemistry of untreated and treated mice retina showing localization of LC3B and Rab7. Arrows indicate co-localization observed only in HCQ-treated retina (vi). (f and g) Representative immunofluorescence images of rat Muller cells treated with 20 μM of HCQ for 24 h. along with untreated controls. (f) Localization of Lamp1 and LC3B proteins with arrows showing higher expression of LC3B (v) and arrowheads showing reduced co-localization in HCQ-treated cells (iii and vi). (g) Localization of Rab7 and LC3B proteins with arrows indicating higher expression of LC3B (v) and arrowheads indicating higher co-localization in HCQ-treated cells (vi). (Values are mean ± SEM; t-test; *p < 0.05, **p < 0.01, ***p < 0.001)
To further understand how HCQ affects the autophagosome–lysosome pathway, we used cultured rat Muller glial cells and treated them with HCQ. Similar to what we observed in the mouse retina, there was an increase in p62, Rab5, Rab7 and LC3B protein levels in the treated cells, along with a significant increase of LC3BII over LC3BI, as observed by the decrease in LC3BI/II ratio (Supplementary Fig. S2a and b). These data suggest that the Muller glial cells respond to HCQ treatment in a similar way to that observed in the mouse retina. By immunocytochemical analysis, we observed an increase in the LC3B protein levels (Fig. 4f ii and v, arrows) and a decrease in co-localization of LC3B and LAMP1 in HCQ-treated Muller cells (Fig. 4f iii and vi, arrowheads). Interestingly, an increased co-localization of LC3B with Rab7 was observed in the HCQ-treated cell compared to the untreated controls (Fig. 4g iii and vi, arrowheads).
Taken together, our data from both in vivo and in vitro studies suggest that HCQ treatment affects the autophagosome–lysosome pathway, possibly autophagosome–lysosome fusion. It might also affect the entire autophagy process of the retinal cells leading to structural and functional defects.
Retinal sphingolipid homeostasis is altered by HCQ treatment
Lysosomes play an integral role in cellular sphingolipid homeostasis [32]. In the central hub of cellular sphingolipid metabolism sits the bioactive lipid, ceramide that plays an important role in various cellular processes [33, 34]. Lysosomes hydrolyze higher order sphingolipids, namely sphingomyelin (SM), glycosphingolipids and gangliosides, to generate ceramide [35], which is further hydrolyzed into sphingosine (Sph) and free fatty acids, that are recycled back to the cytoplasm (Fig. 5a) [32, 35, 36].
Fig. 5. Chronic HCQ exposure alters sphingolipid homeostasis.
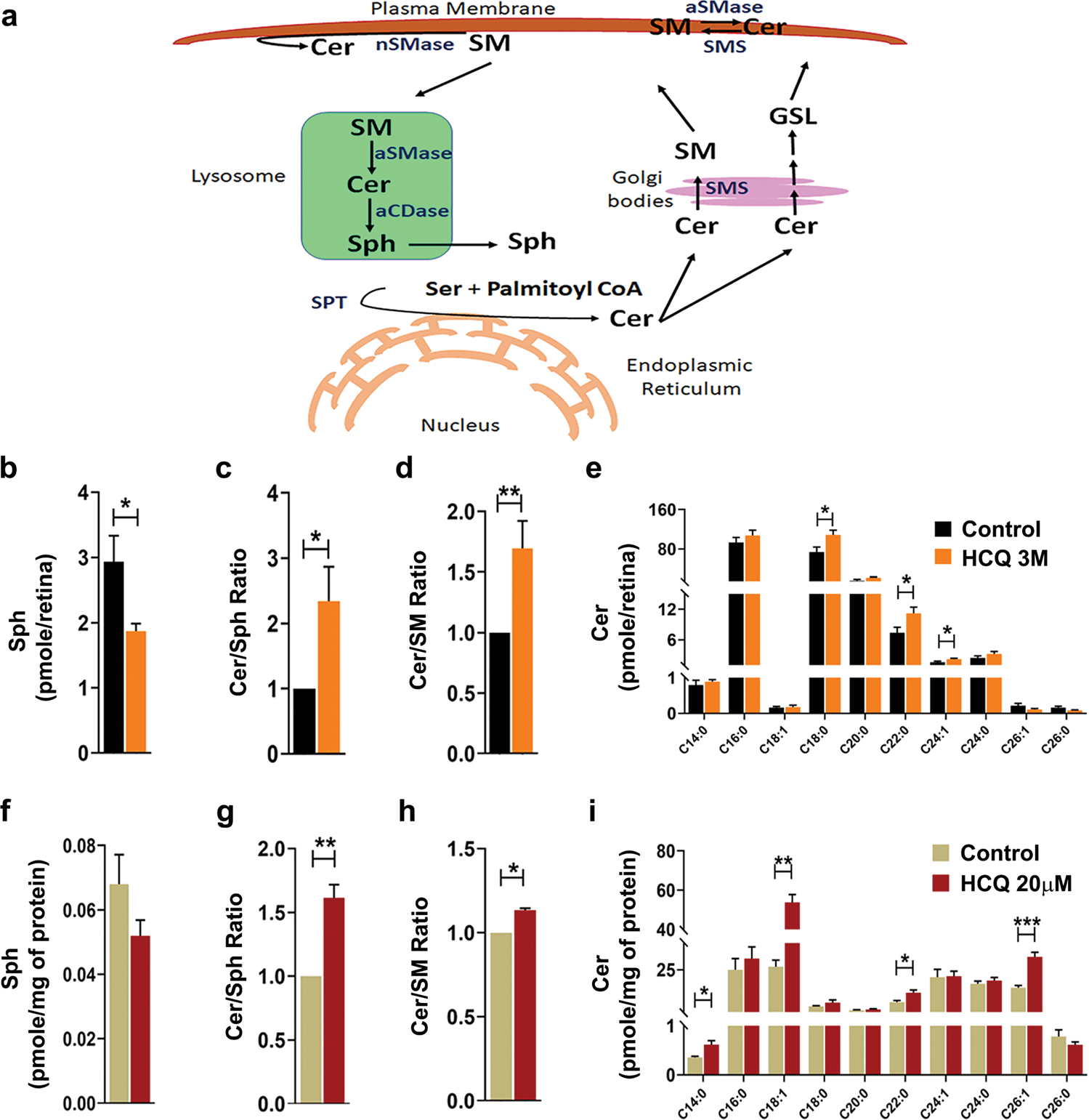
(a) Schematic diagram of sphingolipid metabolism pathway in mammalian cell. Ceramide (Cer) is synthesized by de novo biosynthesis pathway in ER by condensation of serine and palmitoyl CoA catalyzed by Serine palmitoyl transferase (SPT). In the Golgi, Cer is converted to Sphingomyelin (SM) by SMsynthase (SMS) and glycosphingolipids (GSL). In the plasma membrane, interconversion of Cer and SM are catalyzed by neutral sphingomyelinase (nSMases), acidic sphingomyelinase (aSMase) and SMS. In the lysosome, through salvage pathway SM is converted to Cer by aSMase which is further hydrolyzed to Sphingosine (Sph) by acid ceramidase (aCDase). (b-e) Analysis of sphingolipid levels in mice retina (n = 7) showing total sphingosine level (b), ratio of total ceramide to sphingosine and total ceramide to sphingomyelin levels (c and d, respectively) and (e) individual species of ceramide. (f-i) Analysis of sphingolipid levels in rat Muller cells treated with HCQ for 24 h. (n = 4/group) showing levels of total sphingosine (f), ratio of total ceramide to sphingosine and total ceramide to sphingomyelin (g and h, respectively) and individual species of ceramide (i). (Values are mean ± SEM; t-test; *p < 0.05, **p < 0.01, ***p < 0.001)
Since HCQ is known to enter into the lysosomes and to be de-protonated, it is unable to diffuse out and thereby accumulates in the lysosome [37]. With its high pKa (> 8.0), it increases lysosomal pH from its normal range of 4.5–5.0 and thus affects its normal function [37–39]. We hypothesized that HCQ exposure would affect the sphingolipid homeostasis in the retina. Using LC–MS we compared the levels of major cellular sphingolipids in the retina of mice treated with HCQ for 3 months along with untreated controls. We found a significant decrease in the Sph level (reduced by approx. 40%) in the treated mice (Fig. 5b). There is also a significant increase in the ratio of Cer:Sph and Cer:SM, indicating an accumulation of cellular ceramide (Fig. 5c, d). This was further confirmed by the increase observed in the levels of individual ceramide species, namely C18:0, C22:0 and C24:1 (Fig. 5e). We further tested the mRNA levels of a group of sphingolipid metabolic genes and observed a significantly reduced expression of N-acylsphingosine amidohydrolase 1 (Asah1), Sphingomyelin phosphodiesterase 2 (Smpd2) and a significant increase in Sphingosine kinase 2 (Sphk2) genes in the retina of the treated animals. However, no significant changes in expression were detected in Sphingomyelin phosphodiesterase 1 (Smpd1) and Sphingosine kinase 1 (Sphk1) genes (Fig. 6).
Fig. 6. HCQ treatment alters expression of genes involved in sphingolipid metabolism.
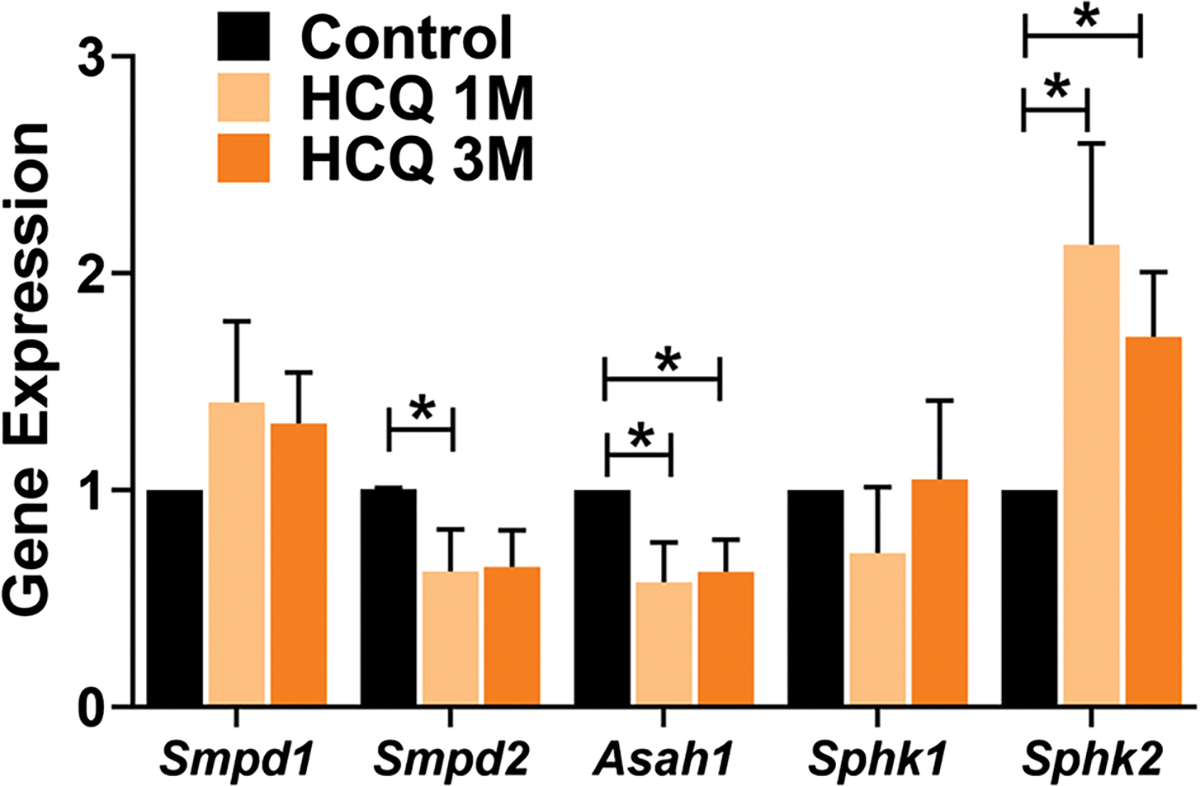
Expression analysis of the relevant sphingolipid metabolism genes by qRT-PCR from untreated and treated mice retina (n = 8). Significant differences were observed in the mRNA levels of neutral sphingomyelinase (Smpd2), that converts sphingomyelin to ceramide (Cer), acid ceramidase (Asah1), which hydrolyzes Cer to sphingosine (Sph) and sphingosine kinase 2 (Sphk2), responsible for phosphorylation of Sph to generate sphingosine-1-phosphate, in the treated retinas. (Values are mean ± SEM, *p < 0.05)
Similar to the HCQ-treated animals, Muller glial cells also showed an altered sphingolipid homeostasis when treated with HCQ. We observed a decrease in total sphingosine level and a significant increase in the Cer:Sph and Cer:SM ratio in the HCQ-treated cells (Fig. 5f–h). This indicates enhanced ceramide accumulation due to HCQ treatment in these cells, which was further established by a significant increase in the levels of individual ceramide species, namely C14:0, C18:1, C22:0 and C26:1 (Fig. 5i).
In summary, our results suggest that in addition to compromising lysosomal functions, HCQ also alters sphingolipid homeostasis of cells, leading to an accumulation of the toxic bioactive lipid, ceramide, which in turn has been shown to lead to retinal cell death in multiple studies [33, 40–42]. Our findings regarding lysosomal dysfunction and alteration of sphingolipid homeostasis and ceramide accumulation suggest two possible mechanisms by which retinal function may be altered in patients undergoing HCQ therapy.
Discussion
HCQ is a drug widely used for malaria, RA and SLE. Although structurally very similar to its predecessor CQ, HCQ is significantly less toxic [43]. However, long-term use of HCQ can also render severe side effects, the most common of which is retinal toxicity leading to loss of central vision, called bull’s-eye maculopathy. The resulting pathology leads to progressive RPE atrophy around the macula, leading to loss of central vision [10–12]. Recent clinical studies suggest that HCQ affects neuronal retina long before initiating toxicity to RPE cells [14, 44]. We conducted a clinical study using a retrospective/prospective mixed model and found that HCQ indeed has a progressive adverse effect on central retinal cells/neurons as revealed by SD-OCT image analysis. HCQ is thought to act through lysosomal basification, which can particularly affect RPE cells as they function to digest large amounts of photoreceptor outer segments each day through the autolysosomal pathway [45, 46]. Alterations in lysosomal breakdown have been implicated in many RPE-related disease conditions [47–49]. However, lysosomal breakdown is a ubiquitous cellular process and is required for protein and lipid breakdown and recycling in many cell types. While the RPE certainly suffers when lysosome function is reduced, it is unsurprising that other cells in the inner retina are also affected. We show here clear evidence that long-term HCQ dosing is inversely related to inner retinal thickness in humans prior to evidence of RPE toxicity (Fig. 1). Studies analyzing visual fields in HCQ patients show central scotoma and vision loss in the central retina before developing RPE toxicity or BEM. Prior reports have shown that lifetime HCQ doses under 1.0 kg did not result in inner retinal thinning or RPE toxicity [28]. Furthermore, progression of damage seen with higher doses can be halted when patients are taken off of the drug, but the damage is not reversed [14]. Thus, HCQ toxicity is cumulative and irreversible. These observations suggest that early monitoring of inner retinal function by visual fields and structural assay using OCT should be considered at least once a year as a normal follow-up in all patients taking HCQ.
CQ and HCQ are lysomotropic agents, and their accumulation in lysosomes leads to increases in lysosomal pH that can affect multiple lysosomal functions [1, 30, 37–39]. However, the mechanism of HCQ toxicity in retinal cells is not known and so far, and there has been no report of an animal model of HCQ toxicity in the retina. In this study, we established a mouse model that mimics the retinal changes observed in the human subjects under long-term HCQ treatment. In this model, we observed compromised visual and retinal functions and defects in the autophagosome–lysosome pathway in the retina that would be expected to affect the autolysosomal degradation and recycling of proteins and lipids. With chronic HCQ treatment, we also found alterations in retinal sphingolipid homeostasis causing upregulation of cytotoxic ceramide. Thus, we propose that lysosomal dysfunction and modulation of sphingolipid metabolism in retinal cells represents a major mechanism by which retinal cell dysfunction and death occurs during long-term HCQ treatment.
Our mouse model of chronic HCQ exposure shows a reduction in the IPL-GC layer of the retina similar to that observed in the human patients (Fig. 2a). There is also a prominent reduction in RGC numbers in mouse retina that received 3 months of HCQ (Fig. 2c). Previous studies showed systemic CQ can affect the central retina and cause RGC death in animal models [17, 20]; however, toxicity of HCQ was not accessed. We report for the first time that HCQ can also affect and cause RGC death at early stages, which could explain why HCQ patients develop central scotoma before developing RPE toxicity or BEM [50]. Interestingly, RGC death in many forms of lipid storage diseases develop ‘cherry-red spot in the macula,’ which has a resemblance to early pathology of BEM [51]. Testing for visual field and multifocal ERG could therefore be integrated into to the ophthalmic care of patients receiving HCQ, at least once in a year, in order to detect early indications of inner retinal toxicity.
The mice were also severely compromised in their visual acuity but not in contrast sensitivity, which may indicate a compromised cone function (Fig. 2f and g). The ERG analysis recapitulated this indication as we detected a significant decrease in scotopic b-wave at higher flash intensity but not in a-wave. Also, we observed a significant decrease in cone photoreceptor responses to flickers (Fig. 2h–j). The significant reduction in the ERG oscillatory potential (OP) we observed clearly indicates a compromised visual signal transmission through retinal secondary neurons (Fig. 2l). Although the specific cell types responsible for generating OPs in retina were not identified, previous studies indicate OPs are generated from bipolar cells or IPL [52] and have a different retinal depth profile for individual peaks [53]. Clinically, OPs indicate early disturbances in the function of the retinal neurons. In addition to visual function, the gene expression profile of the markers for different retinal cells indicates the HCQ effect is more pronounced for cone and bipolar cells. Early effects on cones may explain why BEM in humans develops in the cone dominated macula. Also, a perifoveal ring-like opacity in retinal fundus images that gives the typical bull’s-eye appearance of the macula may be due to RGC loss in the perifoveal region, where their concentration is the highest in the entire retina [54, 55]. We also observed induction of apoptosis in RPE cells, in agreement with the classical theory of RPE toxicity and death resulting in BEM. In conclusion, we developed a mouse model of chronic HCQ with features that resemble human HCQ toxicity. This model may offer important opportunities for determining new parameters for dosage and duration of HCQ treatment, based on specific early and late effects on retinal neurons, glial and RPE cells. The mouse model could provide the basis for unraveling the mechanisms of HCQ toxicity in the retina.
Our current study suggests an alteration in the autophagosomal–lysosomal pathway in the retinal cells plays a key role in HCQ toxicity. Since the ATG4b protein is involved in the early steps of autophagosome formation [56], the increase in Atg4b mRNA level after HCQ treatment (Fig. 4a) might indicate an increase in autophagosome formation in the retinal cells. LC3BI and II protein levels were altered both in in vivo animal models of neuronal ceroid lipofuscinosis (NCL), an inherited lysosomal storage disease (LSD), as well as in in vitro model due to CQ treatment [57–59]. In human RPE-derived ARPE19 cells, CQ treatment leads to increases in LAMP1 protein level [48] and inhibition of autophagic flux in U2OS cells through dysfunction of autophagosome–lysosome fusion [59]. Similarly, with HCQ treatment we observed an increased protein expression of LC3B, particularly the phosphatidylethanolamine (PE) conjugated form LC3BII, SQSTM1/p62 and LAMP1 (Fig. 4b–d). LC3BII is found on the membranes of autophagosomes, and its level is closely correlated with the number of autophagosomes [58, 60]. Both p62 and LC3BII proteins are degraded in autophagy [58, 61], so an increase in LC3BII over LC3BI along with increase in p62 protein levels indicates accumulation of autophagosomes [58, 60, 62–64]. Since LAMP1 protein level is also increased, we can assume that HCQ treatment does not affect the lysosomal biogenesis and the accumulation of autophagosomes might be due to a defect in the fusion process, not due to unavailability of lysosomes [48, 59]. HCQ exposure also led to an increase in the protein levels of both Rab5 and Rab7 (Fig. 4d). Rab proteins are small GTPases and play an important role in maturation of endosomes, trafficking of cargos along microtubules and the final fusion step of late endosome with lysosomes [31]. Since Rab7 become associated with autophagosomal membrane and thus direct their fusion to lysosomes [31, 65], its accumulation and increased co-localization with LC3B (Fig. 4e–g) support the notion of accumulation of autophagosomes. Fusion of autophagosomes with lysosomes co-localizes the lysosomal membrane protein LAMP1 and autophagosomal membrane protein LC3B [66, 67]. Our data showing a reduction in their co-localization (Fig. 4f) further establish the possibility of impairment of autophagosome–lysosome fusion. Together these results indicate a similar effect of HCQ on the autophagosome–lysosome pathway both in mice retina and in cultured retinal cells. The cytotoxic effect of autophagosome accumulation might lead to the death of RGC observed in mice after HCQ treatment (Fig. 2c–d) and could be one of the possible mechanisms of HCQ-induced retinal cell death.
Our study of mouse model provides novel information regarding the modulation of sphingolipid metabolism in vivo and in vitro as a result of HCQ exposure. Sphingolipids are a class of cellular lipids that are involved in multiple aspects of cellular physiology [33, 68–70]. In the last two decades, several studies have established certain sphingolipids as a key signaling molecule in the retina that can modulate the physiology and function of retinal cells and contribute to multiple retinal pathologies [33, 71, 72]. At the cellular level, lysosomes play a significant role in maintaining sphingolipid homeostasis [35, 73]. Defects in lysosome function result in a group of inherited metabolic disorders collectively known as LSD [74–76], with about half of them also having alterations in sphingolipid metabolism [77, 78]. Additionally, several LSDs that affect humans, such as Tay–Sachs, Sandhoff, Fabry, Gaucher, Niemann–Pick diseases types A and B, and Farber, have a common manifestation in the eye known as Macular Cherry-red Spot (MCR) [51, 79, 80] whose clinical and ultra-structural analysis suggests membranous accumulation, and ultimate degeneration of inner retinal neurons, particularly the RGC [81]. The fact that HCQ affects lysosomal function and triggers retinal toxicity also suggests a possible connection between HCQ-mediated retinal toxicity, modulation of sphingolipid homeostasis and a macular phenotype, very similar to MCR.
Our findings suggest a connection between the lysosomal disfunction reported due to HCQ toxicity and an alteration of sphingolipid homeostasis in the retina. We observed a decrease in Sph and SM along with an increase in ceramide in the treated retina (Fig. 5b–e), which clearly indicates accumulation of this toxic lipid in the retinal cells. Cellular free ceramides are known to be deadly 2nd messengers of the cells, since they can induce cell death through various mechanisms such as apoptosis, parthanatos etc. [82–84]. Ceramide accumulation has been shown to be the causative factor for retinal cell death in several in vitro and in vivo studies [42, 82, 83]. An accumulation of ceramide and decrease in Sph levels indicate a possible reduction in the hydrolyzation of ceramide within lysosomes. In lysosomes, ceramide is hydrolyzed to Sph by the hydrolase, N-acylsphingosine amidohydrolase 1 (ASAH1), which is expressed abundantly in the retina [85]. The enzymatic activity of ASAH1 is reduced in alkaline pH [86]; therefore, the reduced hydrolyzation of ceramides in the lysosomes could possibly be due to reduced ASAH1 activity as a result of an increased lysosomal pH caused by HCQ treatment. Lipidomic analysis of the retina-specific deletion of Asah1 gene in mice showed an increased level of ceramide [87]. In the LSD, Farber disease, deficiency of ASAH1 restricts the production of Sph from ceramide [88]. Importantly, since the only source of intracellular Sph is through hydrolyzation of Cer in the lysosomes by ASAH1 [89], reduction of Sph may affect the levels of cellular pro-survival factor, sphingosine-1-phosphate (S1P) [90]. In addition to increased ceramide, the reduction in Sph and S1P level in the HCQ-treated retina could also contribute to retinal neuronal cell pathology and death. Further studies will be necessary to dissect the role of individual bioactive sphingolipids in HCQ-mediated retinal toxicity.
We also observed that ceramides with saturated fatty acid (SFA), including C14:0, C18:0 and C22:0, showed a significant increase after HCQ treatment (Fig. 5e and i). These relatively shorter chain ceramides are known to be highly toxic to cells and have been studied as potential drug candidates for use against various cancer cell lines [91]. Thus, the imbalance in sphingolipid homeostasis seen after HCQ exposure might play a role in the retinal pathology and visual deficits observed both in our mouse model as well as in human patients.
Our study findings must be considered in light of its limitations including number of human patients on HCQ were very low, as well as the number of patients with HCQ toxicity, which limited us from doing multiple comparisons. Though our mouse model is one of the initial demonstration of HCQ toxic effect on mammalian retina, especially on the functional aspects of retinal photoreceptors and effect on various biochemical and molecular parameters, the timeline we exposed the mice to HCQ (3 months) did not cause noticeable death in photoreceptor and other inner neuronal cells, except RGCs. A longer-term treatment and a detail study is needed to understand the cell-specific effect of HCQ in the retina. Though our biochemical and cellular readouts suggested HCQ affected the photoreceptors and RGCs in the mouse model, we limited our in vitro (cell culture) assay to Muller cells, mainly due to unavailability of established and well-characterized cell lines from photoreceptors and RGCs that maintain their properties in vitro. Thus, the results obtained from the in vitro studies that provided important information on the HCQ effect on the cellular physiology cannot be generalized for all the cell types of the retina.
In summary, we have confirmed the appearance of early inner retinal pathology in humans treated with chronic HCQ and were able to establish an animal model that exhibits the same pattern of neuro-retinal toxicity observed in chronic HCQ exposure in humans. Our findings provide insights into the molecular mechanisms that may lead to the toxicity of retinal neuronal cells. Our findings suggest that HCQ treatment reduces autophagolysosomal activity in the retina and alters sphingolipid homeostasis. The independent or additive effect of these HCQ-induced alterations could contribute to the retinal cell death and loss of visual function observed in patients.
Supplementary Material
Acknowledgements
This work was supported by the National Eye Institute grants [EY022071, R01 EY031316] (NM); the US Department of Defense office of the Congressionally Directed Medical Research Programs (CDMRP), Vision Research Program grant (W81XWH-20-1-0900) (NM); the Veterans’ Administration (VA Merit Review Award I01BX004893 (NM); the Veterans’ Administration (VA Merit Review, I BX001792 (CEC) and a Senior Research Career Scientist Award, IK6BX004603 (CEC)); the National Institute of Allergy and Infectious Diseases grant (R01 AI139072 (CEC); and the National Institute of General Medical Sciences grant (R01 GM137578 (CEC) and R01 GM137394 (CEC), grants from Research to Prevent Blindness Inc., USA (NM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the Department of Veterans Affairs, or the US government. UTHSC Plough Center funding (NM) and Neuroscience Institute for Postdoctoral Award (KM) are duly acknowledged. Special thanks to Dr. Dianna Johnson from Ophthalmology, UTHSC, for her help with manuscript editing
Funding
1. US Department of Defense office of the Congressionally Directed Medical Research Programs (CDMRP), Vision Research Program grant (W81XWH-20-1-0900) (NM). 2. US Veterans’ Administration (VA Merit Review Award I01BX004893) (NM). 3. US Veterans’ Administration (VA Merit Review, I BX001792 (CEC) and a Senior Research Career Scientist Award, IK6BX004603 (CEC). 4. National Eye Institute grants [EY022071, R01 EY031316] (NM). 5. National Institute of Allergy and Infectious Diseases grant (R01 AI139072 (CEC). 6. National Institute of General Medical Sciences grant (R01 GM137578 (CEC) and R01 GM137394 (CEC). 7. Research to Prevent Blindness Inc., USA (NM)
Footnotes
Conflict of Interests None.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s12035-022-02825-3.
Ethics Approval and Consent to Participate Human study was approved by University of Oklahoma Institutional Review Board for the Protection of Human Subjects; IRB#: 5218; Approval Date: 02/27/2015
Study Title: Sphingolipids, Autoimmune Diseases and Retinopathies.
Consent for Publication Human study and consent to publish was approved by University of Oklahoma; Institutional Review Board for the Protection of Human Subjects; IRB#: 5218; Approval Date: 02/27/2015.
Author Consent All the authors have seen the manuscript and approved for publication.
Research involving Human Participants Human study was approved by University of Oklahoma; Institutional Review Board for the Protection of Human Subjects; IRB#: 5218; Approval Date: 02/27/2015; Study Title: Sphingolipids, Autoimmune Diseases and Retinopathies.
Animals: Animal studies approved by UTHSC IACUC committee. Approval # 19-0104.
Informed Consent Obtained for all participants according to the approved IRB#: 5218; protocol at the University of Oklahoma. Approval Date: 02/27/2015.
Study Title: Sphingolipids, Autoimmune Diseases and Retinopathies.
Data Availability
All materials and data will be available from the corresponding author following the University of Tennessee’s policy of sharing research materials and data.
References
- 1.Jorge A, Ung C, Young LH, Melles RB, Choi HK (2018) Hydroxychloroquine retinopathy - implications of research advances for rheumatology care. Nat Rev Rheumatol 14(12):693–703. 10.1038/s41584-018-0111-8 [DOI] [PubMed] [Google Scholar]
- 2.Mitjà O, Corbacho-Monné M, Ubals M, Alemany A, Suñer C, Tebé C, Tobias A, Peñafiel J et al. (2020) A Cluster-Randomized Trial of Hydroxychloroquine for Prevention of Covid-19. N Engl J Med 384(5):417–427. 10.1056/NEJMoa2021801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saag MS (2020) Misguided Use of Hydroxychloroquine for COVID-19: The Infusion of Politics Into Science. JAMA 324(21):2161–2162. 10.1001/jama.2020.22389 [DOI] [PubMed] [Google Scholar]
- 4.Furst DE (1996) Pharmacokinetics of hydroxychloroquine and chloroquine during treatment of rheumatic diseases. Lupus 5(Suppl 1):S11–15 [PubMed] [Google Scholar]
- 5.Wallace DJ (1996) The history of antimalarials. Lupus 5 (1_suppl):2–3. 10.1177/0961203396005001021 [DOI] [PubMed] [Google Scholar]
- 6.Abraham R, Hendy RJ (1970) Irreversible lysosomal damage induced by chloroquine in the retinae of pigmented and albino rats. Exp Mol Pathol 12(2):185–200. 10.1016/0014-4800(70)90049-3 [DOI] [PubMed] [Google Scholar]
- 7.Ginsburg H, Geary TG (1987) Current concepts and new ideas on the mechanism of action of quinoline-containing antimalarials. Biochem Pharmacol 36(10):1567–1576. 10.1016/0006-2952(87)90038-4 [DOI] [PubMed] [Google Scholar]
- 8.Pandya HK, Robinson M, Mandal N, Shah VA (2015) Hydroxychloroquine retinopathy: A review of imaging. Indian J Ophthalmol 63(7):570–574. 10.4103/0301-4738.167120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pasadhika S, Fishman GA, Choi D, Shahidi M (2010) Selective thinning of the perifoveal inner retina as an early sign of hydroxychloroquine retinal toxicity. Eye (Lond) 24 (5):756–762; quiz 763. 10.1038/eye.2010.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yusuf IH, Sharma S, Luqmani R, Downes SM (2017) Hydroxychloroquine retinopathy. Eye (Lond) 31(6):828–845. 10.1038/eye.2016.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melles RB, Marmor MF (2014) The risk of toxic retinopathy in patients on long-term hydroxychloroquine therapy. JAMA Ophthalmol 132(12):1453–1460. 10.1001/jamaophthalmol.2014.3459 [DOI] [PubMed] [Google Scholar]
- 12.Latasiewicz M, Gourier H, Yusuf IH, Luqmani R, Sharma SM, Downes SM (2017) Hydroxychloroquine retinopathy: an emerging problem. Eye (Lond) 31(6):972–976. 10.1038/eye.2016.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allahdina AM, Chen KG, Alvarez JA, Wong WT, Chew EY, Cukras CA (2019) Longitudinal Changes in Eyes with Hydroxychloroquine Retinal Toxicity. Retina 39(3):473–484. 10.1097/IAE.0000000000002437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Sisternes L, Hu J, Rubin DL, Marmor MF (2015) Localization of damage in progressive hydroxychloroquine retinopathy on and off the drug: inner versus outer retina, parafovea versus peripheral fovea. Invest Ophthalmol Vis Sci 56(5):3415–3426. 10.1167/iovs.14-16345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kan E, Yakar K, Demirag MD, Gok M (2018) Macular ganglion cell-inner plexiform layer thickness for detection of early retinal toxicity of hydroxychloroquine. Int Ophthalmol 38(4):1635–1640. 10.1007/s10792-017-0635-y [DOI] [PubMed] [Google Scholar]
- 16.Ulviye Y, Betul T, Nur TH, Selda C (2013) Spectral domain optical coherence tomography for early detection of retinal alterations in patients using hydroxychloroquine. Indian J Ophthalmol 61(4):168–171. 10.4103/0301-4738.112161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ivanina TA, Zueva MV, Lebedeva MN, Bogoslovsky AI, Bunin AJ (1983) Ultrastructural alterations in rat and cat retina and pigment epithelium induced by chloroquine. Graefes Arch Clin Exp Ophthalmol 220(1):32–38. 10.1007/BF02307013 [DOI] [PubMed] [Google Scholar]
- 18.Ivanina TA, Sakina NL, Lebedeva MN, Borovyagin VL (1989) A study of the mechanisms of chloroquine retinopathy. I. Chloroquine effect on lipid peroxidation of retina. Ophthalmic Res 21(3):216–220. 10.1159/000266810 [DOI] [PubMed] [Google Scholar]
- 19.Ivanina TA, Sakina NL, Lebedeva MN, Borovjagin VL (1989) A study of the mechanisms of chloroquine retinopathy. II. Chloroquine effect on protein synthesis of retina. Ophthalmic Res 21(3):272–277. 10.1159/000266819 [DOI] [PubMed] [Google Scholar]
- 20.Rosenthal AR, Kolb H, Bergsma D, Huxsoll D, Hopkins JL (1978) Chloroquine retinopathy in the rhesus monkey. Invest Ophthalmol Vis Sci 17(12):1158–1175 [PubMed] [Google Scholar]
- 21.Pasadhika S, Fishman GA (2010) Effects of chronic exposure to hydroxychloroquine or chloroquine on inner retinal structures. Eye (Lond) 24(2):340–346. 10.1038/eye2009.65 [DOI] [PubMed] [Google Scholar]
- 22.Qi H, Cole J 2nd, Grambergs RC, Gillenwater JR, Mondal K, Khanam S, Dutta S, Stiles M et al. (2019) Sphingosine Kinase 2 Phosphorylation of FTY720 is Unnecessary for Prevention of Light-Induced Retinal Damage. Sci Rep 9(1):7771. 10.1038/s41598-019-44047-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paranjpe V, Tan J, Nguyen J, Lee J, Allegood J, Galor A, Mandal N (2019) Clinical signs of meibomian gland dysfunction (MGD) are associated with changes in meibum sphingolipid composition. Ocul Surf 17(2):318–326. 10.1016/j.jtos.2018.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugano E, Edwards G, Saha S, Wilmott LA, Grambergs RC, Mondal K, Qi H, Stiles M et al. (2019) Overexpression of acid ceramidase (ASAH1) protects retinal cells (ARPE19) from oxidative stress. J Lipid Res 60(1):30–43. 10.1194/jlr.M082198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Easterbrook M (1992) Long-term course of antimalarial maculopathy after cessation of treatment. Can J Ophthalmol 27(5):237–239 [PubMed] [Google Scholar]
- 26.Modi YS, Au A, Parikh VS, Ehlers JP, Schachat AP, Singh RP (2016) Volumetric single-layer inner retinal analysis in patients with hyrdroxychloroquine toxicity. Retina 36(10):1941–1950. 10.1097/iae.0000000000001036 [DOI] [PubMed] [Google Scholar]
- 27.Elder M, Rahman AM, McLay J (2006) Early paracentral visual field loss in patients taking hydroxychloroquine. Arch Ophthalmol 124(12):1729–1733. 10.1001/archopht.124.12.1729 [DOI] [PubMed] [Google Scholar]
- 28.Lee DH, Melles RB, Joe SG, Lee JY, Kim JG, Lee CK, Yoo B, Koo BS et al. (2015) Pericentral hydroxychloroquine retinopathy in Korean patients. Ophthalmology 122(6):1252–1256. 10.1016/j.ophtha.2015.01.014 [DOI] [PubMed] [Google Scholar]
- 29.Ruberto G, Bruttini C, Tinelli C, Cavagna L, Bianchi A, Milano G (2018) Early morpho-functional changes in patients treated with hydroxychloroquine: a prospective cohort study. Graefes Arch Clin Exp Ophthalmol 256(11):2201–2210. 10.1007/s00417-018-4103-9 [DOI] [PubMed] [Google Scholar]
- 30.Yoon YH, Cho KS, Hwang JJ, Lee SJ, Choi JA, Koh JY (2010) Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest Ophthalmol Vis Sci 51(11):6030–6037. 10.1167/iovs.10-5278 [DOI] [PubMed] [Google Scholar]
- 31.Hyttinen JM, Niittykoski M, Salminen A (1833) Kaarniranta K (2013) Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim Biophys Acta 3:503–510. 10.1016/j.bbamcr.2012.11.018 [DOI] [PubMed] [Google Scholar]
- 32.Coant N, Sakamoto W, Mao C, Hannun YA (2017) Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv Biol Regul 63:122–131. 10.1016/j.jbior.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Victoria SM, Basu SK, Bano Q, Richard G, Rotstein NP, Nawajes M (2021) Sphingolipids as critical players in retinal physiology and pathology. J Lipid Res:100037. 10.1194/jlr.TR120000972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mandal N, Grambergs R, Mondal K, Basu SK, Tahia F, Dagogo-Jack S (2021) Role of ceramides in the pathogenesis of diabetes mellitus and its complications. J Diabetes Complications 35(2):107734. 10.1016/j.jdiacomp.2020.107734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pralhada Rao R, Vaidyanathan N, Rengasamy M, Mammen Oommen A, Somaiya N, Jagannath MR (2013) Sphingolipid Metabolic Pathway: An Overview of Major Roles Played in Human Diseases. J Lipids 2013:178910. 10.1155/2013/178910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gault CR, Obeid LM, Hannun YA (2010) An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol 688:1–23. 10.1007/978-1-4419-6741-1_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaufmann AM, Krise JP (2007) Lysosomal Sequestration of Amine-Containing Drugs: Analysis and Therapeutic Implications. J Pharm Sci 96(4):729–746. 10.1002/jps.20792 [DOI] [PubMed] [Google Scholar]
- 38.Ohkuma S, Poole B (1978) Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci 75(7):3327–3331. 10.1073/pnas.75.7.3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohkuma S, Chudzik J, Poole B (1986) The effects of basic substances and acidic ionophores on the digestion of exogenous and endogenous proteins in mouse peritoneal macrophages. J Cell Biol 102(3):959–966. 10.1083/jcb.102.3.959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barak A, Morse LS, Goldkorn T (2001) Ceramide: A Potential Mediator of Apoptosis in Human Retinal Pigment Epithelial Cells. Invest Ophthalmol Vis Sci 42(1):247–254 [PubMed] [Google Scholar]
- 41.Strettoi E, Gargini C, Novelli E, Sala G, Piano I, Gasco P, Ghidoni R (2010) Inhibition of ceramide biosynthesis preserves photoreceptor structure and function in a mouse model of retinitis pigmentosa. Proc Natl Acad Sci 107(43):18706–18711. 10.1073/pnas.1007644107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen H, Tran J-TA, Brush RS, Saadi A, Rahman AK, Yu M, Yasumura D, Matthes MT et al. (2012) Ceramide signaling in retinal degeneration. Adv Exp Med Biol 723:553–558. 10.1007/978-1-4614-0631-0_70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verbaanderd C, Maes H, Schaaf MB, Sukhatme VP, Pantziarka P, Sukhatme V, Agostinis P, Bouche G (2017) Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anticancer agents. Ecancermedicalscience 11:781. 10.3332/ecancer.2017.781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahon GJ, Anderson HR, Gardiner TA, McFarlane S, Archer DB, Stitt AW (2004) Chloroquine causes lysosomal dysfunction in neural retina and RPE: Implications for retinopathy. Curr Eye Res 28(4):277–284. 10.1076/ceyr.28.4.277.27835 [DOI] [PubMed] [Google Scholar]
- 45.Mazzoni F, Safa H, Finnemann SC (2014) Understanding photoreceptor outer segment phagocytosis: use and utility of RPE cells in culture. Exp Eye Res 126:51–60. 10.1016/j.exer.2014.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kevany BM, Palczewski K (2010) Phagocytosis of retinal rod and cone photoreceptors. Physiology (Bethesda) 25(1):8–15. 10.1152/physiol.00038.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sinha D, Valapala M, Shang P, Hose S, Grebe R, Lutty GA, Zigler JS Jr, Kaarniranta K, Handa JT (2016) Lysosomes: Regulators of autophagy in the retinal pigmented epithelium. Exp Eye Res 144:46–53. 10.1016/j.exer.2015.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen PM, Gombart ZJ, Chen JW (2011) Chloroquine treatment of ARPE-19 cells leads to lysosome dilation and intracellular lipid accumulation: possible implications of lysosomal dysfunction in macular degeneration. Cell Biosci 1(1):10–10. 10.1186/2045-3701-1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Golestaneh N, Chu Y, Xiao Y-Y, Stoleru GL, Theos AC (2018) Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis 8(1):e2537–e2537. 10.1038/cddis.2016.453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lally DR, Heier JS, Baumal C, Witkin AJ, Maler S, Shah CP, Reichel E, Waheed NK et al. (2016) Expanded spectral domain-OCT findings in the early detection of hydroxychloroquine retinopathy and changes following drug cessation. Int J Retina and Vitreous 2(1):18. 10.1186/s40942-016-0042-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen H, Chan AY, Stone DU, Mandal NA (2014) Beyond the cherry-red spot: Ocular manifestations of sphingolipid-mediated neurodegenerative and inflammatory disorders. Surv Ophthalmol 59(1):64–76. 10.1016/j.survophthal.2013.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tzekov R, Arden GB (1999) The electroretinogram in diabetic retinopathy. Surv Ophthalmol 44(1):53–60. 10.1016/s0039-6257(99)00063-6 [DOI] [PubMed] [Google Scholar]
- 53.Ogden TE (1973) The oscillatory waves of the primate electroretinogram. Vision Res 13(6):1059–1074. 10.1016/0042-6989(73)90144-2 [DOI] [PubMed] [Google Scholar]
- 54.Medeiros NE, Curcio CA (2001) Preservation of Ganglion Cell Layer Neurons in Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci 42(3):795–803 [PubMed] [Google Scholar]
- 55.Curcio CA, Allen KA (1990) Topography of ganglion cells in human retina. J Comp Neurol 300(1):5–25. 10.1002/cne.903000103 [DOI] [PubMed] [Google Scholar]
- 56.Maruyama T, Noda NN (2018) Autophagy-regulating protease Atg4: structure, function, regulation and inhibition. J Antibiot 71(1):72–78. 10.1038/ja.2017.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.von Eisenhart-Rothe P, Grubman A, Greferath U, Fothergill LJ, Jobling AI, Phipps JA, White AR, Fletcher EL et al. (2018) Failure of Autophagy-Lysosomal Pathways in Rod Photoreceptors Causes the Early Retinal Degeneration Phenotype Observed in Cln6nclf Mice. Invest Ophthalmol Vis Sci 59(12):5082–5097. 10.1167/iovs.18-24757 [DOI] [PubMed] [Google Scholar]
- 58.Jiang P, Mizushima N (2015) LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 75:13–18. 10.1016/j.ymeth.2014.11.021 [DOI] [PubMed] [Google Scholar]
- 59.Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, Coppes RP, Engedal N et al. (2018) Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14(8):1435–1455. 10.1080/15548627.2018.1474314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mizushima N, Yoshimori T (2007) How to Interpret LC3 Immunoblotting. Autophagy 3(6):542–545. 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- 61.Tanida I, Ueno T, Kominami E (2008) LC3 and Autophagy. Methods Mol Biol 445:77–88. 10.1007/978-1-59745-157-4_4 [DOI] [PubMed] [Google Scholar]
- 62.Islam MA, Sooro MA, Zhang P (2018) Autophagic Regulation of p62 is Critical for Cancer Therapy. Int J Mol Sci 19 (5). 10.3390/ijms19051405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim JS, Bae GE, Kim KH, Lee SI, Chung C, Lee D, Lee TH, Kwon IS et al. (2019) Prognostic Significance of LC3B and p62/SQSTM1 Expression in Gastric Adenocarcinoma. Anticancer Res 39(12):6711. 10.21873/anticanres.13886 [DOI] [PubMed] [Google Scholar]
- 64.Runwal G, Stamatakou E, Siddiqi FH, Puri C, Zhu Y, Rubinsztein DC (2019) LC3-positive structures are prominent in autophagy-deficient cells. Sci Rep 9(1):10147. 10.1038/s41598-019-46657-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ganley Ian G, Wong P-M, Gammoh N, Jiang X (2011) Distinct Autophagosomal-Lysosomal Fusion Mechanism Revealed by Thapsigargin-Induced Autophagy Arrest. Mol Cell 42(6):731–743. 10.1016/j.molcel.2011.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eskelinen EL (2006) Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med 27(5–6):495–502. 10.1016/j.mam.2006.08.005 [DOI] [PubMed] [Google Scholar]
- 67.Pugsley HR (2017) Assessing Autophagic Flux by Measuring LC3, p62, and LAMP1 Co-localization Using Multispectral Imaging Flow Cytometry. J Vis Exp 125:55637. 10.3791/55637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dressler KA, Mathias S, Kolesnick RN (1992) Tumor necrosis factor-alpha activates the sphingomyelin signal transduction pathway in a cell-free system. Science (New York, NY) 255(5052):1715–1718 [DOI] [PubMed] [Google Scholar]
- 69.Hannun YA, Loomis CR, Merrill AH, Bell RM (1986) Sphingosine inhibition of protein kinase C activity and of phorbol dibutyrate binding in vitro and in human platelets. J Biol Chem 261(27):12604–12609 [PubMed] [Google Scholar]
- 70.Zhang H, Desai NN, Olivera A, Seki T, Brooker G, Spiegel S (1991) Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J Cell Biol 114(1):155–167. 10.1083/jcb.114.1.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grambergs R, Mondal K, Mandal N (2019) Inflammatory Ocular Diseases and Sphingolipid Signaling. Adv Exp Med Biol 1159:139–152. 10.1007/978-3-030-21162-2_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mondal K, Mandal N (2019) Role of Bioactive Sphingolipids in Inflammation and Eye Diseases. In: Adv Exp Med Biol, vol 1161. Springer; New York LLC, pp 149–167. 10.1007/978-3-030-21735-8_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Petersen NHT, Kirkegaard T, Olsen OD, Jäättelä M (2010) Connecting Hsp70, sphingolipid metabolism and lysosomal stability. Cell Cycle 9(12):2305–2309. 10.4161/cc.9.12.12052 [DOI] [PubMed] [Google Scholar]
- 74.Platt FM (2014) Sphingolipid lysosomal storage disorders. Nature 510(7503):68–75. 10.1038/nature13476 [DOI] [PubMed] [Google Scholar]
- 75.Vitner EB, Platt FM, Futerman AH (2010) Common and uncommon pathogenic cascades in lysosomal storage diseases. J Biol Chem 285(27):20423–20427. 10.1074/jbc.R110.134452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schultz ML, Tecedor L, Chang M, Davidson BL (2011) Clarifying lysosomal storage diseases. Trends Neurosci 34(8):401–410. 10.1016/j.tins.2011.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kolter T, Sandhoff K (2006) Sphingolipid metabolism diseases. Biochimica et Biophysica Acta (BBA). Biomembranes 1758(12):2057–2079. 10.1016/j.bbamem.2006.05.027 [DOI] [PubMed] [Google Scholar]
- 78.Grassi S, Chiricozzi E, Mauri L, Sonnino S, Prinetti A (2019) Sphingolipids and neuronal degeneration in lysosomal storage disorders. J Neurochem 148(5):600–611. 10.1111/jnc.14540 [DOI] [PubMed] [Google Scholar]
- 79.Ferreira CR, Gahl WA (2017) Lysosomal storage diseases. Transl Sci Rare Dis 2(1–2):1–71. 10.3233/TRD-160005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hopf S, Pfeiffer N, Liesenfeld M, Mengel KE, Hennermann JB, Schmidtmann I, Pitz S (2019) A comprehensive monocentric ophthalmic study with Gaucher disease type 3 patients: vitreoretinal lesions, retinal atrophy and characterization of abnormal saccades. Orphanet J Rare Dis 14(1):257. 10.1186/s13023-019-1244-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heroman JW, Rychwalski P, Barr CC (2008) Cherry red spot in sialidosis (mucolipidosis type I). Arch Ophthalmol 126(2):270–271. 10.1001/archophthalmol.2007.31 [DOI] [PubMed] [Google Scholar]
- 82.Prado Spalm FH, Vera MS, Dibo MJ, Simon MV, Politi LE, Rotstein NP (2019) Ceramide Induces the Death of Retina Photoreceptors Through Activation of Parthanatos. Mol Neurobiol 56(7):4760–4777. 10.1007/s12035-018-1402-4 [DOI] [PubMed] [Google Scholar]
- 83.German OL, Miranda GE, Abrahan CE, Rotstein NP (2006) Ceramide is a mediator of apoptosis in retina photoreceptors. Invest Ophthalmol Vis Sci 47(4):1658–1668. 10.1167/iovs.05-1310 [DOI] [PubMed] [Google Scholar]
- 84.Mullen TD, Obeid LM (2012) Ceramide and apoptosis: exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer Agents Med Chem 12(4):340–363. 10.2174/187152012800228661 [DOI] [PubMed] [Google Scholar]
- 85.Mandal MA, Agbaga MP, Tran JT, Henry K, Zheng L, Brush RS (2008) Comprehensive Evaluation of Sphingolipid Gene Expression in Normal and Light-Stressed Retina and Oxidant-Stressed 661W Cells. Invest Ophthalmol Vis Sci 49(13):5173–5173 [Google Scholar]
- 86.Schulze H, Schepers U, Sandhoff K (2007) Overexpression and mass spectrometry analysis of mature human acid ceramidase. Biol Chem 388(12):1333–1343. 10.1515/bc.2007.152 [DOI] [PubMed] [Google Scholar]
- 87.Yu FPS, Sajdak BS, Sikora J, Salmon AE, Nagree MS, Gurka J, Kassem IS, Lipinski DM et al. (2019) Acid Ceramidase Deficiency in Mice Leads to Severe Ocular Pathology and Visual Impairment. Am J Pathol 189(2):320–338. 10.1016/j.ajpath.2018.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Klein A, Henseler M, Klein C, Suzuki K, Harzer K, Sandhoff K (1994) Sphingolipid activator protein D (sap-D) stimulates the lysosomal degradation of ceramide in vivo. Biochem Biophys Res Commun 200(3):1440–1448. 10.1006/bbrc.1994.612 [DOI] [PubMed] [Google Scholar]
- 89.Rother J, van Echten G, Schwarzmann G, Sandhoff K (1992) Biosynthesis of sphingolipids: dihydroceramide and not sphinganine is desaturated by cultured cells. Biochem Biophys Res Commun 189(1):14–20. 10.1016/0006-291x(92)91518-u [DOI] [PubMed] [Google Scholar]
- 90.Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S (1996) Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 381(6585):800–803. 10.1038/381800a0 [DOI] [PubMed] [Google Scholar]
- 91.Stover T, Kester M (2003) Liposomal delivery enhances short-chain ceramide-induced apoptosis of breast cancer cells. J Pharmacol Exp Ther 307(2):468–475. 10.1124/jpet.103.054056 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All materials and data will be available from the corresponding author following the University of Tennessee’s policy of sharing research materials and data.