


Abstract
Stereochemical control of DNA biosynthesis was studied using several DNA-synthesizing complexes containing, in each case, a single substitution of a 2′-deoxy-d-nucleotide residue by an enantiomeric l-nucleotide residue in a DNA chain (either in the primer or in the template) as well as 2′-deoxy-l-ribonucleoside 5′-triphosphates (l-dNTPs) as substrates. Three template-dependent DNA polymerases were tested, Escherichia coli DNA polymerase I Klenow fragment, Thermus aquaticus DNA polymerase and avian myeloblastosis virus reverse transcriptase, as well as template-independent calf-thymus terminal deoxynucleotidyl transferase. Very stringent control of stereoselectivity was demonstrated for template-dependent DNA polymerases, whereas terminal deoxynucleotidyl transferase was less selective. DNA polymerase I and reverse transcriptase catalyzed formation of dinucleoside 5′,5′-tetraphosphates when l-dTTP was used as substrate. Comparison between models of template–primer complexes, modified or not by a single l-nucleotide residue, revealed striking differences in their geometry.
INTRODUCTION
Stereochemical control of DNA biosynthesis and stereochemical preferences of the active center of different DNA polymerases are not clear yet. Some contradictions have been reported in the literature. In accordance with the main property of the active center of template-dependent DNA polymerases, their substrates (dNTP) have to bind by at least two separated parts of their molecules, namely the base, with formation of regular Watson–Crick pairs, and the triphosphate residue as reactive group. Therefore, guaranteeing a stringent stereospecificity of the multiple repeating process of DNA chain elongation has to correspond to stereochemical homogeneity of the DNA template, the growing DNA chain and dNTP substrates. This means that each of the components involved in the polymerization process should have nucleotide residues containing 2′-deoxy-β-d-ribofuranose. There is as yet no known data concerning the existence in cells of another stereoregular DNA analog (for example, with 2′-deoxy-β-l-, 2′-deoxy-α-d- or 2′-deoxy-α-l-ribofuranose moities) and its synthesis by known DNA polymerases seems unlikely. Among the main reasons, one should mention the necessity of correspondence between the geometries of DNA polymerase active centers and 2′-deoxy-β-d-ribofuranose residues in dNTPs.
Nevertheless, studies over the past years in non-major limits have deviated from this paradigm. A large number of examples have demonstrated that several sugar-modified 2′,3′-dideoxy-β-l-ribonucleoside 5′-triphosphates (β-l-ddNTP) are bound by active centers of several DNA polymerases and corresponding l-nucleotide residues can be incorporated at the 3′-terminus of the primer DNA chain with kinetic constants rather similar to those of natural β-d-dNTPs (1–10). This molecular mechanism occurs in anti-HIV activity of (–)-β-l-2′,3′-dideoxy-3′-thiacytidine, a well-known drug for treatment of AIDS patients.
Conflicting results have been reported for unmodified β-l-dNTP. Some authors observed their limited incorporation in primer chains (1–3 nt residues) under catalysis by several DNA polymerases (11) whereas another group found no such incorporation for template-dependent DNA polymerases (12).
We report here studies of the influence of a single substitution of an enantiomeric 2′-deoxy-β-l-nucleotide residue for a 2′-deoxy-β-d-nucleotide, either in the template or in the primer (Fig. 1) or in both, on the elongation of DNA chains when β-d-dNTP or β-l-dTTP are used as substrates. Reactions were catalyzed by DNA polymerase I, avian myeloblastosis virus (AMV) reverse transcriptase, calf thymus terminal deoxynucleotidyl transferase (TDT) and, in a separate experiment, Taq DNA polymerase. It is demonstrated that: (i) when the first nucleotide residue downstream of the primer site in the template is a 2′-deoxy-β-l-nucleotide, unmodified primer is extended neither with β-l-dTTP nor with β-d-dNTP; (ii) the primer with a 3′-terminal 2′-deoxy-β-l-nucleotide residue cannot be extended in the presence of the same substrates in complexes formed with either template containing one 2′-deoxy-β-l-nucleotide residue or natural template; (iii) template-independent TDT extends the β-l-substituted primer for one or two steps in the presence of β-d-dNTP, but no elongation occurs in the presence of β-l-dTTP. These observations obviously illustrate the stringent stereochemical control of DNA biosynthesis catalyzed by template-dependent DNA polymerases.
Figure 1.

Structure of templates and primers used in this study. Two different priming sites in Tp1 were used. Primers PrI and PrII are complementary to sites 1–14 (underlined with a continuous line) whereas PrIII and PrIV are complementary to site 15–29 (underlined with a broken line). LA and LT stand for enantiomeric deoxy-2′-l-adenosine and l-thymidine, respectively.
MATERIALS AND METHODS
Chemicals
dNTP, rNTP and ddTTP were purchased from Boehringer Mannheim, d4TTP was synthesized as in Dyatkina et al. (13) and β-l-dTTP as in Semizarov et al. (12). dT(5′-pppp-5′)dA (I in Fig. 7b) was a kind gift of Dr A. Shipitsin (Engelhardt Institute of Molecular Biology, Moscow, Russia), [α-32P]dATP and [γ-32P]ATP (3000 Ci/mmol) were from Radiopreparat (Russia).
Figure 7.
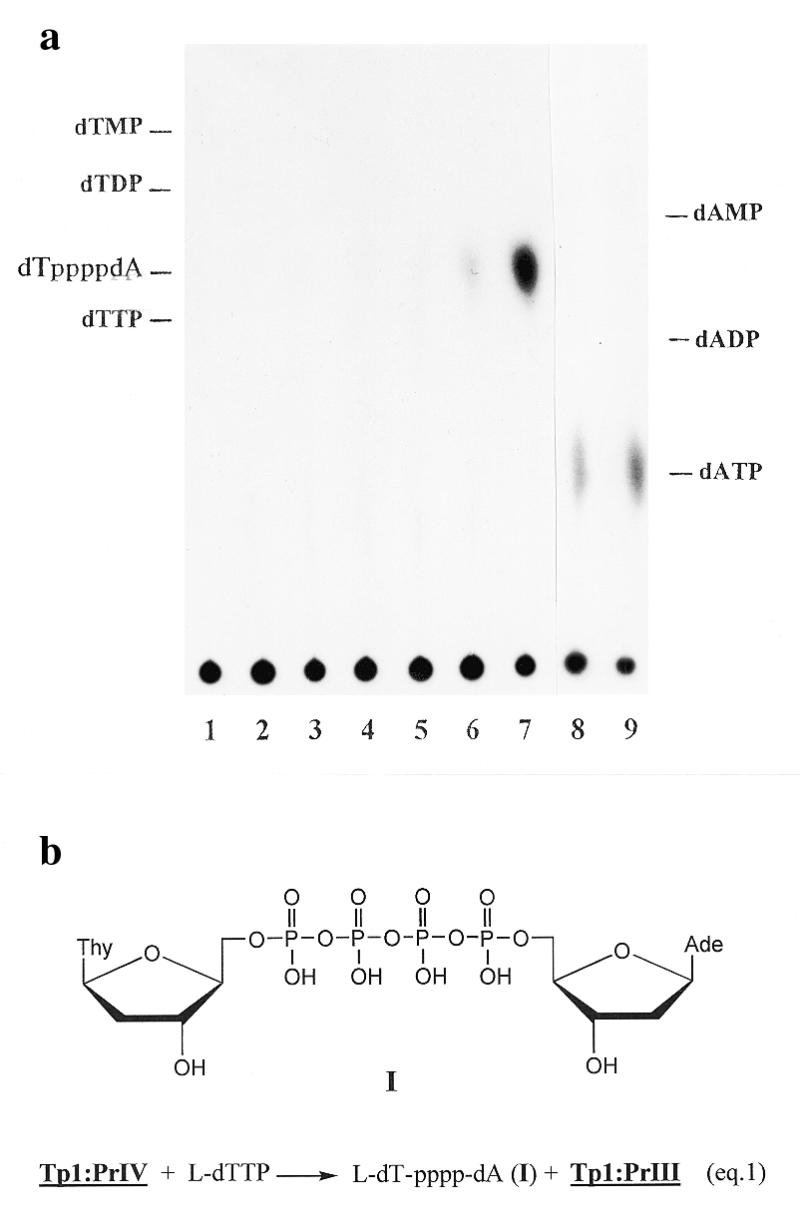
(a) TLC of reaction products of complex Tp1:PrIV with dCTP and β-l-dTTP, when catalyzed by DNA polymerase I. Lane 1, complex Tp1:PrIV + enzyme; lanes 2–4, as 1 + dCTP, 5 (2), 50 (3) and 500 µM (4); lanes 5–7, as 1 + β-l-dTTP, 5 (5), 50 (6) and 500 µM (7); lanes 8 and 9 as 1 + PPi, 0.5 (8) and 1 mM (9). (b) Structure of l-dT-pppp-dA formed during pyrophosphorolysis of PrIV according to equation 1.
Enzymes and DNA
The following enzymes were used: Escherichia coli DNA polymerase I Klenow fragment and calf intestine alkaline phosphatase (Boehringer Mannheim); AMV reverse transcriptase (Promega); calf thymus TDT, T4 polynucleotide kinase and exonuclease III (Amersham Corp.); Taq DNA polymerase (Pharmacia Biotech); snake venom phosphodiesterase (Worthington).
Single-stranded M13mp10 phage DNA (Tp1, Fig. 1) was isolated from the culture medium of the recipient E.coli K12XL1 strain as described (14). Oligonucleotides Tp2 and PrII were synthesized as in Blommers et al. (15) and PrI and PrIII were kind gifts of Dr S. Surzhykov (Engelhardt Institute of Molecular Biology, Moscow, Russia).
Tetradecanucleotide primers PrI, PrII and PrIII were labeled at the 5′-terminus using [γ-32P]ATP and T4 polynucleotide kinase (16) with subsequent enzyme inactivation (10 min, 65°C). The appropriate template–primer complexes were prepared by incubation of 5′-labeled primers and corresponding templates in buffer (10 mM Tris–HCl, pH 8.0, and 5 mM MgCl2) at 65°C for 10 min and cooling to 30°C for 60 min. Then they were purified by filtering through a Biogel A-1.5m 1 ml column in 10 mM Tris–HCl, pH 7.6, containing 1 mM EDTA, and were stored at –20°C for up to 20 days.
Complex Tp1:PrIV was prepared by incubation of complex TpI:PrIII with [α-32P]dATP in a reaction catalyzed by DNA polymerase I under standard conditions. The complex was purified on a Biogel A-1.5m column as above.
Primer extention and pyrophosphorolysis assays for DNA polymerases, AMV reverse transcriptase and TDT
For DNA polymerase I, the assay mixture (6 µl) contained 10 mM Tris–HCl, pH 7.9, 5 mM MgCl2, 1 mM DTT, 0.3 U enzyme, 40–50 nM template–primer complex and variable concentrations of substrates; the reaction was carried out for 15 min at 25°C. The pyrophosphorolysis reaction with the same enzyme was performed in a mixture (6 µl) containing 50 mM Tris–HCl, pH 7.9, 10 mM MgCl2, 50 mM KCl, 1 mM DTT, 0.8 U enzyme, 50 mM template–primer complex and 0.5–1 mM inorganic pyrophosphate (PPi); the mixture was incubated for 1–10 min at 25°C. For Taq DNA polymerase the assay (6 µl) contained 100 mM Tris–HCl, pH 9.0, 1.5 mM MgCl2, 50 mM KCl, 1 mM DTT, 0.5 U enzyme, 50 nM template–primer complex and substrates; the mixture was incubated for 30 min at 60°C. For AMV reverse transcriptase the assay (6 µl) contained 10 mM Tris–HCl, pH 8.2, 5 mM MgCl2, 40 mM KCl, 1 mM DTT, 3 U enzyme, 40–50 nM template–primer complex and substrates; the mixture was incubated for 30 min at 37°C. For TDT the assay (6 µl) contained 100 mM sodium cacodylate, pH 7.2, 2 mM CoCl2, 0.1 mM DTT, 2 U enzyme, 20 nM 5′-labeled primer and substrates; the mixture was incubated for 30 min at 37°C.
All reactions were stopped by adding 3 µl of deionized formamide containing 0.5 mM EDTA and 0.1% bromophenol blue and xylene cyanol. The products were heated at 100°C for 2 min and separated by electrophoresis in a 14–20% denaturating polyacrylamide gel. The gels were autoradiographed with Kodak RX films at –20°C.
Reactions catalyzed by alkaline phosphatase or phosphodiesterase
Reactions catalyzed by alkaline phosphatase (in 100 mM Tris–HCl, pH 9.2, 10 mM MgCl2) contained, in a volume of 6–10 µl, 2–5 µM unlabeled substrates and 0.1 U enzyme (or 3–30 nM 32P-labeled substrates and 0.001 U enzyme) were incubated for 2–15 min at 37°C. Reactions catalyzed by phosphodiesterase were carried out for 30 min at 37°C in a volume of 10 µl in the presence of 10 mM Tris–HCl, pH 8.6, 30 mM MgCl2, 3 mM KCl and 0.4 mM DTT under two conditions: with 2 µM unlabeled substrates and 0.5–1 U enzyme or with 5 nM 32P-labeled substrates and 0.05 U enzyme. Both reactions were stopped by adding 1 µl of 0.5 M EDTA, pH 8.0, and heated at 100°C for 3 min. The reaction products were separated by chromatography on PEI Cellulose F plastic sheets (Merck) in 0.2 M potassium phosphate, pH 8.0, containing 10% ethanol.
Reactions catalyzed by exonuclease III
The assay mixture (volume, 6 µl) containing 5 mM Tris–HCl, pH 7.9, 10 mM MgCl2, 50 mM KCl, 1 mM DTT, 40 nM template–primer complexes (or 20 mM primers) and 6 U enzyme was incubated at 37°C for the indicated times. The reaction was stopped by adding 3 µl of deionized formamide containing 0.5 mM EDTA, 2% bromophenol blue and xylene cyanol, heated at 100°C for 2 min and separated by electrophoresis in a 14% denaturating polyacrylamide gel. The gels were autoradiographed with Kodak RX films at –20°C.
Computer calculations and figures of Tp1:PrI, Tp1:PrII and Tp2:PrI structures were performed with the program Hyper Chem 4.5.
RESULTS
M13mp10 DNA was used as the template in complexes Tp1:PrI, Tp1:PrII, Tp1:PrIII and Tp1:PrIV; the structure of its corresponding fragment is shown in Figure 1. The template Tp2 in complexes Tp2:PrI and Tp2:PrII contains a single l-nucleotide residue at position 15. The primers in complexes Tp1:PrI, Tp2:PrI, Tp1:PrIII and Tp1:PrIV contain only d-nucleotides whereas in complexes Tp1:PrII and Tp2:PrII an l-nucleotide was present at the first position downstream of the primer site.
Figure 2 depicts the results of primer elongation in complexes Tp1:PrI and Tp1:PrII catalyzed by AMV reverse transcriptase. As one can see from lanes 2–5, Series A, elongation of complex Tp1:PrI occured in accordance with the template structure and stopped at the first step when carried out solely in the presence of the chain terminators ddTTP and d4TTP (lanes 6 and 7). At the same time no primer elongation was observed in complex Tp1:PrII (Series B) in the presence of dNTPs at concentrations up to 200 µM (lanes 3–5) or in the presence of the terminating substrates ddTTP and d4TTP up to 100 µM (lanes 6–9). It should be mentioned that primer PrII contained insignificant impurities which did not change during the reaction (Series B, lanes 1–9). A similar absence of elongation was observed for complexes Tp2:PrI and Tp2:PrII (data shown in Table 1), thus the presence of an l-nucleotide residue at the 3′-terminus of a primer does not allow extension by AMV reverse transcriptase. Similarly, the primers in all the complexes were not extended with β-l-dTTP as the substrate.
Figure 2.
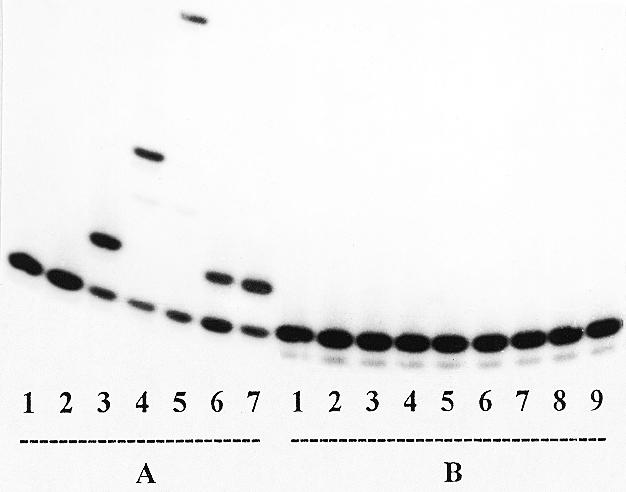
Extension of the primers in complexes Tp1:PrI (Series A) and Tp1:PrII (Series B) catalyzed by AMV reverse transcriptase. Series A: lane 1, primer (control); lane 2, primer + enzyme (control); lanes 3–5, as in lane 2 + 10 µM dTTP (3), dTTP + dGTP (4) and dTTP + dGTP + dATP (5); lane 6, as in 2 + 10 µM ddTTP; lane 7, as 2 + 10 µM d4TTP. Series B: lane 1, primer (control); lane 2, primer + enzyme (control); lanes 3–5, as in lane 2 + 200 µM dTTP (3), dTTP + dGTP (4) and dTTP + dGTP + dATP (5); lanes 6 and 7, as in 2 + 10 µM (6) and 100 µM (7) ddTTP; lanes 8 and 9, as in 2 + 10 (8) and 100 µM (9) d4TTP, respectively.
Table 1. DNA polymerase catalyzed primer elongation in template–primer complexes containing a single substitution of enantiomeric l-nucleotide for the corresponding natural d-nucleotide.

*+, incorporation observed; –, no incorporation. Concentrations of substrates (µM) are shown in parentheses. Refer to Figure 1 for the structure of the template–primer complexes.
Also, primers were not extended with dTTP or with mixtures of dTTP + dGTP or dTTP + dGTP + dATP in complexes Tp1:PrII, Tp2:PrI and Tp2:PrII when reactions were catalyzed by DNA polymerase I (Table 1). Also, primer elongation in complexes Tp1:PrI, Tp1:PrII and Tp2:PrI in the presence of β-l-dTTP was not observed. A similar absence of chain elongation was shown for Taq DNA polymerase for complex Tp2:PrI (Table 1). Results of TDT-catalyzed extension of the PrI and PrII oligodeoxynucleotides are shown in Figure 3. As seen in lanes 2–5 (Series A), primer PrI is effectively elongated at a dNTP concentration of 5 µM to give extended products. At the same time, primer PrII is only extended by one or two units even at higher substrate concentrations (lanes 2–9, Series B). One can assume that the presence of an l-nucleotide may distort the primer 3′-terminus orientation and addition of the next one or two nucleotides enhances this distortion. As a result, complex PrII–enzyme is non-productive and elongation is terminated. In addition, the effectiveness of PrII elongation is affected by the nature of the substrate base (lanes 2–9, Series B). Both primers are elongated by one nucleotide residue in the presence of ddTTP, but in the case of PrII the elongation is less effective (lane 6, Series A versus lanes 10 and 11, Series B). Unlike for primer PrI, no extension of primer PrII by d4TTP was observed. The results obtained are summarized in Table 2.
Figure 3.

Extension of the primers PrI (Series A) and PrII (Series B) catalyzed by TDT. Series A: lane 1, primer + enzyme (control); lanes 2–5, as in lane 1 + 5 µM dATP (2), dGTP (3), dCTP (4) and dTTP (5); lane 6, as in lane 1 + 1 µM ddTTP (6). Series B: lane 1, primer + enzyme (control); lanes 2–9 as in lane 1 + 100 or 10 µM dATP (2 and 3), dGTP (4 and 5), dCTP (6 and 7), dTTP (8 and 9) and ddTTP (10 and 11), respectively.
Table 2. Specificity of TDT + primer PrI or PrII towards substrates and their analogs.

*+++, complete primer extension; ++, extension of >50% of primer; +, extension of <50% of primer; –, no extension.
Comparison of primers PrI and PrII hydrolysis by E.coli exonuclease III (Fig. 4) shows a higher rate of removal of the 3′-terminal nucleotide residue for primer PrII (Series D) than PrI (Series C). Probably, in the case of primer PrII the phosphodiester link between the 3′-terminal and foregoing nucleotide residues (l-dTMP and d-dGMP, respectively) is more sterically accessible to exonuclease III than the analogous (d-dTMP)–(d-dGMP) link of primer PrI. Similar experiments for the Tp1:PrI (Series A) and Tp1:PrII (Series B) complexes did not show significant differences in the hydrolysis rate.
Figure 4.
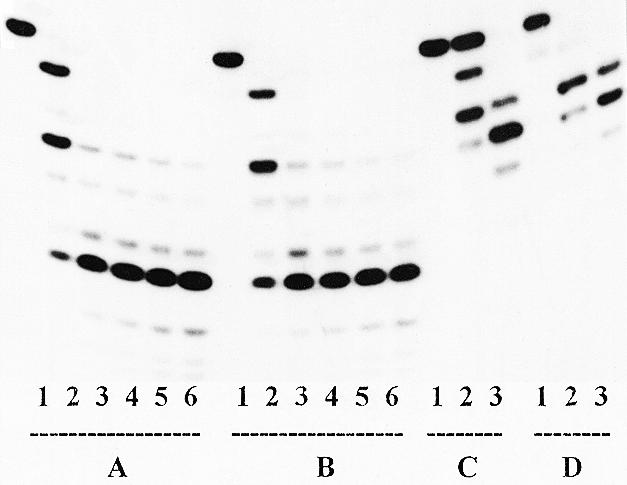
Hydrolysis of the primers in complexes Tp1:PrI (Series A) and Tp1:PrII (Series B) and primers PrI (Series C) and PrII (Series D) catalyzed by exonuclease III. Series A and B: lane 1, primer + template (control); lanes 2–6, as in lane 1 + enzyme for 1 (2), 5 (3), 15 (4), 30 (5) and 60 min (6). Series C and D: lane 1, primer (control); lanes 2–3, as in lane 1 + enzyme for 5 (2) and 30 min (3).
As one can see from Figure 5, pyrophosphorolysis of primer PrII in the Tp1:PrII complex in the presence of PPi and DNA polymerase I did not occur (Series B, lanes 5–7), whereas for the Tp1:PrI complex this reaction was completed for one step and even a little more under the same conditions over 1–10 min (lanes 5–7, Series A). This means that the 3′-terminal l-thymidine residue in the Tp1:PrII complex cannot form a productive complex for the pyrophosphorolysis reaction.
Figure 5.
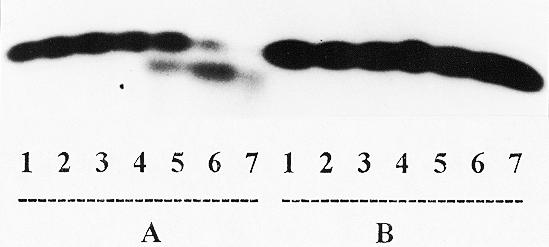
Pyrophosphorolysis of the primers in complexes Tp1:PrI (Series A) and Tp1:PrII (Series B) catalyzed by DNA polymerase I. Lane 1, primer + template (control); lanes 2–4, as in lane 1 + enzyme for 1 (2), 3 (3) and 10 min (4), respectively; lanes 5–7, as in lane 1 + enzyme + 1 mM pyrophosphate for 1 (5), 3 (6) and 10 min (7), respectively.
Transfer of the 3′-terminal [32P]deoxyadenylate from primer PrIV in complex Tp1:PrIV to l-dTTP catalyzed by AMV reverse transcriptase or DNA polymerase I is illustrated in Figure 6. From comparison of lanes 10 and 17 with lane 2 as a control, a shortening of the primer is evident, one step of which is nearly completed at 500 µM β-l-dTTP. From comparison of lanes 5–8 and 12–15 it is obvious that other tested nucleoside 5′-triphosphates (dCTP and rCTP) do not elicit primer shortening. Analysis of the reaction product is presented in Figure 7a. At 50 (lane 6) and 500 µM (lane 7) β-l-dTTP, a concentration-dependent formation of a new radioactive compound with the same mobility (Fig. 7b) as that observed for chemically synthesized dinucleoside tetraphosphate dT(5′-pppp-5′)dA occured and we propose for its structure l-dT-pppp-dA (I). Formation of such a compound was not observed when dCTP was substituted for β-l-dTTP under similar conditions (lanes 2–4). For dTTP and rCTP, shortening of the primer and formation of new compounds were also not observed (data not shown). For comparison one can see results of the pyrophosphorolysis reaction for the Tp1:PrIV complex with PPi (lanes 8 and 9). The structure of I was proved by direct comparison with a synthetic dT(5′-pppp-5′)dA sample. In addition, I was stable to hydrolysis by alkaline phosphatase and was hydrolyzed by phosphodiesterase. Under similar conditions, [γ-32P]ATP was hydrolyzed by both enzymes.
Figure 6.
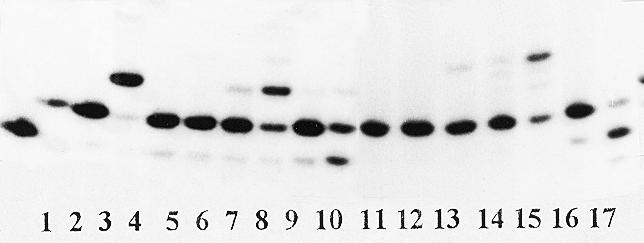
Reaction of complex Tp1:PrIV with dCTP, rCTP or β-l-dTTP catalyzed by AMV reverse transcriptase (lanes 2–10) and DNA polymerase I (lanes 11–17). Lane 1, complex Tp1:PrIII (control); lane 2, complex Tp1:PrIV (control); lanes 3 and 11, complex Tp1:PrIV + enzyme (controls); lane 4, as 3 + 5 µM dGTP; lanes 5 and 6 and 12 and 13, as 3 and 11 + dCTP (50 µM for 5 and 12 and 500 µM for 6 and 13, respectively); lanes 7 and 8 and 14 and 15, as 3 and 11 + rCTP (50 µM for 7 and 14 and 500 µM for 8 and 15, respectively); lanes 9 and 10 and 16 and 17, as 3 and 11 + β-l-dTTP (50 µM for 9 and 16 and 500 µM for 10 and 17, respectively).
DISCUSSION
All data shown in the study can be summarized as follows: (i) elongation of a primer with a 3′-terminal l-nucleotide residue is completely prevented for template-dependent DNA polymerases whereas it occurs in the presence of template-independent TDT, but only for one or two nucleotide residues and at high dNTP concentrations; (ii) elongation of the primer is stopped just before the l-nucleotide residue in the template; (iii) l-dNTPs do not seem to be substrates for any of the seven template-dependent DNA polymerases studied by us here and previously (12). Moreover, the orientation of the 3′-terminal l-deoxythymidylate residue in a primer being complexed with any template should be different from that of the natural d-deoxythymidylate residue in the same primer and prevents productive complex formation. This is obvious from the absence of pyrophosphorolysis of the PrII:TpI complex catalyzed by DNA polymerase I (Fig. 5). Therefore, one may conclude that stereochemical control of DNA biosynthesis by template-dependent DNA polymerases is very stringent and excludes incorporation into the DNA chain or a stop of DNA synthesis by l-dNTPs possibly formed in cells.
Moreover, l-dNTPs are excluded from the reaction mixture by their conversion to dinucleoside 5′,5′-tetraphosphate (to l-dT-pppp-dA, I in our case) according to equation 1 in Figure 7b. A similar reaction does not occur in parallel experiments with nucleoside triphosphates non-complementary to the next nucleotide template residue dCTP, rCTP (Figs 6 and 7a) and dTTP (data not shown). The same results were found for AMV reverse transcriptase. When this paper was in preparation, a similar reaction for NppppN formation was published (17). This reaction was catalyzed by three reverse transcriptases, namely from HIV, AMV and Moloney murine leukemia virus (M-MuLV); the primer in the initial complex was terminated by a ddAMP residue and ATP, its pyrophosphonate analogs, CTP, UTP, GTP, dATP, dCTP, dGTP, ddGTP and ADP were used as substrates in the pyrophosphorolysis reaction. These and our data demonstrate that, firstly, this reaction can be catalyzed by several DNA polymerases and, secondly, reverse transcriptase seems to be less specific towards triphosphate substrates in this pyrophosphorolysis reaction. It is more difficult to explain the biological significance of this reaction. In Meyer et al. (17) it was supposed that, in this way, the 3′-terminal nucleotide residue is removed in extreme cases, for example when termination of newly synthesized DNA chains takes place. In the case of l-dNTP, we speculate that DNA polymerases are capable of modifying l-dNTPs in such a way as to exclude them from competition with natural dNTPs.
Nevertheless, incorporation of 3′-modified l-nucleotide residues in primers has been well demonstrated (1–10). Therefore one can propose that the C3′ OH in l-dNTPs prevents formation of a productive complex of l-dNTP in the DNA polymerase active center which seems to be critical for stereochemical control of DNA biosynthesis. Such an idea was put forward by us earlier (18). The primer C3′ OH of the 3′-terminal l-nucleotide residue in complex Tp1:PrII is orientated differently than the C3′ OH in the Tp1:PrI complex; this is obvious from Figure 8a. The stacking between bases of the l-nucleotide residue and its neighbor in the 5′-direction of the template in the Tp2:PrI complex is significantly modified as compared with stacking between the corresponding d-nucleotide residues (Fig. 8b). This hardly changes both primer and template chain geometry.
Figure 8.

(a) Putative structure of the last two nucleotide units at the 3′-terminus of primers in complexes Tp1:PrI and Tp1:PrII with characteristic internucleoside distances. (b) Angles α between nucleobase axes of p-D-dA-14 and p-D-dA-15 of template Tp1 in complex Tp1:PrI and of p-D-dA-14 and p-L-dA-15 of template Tp2 in complex Tp2:PrI. In this legend, numbers indicate the positions of corresponding nucleotide residues in templates (see Fig. 1).
Acknowledgments
ACKNOWLEDGEMENTS
The authors are indebted to Dr Alexander V. Shipitsin for synthesis of the control sample dTppppdA and to Dr Ian Robbins for critical reading of the manuscript. F.S. thanks the Agence Nationale pour la recherche sur le SIDA (ANRS, France) for the award of a research studentship. The research was supported by a PICS grant (no. 539-Russie, entitled ‘Design of new antiviral drugs among stereoisomeric triphosphate derivatives’) 98-04-22017, Russian Basic Research Foundation Grant no. 99-04-48315 and a Grant of Leading Schools no. 97646.
REFERENCES
- 1.Hart G.J., Orr,D.C., Penn,C.R., Figueiredo,H.T., Gray,N.M., Boehme,R.E. and Cameron,J.M. (1992) Antimicrob. Agents Chemother., 36, 1688–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang C.-N., Skalski,V., Zhou,J.H. and Cheng,Y.-C. (1992) J. Biol. Chem., 267, 22414–22420. [PubMed] [Google Scholar]
- 3.Skalski V., Chang,C.-N., Dutschman,G. and Cheng,Y.-C. (1993) J. Biol. Chem., 268, 23234–23238. [PubMed] [Google Scholar]
- 4.Furman P.A., Davis,M., Liotta,D.C., Paff,M., Frick,L.W., Nelson,D.J., Dornsife,R.E., Wurster,J.A., Wilson,L.J., Fyfe,J.A., Tuttle,J.V., Miller,W.H., Condreay,L., Averett,D.R., Schinazi,R.F. and Painter,G.R. (1992) Antimicrob. Agents Chemother., 36, 2686–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson J.E., Martin,J.L., Borroto-Esoda,K., Hopkins,S., Painter,G., Liotta,D.C. and Furman,P.A. (1993) Antimicrob. Agents Chemother., 37, 1720–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coe D.M., Roberts,S.M. and Storer,R. (1992) J. Chem. Soc. Perkin Trans. 1, 2695–2704. [Google Scholar]
- 7.Merlo V., Roberts,S.M., Storer,R. and Bethell,R.C. (1994) J. Chem. Soc. Perkin Trans. 1, 1477–1481. [Google Scholar]
- 8.Semizarov D.G., Victorova,L.S., Dyatkina,N.B., von Janta-Lipinsky,M. and Krayevsky,A.A. (1994) FEBS Lett., 354, 187–190. [DOI] [PubMed] [Google Scholar]
- 9.Kukhanova M., Liu,S.-H., Mozzherin,D.J., Lin,T.-S., Chu,C.K. and Cheng,Y.-C. (1995) J. Biol. Chem., 270, 23055–23059. [DOI] [PubMed] [Google Scholar]
- 10.Kukhanova M., Li,X.-Y., Chen,S.-H., King,I., Doyle,T., Prusoff,W. and Cheng,Y.-C. (1998) Mol. Pharmacol., 53, 801–807. [PubMed] [Google Scholar]
- 11.Focher F., Maga,G., Bendiscioli,A., Capobianco,M., Colonna,F., Garbesi,A. and Spadari,S. (1995) Nucleic Acids Res., 23, 2840–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semizarov D.G., Arzumanov,A.A., Dyatkina,N.B., Meyer,A., Vichier-Guerre,S., Gosselin,G., Rayner,B., Imbach,J.L. and Krayevsky,A.A. (1997) J. Biol. Chem., 272, 9556–9560. [DOI] [PubMed] [Google Scholar]
- 13.Dyatkina N.B., von Janta-Lipinsky,M., Kukhanova,M.K., Krayevsky,A.A., Chidgeavadze,Z.G. and Beabaelashvilli,R.S. (1987) Bioorg. Khim., 13, 1366–1374. [DOI] [PubMed] [Google Scholar]
- 14.Kraev A.S. (1988) Mol. Biol (Mosk.), 22, 1164–1197. [PubMed] [Google Scholar]
- 15.Blommers M.J.J., Tondelli,L. and Garbesi,A. (1994) Biochemistry, 33, 7886–7896. [DOI] [PubMed] [Google Scholar]
- 16.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 17.Meyer P.R., Matsuura,S.E., So,A.G. and Scott,W.A. (1998) Proc. Natl Acad. Sci. USA, 95, 13471–13476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maga G., Amacker,M., Hübscher,U., Gosselin,G., Imbach,J.-L., Mathé,C., Faraj,A., Sommadossi,J.-P. and Spadari,S. (1999) Nucleic Acids Res., 27, 972–978. [DOI] [PMC free article] [PubMed] [Google Scholar]