

SUMMARY
The complex functions of neuronal synapses depend on their tightly interconnected protein network, and their dysregulation is implicated in the pathogenesis of autism spectrum disorders and schizophrenia. However, it remains unclear how synaptic molecular networks are altered biochemically in these disorders. Here, we applied multiplexed imaging to probe the effects of RNAi knockdown of 16 autism- and schizophrenia-associated genes on the simultaneous joint distribution of ten synaptic proteins, observing several protein composition phenotypes associated with these risk genes. We applied Bayesian network analysis to infer hierarchical dependencies amongst eight excitatory synaptic proteins, yielding predictive relationships that can only be accessed with single-synapse, multiprotein measurements performed simultaneously in situ. Finally, we found that central features of the network are affected similarly across several distinct gene knockdowns. These results offer insight into the convergent molecular etiology of these widespread disorders and provide a general framework to probe subcellular molecular networks.
INTRODUCTION
The functional complexity of the brain is enabled by trillions of chemical synapses that form connections amongst its ~1011 neurons. Each synapse is capable of analog computation that integrates its activity history, chemical environment, and the state of its pre- and post-synaptic neurons to modulate communication through synaptic plasticity. This computation is achieved1 in large part by the synaptic molecular network, a dynamic, compartmentalized biomolecular system of hundreds of proteins2–4 that includes constantly varying levels and activity states of receptors, scaffolding proteins, kinases, and other protein types. This proteomic diversity likely underlies the remarkable cell- and context-specific functional diversity even amongst synapses of the same type5,6. Numerous studies have revealed mechanistic connections between two or three synaptic components at a time, providing the foundation to integrate these connections into a broader context of many-component networks. However, due to the high complexity and interconnectedness of such networks, this integration requires simultaneous single-synapse measurement of numerous proteins, a technique we previously developed and applied to analyze synapse compositions7.
The synaptic molecular network is tightly connected to cognitive disorders, with synaptogenesis and plasticity increasingly appreciated as molecular targets for psychiatric treatments8–10. Accumulating evidence also points to synaptic biochemistry as a focal point of the pathophysiology of psychiatric, neurodevelopmental, and neurodegenerative diseases11–17. Autism spectrum disorder (ASD) and schizophrenia (SCZ) are two such conditions that manifest in a range of specific higher cognitive symptoms that range in intensity from healthy neurodiversity to debilitating brain dysfunction. These latter conditions typically include changes in social and communication behavior18,19, altered perception and sensory habituation, including self-stimulatory behavior19–22, adherence to patterns and focused interests19, impaired language acquisition and use18,23, as well as general intellectual disability24,25 and psychosis26. While divergent in symptom presentation, they are often studied genetically in the same context due to similarities in risk genes and possible functional and pathological associations27–29.
Both ASD and SCZ are highly heritable30,31 and genetically heterogeneous32–34, with many identified risk genes, including rare, highly penetrant de novo mutations35 as well as many common variants that contribute small increases in risk. Thus, a central question is whether the genetic variants that increase risk for ASD or SCZ share similar downstream molecular etiologies, and if so, what are their mechanisms. Genome-wide association studies and rare variant sequencing studies have revealed the prevalence of synaptic genes11,12,17,29,36–41, including adhesion, scaffolding, ion channel, and local translation control proteins, as well as transcription factors upstream of them, among those associated with ASD and SCZ42–46. Additional evidence points to consistent changes in synaptic structural and functional features including dendritic spine morphology47 excitation/inhibition ratios48–51, and global features of gene expression and protein interaction networks11,17,52,53, which are common to different genetic models. Research that implicates perturbations in brain-wide connectivity patterns54,55, possibly related to deficits in predictive processing capacity56, supports the notion that a fundamental synaptopathology expressed variably throughout the brain may contribute to these disorders13. Physical protein-protein interaction approaches such as Y2H and CoIP52,53 have established interaction networks involving synaptic proteins and their changes in vivo in autistic individuals and autism models, demonstrating the promise of studying the synaptic molecular network as a focal point of autism pathogenesis. However, these studies fall short of measuring changes to joint distributions of protein expression levels or identifying perturbed causal connections between relevant proteins.
To characterize how synaptic molecular composition is affected across genetic perturbations associated with ASD and SCZ, we applied RNAi-mediated knockdown of 16 canonical, highly penetrant risk genes associated with either ASD, SCZ, or both, at the onset of synaptogenesis. Once a mature and stable synapse population was established in each genetic context, we measured the amounts of each of ten synaptic proteins across individual synapses using Probe-based Imaging for Sequential Multiplexing (PRISM)7,57,58. PRISM is a recently introduced multiplexed imaging technique that uses single-stranded DNA (ssDNA)-conjugated antibodies or peptides against desired targets, that are confocally imaged sequentially using fluorescently labeled single-stranded locked nucleic acid (ssLNA) imaging probes. The affinity of imaging probes for their targets depends on ionic strength, allowing sequential rounds of imaging of multiple proteins in the same sample and fields of view by exchanging imaging strands using high- and low-salt buffers. Thus, this imaging method provides a combination of extensive multiplexing, moderate throughput, minimal disruption to delicate synapse structures, and single-synapse resolution.
The PRISM imaging output consists of images of the same synaptic puncta over numerous protein channels. Integrating fluorescence intensity over individual puncta and assigning puncta across channels to the same synapse yields individual protein measurements per synapse7,57. We refer to the total integrated fluorescence intensity per protein at a given synapse as the local synaptic protein level. With this approach, in a single experiment, we can generate a type-resolved systematic view of protein level changes caused by different genetic knockdowns, as well changes to distinct, compositionally defined synaptic populations, as previously performed in response to chemical perturbations57. In the current study these changes included a global synaptic protein increase with knockdown of Pten, differential changes in response to Cul1 knockdown, a potentially compensatory increase in synaptic PSD95 with knockdown of Grin2a, and several unique synaptic phenotypes resulting from Dyrk1a knockdown.
Leveraging the unique ability of single-synapse, multiprotein measurements from PRISM to provide a high-dimensional joint probability distribution (PD) of synapses in composition-space, we additionally sought to infer the synaptic protein influence networks that generated the measured protein distributions. Toward this end, we used Bayesian network (BN) inference, a tool previously used to reconstruct entire signaling pathways from multiplexed single-cell data59. BNs is a framework to factor a joint PD into a product of individual conditional PDs. This can be represented by a directed acyclic graph between measured nodes, in which graph edges represent direct conditional dependencies between individual nodes, i.e., retaining only those connections that cannot be explained by mutual dependence on a third node, as well as the estimated causal direction of the pairwise dependencies. In this study, each node refers to the local synaptic level of a certain protein. Substructures in the resulting model generated testable predictions of causal connections (e.g., the hierarchy in which perturbing A affects B and C) between protein levels, some of which were consistent with known protein roles and interactions, and others that were discovered anew to the best of our knowledge.
In particular, the causal chain by which F-actin determines post-synaptic PSD95 levels, which in turn determines post-synaptic SHANK3 levels, we validated independently via direct perturbations, thereby establishing a new causal hypothesis that shapes synaptic protein distributions and, by confirming a novel prediction presented by the model, generating confidence in the new model as a whole. Finally, we present evidence for convergent changes in the inferred synaptic molecular network that are caused by distinct genetic knockdowns, specifically in the strengths of trans-synaptic and intra-postsynaptic edges, offering evidence for a convergent molecular etiology across ASD/SCZ-associated genes.
RESULTS
Effects of ASD- and SCZ-associated gene knockdowns on the synaptic molecular system
The following core synaptic proteins were characterized using PRISM to provide snapshots of the synaptic molecular sub-network (Figure 1A). Synapsin1 was used to define all synapses, with vGluT1 and vGAT used to differentiate glutamatergic from GABAergic synapses. Other proteins included Bassoon, a central presynaptic scaffolding protein which served as a proxy for active zone size60, and the AMPA receptor subunit GluR2, which served as an indicator of synapse strength61. Filamentous β-actin (F-actin), measured via phalloidin, was included as the core of the dendritic spine cytoskeleton that is locally regulated by several ASD/SCZ associated genes (e.g., Trio, Pten and Dyrk1a) and whose dysregulation is implicated in various synaptopathologies62,63. Finally, four scaffolding proteins were included that have crucial roles in shaping the post-synaptic density: PSD95, Homer1a, and SHANK3 in excitatory synapses, and Gephyrin in inhibitory synapses. MAP2 staining via conventional immunofluorescence was also used to trace dendrites to the degree feasible in dense cultures and constrain puncta assignments to synapses, as well as to align images from different imaging probe exchange rounds7,57.
Figure 1: Genes and targets.
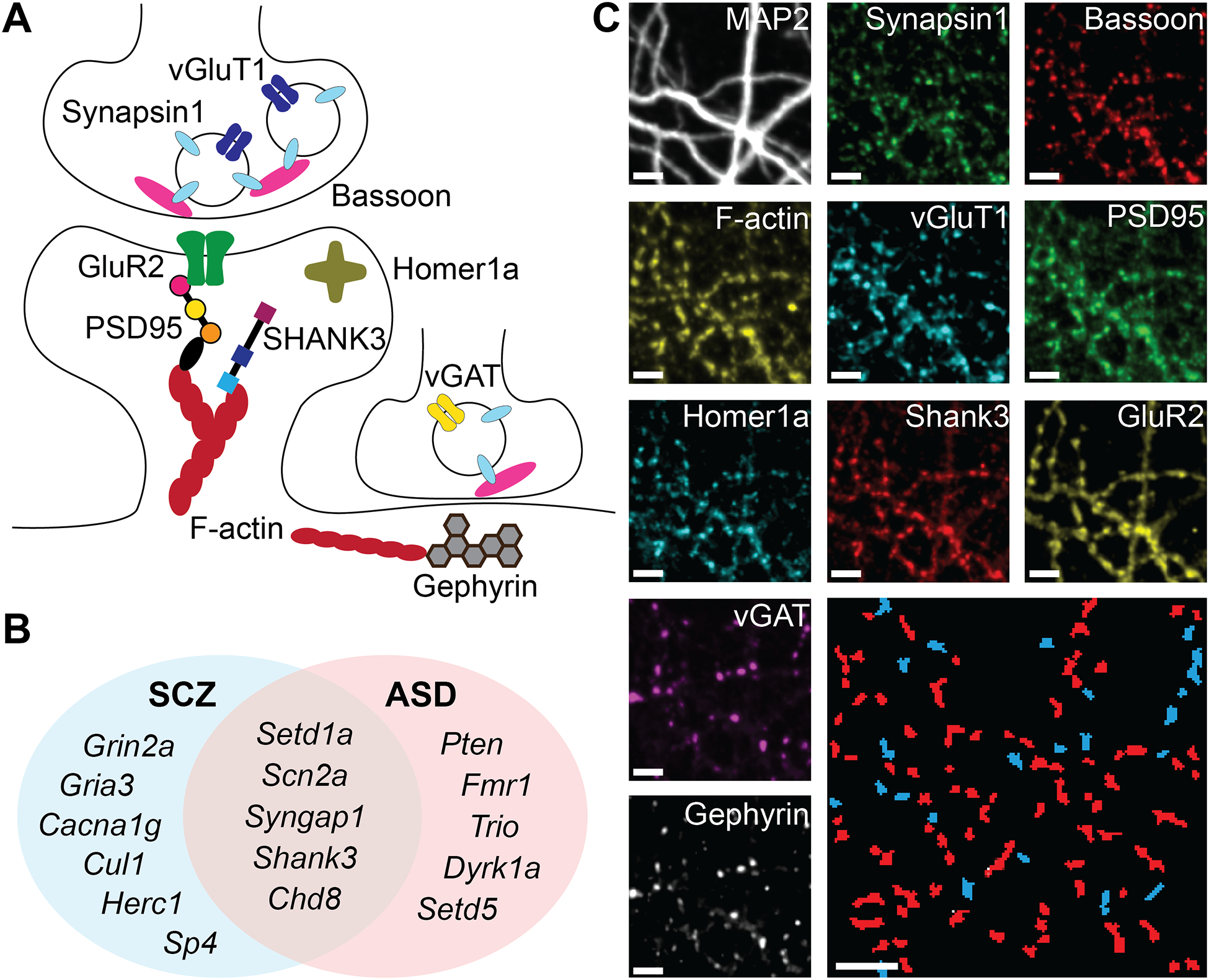
A) Schematic summarizing approximate synaptic context of the 10 imaged targets. B) Venn diagram of gene knockdowns. C) Representative images of the same neuronal culture in different imaging rounds, showing colocalized puncta of each protein. Bottom right – automatically identified and segmented excitatory (red) and inhibitory (blue) synapses. Scale bars are 5μm.
For genetic perturbations, ten autism genes (Figure 1B) were chosen as the best-scoring targets in the Simons Foundation Autism Research Initiative database64,65 (“SFARI score”), half of which are implicated in SCZ as well. Six additional SCZ-specific genes were chosen from the highly penetrant de-novo mutations identified by Singh et al.35 A mixture of four siRNA reagents was used for each gene, and each siRNA treatment was separately validated by RTqPCR for reduction in mRNA levels in cultured hippocampal neurons (figure S1A). The siRNA reagents appeared to act more similarly to a small molecule treatment than a transfected gene, with transfection universal (>99% of cells) and concentration-dependence occurring even at low doses (figure S1D–F). A non-targeting siRNA mix (“NonT”) was included as a negative control and used for treatment comparisons throughout results.
To measure the effect of each gene knockdown on synaptic protein distribution we treated hippocampal neuronal cultures at day-in-vitro (DIV) 6 with one of the corresponding siRNA reagent mixes and fixed the cultures at DIV 19 to image using PRISM (see STAR Methods). We integrated an automated liquid handling platform for probe exchange to complete seven imaging rounds of the same 60 cultures (3–4 per treatment group) in under 12 hours. The resulting fluorescent images of the same synapses across imaging rounds (figures 1C and 2A) were automatically segmented, classified, and quantified using CellProfiler66,67 (see STAR Methods). We combined data from N=4 such experiments, where each used E18 embryonic neurons from a different pregnant rat, for a total of 3.5×106 11-protein synaptic measurements across 220 separate neuronal cultures in 18 treatment groups. To account for variability in staining and imaging conditions between experiments, images in each experiment were manually adjusted to the average intensity of that channel in untreated cultures of the same plate.
Figure 2: Synapse effects of siRNA knockdowns.
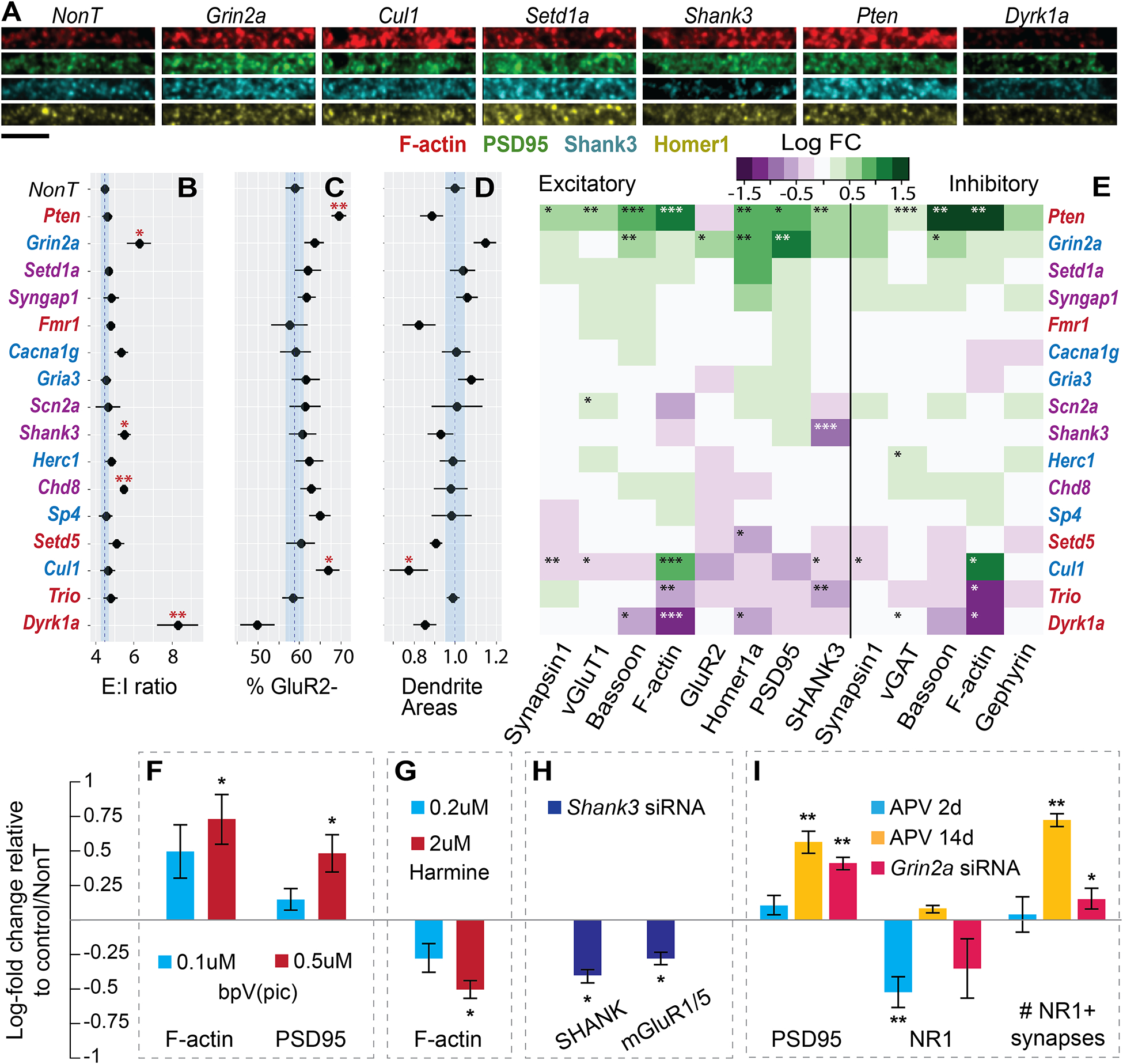
A) Representative images from medium-sized dendrites across 4 channels. Bottom left scale bar is 5um. B) Excitatory (Syn+, vGlut+, vGAT−) to inhibitory (Syn+, vGlut−, vGAT+) count ratios. C) Percent of GluR2 negative synapses from all excitatory synapses. D) Estimates of total dendrite proliferation from MAP2 staining, normalized to NonT. B-D blue lines indicate mean and SEM of NonT measurements. E) Log fold change (relative to NonT) of mean levels of each protein in excitatory and inhibitory synapses. Coloring of genes in B-E: red, only ASD, blue, only SCZ, purple, both. F-I) Validation experiments with chronic treatments (DIV 6–19) measured by conventional IF. Mean synaptic protein levels or other measurements depicted as Log2-fold change from untreated control (for chemical treatment experiments) or NonT siRNA (for RNAi) treated wells. F) Treatment with bpV(pic), a PTEN inhibitor. G) Treatment with Harmine, a Dyrk1a inhibitor. H) SHANK and mGluR1/5 after knockdown of Shank3. I) PSD95, NR1, and density of NR1-positive synapses after short and chronic NMDAR blockade with D-APV, or chronic RNAi knockdown of Grin2a. Bars are mean ± s.e.m. across wells. *p<0.05, **p<0.01, ***p<0.001, two-sided t-test.
Automatically segmented puncta, identified as above-threshold intensity peaks within a size range (see STAR Methods) in each protein channel, were assigned to specific individual synapses based on overlap with Synapsin1 puncta (see STAR Methods). The existence of a punctum in a protein channel assigned to a specific synapse was taken as the presence of that protein in the synapse, and the integrated fluorescence intensity of a certain protein channel across its synapse-associated punctum was assumed to be proportionate to the total level of that protein in the synapse. Based on these data, we first examined the individual effects of different siRNA treatments on three global parameters—excitatory:inhibitory (E:I) synapse ratio (figure 2B), fraction of GluR2-negative excitatory synapses68 (figure 2C), and dendrite growth, measured as overall area stained by MAP2 (figure 2D). E:I synapse ratio, implicated previously to be dysregulated in ASD and SCZ48, was calculated as the ratio of (+vGluT1, −vGAT) to (−vGluT1,+vGAT) synaptic puncta. This ratio was on average ~5:1 and was significantly increased in knockdown of Dyrk1a, consistent with reports of inverse correlation of Dyrk1a expression to E:I ratio in vivo49. It also increased in knockdown of Grin2a, Shank3 and Chd8.
Next, by examining the average levels of each measured protein across synapse populations, we created a map of how the synaptic levels of each protein were on average affected by each treatment (figure 2E). Beyond this overall characterization, we observed several novel synaptic phenotypes, the strongest of which included: (a) two-fold increase in Homer1a under knockdown of Setd1a, a nuclear regulatory lysine methyltransferase, while knockdown of Setd5, a gene of the same family, decreased Homer1a; (b) 80% increase in synaptic F-actin after knockdown of Cul1; other proteins including Synapsin1 are decreased; (c) knockdown of Grin2a leading to a ~70% increase in PSD95, as well as increases in other proteins including Homer1a, GluR2 and Bassoon; (d) A twofold decrease in synaptic F-actin following knockdown of Dyrk1a, accompanied by decreases in other proteins including Bassoon and Homer1a; and (e) knockdown of Trio leading to a decrease in synaptic F-actin and SHANK3.We did not identify significant differential effects on the same protein in the context of excitatory versus inhibitory synapses.
The same images could also be used to assess changes in global protein expression (figure S2), either somatic (i.e., fluorescence in synapse-excluded neuronal bodies) or total (averaged fluorescence over the entire image). Several notable differences from synapse-specific phenotypes appeared. For example, Cul1 knockdown showed a significant increase in synaptic F-actin, but not in global somatic levels, in parallel with both synaptic and (stronger) somatic reduction in other proteins, raising the possibility of a compensatory connection, in either direction, between an increase in F-actin and reduction in other postsynaptic proteins via an unknown mechanism, possibly related to the role of Cul1 in regulating SPAR turnover69.
Some treatment effects in 2E were reported previously or were expected based on pre-existing knowledge of mechanisms involved. For example, we confirmed that siRNA knockdown of Shank3 led to a marked reduction in SHANK3 levels at synapses. Knockdown of Pten led to a broad increase in nearly all synaptic markers, consistent with its role as a negative regulator of PI3K-dependent neurite and synapse proliferation70.
To support the validity of RNAi with PRISM to reveal phenotypes directly connected to gene function, we performed chemical inhibition and knockdown experiments using conventional immunofluorescence (figure 2F–I), showing that: (i) treatment with bpV(pic), a PTEN phosphatase inhibitor, increased synaptic F-actin and PSD95, mimicking Pten knockdown in a dose-dependent manner (2F); (ii) treatment with Harmine, a Dyrk1a inhibitor, decreased F-actin, mimicking Dyrk1a knockdown in a dose-dependent manner (2G) and also increased the fraction of GluR2-positive synapses (figure S2D–F); (iii) knockdown of Shank3 reduced synaptic SHANK as well as mGluR1/5, as previously reported71 (2H); and (iv) knockdown of Grin2a increased PSD95 levels and density of NMDAR-containing synapses (2I). We also noted that the effects of Dyrk1a inhibition on synaptic F-actin and the fraction of GluR2-positive synapses were similar with and without application of heat-induced antigen retrieval (figure S2D–E), suggesting that the observed phenotypes are likely due to changes in protein content and not antigen accessibility. Finally, we noted that the effect on GluR2-positive fraction was similar whether staining for total or only externalized GluR2 (figure S2F), indicating that although GluR2-negative synapses identified in our screen are not necessarily silent68 and vice-versa (because GluR2 may be present but below threshold, or present but not externalized), they may act as a reasonable proxy for changes in silent synapse populations.
To test the hypothesis that PSD95 levels respond to mitigate decreased NMDAR presence or activity, we treated cultures for either a short time (48 hours) or chronically with NMDAR blocker D-APV72 (2I). After the short treatment, NMDAR levels per synapse decreased ~30%. This effect disappeared after two weeks, accompanied by an increase in PSD95 and excitatory synapse density that mimics Grin2a knockdown, consistent with a compensatory response.
Multiplexed imaging reveals clusters of hierarchical synaptic protein compositions
We applied Uniform Manifold Approximation and Projection (UMAP)73,74 on the 11-dimensional dataset of synapse protein levels, yielding the 2D projection in figure 3A of different synapse compositions. The distribution of synapses shows distinct clusters defined combinatorially by the presence or absence of certain proteins, similar to our previous observations57. These included two inhibitory clusters and several excitatory clusters. Protein absences that defined certain clusters may have resulted from ‘true’ complete absences or merely from levels below threshold. However, we observed similar distributions when changing threshold levels for synapse identification (to 75% and 133% of defined levels, figure S3), indicating that the clusters arose at least in part from qualitatively different synapse compositions.
Figure 3: Synaptic multiprotein distributions.
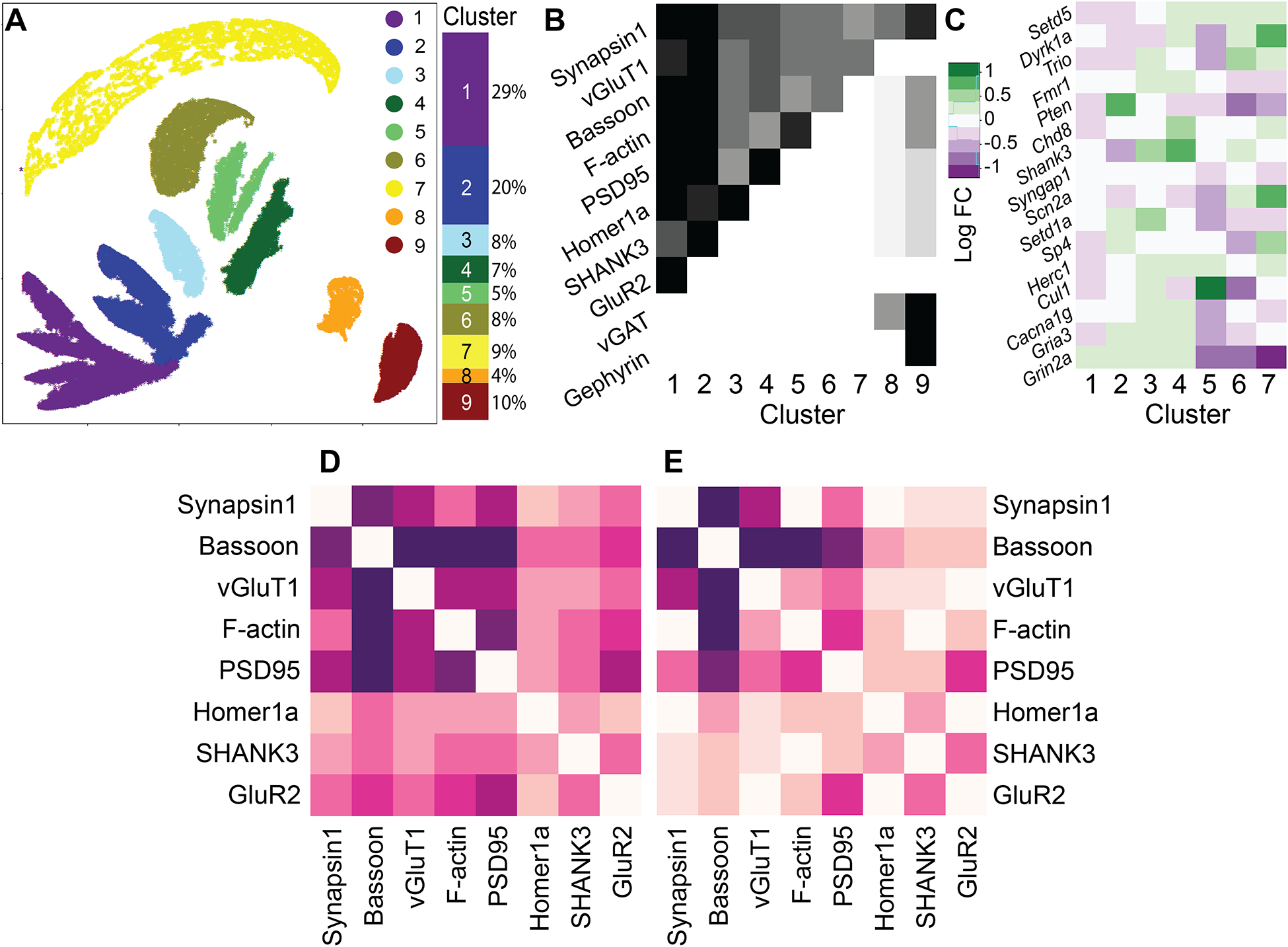
A–C) Composition-defined synaptic subtypes. A) UMAP projection of scaled synaptic measurements. B) Row-normalized (across all clusters) mean levels of each specific protein in specific clusters identified in A. C) Log fold-change of excitatory cluster populations under each treatment. Right: overall composition of the synapse population by cluster. D) Direct correlations between proteins in excitatory synapses. E) Correlations in each pair, controlling for all other 6 proteins.
All treatment groups had synapses in all 9 clusters (figure S4), with some population changes between clusters observed as a result of different gene knockdowns (figure 3C and S4). These changes were consistent with the changes observed in mean protein levels in figure 2E. For example, Pten knockdown, which increased all excitatory proteins save for GluR2, was seen here to enrich cluster #2 (GluR2-negative, positive for all others) at the expense of clusters #1 (GluR2-positive) and #6 (F-actin-negative). Nevertheless, they provided more detailed information about the specific synapse population changes that occurred as a result of gene knockdown.
In addition, we were able to use spatial information to aid in interpreting protein combinations. A small fraction of puncta was identified as vGluT1-positive, vGAT-negative, and Gephyrin-positive (figure S5A,B), despite Gephyrin being well established as an inhibitory synaptic protein75. However, upon closer examination we observed that Gephyrin-Synapsin puncta distances in that subset were 50% greater than expected (figure S5C), leading us to infer that these puncta were probably not associated physically with the other excitatory markers, and to exclude them from future analyses.
Finally, we observed that not every combination of proteins was present (figure 3B). For example, synapses that were negative for Bassoon or β-actin typically lacked or had very low levels of other post-synaptic proteins, indicating a hierarchy in protein dependencies on one another, which we sought to characterize systematically using Bayesian network inference as previously applied to signaling networks59.
Bayesian network inference of the glutamatergic synapse
All excitatory synaptic proteins were positively correlated with one another (figure 3D). However, when we examined each pair individually by measuring their correlation controlled via stratification for all other six proteins (see STAR Methods), some correlations disappeared (figure 3E). This revealed protein pairs that were not directly connected but only correlated through their inter-dependence on a third protein, which could be either a common effector (A←C→B) or an intermediate (A→C→B). To systematically map which causal connections among excitatory synaptic proteins were direct versus indirect, and to establish the directionality of their inter-dependence, we derived a Bayesian network from the 8-dimensional distribution of protein levels in excitatory synapses.
This approach was first tested on simulated PRISM-like Bayesian networks—high-dimensional distributions that mirrored real PRISM data but with the distributions of variables conditional on one another in predetermined ways (figure 4A,E, see STAR Methods for details on network simulation).
Figure 4: Bayesian network inference on simulated networks.
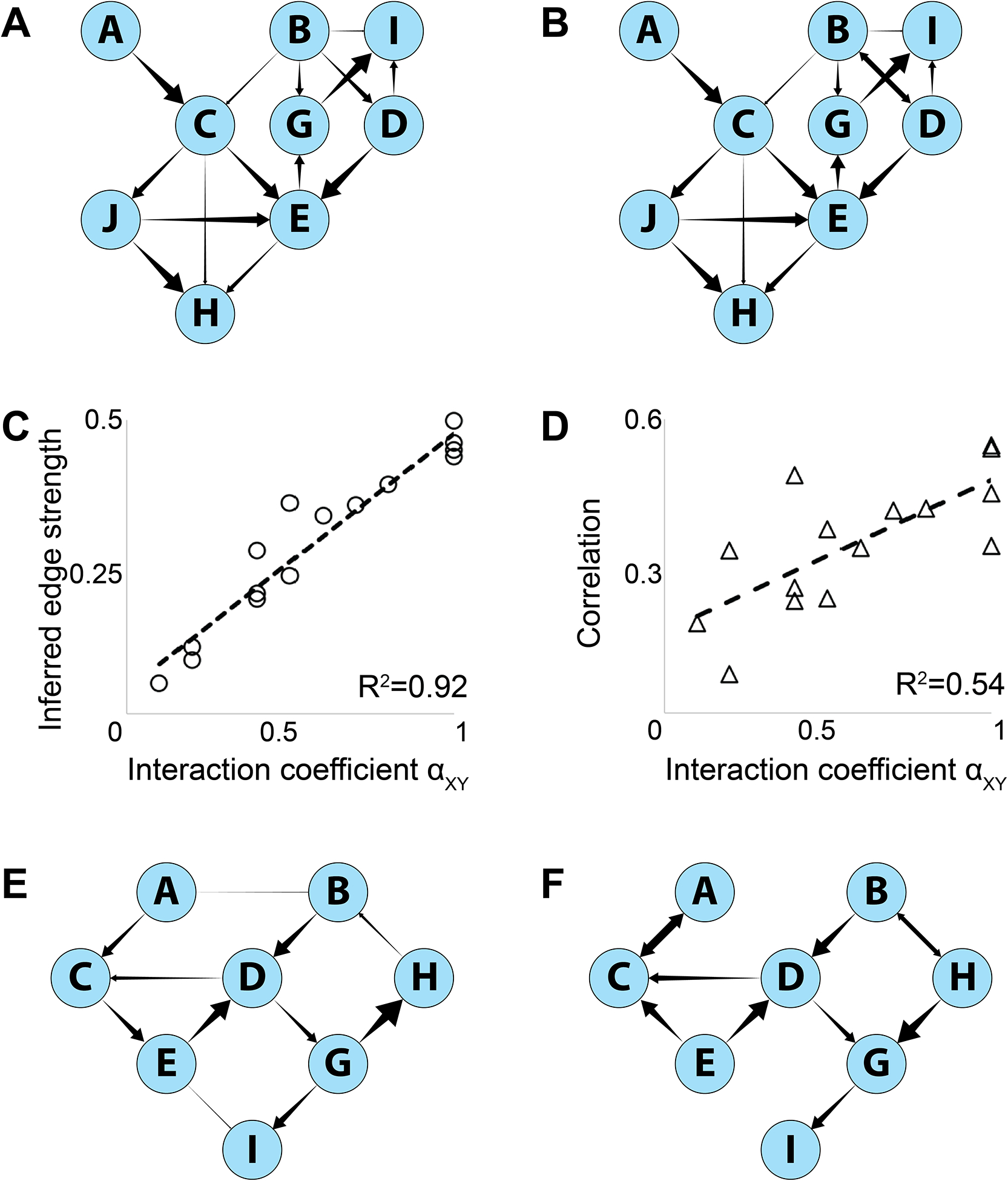
A) Simulated Bayesian network. B) Reconstructed network. C,D) Calculated edge strengths of the edges in (B) versus defined interaction coefficients in (C). C – edge strengths calculated by parent-controlled correlations. F – total (uncorrected) correlations. E) Simulated non-Bayesian network with cycles (CED and BDGH). F) Reconstructed Bayesian network. Cycles cannot be represented but the overall structure and relative edge strengths are preserved. Arrowhead sizes represent predefined interaction parameters αXY in A,E and inferred edge strengths in B,F.
To derive the Bayesian networks (figure 4B,F), we applied the ‘tabu’ algorithm of the BNLEARN package76 in R that searches model-space in a Monte-Carlo-like manner, maximizing an overall score that is based on the likelihood of the data given the model76–79. Additional costs to the score were imposed on each edge to prefer simpler, more parsimonious models76,79. To estimate confidence levels on the presence of edges and their directions, we applied a bootstrapping method that re-derived separate Bayesian networks for 50 independent samplings of 10,000 points in the simulated distribution. An edge was considered present (and is shown in 4B,F) if it appeared in >80% of bootstrapped networks, and a direction indicated (as a unidirectional arrow in 4B,F) if that direction appeared in >60% of bootstrapped networks where the edge was present. To derive edge strengths, shown as arrowhead sizes in figures 4B and 4F, we calculated the correlations between the parent and child node when controlling by stratification for all other parents of that node79,80. In bidirectional edges, both nodes were considered child nodes for this purpose.
This method reconstructed predefined networks with high fidelity. Notably, even when a distribution was not generated by a Bayesian network (i.e., it contained cycles, which cannot be reflected in a directed acyclic graph but can occur in reality as feedback loops) the inference algorithm still reconstructed the general network structure and edge strengths with reasonable fidelity, reversing some intra-cycle edges to avoid loops but preserving extracyclical edges. Edge strength calculation recovered the original interaction parameters (figure 4E) better than using uncontrolled, total correlations (4F).
We applied the above approach to the entire 3.5-million synapse dataset across all treatments yielding the network shown in figure 5A. This network exhibited several features that were anticipated given our knowledge of the function and connectivity of these proteins. For example, the presynaptic proteins Bassoon, Synapsin1 and vGluT1 (the latter two co-localized in the same synaptic vesicles) appear tightly interconnected, and their correlation was independent of the postsynaptic proteins. Of the three presynaptic proteins, Bassoon levels were most directly connected to those of the postsynaptic proteins, possibly by acting as a proxy for the size of the active zone. Of the postsynaptic proteins, GluR2 was directly downstream of PSD95, in accordance with the latter’s role as a dynamic anchor for the receptor whose levels dictate the number of sites that can capture diffusing AMPARs on the postsynaptic density81. Finally, levels of synaptic F-actin were upstream determinants of all other postsynaptic proteins, likely due to the cytoskeletal protein acting as a proxy for the size of the dendritic spine, which may limit other protein amounts. Additional, unanticipated features included presynaptic Bassoon appearing to influence levels of postsynaptic F-actin, PSD95, and Homer1 more than the latter influenced one another, as inferred from network edge strengths as defined above.
Figure 5: Bayesian network of 8 excitatory synaptic proteins.
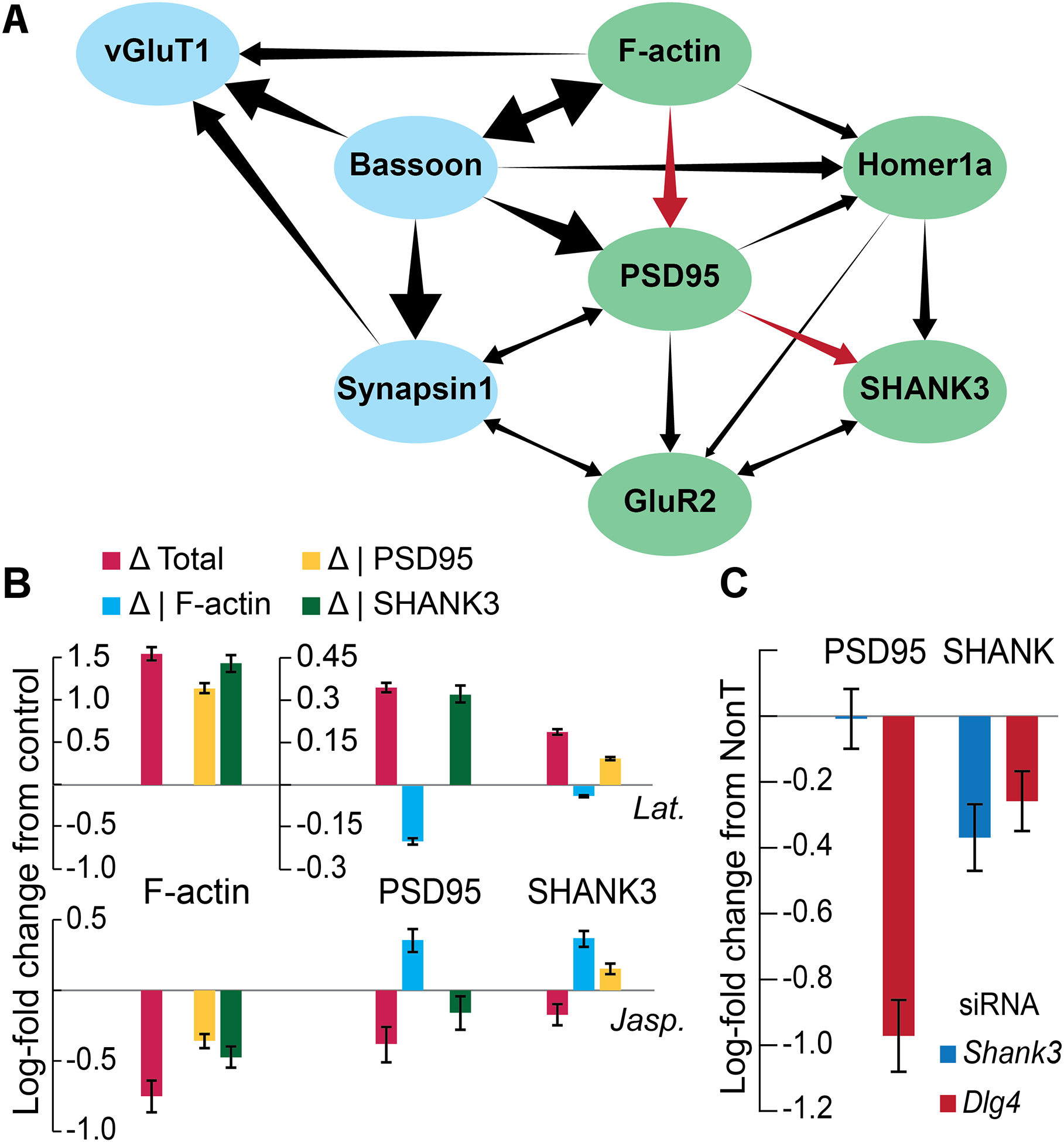
A) The inferred network. Presynaptic proteins in blue, postsynaptic in green. Red arrows indicate substructure probed in B and C. B) Log fold-change in F-actin, PSD95 and SHANK3 after treatment with two actin polymerization perturbations, total and controlled for each protein. C) Log fold-change in PSD95 and Shank3 after treatment with 3 siRNAs: nontargeting, against Dlg4 (PSD95) and against Shank3. Bars are mean ± s.e.m. across wells.
Importantly, our model also lends itself to testable hypotheses of causal connections derived from sub-structures of the network. For example, the position of SHANK3 downstream of the other components predicted that they would not be affected significantly by the direct perturbation of SHANK3, e.g., by siRNA-induced knockdown. Our screen confirmed this (figure 2E, Shank3 line and 2H), consistent with a previous study that used shRNA knockdown of Shank3 to show that mGluR5 levels were reduced but other postsynaptic proteins were unaffected71. To prevent circular logic (i.e., the presence of the Shank3 knockdown data in the network generating this prediction) we derived the same Bayesian network when excluding the Shank3 treatment group (figure S6), showing that BN inference can predict the effects of perturbations that are excluded from the training data.
A more complex, predictive sub-structure of the network is the inter-dependence chain: F-actin → PSD95 → SHANK3. This structure predicts that a) perturbing F-actin should affect both PSD95 and SHANK3, but the effect on SHANK3 should decrease or disappear when controlling for PSD95, while the effect on PSD95 should be relatively independent of SHANK3, and b) perturbing PSD95 should affect SHANK3, but not vice-versa. To test the first prediction, we treated hippocampal cultures with Jasplakinolide (Jasp.) or Latrunculin B (Lat.) at DIV 6, fixing at DIV 19 and staining for Synapsin1, F-actin, PSD95 and SHANK3 (figure 5B). Interestingly, although Jasplakinolide and Latrunculin B are generally inhibitors of F-actin depolymerization and polymerization, respectively, their effects on synaptic F-actin after chronic treatment were reversed. Effects on PSD95 and SHANK3 were consistently in the same direction as the effects on F-actin (decreased for Jasp. and increased for Lat.) and completely disappeared or even reversed when controlling for F-actin. In addition, controlling for PSD95 greatly diminished or reversed the effect on SHANK3, as predicted, while the reverse was not true. To test the second prediction, we treated DIV 6 cultures with siRNA mixes against Shank3, Dlg4 (the gene coding for PSD95), or a non-targeting siRNA mix (NonT), and fixed and stained at DIV 21 for MAP2, Synapsin1, SHANK3, and PSD95 (figure 5C). Synapsin-controlled SHANK3 levels were reduced compared to NonT in both Dlg4 and Shank3-treated synapses, but PSD95 was reduced only in Dlg4-treated synapses, establishing the PSD95>SHANK3 hierarchy. Taken together, these observations support the conditional dependency chain predicted by the network.
Network inference on real PRISM data may be sensitive to experimental and image analysis artifacts, such as thresholding, image quality, and rules for synapse identification, as these may impose artifacts on correlations between protein measurements. We therefore performed several quality controls to ensure that our model did not result in such artifacts (figure S6). In one, we limited network inference only to synapses positive for all protein components. In another, we varied the thresholds for puncta identification in CellProfiler to 75% or 133% of their values used in the primary analysis. In a third, we used puncta of postsynaptic proteins (F-actin and PSD95) to assign synapse identity, instead of Synapsin1 and vGluT1. All these manipulations yielded network structures that were largely similar to that in 5A, especially in presence, relative strengths, and directionalities of trans-synaptic and intra-postsynaptic edges. The few inconsistencies included interchanges in the relative positions of Synapsin1 and vGluT1 in the network. Finally, we derived a network based on different eight-protein measurements in a previous study57 (figure S6). Although that dataset was smaller with fewer perturbations than the current one, and thus with reduced confidence in edge presence and directionality, we observed that the six proteins common to both studies exhibited similar connectivity patterns. That the network features were robust against these manipulations, replicated across experimental conditions, and include substructures that were directly validated in perturbation experiments, strengthened our confidence that these features represent real underlying biology.
Convergent effects of siRNA treatments on network structure
Next, we sought to investigate the effects of siRNA treatments on underlying protein networks to investigate whether convergent synaptic phenotypes resulted from distinct genetic perturbations. When analyzing the effects of a chemical or genetic perturbation on a biomolecular distribution, it is often difficult to distinguish direct from secondary, or downstream effects. Informed by the network model, our multiplexed imaging enables this distinction. We analyzed the effects of each siRNA treatment on each protein of excitatory synapses while controlling for all parent nodes of that protein in the network, thus retaining only effects that were direct and not mediated by a parent node (figure 6A). Isolating direct effects revealed that Pten knockdown affected all postsynaptic protein levels that we examined nearly exclusively through the increased localization of F-actin within dendritic spines, in accordance with what is known of its regulatory role70. This also allowed us to discover direct effects that were hidden under second-order effects in the opposite direction, for example that knockdown of Pten caused a strong specific decrease in synaptic GluR2 in parallel to a general increase of all post-synaptic proteins.
Figure 6: Network-informed analysis of the genetic screen.
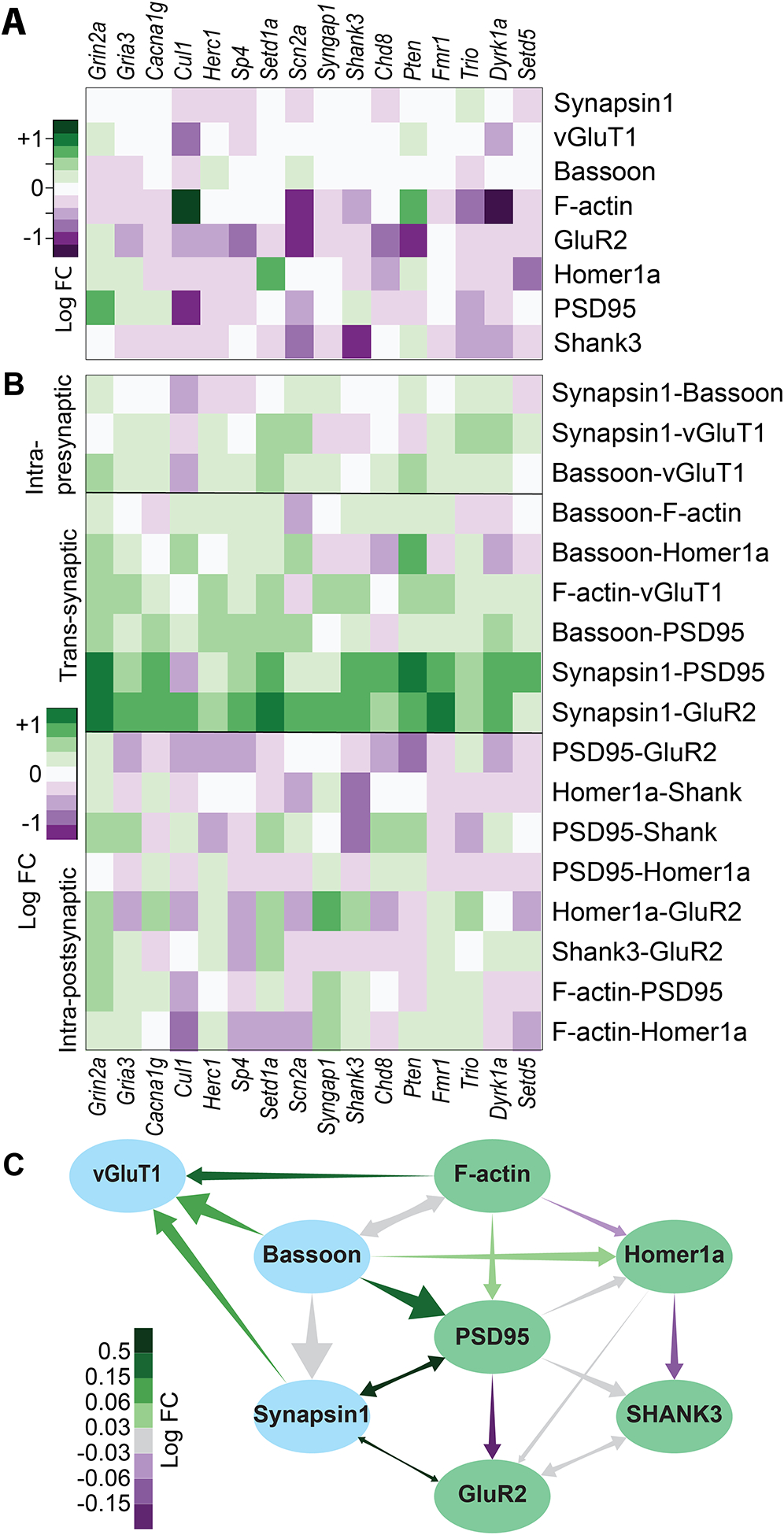
A) ‘Direct’ effects of each treatment on each protein separately. Like 2F but controlled for the parent nodes of each protein. B) Effect of each treatment on the strength of each network edge. C) Network from 4A, each edge colored by the average change in strength across treatments. All colors are log2-fold-change relative to nontargeting siRNA control.
Finally, we looked for quantitative effects of siRNA gene knockdowns on the Bayesian network structure itself, particularly on edge strengths that indicate inter-dependencies of distinct synaptic protein components. We quantified the strengths of the 17 edges in the network for each culture (figure 6B,C). We discovered that some edges—particularly centered around PSD95 and GluR2—were uniformly strengthened or weakened by nearly all treatments, regardless of direct effects on the proteins themselves, even in treatments that had weak if any effects on the protein levels. For example, Synapsin1-PSD95 and Synapsin-GluR2 edges were strengthened in nearly all gene knockdowns compared with the NonT groups, and not weakened in any, even though different treatments increased or decreased each protein on its own. Conversely, the PSD95-Homer1, Homer1-GluR2 or PSD95-GluR2 edges were weakened in most treatments compared with NonT.
Independent treatment effects on different proteins were likely to reduce edge strengths indiscriminately, indicating that that there might be an underlying molecular process by which knockdowns of many different genes all serve to, for example, weaken the extent by which PSD95 determines GluR2 and strengthen the extent by which PSD95 and Synapsin1 influence each other, where these effects are superimposed on any direct effects they may also have on these proteins.
To the best of our knowledge, this is the first time that a direct measurement of synaptic protein networks has yielded molecular phenotypes common to many different ASD- and schizophrenia-associated mutations, which may be reflective of shared synaptic pathogenesis.
DISCUSSION
Detailed parallel phenotyping of synaptic biochemistry in ASD and SCZ models
We used simultaneous measurement of multiple proteins at single-synapse resolution to infer causality relationships among protein numbers in the synaptic molecular network, and systematically map how it is affected by the perturbation of ASD- and SCZ-associated genes. PRISM, supported by automated probe exchange, image analysis, and synapse segmentation and quantification, can be used to measure multiple synapse-specific phenotypes across many treatment groups in a single experiment, as well as to identify synapse types and population changes that are more intricate than bulk effects on a protein due to its ability to resolve synapse-to-synapse heterogeneity in multiple protein levels.
We observed phenotypes that were anticipated based on previous studies or their known activity, such as Pten70 and Shank371. We also observed phenotypes that to the best of our knowledge have not been previously reported, such as Cul1 on F-actin, Setd1a on Homer, and Grin2a on PSD95. Follow-up on the latter yielded tentative evidence for a long-term compensatory mechanism that responds to NMDAR deficiency (by gene knockdown) or reduced activity (by APV blockade, which also reduces NMDAR levels), with increased excitatory synapse density and levels of NMDAR anchoring protein PSD95, possibly a compensatory effect. Of the overall molecular signatures, Dyrk1a exhibited the most unusual pattern, combining a markedly higher E:I ratio, lower levels of F-actin and downstream postsynaptic proteins, but not of GluR2 (and generally a lower percentage of silent synapses). This is congruent with reports of Dyrk1a-associated autism presenting a unique neurological character82,83.
Another important consequence of the ability to measure multiple proteins simultaneously is that it can facilitate deconvolution of direct causal relationships from those mediated by other processes. Instead of having to experimentally constrain any possible confounding variables, multiplexed imaging allows measuring them simultaneously, while moderate throughput offers a large enough number of data points (>3×106 in this study) to directly control for possible confounding variables via stratification. The results of such intra-dataset controls must be interpreted carefully due to potential artifacts arising from comparing different synapse populations. Nevertheless, when using information about causal connections between variables, either from prior knowledge or, as in this case, inferred directly from the probability distributions, such a controlled analysis can help identify when a certain effect is entirely mediated by other variables (as with Pten or Dyrk1a on postsynaptic proteins, mediated by F-actin) or when a certain variable deviates significantly due to a hidden direct effect from what is expected given its upstream network connections (as with Pten on GluR2). The network-based prediction that Dyrk1a effects are mediated via F-actin is supported by an in-depth investigation84 of changes to dendritic spine formation in heterozygous Dyrk1a truncation mutants.
It is important to note that although parallel measurements of many proteins may serve as a hypothesis generator for synapse level molecular mechanisms, this study is fundamentally a phenotypic assay and does not purport to test or establish any specific mechanism definitively. Some gene perturbation effects may be the result of transcriptional changes in upstream gene expression networks (for example, by histone modifying enzymes), to which our assay is agnostic.
Interpreting inter-protein dependencies from Bayesian network structure and edge strengths
In the inferred Bayesian network, an edge from node A to B indicates that the PD of B depends on A even after accounting for all other measured components. This dependence can be direct or via a hidden (unmeasured) component. Conversely, lack of a direct edge between two correlated nodes indicates that any correlation between them can be explained by the other known components. Thus, while the overall structure is dependent on the set of proteins measured, we expect ‘non-edges’, such as F-actin-SHANK3, to be a conserved feature even as the network expands to include more proteins. The structure we observed hints at some general rules governing synaptic molecular composition, which have also been observed in the literature: relative independence of the presynaptic active zone assembly and molecular composition85,86, and receptor levels driven by scaffolding proteins rather than the reverse.81,87,88
The interpretation of changing edge strengths is not mechanistically obvious. As a rule, injecting perturbative noise into a system weakens correlations by default. Thus, a weakened edge may correspond to a loss of correlation that can occur if only one protein of a pair is perturbed, or if they are perturbed in different directions. However, this is not always the case—for example, knockdown of Cul1 reduces both PSD95 and GluR2 to a similar extent, but also weakens the PSD95-GluR2 edge. In general, an edge from a parent node to a child node is considered in the context of all the other parents of that node. Thus, an effect that perturbs the mechanism by which one parent node affects the distribution of the child will weaken the corresponding edge but may strengthen the edge from another parent, and vice-versa. It is thus interesting to observe that trans-synaptic edges are strengthened by the genetic knockdowns in this study, while intra-postsynaptic edges are generally weakened.
Comparison with other protein network models
Our results add to a growing body of genomic and proteomic observations of convergent changes at the protein network level in ASD. These are based on physical interaction networks derived from yeast two-hybrid (Y2H) tests52 or bulk quantitative multiplexed co-immunoprecipitation (QMI)53. It is important to note differences between physical interaction networks as obtained from Y2H screens or coimmunoprecipitation, and our network, which does not provide information about direct biochemical protein-protein associations, but rather provides constraints on the multiprotein joint PD that reports on synaptic-level protein co-localizations. When many parallel biochemical interaction pathways exist, the Bayesian network enables us to infer those that causally determine synapse protein levels. For example, the F-actin → PSD95 → SHANK3 causal chain we established implies that it is likely an interaction chain from actin to PSD95 (possibly via ARPC489) that drives PSD95 (and therefore SHANK3) levels. It must be noted that for F-actin, our measurement mixes together signal from both pre- and post-synaptic β-actin filaments. However, because postsynaptic F-actin is thought to be more abundant, we believe that the observed conditional dependencies are due mostly to postsynaptic F-actin levels.
Causal correlations may also show up if a certain target is the best available proxy for a different measure that drives protein levels such as Bassoon for the active zone or F-actin for the dendritic spine. For example, knockdown of Bassoon itself does not seem to affect postsynaptic protein localization and ultrastructure. Rather, we hypothesize that the edges from Bassoon to F-actin, PSD95 and Homer represent different paths, through unmeasured targets, by which the size of the active zone affects levels of different post-synaptic proteins. Finally, our network so far only measured protein abundances, and not phosphorylation activation or other modification changes (or splice isoforms), which are often influenced by interaction partners.
The two frameworks thus complement each other: Physical interaction networks provide mechanistic biochemical associations albeit in some cases in unphysiological conditions such as in yeast and, by casting a wider, less biased net, identify new targets to measure, whereas Bayesian network analysis integrates these details into a global picture of what shapes the overall synapse protein composition. This, in turn, provides insight into possible dynamical processes in the synaptic molecular network, which are tested by once again returning to mechanistic connections between components. Based on recent advances in automated, multiplexed superresolution imaging such as maS3TORM90, Bayesian network analysis could be, in the future, directly combined with single-synapse-level knowledge of physical synaptic protein organization.
The steady-state of the synaptic molecular network in ASD and SCZ
We propose a theoretical framework for future exploration into the convergence of phenotypes of different disease-associated mutations at the level of the synaptic molecular network. A dynamical system like the synapse with many components generally exists in a high-dimensional space of component abundances and activation states. However, with sufficient interactions and feedback loops such systems invariably settle into a much lower-dimensional space of allowable stable or metastable states. In the case of the synapse, these are guaranteed by the high interconnectivity among synaptic proteins, and feedback constraints such as homeostatic plasticity. In other words, since synaptic structural dynamics (e.g., LTP and LTD) occur at longer timescales than the biochemical feedback interactions that establish the allowed states, the former navigate a comparatively narrow landscape of only those states that the dynamical system can consistently sustain, and the synapse population distribution reflects this landscape.
A similar constraint-induced dimensionality reduction is considered to stabilize symmetric phenotypes in genotype-phenotype maps91. In the context of ASD- and schizophrenia-associated genotypes mapping to a synaptic phenotype space with reduced dimensionality caused by interaction-based constraints, this means that a multitude of seemingly unrelated disease-associated mutations, can drive similar perturbations to the lower-dimensional stable/allowed-state landscape, as measured in synaptic protein networks.
Thorough investigation of such a mechanism in ASD and SCZ will require characterization of this space of allowed synaptic states and how it changes under different mutations, a characterization for which this work and others provide initial outlines. This characterization should also provide additional starting points to investigate the downstream effects of convergent synapse structure phenotypes on the architecture of the synaptome and specific neuronal circuits.
Given the potential of PRISM with automated probe exchange for increasingly higher throughput, along with the theoretically unlimited multiplexing capacity, our method of multiplexed imaging with single-synapse analysis is well-poised to investigate such hypotheses, as well as any other processes in the synaptic molecular network. Specifically, the properties of BN inference mean that additional measured nodes would increase both the scope of, and confidence in, network structures, as well as help establish differences in network structure underlying qualitatively different functionality in excitatory synapses connecting different neuron types6. Finally, the tool presented here needs not be limited to synapses, and can be applied to any subcellular structure which can be identified individually in fluorescent microscopy, including mitochondria, phagosomes, nuclear compartments, etc. Thus, PRISM, supported by Bayesian network analysis may come to serve as a general hypothesis-generating tool for understanding complex protein networks in situ in cells, organelles, and subcellular structures.
Limitations of the study
The objective of this study was to perform high-dimensional in situ synaptic protein phenotyping in an ASD- and SCZ-associated genetic screen and search for similarities in synaptic protein compositions and convergent synapse phenotypes across disease-associated genes. One major limitation of this study was the use of self-transfecting siRNA knockdown in 2D rodent neuronal culture as a model for ASD and SCZ genotypes. While this model system often reproduces qualitative effects of haploinsufficiency or partial loss of function (PLOF), they may be fundamentally different from, for example, complete knockout or gain-of-function models, some of which have observed different synaptic phenotypes92,93. Even for the former, gene dosage differences may produce qualitatively different results, with our treatments variably reducing mRNA levels by 40–80%, which may be different from the gene dosage produced by heterozygous mutants or PLOF. As such, this study should be treated as a starting point to investigate convergent gene-proteome connections rather than a definitive description of protein changes in the disease itself.
Another potential limitation is the exclusive use of immunofluorescence for protein quantification, which offers the possibility of artifacts due to variability in epitope accessibility as well as non-specific binding that may confound results. While we have performed extensive validations in this and previous studies to establish the quantitative validity of PRISM, the possibility of such artifacts remains. Finally, the network inference approach presented is based on one imaging dataset and may additionally contain artifactual edges and substructures, despite care to account for possible statistical artifacts. Future work that incorporates additional protein nodes and a larger variety of cellular conditions will help to further establish the biological validity of the present work.
STAR METHODS
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mark Bathe (mbathe@mit.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Single-synapse measurements as processing output of CellProfiler applied on imaging data have been deposited to Mendeley Data as CSV files under the paper title.
Original code for data analysis, network simulation, and network inference, as well as original CellProfiler pipelines, have been deposited to github.com/lcbb/PRISM-CellProfiler-Analysis-Pipelines.
Raw and processed imaging data is locally stored due excessive file sizes and will be delivered upon request.
Experimental model and subject details
All experiments were performed on dissociated hippocampal neurons of embryonic day 18 Sprague-Dawley rats. Each experiment included neuronal cultures of pooled neurons from 1–3 embryos of a single pregnant rat, and each experiment used a separate pregnant rat. Embryos were not sexed prior to dissection and culturing.
Culturing conditions (for detailed conditions see method details section): Embryo hippocampi were dissected in 4°C Hibernate E supplemented with B27. Hippocampal tissues were digested in Hibernate E using papain at 37°C. Neurons were plated at 15,000 cells/well in NbActiv1 supplemented with 25mM glutamate on poly-d-lysine coated 96-well-plates, treated at DIV 2 with AraC and at DIV 6 with the siRNA/chemical treatment and left to develop in NbActiv4 until fixation and staining at DIV 19.
Procedures for rat neuronal culture were reviewed and approved for use by the Broad Institutional Animal Care and Use Committee, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Method details
Antibody details:
Table S1 details the antibodies used for multiplexed imaging, including the conjugation strategy used for each. The Key Resources Table provides additional details, and information on all other antibodies used for follow-up validation experiments.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-MAP2, chicken polyclonal | Novus Biologicals | Cat#NB300–213 |
| anti-PSD95, rabbit monoclonal | Cell Signaling Technology | Cat#3450 Clone: D27E11 |
| anti-Gephyrin, rat monoclonal IgG1 | Synaptic Systems | Cat#147208 Clone: rtmAb7a |
| anti-GluR2, N-terminus, guinea pig polyclonal | Synaptic Systems | Cat#185105 |
| anti-NR2A, mouse monoclonal IgG2a | Neuromab | Cat#75–288 Clone: N327/95 |
| anti-Shank3, mouse monoclonal IgG1 | Synaptic Systems | Cat#162311 Clone: 144b12 |
| anti-vGAT, mouse monoclonal IgG3 | Synaptic Systems | Cat#131011 Clone: 117G4 |
| anti-Synapsin1, mouse monoclonal IgG1 | Synaptic Systems | Cat#106011 Clone: 46.1 |
| anti-Homer1, mouse monoclonal IgG1 | Synaptic Systems | Cat#160011 Clone: 2G8 |
| anti-vGluT1, camelid sdAb | Synaptic Systems | Cat#N1602 |
| anti-mGluR5, rabbit polyclonal | Sigma Aldrich | Cat#AB5675 |
| anti-Shank1/2/3, mouse monoclonal IgG2a | Synaptic Systems | Cat#162111 Clone: 151E3 |
| anti-NR1, mouse monoclonal IgG2a | Millipore | Cat#MAB363 Clone: 54.1 |
| anti-GluR2, C-terminus, mouse monoclonal IgG2a | Millipore | Cat#MAB397 Clone: 6C4 |
| anti-Synapsin1, goat polyclonal | Santa Cruz biotechnologies | Cat#sc-7379 |
| anti-Rabbit, goat polyclonal | Invitrogen | Cat#A16126 |
| anti-Mouse IgG1, goat polyclonal | Abcam | Cat#ab98689 |
| anti-Mouse IgG2, goat polyclonal | Novus Biologicals | Cat#NB7513 |
| anti-Guinea Pig, goat polyclonal | Invitrogen | Cat#A18777 |
| anti-Rat, goat polyclonal | Invitrogen | Cat#A18873 |
| anti-Goat 405, donkey polyclonal | Abcam | ab175665 |
| anti-Goat 488, donkey polyclonal | Invitrogen | A-11055 |
| anti-Chicken 405, goat polyclonal | Abcam | ab175674 |
| anti-Chicken 488, goat polyclonal | Invitrogen | A-11039 |
| anti-Mouse 488, goat polyclonal | Abcam | ab150113 |
| anti-Mouse 568, donkey polyclonal | Invitrogen | A10037 |
| anti-Mouse 647, donkey polyclonal | Abcam | ab150107 |
| anti-Rabbit 488, goat polyclonal | Abcam | ab150077 |
| anti-Rabbit 568, donkey polyclonal | Abcam | ab175470 |
| anti-Rabbit 647, donkey polyclonal | Invitrogen | A-31573 |
| Biological Samples | ||
| E18 Rat Hippocampal Neurons | Privately sourced at the Broad Institute | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4-(N-Maleimidomethyl)cyclohexane-1-carboxylic acid N-hydroxysuccinimide (SMCC) | Sigma Aldrich | Cat#573115 CAS 92921-24-9 |
| Phalloidin 7-Ornithine | Bachem | Cat#H7643 |
| Harmine | Tocris | Cat#5075 CAS 442-51-3 |
| bpV(pic) | Sigma Aldrich | Cat#SML0885 CAS 148556-27-8 |
| D-AP5 | Alomone Labs | Cat#D-145 CAS 79055-68-8 |
| RNase A | Invitrogen | Cat#EN0531 |
| RNase T1 | Invitrogen | Cat#EN0542 |
| Hibernate E | Thermo Fisher | Cat#A1247601 |
| NbActiv1 | Brainbits LLC (now Transnetyx) | Cat#NB1 |
| NbActiv4 | Brainbits LLC (now Transnetyx) | Cat#NB4 |
| B27 Supplement | Thermo Fisher | Cat#17504044 |
| Arabinofuranosyl Cytidine (AraC) | Sigma Aldrich | Cat#C6645 CAS 69-74-9 |
| Atto 565 NHS | Atto-Tec GmbH | Cat#AD 565–31 |
| Atto 655 NHS | Atto-Tec GmbH | Cat#AD 655–31 |
| Critical Commercial Assays | ||
| LightCycler 480 Probes Master | Roche | Cat#04707494001 |
| TaqManTM Gene Expression Assays | Life Technologies | See table S3 |
| SiteClick Azido Antibody Modification Kit | Thermo Fisher | Cat#S10900 |
| Experimental Models: Organisms/Strains | ||
| Rattus Norvegicus, Sprague-Dawley, wild-type | Broad Institute | |
| Oligonucleotides | ||
| Thiol-modified ssDNA docking strands | Integrated DNA Techologies | See table S2 |
| DBCO-modified ssDNA docking strands | Qiagen | See table S2 |
| Amine-modified LNA imaging strands | Qiagen | See table S2 |
| Software and Algorithms | ||
| CellProfiler version 3.0 | McQuin, C. et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 16, (2018). | |
| R package: umap, version 0.2.7.0 | Tomasz Konopka. umap: Uniform Manifold Approximation and Projection. R package version 0.2.7.0 https://CRAN.R-project.org/package=umap. (2020). | |
| R package: bnlearn, version 4.7 | Scutari, M. Learning Bayesian networks with the bnlearn R Package. J. Stat. Softw. 35, 1–22 (2010). | |
| CellProfiler Synapse Analysis Pipeline | This study | |
| R markdown document: Workup and analysis of PRISM data | This study | |
| R markdown document: Network simulation and inference | This study | |
Antibody-docking strand conjugation – SMCC:
Most antibodies were conjugated using 4-(N-Maleimidomethyl)cyclohexane-1-carboxylic acid N-hydroxysuccinimide (SMCC), a heterobifunctional molecule which binds exposed amines on the antibody via an N-hydroxysuccinimide (NHS) moiety on one end and binds thiol-oligonucleotides via a maleimide on the other end. Antibodies were purchased as formulations without serum proteins as listed below (Table S1), 0.1–1 mg of antibody were purified into PBS using Zeba spin columns (7 kDa, Thermo Fisher Scientific). Subsequently, antibodies were concentrated to 1 mg/ml using Amicon Ultra centrifugal filters (100 kDa, 4000 g, EMD Millipore). The initial concentration of anti-vGAT was 2 mg/ml. From a freshly prepared stock at 2 mM in DMF, SMCC (Sigma Aldrich) was added to the antibody at 7.5x molar excess. The reaction mixture was protected from light and incubated for 3 h at 4°C on a shaker. Excess SMCC was removed by purification into PBS using Zeba spin columns (7 kDa, Thermo Fisher Scientific). In parallel, 25 nmol 5’ thiol-modified ssDNA (Integrated DNA Technologies, modification catalog no. /5-ThioMC6-D/) was dissolved in 25 ul water and 55 ul PBS with 2 mM EDTA at pH 8.0 (Table S2). After the addition of 20 ul of a freshly prepared stock of 500 mM DTT in PBS with 2 mM EDTA at pH 8.0, the reaction mixture was protected from light and incubated for 2 h at 25°C on shaker. The reduced 5’ thiol-modified ssDNA was purified into water using NAP-5 columns (GE Life Sciences). Fractions containing ssDNA were identified using absorbance measurements at 260 nm and DTT was monitored calorimetrically using bicinchoninic acid. The reduced 5’ thiol-modified ssDNA was immediately added to the antibody-SMCC conjugate at 15x molar excess, the reaction mixture was protected from light and incubated overnight at 4°C on a shaker. Antibody-ssDNA conjugates were purified into PBS using Amicon Ultra centrifugal filters (50 kDa, 4000 g, EMD Millipore). Amino-modified phalloidin (Bachem) was conjugated using the procedure described above, but with the following changes: the molar excess of SMCC was 10x and the molar excess of reduced 5’ thiol-modified ssDNA was 1x. HPLC purification was employed to remove unreacted SMCC and 5’ thiol-modified ssDNA, respectively (Waters, BEH C18 column, gradient for phalloidin-SMCC: from 80% TFA in water and 20% acetonitrile to 20% TFA in water and 80% acetonitrile over 10 min, gradient for phalloidin-ssDNA: from 90% 0.1 M TEAA in water and 10% acetonitrile to 60% 0.1 M TEAA in water and 40% acetonitrile over 10 min). Antibody concentration were determined by absorbance measurements at 280 nm. Conjugation efficiency was estimated by MALDI-TOF mass spectrometry and ranged from 1 to 3, depending on the antibody. Antibody-ssDNA conjugates were stored at −20 C in PBS with 50% glycerol.
Antibody-docking strand conjugation - SiteClick:
For Homer1, ssDNA-antibody conjugates were synthesized using the SiteClick™ (Invitrogen) conjugation technique following the manufacturer’s protocol. This technique replaces the Fc galactoses on the antibody with azide-modified sugars, which then react with a DBCO-modified oligonucleotide. 200 μg of the anti-Homer 1 was concentrated to 2 mg/ml in 1x Tris buffer and incubated with β-galactosidase. Azide-modified, terminal galactosides were attached using β-galactosyltransferase. Azide-modified antibody was purified into 1x Tris buffer using Amicon Ultra centrifugal filters (50 kDa, 4000 g, EMD Millipore). 5’ DBCO-modified ssDNA (Integrated DNA Technologies, modification catalog no. /5-DBCON/) was dissolved in water, added to azide-modified antibody at a molar excess of 30 and incubated overnight at 25°C (Table S2). Antibody-ssDNA conjugates were purified into PBS using Amicon Ultra centrifugal filters (50 kDa, 4000 g, EMD Millipore). Antibody concentrations were determined by absorbance measurements at 280 nm. Conjugation efficiency was estimated by MALDI-TOF mass spectrometry and ranged from 1 to 2, depending on the antibody batch. Antibody-ssDNA conjugates were stored at −20°C in PBS with 50% glycerol.
Imager strands:
25 nmol of 5’/3’ diamino-modified ssLNA (Qiagen) was dissolved in 500 ul PBS with 10% DMSO at pH 8.3 and 250 nmol of NHS-Atto 565 or NHS-Atto 655 (Sigma Aldrich) were added from a 15 mM stock in DMSO (Table S2). Following immediate vortexing, the reaction mixture was protected from light and incubated overnight at 25°C on a shaker. Excess dye was removed using NAP-5 columns (GE Life Sciences). Fractions containing ssDNA were identified using absorbance measurements at 260 nm. Subsequently, 0.1 M TEAA was added ssLNA-dye conjugates and conjugates bearing two dyes were purified by HPLC (Waters, BEH C18 column, gradient for Atto 565: from 80% 0.1 M TEAA in water and 20% acetonitrile to 70% 0.1 M TEAA and 30% acetonitrile over 10 min, gradient for Atto 655: from 90% 0.1 M TEAA in water and 10% acetonitrile to 75% 0.1 M TEAA in water and 25% acetonitrile over 10 min). Peaks corresponding to ssLNA conjugates bearing two, one or no dye were assigned based on absorbance spectra. Solvents were removed in vacuo and ssLNA-dye conjugates were dissolved in water at 10 to 100 μM, depending on the yield. Yields were determined by absorbance measurements using 565 nm or 655 nm wavelengths.
Neuronal culture and treatment:
Procedures for rat neuronal culture were reviewed and approved for use by the Broad Institutional Animal Care and Use Committee, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. In each of N=4 biological repeats, 1–2 Embryonic Day 18 embryos were collected from a separate pregnant Sprague Dawley rat killed by CO2 (Taconic). Embryo hippocampi were dissected in 4°C Hibernate E supplemented with 2% B27 supplements and 100 U/ml penicillin/strep (Thermo Fisher Scientific). Hippocampal tissues were digested in Hibernate E containing 20 U/ml papain, 1 mm L-cysteine, 0.5 mm EDTA (Worthington Biochem), and 0.01% DNase (Sigma-Aldrich) for 8 min. Neurons were centrifugated at 1000rpm by 5min, pellet with cells were then resuspended into NbActiv1 (BrainBits LLC, now TransnetYX) supplemented with 25mM glutamate, and plated at a density of 15,000 cells/well onto poly-d-lysine-coated, black-walled, thin-bottomed 96-well plates (Corning BioCoat). After 48 hours, AraC was added to each culture at a concentration of 1uM, to suppress glia proliferation and minimize well-to-well variability resulting from it. At DIV 5, the media was entirely replaced with warm NbActiv4. At DIV 6, each culture was treated with Accell SMARTpool (Dharmacon/Horizon from Perkin Elmer), a mix of four chemically modified self-transfecting siRNAs, against the relevant gene (Table S3) to a total siRNA concentration of 1uM in NbActiv4. Cultures were then left undisturbed until fixation on DIV 21. Each plate included 60 wells/separate cultures, 3–4 in each treatment group. Across 4 plates, one for each biological repeat, this results in a total of n=11–18 technical repeats in each treatment group. For validation experiments, cultures were treated at DIV 6 with 0.1uM/0.5uM bpV(pic), 0.2uM/2uM Harmine, 20uM/50uM D-AP5, and Nontargeting, Shank3 or Grin2a Accell SMARTpool siRNA, and left undisturbed until fixation on DIV 8 or 19 as described.
RTqPCR knockdown validation:
RTqPCR was performed using Fast Advanced Cells-to-CT kit (Ambion) according to the manufacturer’s protocol. In short, cells were prepared for lysis by washing them with cold PBS 1x, then Stop solution was added following lysis buffer with DNAse I. RT Master Mix using Cells-to-Ct lysate was prepared and reverse transcription was done on a thermal cycler. Lastly, qPCR was done using LightCycler® 480 Probes Master (Roche) with TaqManTM Gene Expression Assays designed for each target (see Table S3 for catalog numbers) and performed on a LightCycler® 480 Instrument. Two TaqManTM Gene Expression Assays (Life Technologies). Actb was used as a reference gene to normalize the results (Life Technologies). For relative quantification of gene expression, the 2− ΔΔCt method was used.
Staining and Imaging:
Cells were fixed and stained as described previously7,57,58. Cells were fixed in fixation solution (4% paraformaldehyde and 4% sucrose in PBS) for 20min at RT, then permeabilized with 0.25% Triton X-100 in PBS for 10. They were then incubated in a mixture of RNases A and T1 to reduce the fluorescent background caused by ssLNA-RNA binding and blocked with 5% Bovine Serum Albumin (BSA). The first round of primary staining was performed using unconjugated primary antibodies (table S1 rows 1–6) diluted in the regular blocking buffer. Cells were blocked with nuclear blocking buffer [5% BSA and 1 mg/ml salmon sperm DNA (Sigma-Aldrich) in PBS] and then incubated with conjugated secondary antibodies (table S1 rows 7–11) diluted in the nuclear blocking buffer. After post-fixation, cells were stained in the third round with conjugated primary antibodies (table S1 rows 12–16) in nuclear blocking buffer and then with DAPI.
High-throughput spinning disk confocal LNA-PRISM imaging was performed using the Opera Phenix High-Content Screening System (PerkinElmer) as described before7,57,66 with the following main changes: first, two colors were used for PRISM in each round, and second, probe introduction, wash and exchange was performed automatically using a Bravo automated liquid handling system. In each round, a pair of imaging probes in two colors (see table S2 for sequences) was freshly diluted to 10 nM in imaging buffer (500 mm NaCl in PBS, pH 8) immediately before imaging. Neurons were incubated with imaging probes for 5 min and then washed twice with imaging buffer to remove unbound probe. The plates were then imaged in 4 wavelengths: 405nm (DAPI), 488nm (MAP2), 561nm (orange probe) and 647nm (red probe). For each field of view, a stack of five images was acquired with an axial step-size of 1 μm. Either four (in one plate) or nine (in the other three plates) lateral fields of view were imaged in each culture. Following each round of imaging, cells were washed two times with wash buffer (0.01 × PBS) for 3 min per round, and then re-imaged to ensure that all PRISM fluorescent signal was removed before introducing the next probe pair. After all imaging rounds, neurons were stained with a 568nm fluorescent nanobody against vGlut1 (table S1 row 17) for 1 hour and imaged again. Although the combined staining with multiple antibodies simultaneously may reduce primary PRISM signal due to crowding, we observed this reduction to be less than 15%, and should be identical across all treatment groups.
For staining externalized GluR2 (figure S2), an N-terminal-specific Guinea Pig anti-GluR2 antibody (see table S1) was used before permeabilization, and a C-terminal-specific Mouse anti-GluR2 antibody was used after permeabilization, along with a labeled anti-vGluT1 nanobody and a chicken anti-MAP2 antibody. For heat-induced antigen retrieval, wells were incubated after permeabilization with pre-heated antigen retrieval buffer (10mM Citric Acid, 0.05% Tween 20, pH 6.0) at 95°C for 30min, and then stained as usually. We also attempted proteolysis-induced epitope retrieval (0.05% Trypsin, 20min at 37°C) which completely abolished Synapsin1 and F-actin signals.
Automated image analysis using CellProfiler:
CellProfiler was used to automatically correct images for uneven illumination, align images across channels, and segment and quantify synapses in images. This tool allows for modular construction of pipelines for image analysis66,67. The pipeline used here is similar to a previous study57 and is available on github.com/lcbb/PRISM-CellProfiler-Analysis-Pipelines. The main steps in the image analysis pipeline are as follows:
By-pixel maximum projections of confocal Z-stacks of all images in each round are calculated separately and loaded into CellProfiler.
MAP2 (488nm) images in each round are used to align images of other channels between rounds.
An illumination profile correction is applied to all images based on background averages across all wells.
For each round and wavelength, the average intensity in untreated wells of a plate is calculated and used to normalize the images in all other wells. This is used to account for between-plate differences in exogenous brightness (staining strength, laser strength, exposure time etc.)
The DAPI image is used to identify nuclei objects. All other images of the same field are then masked by the nuclei to prevent artifacts from non-specific nuclear localization of the antibodies.
The MAP2 image is used to identify dendrite objects.
A white top hat filter with a radius of 4px is applied to all synaptic protein images across all rounds to enhance puncta.
For synapse counting analysis (figure 2B–D), synaptic objects were segmented and identified in images of each channel by applying the RobustBackground tool, which calculates an optimal threshold value for each window individually based on the intensity histogram. For all other analyses, we calculated a per-channel global threshold from the average threshold calculated by RobustBackground across all imaged fields in untreated wells. We then applied this value as a uniform threshold to all images of that channel to ensure that all images are segmented identically.
Synapsin1 puncta are then masked using the dendrites previously identified, to retain only puncta which are within 12px of a dendrite. These are then defined as synapses.
Puncta in all other channels are assigned to synapses if they overlapped with Synapsin1 puncta more than 6.25% (for postsynaptic proteins) or more than 50% (for presynaptic proteins).
Finally, levels of each protein per synapse are calculated as the intensity integral of that protein’s image across its punctum. If a certain protein did not have an identified puncta associated with a synapse, its level was marked as 0.
Synapses were identified as excitatory if they contained only vGlut, inhibitory if they contained only vGAT, and otherwise excluded from further analysis (positive or negative for both vGlut and vGAT). Excluded synapses were 20–30% of all identified synapses. We also performed the same analysis with uniform threshold values of 75% and 133% of the calculated average, which yielded more and less synaptic puncta, respectively, but similar observations in treatment effects, clusters, Bayesian networks and edge strengths. In controls for network inference by alternative synapse identification, instead of Synapsin1 puncta for synapse definition and assignment of all other proteins, we used postsynaptic puncta defined by merging of F-actin, PSD95 and Shank3 puncta.
Network simulation:
Data points for simulated networks were sampled from a modified gamma distribution, which contains a separate probability for a value of 0 (see equation 1). To simulate dependency of one variable on others, the distribution from which the child variable is sampled has p0 and scale parameter θ modified based on the values of the parent variables according to equation 2.
| (1) |
| (2) |
Where X, A, B, C are variable names, is the probability for node X to be 0, is the gamma distribution with shape parameter k and scale parameter θ, and are the values of B and C for a specific datapoint relative to their median, and αAB and αAC are the predefined interaction coefficients of A with B and C, respectively (edge strengths in the B → A ← C subgraph).
20 such networks with 6–12 nodes were generated, sampled, and reconstructed to determine the optimal Bayesian network inference algorithm. To simulate a network with a cyclic dependence, e.g. A→B→C→A, A is separated into two variables A1 and A2 to create a directed acyclic graph which is sampled as above, and the final value of A is A1 + A2.
Bayesian network analysis and controlled edge calculation:
Bayesian network inference was performed on a combined random sample of synapses from each well, while limiting only to excitatory-labeled synapses and 8 excitatory synaptic proteins. Measurements in each protein were discretized into 51 bins in the following way: all measurements of 0 (no puncta of that protein associated with the synapse) were assigned to bin 0, and bins 1–50 were assigned by equal-frequency discretization.
The discretized dataset was then sampled for 3000 points which were used to construct a Bayesian network using the likelihood-score-maximizing ‘tabu’ algorithm76. 50 such samplings and rederivations of the network were used to establish confidence in the presence and direction of edges. Network derivation was done using the tabu and boot.strength functions in the R package bnlearn76, A similar procedure was applied to simulated datasets, data from a previous synaptic scaling study57, and adversarially modified data sets.
Given a network, we define the strength of an edge between two nodes as the average correlation of the two variables across strata where the other parents of the daughter node are held constant79,80. That is, the strength of an edge from A to B, where B also has edges leading to it from n other variables, for example C and D with n=2, as the correlation between A and B when controlling for C and D. To estimate that, we repeated the following algorithm to calculate average correlations between A and B across strata of equal C and D:
Sample a point (A0, B0, C0, D0)
Find set of all points (A, B, C, D) such that where n is the number of variables to control for (2 in this example) and ε is a predetermined tolerance level set at 0.5 (smaller tolerances did not yield significantly different measures)
If the set contains more than 5 points, calculate Pearson’s correlation coefficient cor(A, B) across that set.
Average the resulting correlation measure across 20 · 2n such samplings.
A similar stratification procedure was done to assess the conditional effect of a certain treatment on protein A when controlling for proteins B and C. The treatment and NonT groups were pooled together, a point was sampled at random and a set of all points with similar B and C was found, and the log2-fold difference between the mean levels of A in treated vs NonT synapses was calculated and averaged across many samplings.
Quantification and statistical analysis
Software:
All statistical analysis was performed in R (versions 4.0.0–4.3.0), using the base, stats, umap (version 0.2.7.0), and bnlearn (version 4.7) packages.
Biological and technical repeats:
Each experiment includes pooled neurons from 1–3 embryos of one pregnant rat, split into wells of a 96-well-plate. The wells constitute separately grown, treated and stained cultures. Each well is subsequently considered a technical repeat. All images in different wells from a single experiment are analyzed using the same CellProfiler pipeline for consistency in synapse determination and quantification. The main RNAi screen pools 4 experiments into one dataset by dividing all synaptic protein values of a certain experiment by the average values for untreated wells in that experiment.
Nature of repeats and values per repeat:
For statistical analysis, relevant values are calculated for synapses in individual wells: (i) Average protein intensity integral over synaptic puncta. (ii) Total number of synapses or synapses conforming to a certain condition, (iii) Fraction of synapses conforming to a certain condition, (iv) Total area of identified dendrites, (v) Average protein intensity over whole image, (vi) Average protein intensity integral over soma, (vii) Network edge strengths (i.e., controlled correlation between nodes). By-well values are then used for calculating standard error and significance testing. Significance testing was done using a two-tailed Student’s T-test. The number of wells per treatment group is 11–18 for the main RNAi screen and 4–7 for each validation experiment. All heatmaps and bar graphs present average values across wells, all error bars are standard error of mean across wells.
Data exclusion:
The following subsets of images from the main RNAi screen were excluded from subsequent analysis:
One row in plate #1, for which one of the imaging rounds was out of focus.
All images of NR2A, for which staining was very diffuse and very few puncta could be identified.
All images of cultures treated with siRNA against Xpo7, which exhibited highly irregular staining patterns for Homer1 that could not be reproduced with other batches of that siRNA. We attributed the effect to an issue with the specific siRNA batch used.
UMAP and clustering:
Uniform Manifold Approximation and Projection (UMAP) was performed using the umap R package (version 0.2.7.0). A combined sample was used with 200 points randomly sampled from each well. Each variable was scaled to a standard deviation of 1, and UMAP was applied with min_dist=0. Density-based clustering on the 2D layout was done automatically using HDBSCAN94,95 with parameters that yielded 14 clusters, after which small clusters that accounted for <1% of all points were manually merged into the nearest (by centroid distance) large cluster, resulting in the 9 main clusters shown in figure 3.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIMH R01-MH112694, NIMH R21-MH130624, and NSF-PoLS 1707999. The work was also supported by the Center for Discovery of Therapeutics at the Broad Institute and the use of the PerkinElmer Opera Phenix High-Content/High-Throughput imaging system funded by the S10 Grant NIH OD-026839-01, as well as by MIT CEHS core center grant P30-ES002109 from the National Institute of Environmental Health Science. J.R.C. and K.p.D.A were supported by the Stanley Center for Psychiatric Research. The authors would also like to thank Agnes Walsh for assistance in preparing DNA-conjugated antibodies and Douglas Lauffenburger for advice on Bayesian Network inference.
REFERENCES
- 1.Grant SGN Synapse signalling complexes and networks: Machines underlying cognition. BioEssays 25, 1229–1235 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Grant SGN The synapse proteome and phosphoproteome: A new paradigm for synapse biology. Biochem. Soc. Trans 34, 59–63 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Husi H, Ward MA, Choudhary JS, Blackstock WP & Grant SGN Proteomic analysis of NMDA receptor – adhesion protein signaling complexes. Nat. Neurosci 3, 661–669 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Bayés Á et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat.Neurosci 14, 19–21 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu F et al. Architecture of the Mouse Brain Synaptome. Neuron 99, 781–799.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campagnola L et al. Local connectivity and synaptic dynamics in mouse and human neocortex. Science 375, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo SM et al. Multiplexed and high-throughput neuronal fluorescence imaging with diffusible probes. Nat. Commun 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casarotto PC et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 184, 1299–1313.e19 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fred SM et al. Pharmacologically diverse antidepressants facilitate TRKB receptor activation by disrupting its interaction with the endocytic adaptor complex AP-2. J. Biol. Chem 294, 18150–18161 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomasetti C et al. Treating the synapse in major psychiatric disorders: The role of postsynaptic density network in dopamine-glutamate interplay and psychopharmacologic drugs molecular actions. Int. J. Mol. Sci 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fromer M et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lima Caldeira G, Peça J & Carvalho AL New insights on synaptic dysfunction in neuropsychiatric disorders. Curr. Opin. Neurobiol 57, 62–70 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Guang S et al. Synaptopathology involved in autism spectrum disorder. Front. Cell. Neurosci 12, 1–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson J et al. Targeting the synapse in Alzheimer’s disease. Front. Neurosci 13, 1–8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duman RS & Li N A neurotrophic hypothesis of depression : role of synaptogenesis in the actions of NMDA receptor antagonists. Philos. Trans. R. Soc. B Biol. Sci 367, 2475–2484 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duman RS, Aghajanian GK, Sanacora G & Krystal JH Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med 22, 238–249 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peca J & Feng G Cellular and synaptic network defects in autism. Curr. Opin. Neurobiol 22, 866–872 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prizant BM Language acquisition and communicative behavior in autism: Toward an understanding of the ‘whole’ of it. J. Speech Hear. Disord 48, 296–307 (1983). [DOI] [PubMed] [Google Scholar]
- 19.Schreibman L Diagnostic Features of Autism. J. Child Neurol 3, s57–s64 (1988). [DOI] [PubMed] [Google Scholar]
- 20.Geyer MA, Braff DL & Geyer MA Startle Habituation and Sensorimotor Gating in Schizophrenia and Related Animal Models. Schizophr. Bull 13, 643–668 (1987). [DOI] [PubMed] [Google Scholar]
- 21.Green SA et al. Neurobiology of sensory overresponsivity in youth with autism spectrum disorders. JAMA Psychiatry 72, 778–786 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Javitt DC & Freedman R Sensory processing dysfunction in the personal experience and neuronal machinery of schizophrenia. Am. J. Psychiatry 172, 17–31 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eigsti IM, De Marchena AB, Schuh JM & Kelley E Language acquisition in autism spectrum disorders: A developmental review. Res. Autism Spectr. Disord 5, 681–691 (2011). [Google Scholar]
- 24.Matson JL & Shoemaker M Intellectual disability and its relationship to autism spectrum disorders. Res. Dev. Disabil 30, 1107–1114 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Morgan VA, Leonard H, Bourke J & Jablensky A Intellectual disability co-occurring with schizophrenia and other psychiatric illness: Population-based study. Br. J. Psychiatry 193, 364–372 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Yung AR et al. Prediction of psychosis - A step towards indicated prevention of schizophrenia. Br. J. PSYCHIATRY 14–20 (1998). [PubMed] [Google Scholar]
- 27.Carroll LS & Owen MJ Genetic overlap between autism schizophrenia and bipolar disorder. Genome Med. (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Lacy N & King BH Revisiting the relationship between autism and schizophrenia: Toward an integrated neurobiology. Annu. Rev. Clin. Psychol 9, 555–587 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Li Z, Fan C, Zhang D & Chen J Genetics implicate common mechanisms in autism and schizophrenia: Synaptic activity and immunity. J. Med. Genet 54, 511–520 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Hilker R et al. Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register. Biol. Psychiatry 83, 492–498 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Tick B, Bolton P, Happé F, Rutter M & Rijsdijk F Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry 57, 585–595 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fanous AH & Kendler KS Genetic heterogeneity, modifier genes, and quantitative phenotypes in psychiatric illness: Searching for a framework. Mol. Psychiatry 10, 6–13 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Willsey JJ & State MW Autism spectrum disorders: From genes to neurobiology. Curr. Opin. Neurobiol 30, 92–99 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.An JY & Claudianos C Genetic heterogeneity in autism: From single gene to a pathway perspective. Neurosci. Biobehav. Rev 68, 442–453 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Singh T et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 604, 509–516 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bourgeron T A synaptic trek to autism. Curr. Opin. Neurobiol 19, 231–234 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Yu S, Fu Y & Li X Synaptic proteins and receptors defects in autism spectrum disorders. Front. Cell. Neurosci 8, 1–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zoghbi HY & Bear MF Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol 4, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darnell JC et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spratt PWE et al. The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron 103, 673–685.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ellingford RA et al. Cell-type-specific synaptic imbalance and disrupted homeostatic plasticity in cortical circuits of ASD-associated Chd8 haploinsufficient mice. Mol. Psychiatry (2021) doi: 10.1038/s41380-021-01070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De La Torre-Ubieta L, Won H, Stein JL & Geschwind DH Advancing the understanding of autism disease mechanisms through genetics. Nat. Med 22, 345–361 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iossifov I et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gulsuner S et al. XSpatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanders SJ et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kirov G et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 17, 142–153 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Penzes P, Cahill ME, Jones KA, Vanleeuwen JE & Woolfrey KM Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci 14, 285–293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antoine MW, Langberg T, Schnepel P & Feldman DE Increased Excitation-Inhibition Ratio Stabilizes Synapse and Circuit Excitability in Four Autism Mouse Models. Neuron 101, 648–661.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Souchet B et al. Excitation/inhibition balance and learning are modified by Dyrk1a gene dosage. Neurobiol. Dis 69, 65–75 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Canitano R & Pallagrosi M Autism spectrum disorders and schizophrenia spectrum disorders: Excitation/inhibition imbalance and developmental trajectories. Front. Psychiatry 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foss-Feig JH et al. Searching for Cross-Diagnostic Convergence: Neural Mechanisms Governing Excitation and Inhibition Balance in Schizophrenia and Autism Spectrum Disorders. Biol. Psychiatry 81, 848–861 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sakai Y et al. Protein interactome reveals converging molecular pathways among autism disorders. Sci. Transl. Med 3, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown EA et al. Clustering the autisms using glutamate synapse protein interaction networks from cortical and hippocampal tissue of seven mouse models. Mol. Autism 9, 48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moreau CA et al. Mutations associated with neuropsychiatric conditions delineate functional brain connectivity dimensions contributing to autism and schizophrenia. Nat. Commun 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Menon V Large-scale brain networks and psychopathology: A unifying triple network model. Trends Cogn. Sci 15, 483–506 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Sinha P et al. Autism as a disorder of prediction. Proc. Natl. Acad. Sci. U. S. A 111, 15220–15225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Danielson E et al. Molecular Diversity of Glutamatergic and GABAergic Synapses from Multiplexed Fluorescence Imaging. eNeuro 8, 1–18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tomov ML et al. Resolving cell state in iPSC-derived human neural samples with multiplexed fluorescence imaging. Commun. Biol 4, 1–9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sachs K, Perez O, Pe’er D, Lauffenburger DA & Nolan GP Causal protein-signaling networks derived from multiparameter single-cell data. Science 308, 523–529 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Gundelfinger ED, Reissner C & Garner CC Role of Bassoon and Piccolo in Assembly and Molecular Organization of the Active Zone. Front. Synaptic Neurosci 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Derkach VA, Oh MC, Guire ES & Soderling TR Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci 8, 101–113 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Cingolani LA & Goda Y Actin in action: The interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci 9, 344–356 (2008). [DOI] [PubMed] [Google Scholar]
- 63.Gentile JE, Carrizales MG & Koleske AJ Control of Synapse Structure and Function by Actin and Its Regulators. Cells 11, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Banerjee-Basu S & Packer A SFARI Gene: an evolving database for the autism research community. DMM Dis. Models Mech 3, 131–133 (2010). [DOI] [PubMed] [Google Scholar]
- 65.Abrahams BS et al. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 4, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lamprecht MR, Sabatini DM & Carpenter AE CellProfiler: free, versatile software for automated biological image analysis Short Technical Reports. BioTechniques 42, 71–75 (2007). [DOI] [PubMed] [Google Scholar]
- 67.McQuin C et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 16, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kerchner GA & Nicoll RA Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat. Rev. Neurosci 9, 813–825 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ang XL, Seeburg DP, Sheng M & Harper JW Regulation of Postsynaptic RapGAP SPAR by Polo-like Kinase 2 and the SCFβ-TRCP Ubiquitin Ligase in Hippocampal Neurons. J. Biol. Chem 283, 29424–29432 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Garcia-Junco-Clemente P & Golshani P PTEN: A master regulator of neuronal structure, function, and plasticity. Commun. Integr. Biol 7, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Verpelli C et al. Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. J. Biol. Chem 286, 34839–34850 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rao A & Craig AM Activity Regulates the Synaptic Localization of the NMDA Receptor in Hippocampal Neurons. Neuron vol. 19 801–812 (1997). [DOI] [PubMed] [Google Scholar]
- 73.Konopka Tomasz. umap: Uniform Manifold Approximation and Projection. R package version 0.2.7.0 https://CRAN.R-project.org/package=umap. (2020).
- 74.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. (2021). [Google Scholar]
- 75.Tyagarajan SK & Fritschy JM Gephyrin: A master regulator of neuronal function? Nat. Rev. Neurosci 15, 141–156 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Scutari M Learning Bayesian networks with the bnlearn R Package. J. Stat. Softw 35, 1–22 (2010).21603108 [Google Scholar]
- 77.Yu J, Smith VA & Wang PP Advances to Bayesian network inference for generating causal networks from observational biological data. Bioinformatics 20, 3594–3603 (2004). [DOI] [PubMed] [Google Scholar]
- 78.Wilkinson DJ Bayesian methods in bioinformatics and computational systems biology. Brief. Bioinform 8, (2007). [DOI] [PubMed] [Google Scholar]
- 79.Pe’er D Bayesian Network Analysis of Signaling Networks: A Primer. Sci. Signal (2005). [DOI] [PubMed] [Google Scholar]
- 80.Ebert-Uphoff I Measuring Connection Strengths and Link Strengths in Discrete Bayesian Networks. Ga. Tech Res. Rep (2007). [Google Scholar]
- 81.Chen X et al. PSD-95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc. Natl. Acad. Sci. U. S. A 112, E6983–E6992 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Van Bon BWM et al. Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol. Psychiatry 21, 126–132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Earl RK et al. Clinical phenotype of ASD-associated DYRK1A haploinsufficiency. Mol. Autism 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dang T et al. Autism-associated Dyrk1a truncation mutants impair neuronal dendritic and spine growth and interfere with postnatal cortical development. Mol. Psychiatry 23, 747–758 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Prokop A, Landgraf M, Rushton E & Broadie K Presynaptic Development at the Drosophila Neuromuscular Junction: Assembly and Localization of Presynaptic Active Zones. Neuron vol. 17 617–626 (1996). [DOI] [PubMed] [Google Scholar]
- 86.Owald D & Sigrist SJ Assembling the presynaptic active zone. Curr. Opin. Neurobiol 19, 311–318 (2009). [DOI] [PubMed] [Google Scholar]
- 87.Vessey JP & Karra D More than just synaptic building blocks: Scaffolding proteins of the post-synaptic density regulate dendritic patterning. J. Neurochem 102, 324–332 (2007). [DOI] [PubMed] [Google Scholar]
- 88.Lin Y, Skeberdis VA, Francesconi A, Bennett MVL & Zukin RS Postsynaptic density protein-95 regulates NMDA channel gating and surface expression. J. Neurosci 24, 10138–10148 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fernández E et al. Targeted tandem affinity purification of PSD-95 recovers core postsynaptic complexes and schizophrenia susceptibility proteins. Mol. Syst. Biol 5, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klevanski M et al. Automated highly multiplexed super-resolution imaging of protein nano-architecture in cells and tissues. Nat. Commun 11, 1552 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Johnston IG et al. Symmetry and simplicity spontaneously emerge from the algorithmic nature of evolution. Proc. Natl. Acad. Sci 119, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pan F, Aldridge GM, Greenough WT & Gan W-B Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proc. Natl. Acad. Sci 107, 17768–17773 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Durand CM et al. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol. Psychiatry 17, 71–84 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hahsler M, Piekenbrock M & Doran D dbscan: Fast Density-Based Clustering with R. J. Stat. Softw 91, 1–30 (2019). [Google Scholar]
- 95.Campello RJGB, Moulavi D, Zimek A & Sander J Hierarchical Density Estimates for Data Clustering, Visualization, and Outlier Detection. ACM Trans. Knowl. Discov. Data 10, 5:1–5:51 (2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-synapse measurements as processing output of CellProfiler applied on imaging data have been deposited to Mendeley Data as CSV files under the paper title.
Original code for data analysis, network simulation, and network inference, as well as original CellProfiler pipelines, have been deposited to github.com/lcbb/PRISM-CellProfiler-Analysis-Pipelines.
Raw and processed imaging data is locally stored due excessive file sizes and will be delivered upon request.