

Abstract
Background
N6‐methyladenosine (m6A) modification is the most common modification that occurs in eukaryotes. Although substantial effort has been made in the prevention and treatment of gastric cancer (GC) in recent years, the prognosis of GC patients remains unsatisfactory. The regulatory mechanism between m6A modification and GC development needs to be elucidated. In this study, we examined m6A modification and the downstream mechanism in GC.
Methods
Dot blotting assays, The Cancer Genome Atlas analysis, and quantitative real‑time PCR (qRT‐PCR) were used to measure the m6A levels in GC tissues. Methylated RNA‐immunoprecipitation sequencing and RNA sequencing were performed to identify the targets of m6A modification. Western blotting, Transwell, wound healing, and angiogenesis assays were conducted to examine the role of centromere protein F (CENPF) in GC in vitro. Xenograft, immunohistochemistry, and in vivo metastasis experiments were conducted to examine the role of CENPF in GC in vivo. Methylated RNA‐immunoprecipitation‐qPCR, RNA immunoprecipitation‐qPCR and RNA pulldown assays were used to verify the m6A modification sites of CENPF. Gain/loss‐of‐function and rescue experiments were conducted to determine the relationship between CENPF and the mitogen‐activated protein kinase (MAPK) signaling pathway in GC cells. Coimmunoprecipitation, mass spectrometry, qRT‐PCR, and immunofluorescence assays were performed to explore the proteins that interact with CENPF and elucidate the regulatory mechanisms between them.
Results
CENPF was upregulated in GC and facilitated the metastasis of GC both in vitro and in vivo. Mechanistically, increased m6A modification of CENPF was mediated by methyltransferase 3, and this modified molecule could be recognized by heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1), thereby promoting its mRNA stability. In addition, the metastatic phenotype of CENPF was dependent on the MAPK signaling pathway. Furthermore, CENPF could bind to FAK and promote its localization in the cytoplasm. Moreover, we discovered that high expression of CENPF was related to lymphatic invasion and overall survival in GC patients.
Conclusions
Our findings revealed that increased m6A modification of CENPF facilitates the metastasis and angiogenesis of GC through the CENPF/FAK/MAPK and epithelial‐mesenchymal transition axis. CENPF expression was correlated with the clinical features of GC patients; therefore, CENPF may serve as a prognostic marker of GC.
Keywords: CENPF, epithelial‐mesenchymal transition, FAK, gastric cancer, MAPK, metastasis, N6‐methyladenosine
List of Abbreviations
- m6A
N6‐methyladenosine
- GC
gastric cancer
- MeRIP‐seq
methylated RNA‐immunoprecipitation sequencing
- RNA‐seq
RNA sequencing
- MAPK
mitogen‐activated protein kinase
- CENPF
centromere protein f
- FAK
focal adhesion kinase
- PI3K
phosphatidylinositol 3‐kinase
- HUVECs
human umbilical vein endothelial cells
- IgG
immunoglobulin G
- IHC
immunohistochemistry
- SD
standard deviation
- TCGA
The Cancer Genome Atlas
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- siRNA
small interfering RNA
- HE
hematoxylin‐eosin
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- RIP
RNA immunoprecipitation
- EMT
epithelial‐mesenchymal transition
- METTL3
methyltransferase 3
- HNRNPA2B1
heterogeneous nuclear ribonucleoprotein a2/b1
- METTL14
methyltransferase 14
- WTAP
wilms tumor 1‐associating protein
- FTO
fat mass and obesity‐associated protein
- ALKBH5
alkb homolog 5
- IP
immunoprecipitation
- OS
overall survival
1. BACKGROUND
Gastric cancer (GC) is one of the most prevalent malignancies worldwide, ranking fifth in incidence rate and fourth in cancer‐related mortality globally [1]. The incidence of GC is particularly high in East Asia, especially China [2, 3, 4]. Owing to the rapid progression and high rate of metastasis [5], many GC patients are already in the advanced stage at the time of diagnosis, resulting in poor prognosis [6]. Although many therapeutic methods have been administered to GC patients, such as neoadjuvant chemoradiotherapy, targeted therapy and immunotherapy, the prognosis of patients with GC remains unsatisfactory [7, 8, 9]. Therefore, it is necessary to further explore the specific molecular mechanism underlying GC progression to seek new therapeutic targets and improve the prognosis of GC patients.
N6‐methyladenosine (m6A) modification is the most abundant modification in eukaryotic mRNAs to date [10]. m6A modification is a dynamic reversible process which is usually mediated by m6A methyltransferases (“writers”) and removed by m6A demethylases (“erasers”) [11, 12]. Several RNA‐binding proteins (“readers”) can specifically recognize these m6A sites and affect the function of RNAs [13]. Recent studies have illustrated that m6A modification can affect RNA function by affecting RNA splicing, translation, stabilization, and degradation [14, 15, 16, 17]. Although many studies have been conducted on m6A modification in cancers, few studies have focused on the global methylation level of GC.
Centromere protein F (CENPF) encodes a protein involved in centromere‐kinetochore complex formation and is an important component that participates in the G2 phase of the cell cycle. In recent years, CENPF has been reported to be involved in the malignant progression of many tumors, such as breast cancer [18] and prostate cancer [19]. In addition, CENPF can affect the MAP kinase (MAPK) signaling pathway [20, 21]. Moreover, CENPF was found to be associated with chemotherapy resistance in a recent study [22]. However, the mechanism between CENPF and the MAPK pathway remains to be investigated.
Focal adhesion kinase (FAK) is an important member of integrin‐mediated signal transduction, and it has tyrosine kinase activity and can be self‐phosphorylated. It has been reported that FAK plays an essential role in regulating cell proliferation, adhesion, apoptosis and cell cycle progression. Moreover, FAK can activate the phosphatidylinositol 3‐kinase (PI3K), AKT1 signaling pathway and MAPK signaling pathway via downstream mechanisms [23, 24, 25]. However, the relationship between FAK and CENPF is still unclear.
In this study, we thoroughly investigated the mechanism by which m6A‐modified CENPF promotes the malignant progression of GC by promoting the nuclear export of FAK and ultimately activating the MAPK signaling pathway. These findings are expected to provide a target for GC treatment in the future.
2. MATERIALS AND METHODS
2.1. Tissue specimens
Primary tissue specimens and adjacent normal tissues in this study were collected from GC patients receiving radical gastrectomy at the First Affiliated Hospital of Nanjing Medical University (Nanjing, Jiangsu, China) from January 2014 to December 2016. None of the patients with GC received preoperative chemotherapy or radiotherapy. All specimens were obtained under the administration of the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University and the approval of all participants. The pathological diagnosis was performed according to American Joint Committee on Cancer classification (AJCC 8th edition).
2.2. Cell culture and cell treatment
The human GC cell lines HGC27 and AGS as well as human umbilical vein endothelial cells (HUVEC) were purchased from Shanghai Institutes for Biological Sciences (Shanghai, China). AGS was cultured in F12K medium (Gibco, California, USA) while HGC27 and HUVEC were cultured in RPMI 1640 medium (Gibco). All cell culture media were supplemented with 10% fetal bovine serum (FBS; Invitrogen, California, USA) and 1% penicillin/streptomycin (Gibco). All cells were cultured in a humid environment of 37°C with 5% CO2. The GC cell lines were treated with U0126 (10 μmol/L, Beyotime, Shanghai, China) for 24 h or PND‐1186 (1 μmol/L, MCE, New Jersey, USA) for 2 h.
2.3. Western blotting
Total protein was extracted from cells by using RIPA lysis buffer (Beyotime, Shanghai, China) adding with protease and phosphatase inhibitor cocktail (NCM Biotech, Suzhou, Jiangsu, China). The concentration was calculated according to Bicinchoninic acid protein assay kit (Beyotime). The protein was mixed with sodium dodecyl‐sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) loading buffer (Beyotime) and was then heated in boiled water for 5 min. The samples were separated by 10% or 7.5% SDS‐containing polyacrylamide gels and transferred to a polyvinylidene fluoride or polyvinylidene difluoride (PVDF) membrane (Thermo Fisher Scientific, Waltham, USA). For proteins with a molecular weight greater than 100 kDa, we used 7.5% acrylamide, and for proteins less than 100 kDa, we used 10% acrylamide. After blocking with QuickblockTM Buffer (Beyotime), the membrane was incubated with the primary antibody overnight at 4°C and the secondary antibody for 2 h at room temperature. Afterwards, the protein was visualized by ECL chemiluminescent reagent (Millipore, Billerica, Massachusetts, USA). The antibodies used in this study are listed in Supplementary Table S1.
2.4. Immunoprecipitation
Total protein was extracted from cells by using RIPA lysis buffer (Beyotime) adding with protease and phosphatase inhibitor cocktail (NCM Biotech). The cells were then collected at 12,000 rpm for 10 min at 4°C. Afterwards, the lysates were immunoprecipitated with indicated antibodies and the protein A/G‐agarose beads (Santa Cruz Biotechnology, Dallas, Texas, USA) was utilized for the immunoprecipitation of cell lysates. Western blot was used to analyze the sample and purified immunoglobulin G (IgG) from host species was used as a control. The antibodies used in this study are listed in Supplementary Table S1.
2.5. RNA extraction and quantitative real‑time PCR (qRT‐PCR) assay
Total RNA was extracted by TRIzol reagent (Invitrogen) and was then reverse transcribed into cDNA by the PrimeScript™ RT Master Mix Kit (Takara, Kusatsu, Shiga, Japan). The Universal SYBR Green Master Kit (Roche, Basel, Basel‐Stadt, Switzerland) was used for qRT‐PCR and the process was conducted by means of 7500 Real‐Time PCR System (Applied Biosystems, Waltham, Massachusetts, USA). The 2−ΔΔCT method was used to calculate the relative expression levels of samples, in which GAPDH was used as the internal reference of mRNAs. The primer sequences used in this study are listed in Supplementary Table S2.
2.6. Actinomycin D assay
HGC‐27 and AGS cells were plated in 24‐well plates at a density of 5 × 104 cells per well and cultivated for an overnight period. Following the addition of 2 mg/L Actinomycin D (Act D) (Sigma‐Aldrich, St. Louis, Missouri, USA) to the cells, total RNA was extracted at the specified time intervals (0, 1, 2, 4, 6, and 8 h). Then, qRT‐PCR was performed to examine the stability of the mRNA.
2.7. Transfection and plasmid construction
Small interfering RNA (siRNA) and short hairpin RNA (shRNA) for gene knockdown and plasmids for overexpression were designed and synthesized by GenePharma (Shanghai, China). Transient transfections were carried out for siRNA using 4 μL of Lipofectamine 3000 (Thermo Fisher Scientific) and 5 μL of siRNA, and for plasmids using a plasmid to transfection reagent ratio of 1 μg:1 μL per well in 6‐well plates. All sequences are listed in Supplementary Table S2.
2.8. Transwell assay
Cells were digested and resuspended in bare culture to prepare the cell suspension. Cell suspension (200 μL) was added into the upper chamber while 750 μL of cell culture with FBS was added into the lower chamber. The chambers were removed and fixed with methanol for 30 min after 48 h. Next, cells were stained with 0.1% crystal violet solution (Beyotime) for 20 min, and the upper layer of chambers were wiped off with a cotton swab. Finally, 5 fields were randomly photographed with a microscope and cells were counted.
2.9. Wound healing assay
Spread the cells onto a 6‐well plate with a horizontal line drawn in advance with a marker pen on the back. When cells had grown to optimal density, the tip of the pipetting gun was used to scratch the cells in the center of the hole perpendicular to the labeled horizontal line. The plate was washed with phosphate‐buffered saline (PBS) and bare culture medium was added. The wound healing was observed under a microscope at 0 h and 24 h respectively. Image J software was used to measure the proportion of wound healing.
2.10. In vitro angiogenesis assay
First, 50 μL of matrix gel (BD Biosciences, Franklin Lake, New Jersey, USA) was added to a 96‐well plate and left for 30 min at 37°C. Subsequently, HUVEC cells were resuspended in HGC27 or AGS cell medium supplemented with 1% microvascular growth supplement and evenly spread on Matrigel. After HUVEC cells formed blood vessels, they were photographed to assess the formation of vascular structures and numbers. Image J software (National Institutes of Health, Bethesda, Maryland, USA) was applied to count the numbers and total length of branches to assess the degree of angiogenesis representing in vitro.
2.11. Flow cytometric analysis
A total of 3 × 105 cells were collected, and 1 mL PBS at room temperature was added. Then, cells were slowly added to absolute ethanol (pre‐cooled at ‐20°C), stirred at high speed while adding, and fixed at ‐20°C overnight. On the day of testing, the ethanol was discarded, and 3 mL PBS was added, which was rehydrate for 15 min. Finally, the supernatant was discarded, and 1 mL DNA staining solution (Multisciences, Hangzhou, Zhejiang, China) was added, followed by swirling and mixing for 5‐10 s. After incubation for 30 min, the samples were detected by flow cytometry (Beckman Coulter Life Sciences, Indianapolis, Indiana, USA).
2.12. Methylated RNA‐immunoprecipitation‐qPCR (MeRIP‐qPCR)
The MeRIP assay was performed according to the manufacturer's instructions of Magna MeRIP™ m6A Kit (17‐10499; Millipore, Billerica, Massachusetts, USA). In brief, RNA was chemically fragmented into 100 nucleotides or smaller fragments followed by magnetic immunoprecipitation with a monoclonal antibody (1:1000, MABE1006, Millipore) toward m6A. After washing with IP buffer, the RNA was eluted and precipitated with ethanol. Then, the isolated RNA fragments could be subjected with qRT‐PCR. The antibodies used in this study are listed in Supplementary Table S1.
2.13. RNA immunoprecipitation (RIP) assay
The RIP assay was performed according to the manufacturer's instructions of Magna RIPTM RNA‐Binding Protein Immunoprecipitation Kit (#17‐700, Millipore Sigma). In short, cells were harvested and incubated overnight with magnetic beads coated with HNRNPA2B1 antibody (ab259894, Abcam, Cambridge, Cambridgeshire, UK). Then the lysis buffer was washed several times and proteinase K was added to digest the protein. After RNAs were extracted from the immunoprecipitation and the input, the level of CENPF mRNA was quantified via qRT‐PCR assays, which was normalized to input.
2.14. Dot blot assay
The total RNA was extracted with the TRIzol reagent (Invitrogen) and the mRNA purification was performed using an mRNA purification kit (61006, Thermofisher, USA). Then mRNAs were diluted with RNase‐free water to a specific concentration, which were denatured at 72°C for 5 min and then refrigerated to prevent the re‐formation of mRNA secondary structures. mRNA (2 μL) was loaded onto Amersham Hybond‐N+ membranes suitable for RNA transfer (GE Healthcare, Pittsburgh, Pennsylvania, USA). After drying at room temperature, the membrane was put into the UVP CL‐1000 ultraviolet crosslinking instrument (Analytik Jena US, Upland, California, USA) for 5 min, and then washed with ddH2O in a clean washing dish for 5 min at room temperature. After incubation with 0.02% Methylene blue (M9140‐25g; Sigma‐Aldrich, St. Louis, Missouri, USA) prepared with 0.3 mol/L Sodium Acetate for 5 min, the membrane was washed with ddH2O for several times and can be scanned after the background becomes pale. Currently, total RNA content was displayed. After washing three times with phosphate buffered saline with Tween20 (PBST; Beyotime, Shanghai, China), the membranes were blocked with 5% skim milk powder (Beyotime, Shanghai, China) for 1 h, which followed by incubated with specific m6A antibody (1:1000, MABE1006, Millipore) overnight at 4°C. On the next day, the amount of RNA bound by m6A was developed after the membrane was incubated with specific secondary antibody (Cell signaling Technology, Danvers, Massachusetts, USA) for 1 h.
2.15. RNA‐pulldown assay
RNA was transcribed in vitro using the RNAmax‐T7 Transcription Kit (R11074.2, RIBOBIO, Guangzhou, Guangdong, China) to obtain the Biotin‐labeled RNA. Cells were collected and incubated with labeled CENPF RNA overnight at 4°C. The mixture of probes and cell lysis buffer was combined with the magnetic beads. Purified probe‐bound proteins were then collected using protein extraction kit (QIAGEN, Dusseldorf, North Rhine‐Westphalia, Germany). Finally, the protein was detected by western blot assay and its composition was detected by mass spectrometry. Protein samples were analyzed by MS (OE Biotech, Shanghai, China). Briefly, 2 L aliquots of the resuspended lyophilized peptide fractions were fed into a nanoViper C18 (Acclaim PepMap 100, 75 μm × 2 cm) trap column. Online Chromatography separation was performed using the Easy nLC 1200 system (ThermoFisher, Massachusetts, USA). With a volume of 20 L of 100% solvent A (0.1% formic acid), the trapping and desalting procedures were carried out. An analytical column (Acclaim PepMap RSLC; 75 μm × 25 cm; C18‐2 μm 100 Å) was employed with an elution gradient of 5%‐38% solvent B (80% acetonitrile, 0.1% formic acid) for 60 min. Tandem MS data were acquired using a Thermo Fisher Q Exactive mass spectrometer (Thermo Fisher, Massachusetts, USA) equipped with a Nano Flex ion source using data‐dependent acquisition mass spectrum techniques. Data were collected utilizing an interface heater temperature of 275°C and an ion spray voltage of 1.9 kV. The scan spanned from 350 to 2,000 m/z at a resolution of 70,000 and a maximum injection duration of 100 ms for a full MS survey scan with a target value of 3 × 106. Only spectra with a charge state between 2 and 5 were chosen for the MS2 scan and subjected to high‐energy collision dissociation with a normalized collision energy of 28. The ion trap's rapid mode was used to quickly capture the MS2 spectra, with an AGC target of 8000 and a 50 ms maximum injection duration. For 25 seconds, dynamic exclusion was set. The ProteomeDiscover software (version 2.1) was used to process the data.
2.16. m6A RNA‐sequencing (RNA‐seq)
m6A RNA‐seq was performed by Cloudseq Biotech Inc. (Shanghai, China). Total RNA was isolated from cells and tissues using the Magzol Reagent (Magen, Guangzhou, Guangdong, China), as directed by the manufacturer. A K5500 microspectrophotometer (Beijing Kaiao, Beijing, China) and an Agilent 2200 TapeStation system (Agilent Technologies, Santa Clara, California, USA) were used to assess the quantity and integrity of the RNA yield. Quality control was performed on m6A antibody‐immunoprecipitated RNA using the Qubit (Thermo Fisher Scientific) and Agilent 2200 TapeStation (Agilent Technologies) systems. RNA was fragmented into molecules of approximately 200 bp length. The RNA fragments were then subjected to first‐ and second‐strand cDNA synthesis, adaptor ligation, and enrichment with a low cycle using the NEBNext® Ultra RNA LibraryPrep Kit for Illumina (New England Biolabs, Beverly, Massachusetts, USA). Both the input and m6A IP samples were performed for RNA‐seq library generation with NEBNext® Ultra II Directional RNA Library Prep Kit (New England Biolabs, Beverly, Massachusetts, USA). Sequencing of the Library was performed on an illumina Hiseq instrument (Illumina, San Diego, California, USA) with 150 bp paired end reads.
2.17. Immunofluorescence (IF) analysis
GC cells were seeded on confocal dishes (Solarbio, Beijing, China) and incubated in proper media for one night. Then the cells were fixed with formaldehyde (Biosharp, Hefei, Anhui, China) and permeabilized with 0.2% TritonX‐100 (Beyotime, Shanghai, China), which followed by blocking with 1% bovine serum albumin (BSA; Beyotime, Shanghai, China) for 30 min. Next, cells were incubated with the primary antibody overnight at 4°C and secondary antibody for 2 h on the next day. Finally, cells were detected with a confocal microscope (Leica, Weszler, Hesse, Germany). The reagents required above were obtained from Beyotime. The antibodies used in this study are listed in Supplementary Table S1.
2.18. Immunohistochemical (IHC) analysis
In brief, tumor tissues were fixed with 10% formalin (Biosharp, Hefei, Anhui, China) and embedded in paraffin. IHC staining was carried out on sections from each block of archived tissue following the sectioning of paraffin blocks and antigen retrieval in citrate buffer (pH 6.0; Beyotime, Shanghai, China) for 20 min. The sections were then incubated with primary antibodies at 4°C overnight. The next day, the sections were incubated with the secondary antibodies of the corresponding species at 37°C for 1 h, and then, the color reaction was developed with DAB solution (Beyotime, Shanghai, China). Finally, the sections were counterstained with hematoxylin (Beyotime, Shanghai, China), dehydrated and observed under a microscope (Nikon, Marunouchi, Chiyoda Ward, Tokyo, Japan). IHC score = frequency × intensity. According to the proportion of positive cells, the frequency was classified into grade 0, grade 1, grade 2, grade 3 and grade 4. The proportion of positive cells was divided into <5%, 5%‐25%, 25%‐50%, 50%‐75% and > 75%. Cell intensity staining was divided into 0, 1, 2 and 3, corresponding to negative, weak, medium, and strong, respectively. The antibodies used in this study are listed in Supplementary Table S1.
2.19. Animals
Female BALB/c nude mice aged 5 weeks were acquired from the Nanjing Medical University Laboratory Animal Center (Nanjing, Jiangsu, China) and maintained in an specific pathogen‐free facility with a 12/12 h day/night cycle and unrestricted access to food and water. The Ethics Committee of the Laboratory Animal Management of Nanjing Medical University gave its approval to this animal experiment. If the tumor weight exceeded 10% of the body weight or the body weight loss exceeded 20%‐25%, the animals were terminated. All animals were euthanized.
2.19.1. in vivo tumorigenesis models
Stably transfected GC cells (1 × 106) with sh‐CENPF or CENPF overexpression were mixed in 0.1 mL PBS, and then were injected subcutaneously into the groin of 5‐week‐old BALB/C nude mice (n = 5 each group). After four weeks, the nude mice were sacrificed, and tumor tissues were dissected. Finally, the weight and volume of the tumors were measured. Tumor volume = length × width2 × 1/2.
2.19.2. in vivo liver metastasis assay
A total of 1 × 106 luciferase labeled GC cells were injected into the spleen of 5‐week‐old BALB/C nude mice. After 4‐5 weeks, bioluminescence signals of liver metastases were detected via an IVIS imaging system (PerkinElmer, Norwalk, Connecticut, USA). Liver tissues were removed for hematoxylin‐eosin (HE) (Beyotime, Shanghai, China) stained sections to evaluate metastatic liver lesions.
2.20. Bioinformatics
The expression of CENPF in GC patients was examined using the Cancer Genome Atlas (TCGA; http://www.cancergenome.nih.gov/). Based on the expression of CENPF, the Kaplan‐Meier plotter (http://www.kmplot.com) was utilized to examine the overall survival of GC patients in GSE14210 and GSE15459 datasets. The m6A modification sites of CENPF were predicted using the SRAMP database (http://www.cuilab.cn/sramp/). Dreme software (http://meme.nbcr.net) was used to scan the sequences of m6A peaks to find meaningful motif sequences according to the previous study [26]. MetaPlot software (https://github.com/olarerin/metaPlotR) was applied to describe the m6A peaks in metagenes [27]. KEGG Orthology Based Annotation System (KOBAS, http://kobas.cbi.pku.edu.cn/) was used for gene functional annotation and enrichment [28].
2.21. Statistics analysis
SPSS 22.0 software (IBM, Armonk, New York, USA) was used for the statistical analysis of all data in this study, and the presentation of data results was as follows: mean ± standard deviation (SD) was used for continuous normal distribution data, Mann‐Whitney U test was used for grade data, and independent Student's t test was used for comparison. Analysis of variance was used for comparison between multiple groups, and linear regression was used for correlation analysis. All statistical tests were two‐tailed exact tests with a significance level of P < 0.05.
3. RESULTS
3.1. GC was associated with high levels of m6A mRNA modification
To explore m6A modification in GC, we measured the m6A RNA levels in 15 GC tissues and paired normal gastric mucosa tissues. A dot blotting assay showed that the level of m6A in GC tissues was significantly higher than that in paired normal tissues (Figure 1A). Since the variant m6A level is controlled by m6A writers and erasers, we further measured the mRNA levels of several m6A writers (methyltransferase 3 [METTL3], methyltransferase [METTL14] and wilms tumor 1‐associating protein [WTAP]) and erasers (fat mass and obesity‐associated protein [FTO] and alkb homolog 5 [ALKBH5]) in TCGA database and 60 pairs of GC tissues. We found that these writers and erasers exhibited significantly increased expression in the TCGA database (Figure 1B). However, only METTL3 and WTAP showed higher expression in 60 GC tissues than in paired normal tissues (Figure 1C). Furthermore, we found a positive correlation between m6A level and METTL3 expression in GC tissues (Figure 1D). The above results indicated that the high m6A level in GC tissues was mainly mediated by m6A writers, especially METTL3.
FIGURE 1.
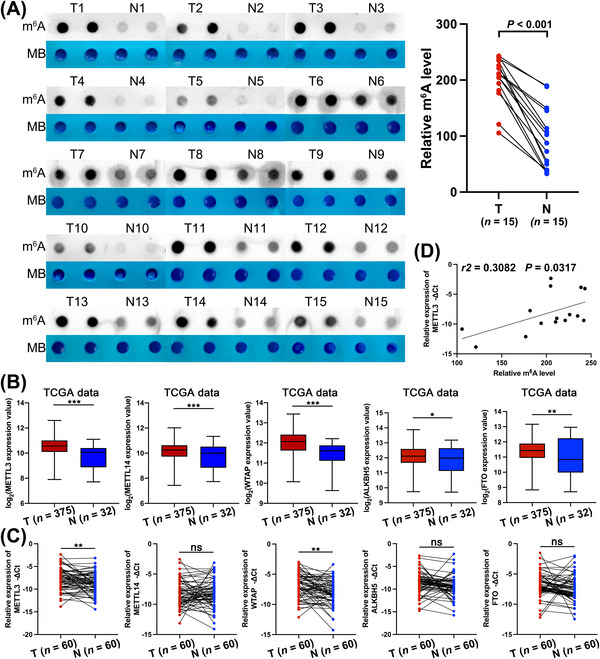
GC was associated with high levels of m6A mRNA methylation, (A) The m6A levels in 15 GC tissues and paired normal gastric mucosa tissues based on the dot blot assay. (B) The relative expression level of m6A writers (METTL3, METTL14, and WTAP) and erasers (FTO and ALKBH5) base on the TCGA database (375 GC tissues and 32 adjacent normal tissues). (C) The relative expression level of m6A writers (METTL3, METTL14, and WTAP) and erasers (FTO and ALKBH5) in 60 paired GC tissues via qRT‐PCR assay. (D) The correlation between m6A level and the mRNA expression of METTL3 in 15 GC tissues and paired normal gastric mucosa tissues based on the dot blot and qRT‐PCR assays. Data are expressed as the mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Abbreviations: TCGA, The Cancer Genome Atlas; METTL3, methyltransferase 3; METTL14, methyltransferase 14; WTAP, wilms tumor 1‐associating protein; FTO, fat mass and obesity‐associated protein; ALKBH5, AlkB homolog 5; SD, standard deviation.
3.2. Differentially expressed genes regulated by m6A in GC
To explore the differentially expressed genes regulated by m6A in GC, we performed m6A‐seq in three GC tissues and paired normal gastric mucosa tissues. m6A modification is predominantly located in RRACH (R = G or A, H = A, C, or U) sequences. Dreme software was used to scan the sequences of these peaks to find meaningful motif sequences [26]. The results from the m6A‐seq suggested that the GGACU motif was the most significant motif in GC tissues and paired normal tissues (Figure 2A). In addition, we measured the region of mRNA where the m6A peak was located, and the results showed that the peaks of m6A modification were mainly enriched in the CDS region (Figure 2B). MetaPlot software was applied to describe the m6A peaks in metagenes [27], and the results showed that the peaks of m6A modification were especially abundant in the stop codons region (Figure 2C). Next, we performed cluster analysis based on the fold change (FC) enrichment of methylated modified genes (Figure 2D). Compared with the normal gastric mucosal tissues, 2,101 hypermethylated m6A peaks and 1,193 hypomethylated m6A peaks were identified in GC tissues (Figure 2E). To further illustrate the mechanism underlying the high m6A level in GC, we also performed RNA‐seq, and the results showed that 649 genes were significantly upregulated, while 654 genes were downregulated (FC ≥ 2, P < 0.05). Next, a scatter plot was drawn to show the distribution of genes according to their differential expression and differential methylation levels (FC ≥ 2, P < 0.05, Figure 2F). Furthermore, KEGG enrichment analysis of these differential genes demonstrated that these genes were mainly related to MAPK signaling pathways (Figure 2G). Finally, we overlapped the RNA‐seq results, m6A‐seq results and TCGA databases analysis results (FC > 10 and TCGA logFC > 1 or < ‐1, Figure 2H). Among these 12 overlapping genes, CENPF was reported to regulate the MAPK signaling pathway and attracted our attention due to its remarkable level of hypermethylation.
FIGURE 2.
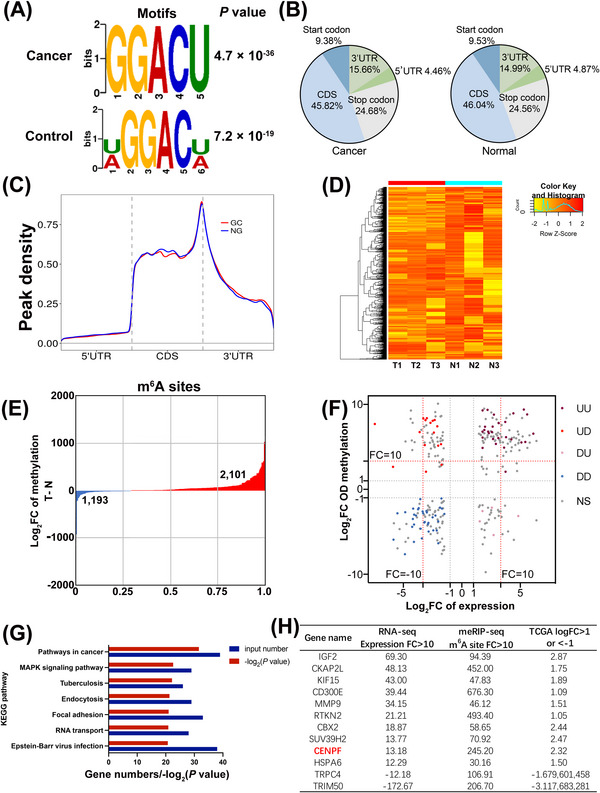
Differentially expressed genes regulated by m6A in GC, (A) The representative m6A motif in GC tissues and paired normal tissues via Dreme software. (B) The distribution of m6A sites in GC tissues and normal tissues via m6A‐seq. (C) The enrichment of m6A peaks in GC tissues and normal tissues via MetaPlot software. (D) The FC enrichment of methylated modified genes in m6A‐seq. (E) The description of hypermethylated and hypomethylated m6A peaks in m6A‐seq. (F) A scatter plot of gene distribution based on their expression levels and methylation levels (FC ≥2; P <0.05). (G) KEGG enrichment analysis of these differential methylated genes. (H) The list of candidate genes based on their expression (FC > 10), m6A level (FC > 10) and TCGA expression (logFC >1 or < ‐1). Data are expressed as the mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Abbreviations: CDS, coding sequence; UTR, untranslational region; UU, upmethylated and upregulated; UD, upmethylated and downregulated; DU, downmethylated and upregulated; DD, downmethylated and downregulated; IGF2, insulin like growth factor 2; CKAP2L, cytoskeleton associated protein 2 like; KIF15, kinesin family member 15; MMP9, matrix metallopeptidase 9; RTKN2, rhotekin 2; CBX2, chromobox 2; SUV39H2, suppressor of variegation 3‐9 homolog 2; CENPF, centromere protein F; HSPA6, heat shock protein family a member 6; TRPC4, transient receptor potential cation channel subfamily C member 4; TRIM50, tripartite motif containing 50; FC, fold change.
3.3. CENPF promoted GC cell migration, invasion, and angiogenesis in vitro
To evaluate the function of CENPF, we first measured its expression in the TCGA database and 60 paired GC tissues. As shown in Figure 3A‐B, the expression of CENPF mRNA was significantly upregulated in GC tissues compared with normal gastric mucosa tissues. The protein level of CENPF was also higher in 12 GC tissues than in paired gastric mucosa tissues, as shown by western blotting assay (Figure 3C). To show the intertumoral distribution of CENPF, we performed tissue IF assay with CENPF and pan‐keratin antibodies and found that CENPF was significantly overexpressed in tumor tissues (Supplementary Figure S1A). Furthermore, we found that high expression of CENPF was relevant to lymphatic invasion according to the clinical features of the above 60 GC patients (Table 1). To explore the relationship between CENPF expression and metastasis phenotypes, we constructed siRNA and overexpression plasmids, the transfection efficiency of which was detected via qRT‐PCR and western blotting assays (Supplementary Figure S1B‐D). As determined by Transwell and wound healing assays, we found that decreased CENPF inhibited migration and invasion, while CENPF overexpression promoted these phenotypes in GC cells (Figure 3D‐E, Supplementary Figure S1E‐F). In addition, angiogenesis assays revealed that CENPF overexpression enhanced angiogenesis, while decreased CENPF suppressed angiogenesis (Figure 3F, Supplementary Figure S1G). Furthermore, as an important component involved in the G2 phase of the cell cycle, we found that CENPF deficiency arrested the cell cycle in the G2 phase, while increased CENPF promoted the cell cycle (Figure 3G, Supplementary Figure S1H). Finally, CENPF knockdown decreased the expression of markers of the cell cycle (CDK1 and cyclin B1), MAPK signaling pathway (phospho‐Erk1/2 and phospho‐Elk1), angiogenesis (CD31 and VEGF) and epithelial‐mesenchymal transition (EMT; N‐cadherin and Snail) in GC cells (Figure 3H). Overall, we found that CENPF knockdown inhibited GC cell migration, invasion, and angiogenesis. Moreover, CENPF deficiency blocked the cell cycle in G2 phase and inhibited the MAPK signaling pathway.
FIGURE 3.
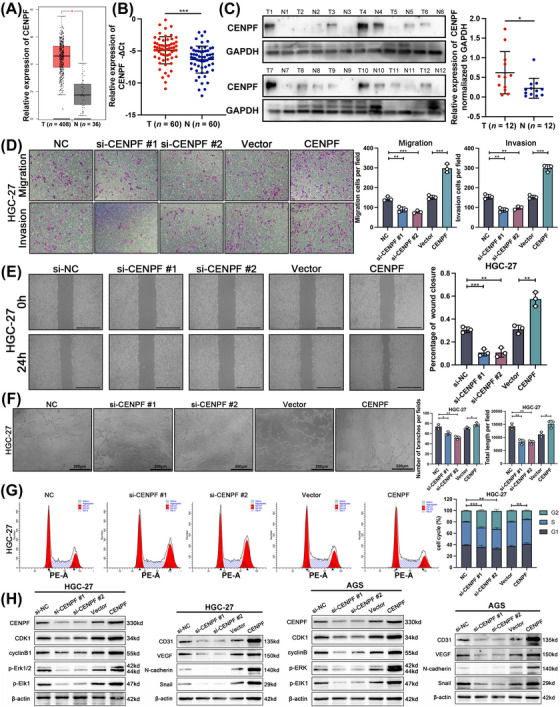
CENPF promoted GC cell migration, invasion, and angiogenesis in vitro angiogenesis, (A) The relative expression level of CENPF base on the TCGA database (375 GC tissues and 32 adjacent normal tissues). (B) The expression of CENPF in 60 paired GC tissues and normal tissues via qRT‐PCR assay. (C) The protein expression of CENPF in 12 GC tissues and normal tissues via western blotting assay. (D) Representative images and quantification of Transwell assay for CENPF‐overexpressing and CENPF‐knockdown HGC27 cells, magnification = 100 × . (E) Representative images and quantification of wound healing assay for CENPF‐overexpressing and CENPF‐knockdown HGC27 cells, scale bars = 100 μm. (F) Representative images and quantification of angiogenesis assay for CENPF‐overexpressing and CENPF‐knockdown HGC27 cells, scale bars = 200 μm. (G) Representative images and quantification of cell cycle for CENPF‐overexpressing and CENPF‐knockdown HGC27 cells via flow cytometric analysis. (H) Protein levels of CENPF, CDK1, cyclinB1, p‐Erk1/2, p‐Elk1, CD31, VEGF, N‐cadherin, and Snail in CENPF‐overexpressing and CENPF‐knockdown GC cells were detected via western blotting. Data are expressed as the mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Abbreviations: CENPF, centromere protein F; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; CDK1, cyclin dependent kinase 1; Erk1/2, extracellular signal‐regulated kinase 1/2; Elk1, ETS‐like gene 1; VEGF, vascular endothelial growth factor; SD, standard deviation.
TABLE 1.
Expression of CENPF in 60 human GC and the clinicopathological characteristics of these patients.
| Characteristic | Total, cases (%) | CENPF expression, cases (%) | P value | |
|---|---|---|---|---|
| Low | High | |||
| Gender | 0.152 | |||
| Female | 17 (28.33) | 11 (36.67) | 6 (20.00) | |
| Male | 43 (71.67) | 19 (63.33) | 24 (80.00) | |
| Age at surgery (years) | 0.519 | |||
| ≥60 | 48 (80.00) | 23 (76.67) | 25 (83.33) | |
| < 60 | 12 (20.00) | 7 (23.33) | 5 (16.67) | |
| T grade | 0.542 | |||
| T1 + T2 | 14 (23.33) | 8 (26.67) | 6 (20.00) | |
| T3 + T4 | 46 (76.67) | 22 (73.33) | 24 (80.00) | |
| Lymphatic invasion | 0.032* | |||
| Negative (N0) | 22 (36.67) | 15 (50.00) | 7 (23.33) | |
| Positive (N1‐N3) | 38 (63.33) | 15 (50.00) | 23 (76.67) | |
| Tumor site | 0.870 | |||
| Up | 26 (43.33) | 12 (40.00) | 14 (46.67) | |
| Middle | 13 (21.67) | 7 (23.33) | 6 (20.00) | |
| Down | 21 (35.00) | 11 (36.67) | 10 (33.33) | |
| Stage | 0.791 | |||
| I‐II | 23 (38.33) | 12 (40.00) | 11 (36.67) | |
| III‐IV | 37 (61.67) | 18 (60.00) | 19 (63.33) | |
| Size (cm) | 0.542 | |||
| <3 | 14 (23.33) | 6 (20.00) | 8 (26.67) | |
| ≥3 | 46 (76.67) | 24 (80.00) | 22 (73.33) | |
| Differentiation | 0.176 | |||
| Highly or moderately differentiated | 21 (35.00) | 13 (43.33) | 8 (26.67) | |
| Poorly differentiated | 39 (65.00) | 17 (56.67) | 22 (73.33) | |
P < 0.05 represents statistical significance (Chi‐squre test).
3.4. CENPF enhanced GC cell growth, metastasis, and angiogenesis in vivo
To further confirm the effect of CENPF in vivo, we constructed stable CENPF‐knockdown GC cells (sh‐CENPF) that were subcutaneously injected into nude mice. Four weeks after tumor growth, we observed that CENPF overexpression in AGS and HGC27 cells increased subcutaneous tumor size and weight, while CENPF knockdown showed the opposite results (Figure 4A‐B). IHC of CD31 showed that tumors from CENPF‐overexpressing HGC‐27 and AGS cells had more vessels, while there were few vessels in the CENPF knockdown groups (Figure 4C). Next, we established a liver metastasis model through splenic injection of HGC‐27 and AGS cells to investigate the role of CENPF in metastasis. As shown in Figure 4D‐E, the CENPF overexpression group exhibited more liver metastatic foci, while the CENPF‐deficient group showed fewer liver metastatic foci. Similar results were also observed for the liver index (Figure 4D‐E). In summary, these results indicated that CENPF could promote GC growth, metastatic potential and the ability of angiogenesis in vivo.
FIGURE 4.
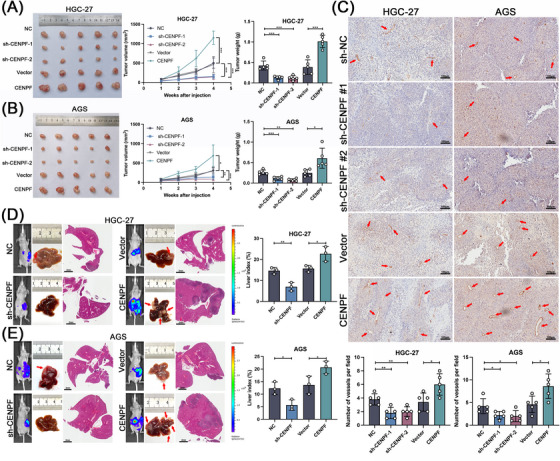
CENPF enhanced GC cell growth, metastasis, and angiogenesis in vivo, (A‐B) Photograph and quantification of subcutaneous tumors via injecting CENPF‐overexpressing and CENPF‐knockdown GC cells. (C) Representative images of CD31 IHC staining in subcutaneous tumors, scale bars = 100 μm (upper panels) and quantification of CD31 IHC staining of subcutaneous tumors in both GC cells (lower panels). (D) Representative specimens, HE staining photographs, bioluminescent images, and liver index quantification of HGC‐27 cell liver metastatic model, scale bars = 2 mm. (E) Representative specimens, HE staining photographs, bioluminescent images, and liver index quantification of AGS cell liver metastatic model, scale bars = 2 mm. Data are expressed as the mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Abbreviations: CENPF, centromere protein F; GC, gastric cancer; IHC, immunohistochemical; HE, hematoxylin‐eosin; SD, standard deviation.
3.5. CENPF was regulated by METTL3 in an m6A‐dependent manner in GC
First, we searched m6A sites of CENPF in m6A sequencing to further explore m6A modification (Figure 5A). Next, we intersected the above m6A sites with the sites predicted in the SRAMP database and labeled them sites 1 to 4 (Figure 5B). Then, we evaluated the m6A modification of CENPF mRNA via MeRIP‐qPCR in HGC27 and AGS cells, which showed that m6A enrichment in the site 2 of CENPF mRNA was the most significant region (Figure 5C, Supplementary Figure S2A). Furthermore, after METTL3 knockdown in GC cells, the m6A enrichment in the site 2 region was most significantly decreased (Figure 5D, Supplementary Figure S2B). It has been reported that m6A readers mediate the recognition of the m6A modification and play multiple roles. We tried to explore the potential readers of CENPF mRNA via an RNA pulldown assay in HGC27 cells (Figure 5E). According to the results of silver staining and mass spectrometry, we found that HNRNPA2B1 was a potential reader of CENPF (Figure 5F). The binding of HNRNPA2B1 and CENPF mRNA was also verified in both HGC27 and AGS cells (Figure 5G). In addition, the direct binding of HNRNPA2B1 and CENPF mRNA in HGC27 and AGS cells was verified via a RIP assay (Figure 5H). After METTL3 deficiency, the connection between CENPF and HNRNPA2B1 was suppressed (Figure 5I). Moreover, we found that HNRNPA2B1 deficiency significantly inhibited the expression level of CENPF (Figure 5J). In the TCGA database and 60 GC tissues, the expression of HNRNPA2B1 was positively correlated with that of CENPF (Figure 5K‐L). Since HNRNPA2B1 acts as an m6A reader, it was reported to play an important role in promoting RNA stability [29]. Actinomycin D assay showed that the knockdown of METTL3 or HNRNPA2B1 promoted the degradation of CENPF mRNA (Figure 5M, Supplementary Figure S2C). In conclusion, we found that the high m6A level of CENPF was mediated by METTL3, which could be recognized by HNRNPA2B1, thereby promoting mRNA stability.
FIGURE 5.
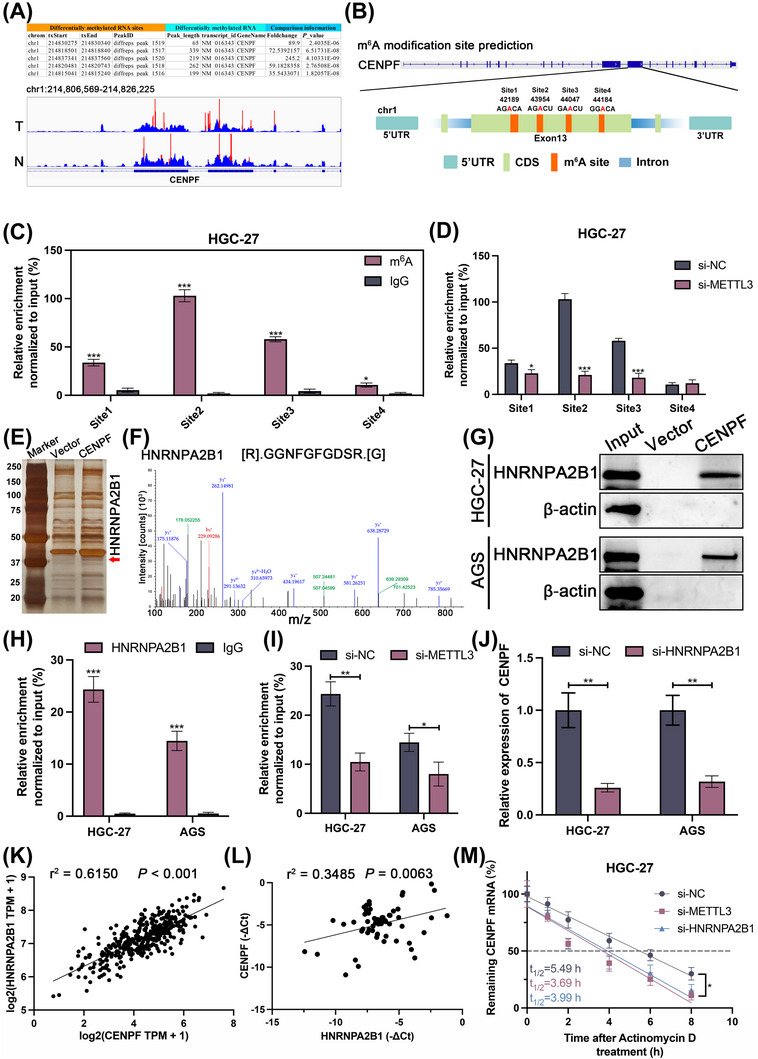
CENPF was regulated by METTL3 in an m6A‐dependent manner in GC, (A) The m6A modification sites of CENPF via m6A‐seq. (B) Schematic of the m6A sites of CENPF based on the m6A‐seq and the prediction from SRAMP databases. (C) The relative enrichment of CENPF mRNA m6A modification level normalized to input (%) in HGC27 cell via MeRIP‐qPCR. (D) The relative enrichment of CENPF mRNA m6A modification level normalized to input (%) after METTL3 knockdown in HGC27 cell via MeRIP‐qPCR. (E) Identification of protein complex binding to CENPF mRNA via incubation of biotinylated CENPF mRNA with protein extracts from HGC27 cells via RNA‐pulldown assay. (F) The potential m6A reader connected to CENPF mRNA by mass spectrometry. (G) Identification of the connection between CENPF mRNA and HNRNPA2B1 in HGC27 and AGS cells via RNA‐pulldown assay. (H) The direct binding between HNRNPA2B1 protein and CENPF was detected in HGC27 and AGS cells via RIP assay. (I) The enrichment between HNRNPA2B1 protein and CENPF mRNA normalized to input (%) was detected in si‐NC versus si‐METTL3 HGC27 and AGS cells via RIP assay. (J) The relative expression of CENPF in si‐NC versus si‐HNRNPA2B1 HGC27 and AGS cells via qRT‐PCR assay. (K) The correlation between CENPF and HNRNPA2B1 mRNA level were detected via TCGA database. (L) The correlation between CENPF and HNRNPA2B1 mRNA level were detected in 60 GC tissues via qRT‐PCR assay. (M) The stabilization of CENPF mRNA were detected in METTL3 or HNRNPA2B1 knockdown HGC27 cells via Actinomycin D treatment assay. Data are expressed as the mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Abbreviations: CENPF, centromere protein F; CDS, coding sequence; UTR, untranslational region; METTL3, methyltransferase 3; HNRNPA2B1, heterogeneous nuclear ribonucleoprotein A2/B1; RIP, RNA immunoprecipitation; SD, standard deviation.
3.6. The metastatic phenotype of CENPF was dependent on the MAPK signaling pathway
To further explore the effect of the CENPF/MAPK axis on the metastatic ability of GC cells, U0126 (a MEK/ERK‐specific inhibitor) was used in a series of rescue experiments. The results indicated that U0126 significantly suppressed the migration, invasion, and angiogenesis of HGC27 cells and reversed the effects of CENPF overexpression on promoting these processes in Transwell, wound healing, and in vivo angiogenesis assays (Figure 6A‐C). In addition, the enhanced effect of increased CENPF in late G2 phase could be rescued by U0126 in HGC27 cells (Figure 6D). Similar results were observed after transfection with the CENPF overexpression plasmid and U0126 in AGS cells (Supplementary Figure S3A‐D). Moreover, as downstream targets of CENPF, the expression levels of proteins related to the cell cycle, MAPK signaling pathway, angiogenesis and EMT were measured by western blotting. The results demonstrated that treatment with U0126 rescued the promoting effect of CENPF overexpression on the protein levels of CDK1, cyclinB1, phoshpo‐Erk1/2, phospho‐EIK1, CD31, VEGF, N‐cadherin, and Snail (Figure 6E, Supplementary Figure S3E). Furthermore, an in vivo liver metastasis assay showed that U0126 inhibited GC cell liver metastasis and restored the effect of CENPF overexpression (Figure 6F, Supplementary Figure S3F). Taken together, the above results suggested that CENPF induced the metastasis and angiogenesis of GC cells through the MAPK signaling pathway.
FIGURE 6.
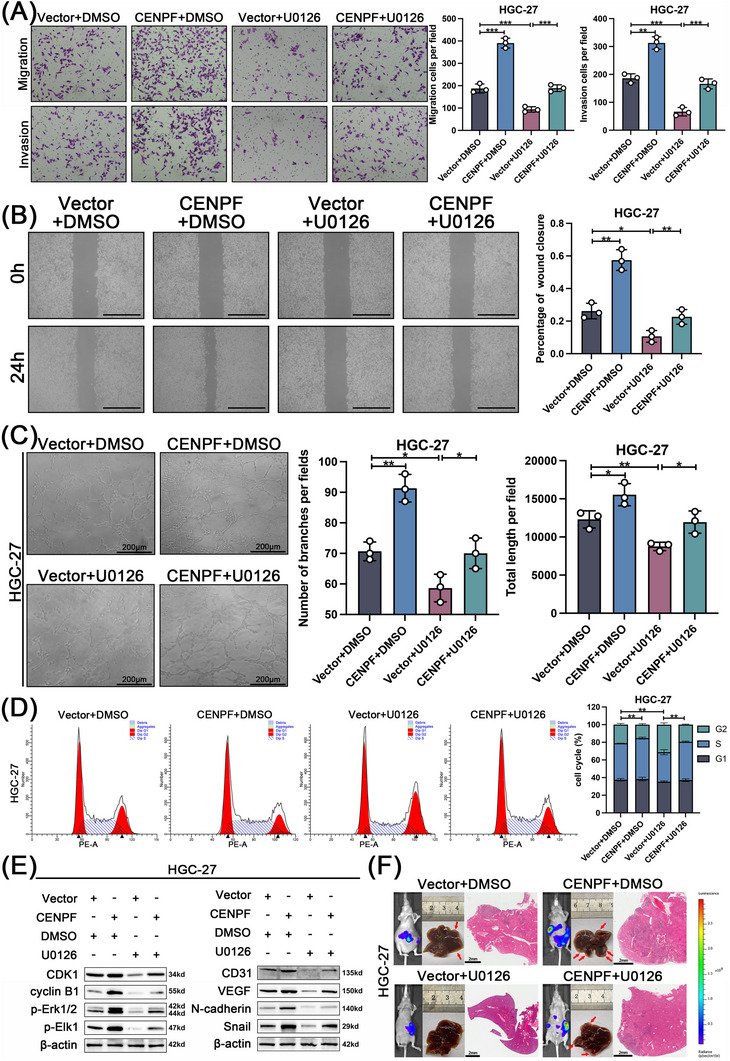
The metastatic phenotype of CENPF was MAPK signaling pathway dependent, (A‐C) HGC27 cells were divided into four groups (vector + DMSO, CENPF + DMSO, vector + U0126, and CENPF + U0126). The migration, invasion, and angiogenesis ability of HGC27 was analyzed through Transwell (A; magnification = 100 ×), wound healing (B; scale bar = 100 μm), and angiogenesis assays (C; scale bar = 200 μm). (D) The cell cycle progression was analyzed by flow cytometry after HGC27 cells were transfected with CENPF overexpression plasmid and/or treated with U0126. (E) The relative expression of CDK1, cyclinB1, p‐Erk1/2, p‐Elk1, CD31, VEGF, N‐cadherin, and Snail at the protein level in HGC27 cells transfected with CENPF overexpression plasmid and/or treated with U0126 was determined by western blot analysis. (F) Representative specimens, HE staining photographs, and bioluminescent images of HGC27 liver metastatic model (scale bar = 2 mm). Data are expressed as the mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Abbreviations: CENPF, centromere protein F; DMSO, dimethyl sulfoxide; CDK1, cyclin dependent kinase 1; Erk1/2, extracellular signal‐regulated kinase 1/2; Elk1, ETS‐like gene 1; VEGF, vascular endothelial growth factor; HE, hematoxylin and eosin; SD, standard deviation.
3.7. CENPF activated the MAPK signaling pathway by promoting FAK nuclear export
To explore the downstream mechanism between CENPF and the MAPK signaling pathway, we generated a plasmid carrying a flagged CENPF to immunoprecipitate CENPF and found that FAK might be the mechanism by which CENPF regulates the MAPK signaling pathway (Figure 7A‐B). The connection between CENPF and FAK was further verified via a co‐IP assay in HGC27 and AGS cells (Figure 7C). However, regardless of the expression of CENPF, the expression of FAK was not changed in the HGC27 cells (Figure 7D). It was reported that CENPF is mainly located in the nucleus but can enter the cytoplasm in late G2 phase [30, 31]. We wondered whether CENPF mediated FAK nuclear export since cytoplasmic FAK could activate the MAPK signaling pathway. Nocodazole, a cell cycle inhibitor, arrests cells in the G2 phase. The effect of Nocodazole in HGC27 and AGS cells was verified via flow cytometry assay (Figure 7E). Moreover, the distribution of CENPF in GC cells was also examined via IF assay, and CENPF partially entered the cytoplasm in late G2 phase (Figure 7F). After nucleoplasm separation and protein extraction of GC cells, it was found that CENPF knockdown indeed reduced the expression of FAK in the cytoplasm, while FAK in the nucleus increased after CENPF knockdown (Figure 7G). IF assays further confirmed that CENPF deficiency significantly restricted FAK in the nucleus (Figure 7H). Based on the report that PND‐1186 is an inhibitor of FAK and that it can restrict FAK in the nucleus and inhibit its activity [32], we verified that PND‐1186 could repress the activation of FAK and block FAK in the nucleus in GC cells (Supplementary Figure S4A‐B). Finally, the effect of CENPF overexpression on the MAPK signaling pathway could be restored by PND‐1186 in HGC27 and AGS cells (Figure 7I). Based on the above results, we proved that CENPF could activate the MAPK signaling pathway by promoting FAK translocation from the nucleus to the cytoplasm.
FIGURE 7.
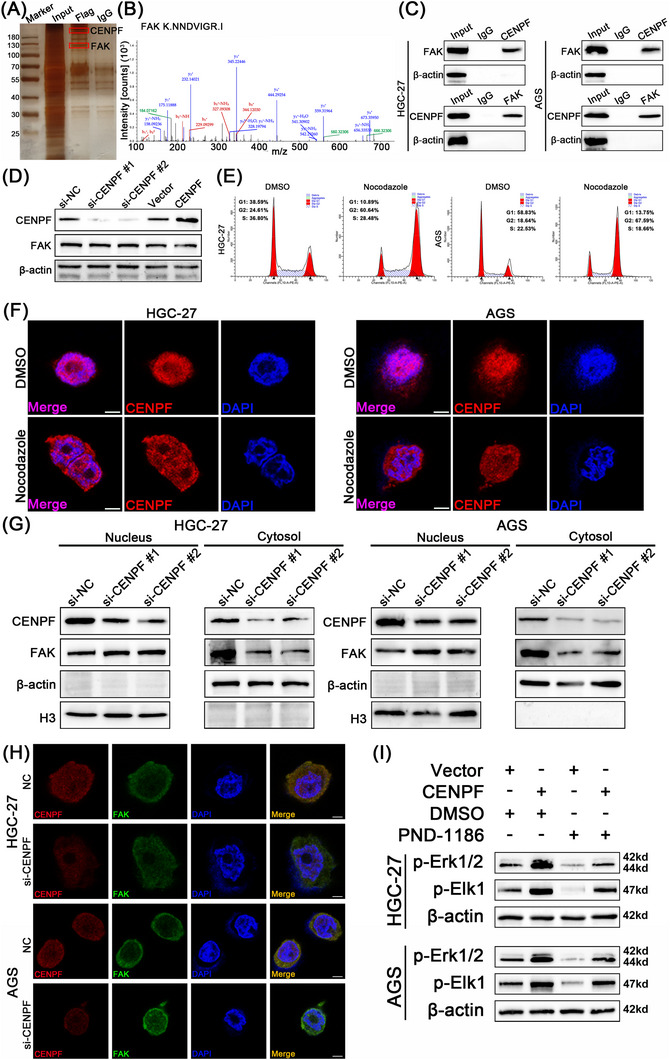
CENPF activated the MAPK signaling pathway by promoting FAK nuclear export, (A) Identification of protein complex binding to CENPF protein via incubation of Flag antibody with protein extracts from HGC27 cell transfected with Flag‐CENPF via IP and MS assay. (B) Secondary mass spectrometry of FAK protein bound to CENPF. (C) The binding between CENPF and FAK was detected in HGC 27 and AGS cells via co‐IP assay. (D) Protein level of CENPF and FAK in CENPF‐overexpressing and CENPF‐knockdown HGC27 cells via western blotting assay. (E) The effect of Nocodazole on cell cycle in HGC27 and AGS cells via Flow Cytometry assay. (F) The location of CENPF in HGC27 and AGS cells treated with Nocodazole via IF assay, scale bars = 5 μm. (G) Protein level of CENPF and FAK in cytoplasm and nucleus respectively after CENPF Knockdown via western blotting assay. (H) The distribution of CENPF and FAK in HGC27 and AGS cells after CENPF knockdown via IF assay, scale bars = 5 μm. (I) The relative expression of p‐Erk1/2 and p‐Elk1 at the protein level in HGC27 or AGS cells transfected withCENPF overexpression plasmid and/or treated with PND‐1186 was determined by western blot analysis. Data are expressed as the mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Abbreviations: IP, immunoprecipitation; MS, mass spectrometry; IF, immunofluorescence; CENPF, centromere protein F; FAK, focal adhesion kinase; DMSO, dimethyl sulfoxide; H3, histone 3; Erk1/2, extracellular signal‐regulated kinase 1/2; Elk1, ETS‐like gene 1; SD, standard deviation.
3.8. The clinical significance of the CENPF/FAK axis in inducing tumor angiogenesis and the MAPK signaling pathway in human GC tissues
Given the discovery of the CENPF/FAK axis in GC cells and the relationship among the MAPK signaling pathway, angiogenesis and EMT demonstrated above, we further assessed the clinical significance of CENPF in a cohort of 60 GC patients and Kaplan‐Meier plotter (http://www.kmplot.com/) to clarify its clinical value. First, survival analysis revealed that higher CENPF expression was related to poorer overall survival (OS) in 60 GC patients (Figure 8A). Based on the GSE14210 dataset, it was found that high expression of CENPF was positively correlated with poor survival (Figure 8B). Although there was no significant P value in the survival analysis of the GSE15459 dataset, there was a tendency for poor survival in the group with high CENPF expression (Figure 8C). Moreover, we detected the expression of CENPF, phospho‐Erk1/2, CD31, and N‐cadherin in 52 GC tissues via IHC staining (Figure 8D). CENPF expression was positively correlated with the expression of phospho‐Erk1/2, CD31 and N‐cadherin (Figure 8E), suggesting that CENPF promoted GC metastasis and angiogenesis through the MAPK signaling pathway (Figure 8F).
FIGURE 8.
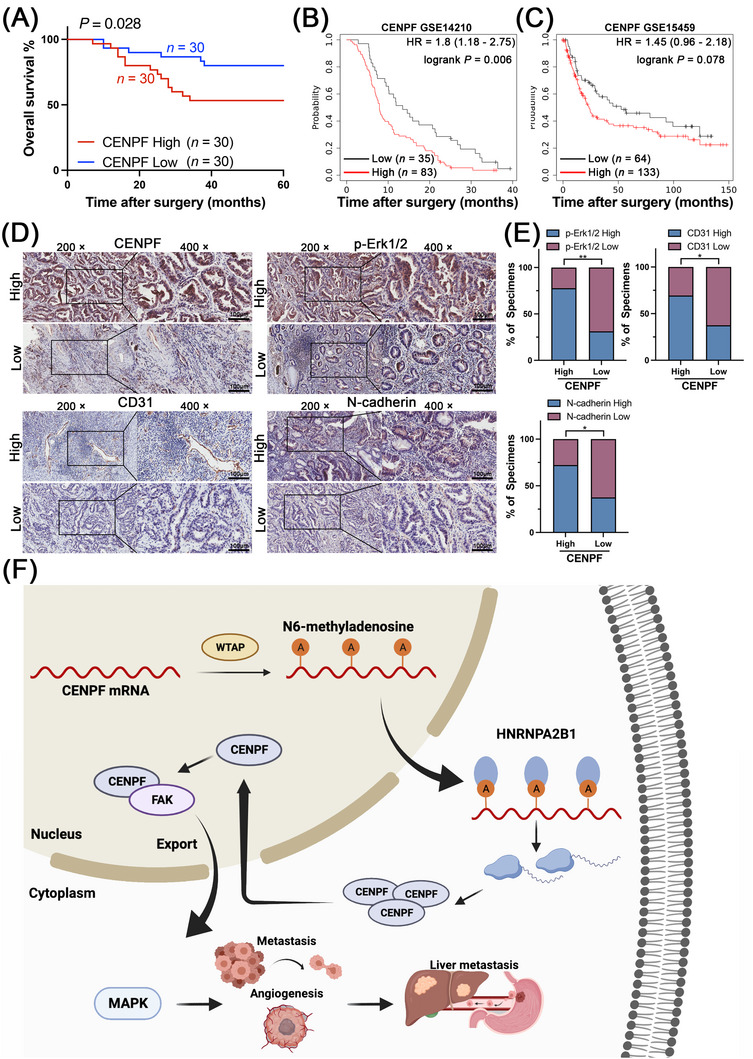
The clinical significance of the CENPF/FAK axis in inducing tumor angiogenesis and MAPK signaling pathway in human GC tissues, (A) The 5‐year overall survival analysis of 60 GC patients base on the expression of CENPF. (B‐C) The Kaplan‐Meier plotter survival analysis of CENPF in GSE14210 and GSE15459 datasets. (D‐E) CENPF levels were significantly associated with the expression of p‐Erk1/2, CD31 and N‐cadherin in 52 primary human GC specimens. (D) Representative cases are shown (scale bar = 10 μm). (E) The percentages of specimens showing low or high CENPF expression relative to the levels of p‐Erk1/2, CD31, and N‐cadherin are shown. (F) A graphic schematic of the mechanism by which m6A methylated CENPF activates MAPK signaling pathway and promotes GC metastasis. Data are expressed as the mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Abbreviations: CENPF, centromere protein F; FAK, focal adhesion kinase; Erk1/2, extracellular signal‐regulated kinase 1/2; HNRNPA2B1, heterogeneous nuclear ribonucleoprotein A2/B1; MAPK, mitogen‐activated protein kinase; IHC: immunohistochemical; SD, standard deviation.
4. DISCUSSION
As the most abundant pretranscription modification in eukaryotes, m6A modification has become a hot research topic in recent years. Although research on m6A modification began in the 1970s, it stalled due to technical limitations. In recent years, under the leadership of Professor Chuan He and other research teams, research on methylation modification of m6A has made considerable progress through continuous technological innovation and overcoming of difficulties. Professor Chuan He was the first to identify FTO as an enzyme capable of removing m6A from RNAs, leading to substantial progress in the research of RNA m6A modification [33]. The level of methylation is mainly regulated by methylases and demethylases to maintain homeostasis, and specific RNA binding proteins (m6A readers) can recognize these methylation sites and affect these modified RNAs [34]. According to the current research progress, most relevant studies mainly focus on methylases and demethylases or readers, and there are few reports on the global methylation level in tumors [35, 36, 37, 38]. This study innovatively started from the phenomenon of hypermethylation in GC tissues and found differentially methylated and expressed genes in GC by m6A‐seq and RNA‐seq. Moreover, these differentially expressed genes in GC were enriched in the MAPK signaling pathway. CENPF was selected for this study according to the above results.
CENPF encodes a protein involved in centromere‐kinetochore complex formation and is an important component involved in the G2 phase of the cell cycle. It has been reported to play essential roles in various diseases, where CENPF has been reported to activate the MAPK signaling pathway [20, 21, 39, 40]. In this study, we first analyzed clinicopathological data and found that CENPF was highly expressed in GC tissues and correlated with lymph node metastasis. Subsequent functional experiments suggested that CENPF could promote GC cell metastasis and angiogenesis and activate the MAPK signaling pathway in vitro and in vivo. The methylation sites of CENPF showed that there were four m6A sites, and site 2 was the most obvious site via MeRIP assay. The m6A reader HNRNPA2B1 bound to CENPF was further identified by RNA‐pulldown assay, which promoted the mRNA stability of CENPF. Although CENPF can activate the MAPK signaling pathway, its specific molecular mechanism has not yet been reported. In this study, we thoroughly explored the molecular mechanism by which CENPF activated MAPK pathway.
FAK is a tyrosine kinase involved in many signaling pathways. It has been reported that FAK is distributed in both the nucleus and cytoplasm [41, 42, 43]. In addition to its typical role as a cytoplasmic kinase downstream of integrin and growth factor receptor signaling, FAK in the nucleus can promote p53 degradation through ubiquitination, leading to the progression of cancer cells [44]. FAK can also regulate the expression of GATA4 and IL‐33, thereby reducing the inflammatory response and immune escape [45]. Therefore, it is of great significance to study the unique effects of FAK in the nucleus and cytoplasm. In this study, we used a co‐IP assay to explore the downstream mechanism between CENPF and the MAPK signaling pathway, and FAK was selected as a candidate target. Further exploration revealed that CENPF could not affect the expression of FAK. Interestingly, we found that CENPF could enter the cytoplasm from the nucleus during G2 phase, and the distribution of FAK in GC cells attracted our attention [30, 31]. After the application of PND‐1186, a FAK inhibitor that can retain FAK in the nucleus and inhibit its activity [32], a molecular mechanism by which CENPF activates the MAPK signaling pathway by promoting FAK entry into the cytoplasm was elucidated. This is the first report on the regulatory relationship between CENPF and FAK.
To further elucidate the clinical significance of CENPF, we performed survival analysis on 60 GC patients, and found that patients with high expression of CENPF had a poor prognosis. In addition, according to the expression of CENPF IHC scores, we found that the scores of phospho‐ERK1/2, CD31 and N‐cadherin were positively correlated with CENPF. In conclusion, this study of CENPF promoting metastasis and angiogenesis in GC may make it a promising diagnostic and therapeutic target in the future.
The present study has some limitations. CENPF is mainly located in the nucleus but can partially enter the cytoplasm during the G2 phase. In addition, we proved that CENPF could activate the MAPK signaling pathway by promoting FAK translocation from the nucleus to the cytoplasm. However, the regulatory mechanism between CENPF and FAK in the G2 phase needs to be further explored. Besides, there are several methylated genes in the m6A sequencing that need to be further explored in GC.
5. CONCLUSIONS
In conclusion, our results indicated that CENPF was highly m6A methylated and highly expressed in GC, and these phenomena were related to the MAPK signaling pathway. CENPF can promote GC cell metastasis and angiogenesis in vitro and in vivo. Mechanistically, the high m6A level of CENPF was induced by METTL3 and recognized by HNRNPA2B1, which can stabilize CENPF mRNA. In addition, CENPF promoted the nuclear export of FAK, thereby activating the MAPK signaling pathway and promoting the metastasis and angiogenesis of GC. Taken together, our study identified a new role of m6A‐methylated CENPF in GC metastasis through the FAK/MAPK axis, which is expected to be a therapeutic target for GC in the future.
DECLARATIONS
AUTHOR CONTRIBUTIONS
Jing Yang and Zetian Chen participated in the experimental design and data analysis. Xing Zhang performed some of the in vitro experiments. Penghui Xu performed the experiments and wrote the manuscript. Yiwen Xia provided guidance for animal experiments of this study. Sen Wang and Weizhi Wang helped with the bioinformatics analysis. Zekuan Xu was responsible for quality control. All authors read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
FUNDING INFORMATION
This work was partially supported by the Special Foundation for National Science and Technology Basic Research Program of China (2019FY101104), National Natural Science Foundation of China (81871946 and 82072708), the Primary Research & Development Plan of Jiangsu Province (BE2016786), the Program for Development of Innovative Research Team in the First Affiliated Hospital of NJMU, the Priority Academic Program Development of Jiangsu Higher Education Institutions (JX10231801), Jiangsu Key Medical Discipline (General Surgery; ZDXKA2016005), and Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Centre for Cancer Personalized Medicine, Nanjing Medical University.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University (permit number: IACUC‐2011012). The tissue samples were obtained with written informed consent from each patient. The experiments concerning animals were approved by the Nanjing Medical University Ethics Committee (permit number: 2020‐SRFA‐174).
CONSENT FOR PUBLICATION
Not applicable.
Supporting information
Supporting information
ACKNOWLEDGMENTS
The authors have nothing to report.
Xu P, Yang J, Chen Z, Zhang X, Xia Y, Wang S, et al. N6‐methyladenosine modification of CENPF mRNA facilitates gastric cancer metastasis via regulating FAK nuclear export. Cancer Commun. 2023;43:685–705. 10.1002/cac2.12443
DATA AVAILABILITY STATEMENT
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–49. [DOI] [PubMed] [Google Scholar]
- 2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–32. [DOI] [PubMed] [Google Scholar]
- 3. Rawla P, Barsouk A. Epidemiology of gastric cancer: global trends, risk factors and prevention. Prz Gastroenterol. 2019;14(1):26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Qiu H, Cao S, Xu R. Cancer incidence, mortality, and burden in China: a time‐trend analysis and comparison with the United States and United Kingdom based on the global epidemiological data released in 2020. Cancer Commun (Lond). 2021;41(10):1037–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maehara Y, Hasuda S, Koga T, Tokunaga E, Kakeji Y, Sugimachi K. Postoperative outcome and sites of recurrence in patients following curative resection of gastric cancer. Br J Surg. 2000;87(3):353–7. [DOI] [PubMed] [Google Scholar]
- 6. Allemani C, Weir HK, Carreira H, Harewood R, Spika D, Wang XS, et al. Global surveillance of cancer survival 1995‐2009: analysis of individual data for 25,676,887 patients from 279 population‐based registries in 67 countries (CONCORD‐2). Lancet. 2015;385(9972):977–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Japanese Gastric Cancer A. Japanese gastric cancer treatment guidelines 2018 (5th edition). Gastric Cancer. 2021;24(1):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang FH, Zhang XT, Li YF, Tang L, Qu XJ, Ying JE, et al. The Chinese Society of Clinical Oncology (CSCO): Clinical guidelines for the diagnosis and treatment of gastric cancer, 2021. Cancer Commun (Lond). 2021;41(8):747–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ajani JA, D'Amico TA, Bentrem DJ, Chao J, Cooke D, Corvera C, et al. Gastric Cancer, Version 2.2022, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2022;20(2):167–92. [DOI] [PubMed] [Google Scholar]
- 10. Zhao BS, Roundtree IA, He C. Post‐transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18(1):31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6‐methyladenosine and its role in cancer. Mol Cancer. 2019;18(1):176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28(6):616–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6‐methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15(5):313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mendel M, Delaney K, Pandey RR, Chen KM, Wenda JM, Vagbo CB, et al. Splice site m(6)A methylation prevents binding of U2AF35 to inhibit RNA splicing. Cell. 2021;184(12):3125–42 e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin X, Chai G, Wu Y, Li J, Chen F, Liu J, et al. RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat Commun. 2019;10(1):2065. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16. Dixit D, Prager BC, Gimple RC, Poh HX, Wang Y, Wu Q, et al. The RNA m6A Reader YTHDF2 Maintains Oncogene Expression and Is a Targetable Dependency in Glioblastoma Stem Cells. Cancer Discov. 2021;11(2):480–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hou G, Zhao X, Li L, Yang Q, Liu X, Huang C, et al. SUMOylation of YTHDF2 promotes mRNA degradation and cancer progression by increasing its binding affinity with m6A‐modified mRNAs. Nucleic Acids Res. 2021;49(5):2859–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun J, Huang J, Lan J, Zhou K, Gao Y, Song Z, et al. Overexpression of CENPF correlates with poor prognosis and tumor bone metastasis in breast cancer. Cancer Cell Int. 2019;19:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aytes A, Mitrofanova A, Lefebvre C, Alvarez MJ, Castillo‐Martin M, Zheng T, et al. Cross‐species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell. 2014;25(5):638–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shahid M, Lee MY, Piplani H, Andres AM, Zhou B, Yeon A, et al. Centromere protein F (CENPF), a microtubule binding protein, modulates cancer metabolism by regulating pyruvate kinase M2 phosphorylation signaling. Cell Cycle. 2018;17(24):2802–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen H, Wu F, Xu H, Wei G, Ding M, Xu F, et al. Centromere protein F promotes progression of hepatocellular carcinoma through ERK and cell cycle‐associated pathways. Cancer Gene Ther. 2022;29(7):1033–42. [DOI] [PubMed] [Google Scholar]
- 22. Guo SH, Ma L, Chen J. Identification of Prognostic Markers and Potential Therapeutic Targets in Gastric Adenocarcinoma by Machine Learning Based on mRNAsi Index. J Oncol. 2022;2022:8926127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song X, Xu H, Wang P, Wang J, Affo S, Wang H, et al. Focal adhesion kinase (FAK) promotes cholangiocarcinoma development and progression via YAP activation. J Hepatol. 2021;75(4):888–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shen M, Jiang YZ, Wei Y, Ell B, Sheng X, Esposito M, et al. Tinagl1 Suppresses Triple‐Negative Breast Cancer Progression and Metastasis by Simultaneously Inhibiting Integrin/FAK and EGFR Signaling. Cancer Cell. 2019;35(1):64‐80 e7. [DOI] [PubMed] [Google Scholar]
- 25. Demircioglu F, Wang J, Candido J, Costa ASH, Casado P, de Luxan Delgado B, et al. Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat Commun. 2020;11(1):1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bailey TL. DREME: motif discovery in transcription factor ChIP‐seq data. Bioinformatics. 2011;27(12):1653–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Olarerin‐George AO, Jaffrey SR. MetaPlotR: a Perl/R pipeline for plotting metagenes of nucleotide modifications and other transcriptomic sites. Bioinformatics. 2017;33(10):1563–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bu D, Luo H, Huo P, Wang Z, Zhang S, He Z, et al. KOBAS‐i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Research. 2021;49(W1):W317–W25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jiang F, Tang X, Tang C, Hua Z, Ke M, Wang C, et al. HNRNPA2B1 promotes multiple myeloma progression by increasing AKT3 expression via m6A‐dependent stabilization of ILF3 mRNA. J Hematol Oncol. 2021;14(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berto A, Doye V. Regulation of Cenp‐F localization to nuclear pores and kinetochores. Cell Cycle. 2018;17(17):2122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Loftus KM, Cui H, Coutavas E, King DS, Ceravolo A, Pereiras D, et al. Mechanism for G2 phase‐specific nuclear export of the kinetochore protein CENP‐F. Cell Cycle. 2017;16(15):1414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jeong K, Murphy JM, Kim JH, Campbell PM, Park H, Rodriguez YAR, et al. FAK Activation Promotes SMC Dedifferentiation via Increased DNA Methylation in Contractile Genes. Circ Res. 2021;129(12):e215–e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6‐methyladenosine in nuclear RNA is a major substrate of the obesity‐associated FTO. Nat Chem Biol. 2011;7(12):885–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shi H, Wei J, He C. Where, When, and How: Context‐Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell. 2019;74(4):640–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3‐mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69(7):1193–205. [DOI] [PubMed] [Google Scholar]
- 36. Chen Y, Peng C, Chen J, Chen D, Yang B, He B, et al. WTAP facilitates progression of hepatocellular carcinoma via m6A‐HuR‐dependent epigenetic silencing of ETS1. Mol Cancer. 2019;18(1):127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu J, Chai P, Xie M, Ge S, Ruan J, Fan X, et al. Histone lactylation drives oncogenesis by facilitating m(6)A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 2021;22(1):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qiu X, Yang S, Wang S, Wu J, Zheng B, Wang K, et al. M(6)A Demethylase ALKBH5 Regulates PD‐L1 Expression and Tumor Immunoenvironment in Intrahepatic Cholangiocarcinoma. Cancer Res. 2021;81(18):4778–93. [DOI] [PubMed] [Google Scholar]
- 39. Hexiao T, Yuquan B, Lecai X, Yanhong W, Li S, Weidong H, et al. Knockdown of CENPF inhibits the progression of lung adenocarcinoma mediated by ERbeta2/5 pathway. Aging (Albany NY). 2021;13(2):2604–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mahmoud AD, Ballantyne MD, Miscianinov V, Pinel K, Hung J, Scanlon JP, et al. The Human‐Specific and Smooth Muscle Cell‐Enriched LncRNA SMILR Promotes Proliferation by Regulating Mitotic CENPF mRNA and Drives Cell‐Cycle Progression Which Can Be Targeted to Limit Vascular Remodeling. Circ Res. 2019;125(5):535–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jeong K, Murphy JM, Ahn EE, Lim SS. FAK in the nucleus prevents VSMC proliferation by promoting p27 and p21 expression via Skp2 degradation. Cardiovasc Res. 2022;118(4):1150–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Canel M, Byron A, Sims AH, Cartier J, Patel H, Frame MC, et al. Nuclear FAK and Runx1 Cooperate to Regulate IGFBP3, Cell‐Cycle Progression, and Tumor Growth. Cancer Res. 2017;77(19):5301–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lees DM, Reynolds LE, Pedrosa AR, Roy‐Luzarraga M, Hodivala‐Dilke KM. Phosphorylation of pericyte FAK‐Y861 affects tumour cell apoptosis and tumour blood vessel regression. Angiogenesis. 2021;24(3):471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, et al. Nuclear FAK promotes cell proliferation and survival through FERM‐enhanced p53 degradation. Mol Cell. 2008;29(1):9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jeong K, Kim JH, Murphy JM, Park H, Kim SJ, Rodriguez YAR, et al. Nuclear Focal Adhesion Kinase Controls Vascular Smooth Muscle Cell Proliferation and Neointimal Hyperplasia Through GATA4‐Mediated Cyclin D1 Transcription. Circ Res. 2019;125(2):152–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.