

Abstract
The Ref-1 (also called APE or HAP1) protein is a bifunctional enzyme impacting on a wide variety of important cellular functions. It acts as a major member of the DNA base excision repair pathway. Moreover, Ref-1 stimulates the DNA-binding activity of several transcription factors (TFs) through the reduction of highly reactive cysteine residues. Therefore, it represents a mechanism that regulates eukaryotic gene expression in a fast way. However, it has been demonstrated that external stimuli directly act on Ref-1 by increasing its expression levels, a time-consuming mechanism representing a paradox in terms of rapidity of TF regulation. In this paper we demonstrate that this is only an apparent paradox. Exposure of B lymphocytes to H2O2 induced a rapid and sustained increase in Ref-1 protein levels in the nucleus as evaluated by both western blot analysis and by pulse–chase experiments. A time course, two color in situ immunocytochemistry indicated that the up-regulation of Ref-1 in the nucleus at <30 min was primarily the consequence of translocation of its cytoplasmic form. This early nuclear accumulation is effective in modulating the DNA-binding activity of the B cell-specific activator protein BSAP/Pax-5. In fact, EMSA experiments demonstrate that a transient interaction with Ref-1 up-regulates the DNA-binding activity of BSAP/Pax-5. Moreover, in a co-transfection experiment, Ref-1 increased the BSAP/Pax-5 activating effect on an oligomerized BSAP/Pax-5 binding site of the CD19 promoter by 5- to 8-fold. Thus, Ref-1 mediates its effect by up-regulating the DNA-binding activity of BSAP/Pax-5, accounting for a new and fast outside/inside pathway of signaling in B cells.
INTRODUCTION
A primary role in regulating the activity of the basal transcriptional machinery is exerted by promoter-specific transcription factors (TFs) (1). In order to adjust the expression of target genes according to external requirements, signaling mechanisms control molecular functions of TFs. The activity of a TF can be regulated at different levels: (i) at the mRNA production level through changes in the activity of its transcription unit (2,3); (ii) at the pre-translational level through alternative splicing (4,5); (iii) at the post-translational level through modifications of the glycosylation (6), acetylation (7), phosphorylation (8) and redox state (9) of the TF itself. Since it acts on pre-existing molecules, regulation at the redox state level represents a fast and cheap way to regulate TF activity and, in recent years, it has been well documented (10). Redox regulation chiefly occurs through reduction/oxidation of specific cysteine residues that are situated in the DNA-binding domains of TFs. Examples of regulation of TFs by redox of reactive cysteine residues in eukaryotes are those of AP-1 and NF-κB (11; for a review see 12). Both proteins require reducing conditions for DNA binding in vitro, while in vivo these factors become activated by oxidative stress-promoting agents such as H2O2 and bleomycin (12). This apparent contradiction has been explained by the finding of the presence of reducing enzymes, such as thioredoxin (TRX) and Ref-1, whose expression is induced by oxidative stress (13,14).
Ref-1 has been identified as a protein capable of either apurinic/apyrimidinic endonuclease DNA repair activity and nuclear redox activity, being able to induce AP-1 DNA-binding activity, as well as that of NF-κB, Myb, members of the ATF/CREB family, HIF-1α (14) and p53 (15). Ref-1 protein expression is selectively induced by non-toxic levels of a variety of reactive oxygen species (ROS), such as the superoxide anion (![]() ), H2O2 and the hydroxyl radical (·OH), which are by-products of respiration. ROS can also be generated by external agents, such as ionizing radiation (16), during pathological states in activated neutrophils and as a useful tuning device for intracellular signal transduction, as is the case of the cascades induced by cytokines such as tumor necrosis factor-α or interleukin-1β (11). The expression levels of the Ref-1 promoter are strictly regulated themselves. The transcriptional activation of Ref-1 by oxidative stress requires a c-jun-containing multiprotein complex (17). c-jun plays a central role in activation of the Ref-1 promoter while being at the same time the target of redox regulation by Ref-1 (14). Therefore, oxidative stress engages an auto-sustaining loop leading to Ref-1 accumulation in the cell. It has been demonstrated that Ref-1 induction by ROS is due to translational mechanisms, being inhibited by treatment of cells with cycloheximide (CH) (18). From a regulatory point of view this is an apparent paradox, since the external signal would use a fast mechanism of regulation (i.e. reduction of cysteine residues) via a time-consuming event such as new synthesis of the regulator (i.e. Ref-1). For this reason we explored the possibility that Ref-1 function is controlled at a different level than synthesis by using the lymphocyte/BSAP model. Lymphocytes are one of the cell types which are subjected to oxidative stress during the inflammatory response and which use ROS as a tuning device for signal transduction (10). BSAP plays a key role in regulating fundamental events in B cells, such as differentiation and isotype switching, which occur during the inflammatory response in particular areas called germinal centers. BSAP (also called Pax-5, therefore here referred to as BSAP/Pax-5) belongs to the Pax family of TFs, which play a developmental role in a wide variety of species from nematodes to vertebrates (19). Pax proteins recognize DNA sequences through a conserved element called the Prd domain. We have recently demonstrated that the DNA-binding activity of the Prd domain (20) is redox sensitive and that the transcriptional activity of Pax-8 is directly regulated by Ref-1 (21).
), H2O2 and the hydroxyl radical (·OH), which are by-products of respiration. ROS can also be generated by external agents, such as ionizing radiation (16), during pathological states in activated neutrophils and as a useful tuning device for intracellular signal transduction, as is the case of the cascades induced by cytokines such as tumor necrosis factor-α or interleukin-1β (11). The expression levels of the Ref-1 promoter are strictly regulated themselves. The transcriptional activation of Ref-1 by oxidative stress requires a c-jun-containing multiprotein complex (17). c-jun plays a central role in activation of the Ref-1 promoter while being at the same time the target of redox regulation by Ref-1 (14). Therefore, oxidative stress engages an auto-sustaining loop leading to Ref-1 accumulation in the cell. It has been demonstrated that Ref-1 induction by ROS is due to translational mechanisms, being inhibited by treatment of cells with cycloheximide (CH) (18). From a regulatory point of view this is an apparent paradox, since the external signal would use a fast mechanism of regulation (i.e. reduction of cysteine residues) via a time-consuming event such as new synthesis of the regulator (i.e. Ref-1). For this reason we explored the possibility that Ref-1 function is controlled at a different level than synthesis by using the lymphocyte/BSAP model. Lymphocytes are one of the cell types which are subjected to oxidative stress during the inflammatory response and which use ROS as a tuning device for signal transduction (10). BSAP plays a key role in regulating fundamental events in B cells, such as differentiation and isotype switching, which occur during the inflammatory response in particular areas called germinal centers. BSAP (also called Pax-5, therefore here referred to as BSAP/Pax-5) belongs to the Pax family of TFs, which play a developmental role in a wide variety of species from nematodes to vertebrates (19). Pax proteins recognize DNA sequences through a conserved element called the Prd domain. We have recently demonstrated that the DNA-binding activity of the Prd domain (20) is redox sensitive and that the transcriptional activity of Pax-8 is directly regulated by Ref-1 (21).
We here demonstrate that the first event of Ref-1 regulation consists of an increase in cytoplasmic to nuclear translocation. By this mechanism BSAP/Pax-5 is activated and, therefore, an outside/inside signaling pathway is generated in B cells.
MATERIALS AND METHODS
Expression vectors
Plasmid pDS56Ref-1 was used for expression of recombinant Ref-1 His-tagged protein, while for eukaryotic expression we used the vector CMV-Ref-1 (22). Plasmid pT7.7Pax-5 was used for recombinant BSAP/Pax-5 protein expression (20,23). For eukaryotic expression of BSAP/Pax-5, we used the vector CMV-BSAP together with a Δ71-BSAP promoter construct, which bears two copies of the CD19 BSAP site in the SalI site of Δ71 upstream of a chloramphenicol acetyltransferase (CAT) reporter gene.
Protein expression and purification
The recombinant Pax-5 protein (rPax-5) was obtained from overexpression in Escherichia coli and then purified to homogeneity by ion exchange chromatography onto a Mono-S column (Pharmacia) following previous published procedures (20). The purified protein gave a single band on an overloaded SDS–PAGE gel. Fractions containing purified proteins were dialyzed against water and then stored at –85°C. When required, in vitro protein oxidation was obtained by prolonged air exposure.
The recombinant Ref-1 protein (rRef-1) was obtained as an hexahistidine tag fusion protein by overexpression in E.coli and then purified by nickel chelate chromatography from bacterial extracts and treated as previously described (22). The protein was dialyzed against a reducing buffer containing 5 mM DTT. For the functional assay the protein was then diluted 1:100 in non-DTT containing buffer.
Proteins concentrations were determined using the Bradford method (24).
EMSA analysis
Double-stranded oligodeoxynucleotides, labeled at the 5′-end with 32P, were used as probes in gel retardation assays. The H2A-2.2 site is a 25mer whose upper strand is 5′-TCTGACGCAGCGGTGGGTGACGACT-3′ (25). The BS2 site is an 18mer whose upper strand is 5′-CTCTAATGGCTTTTTCTC-3′ (20). The EMSA was performed as previously described (20). The gel was dried and then exposed to an X-ray film at –80°C.
Cell lines and transfection
Raji cells were grown in RPMI 1640 medium while HeLa and NS-1 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM). Both media were supplemented with 10% fetal calf serum (FCS), glutamine and antibiotics.
HeLa cells (0.8 × 106 cells/60 mm culture dish) were transfected using a calcium phosphate method (26). The following amounts of plasmids were transfected in each 60 mm dish: CMV-Ref-1, 0.5 µg; CMV-Luc, 0.5 µg; Δ71-BSAP, 0.3 µg; CMV-BSAP, 0.3 µg.
NS-1 cells were transfected by electroporation using a Bio-Rad gene pulser (Bio-Rad Inc.). Aliquots of 1.5 × 107 cells were resuspended in 350 µl of RPMI in a Bio-Rad electroporation cuvette (0.4 cm gap) and pulsed once with 340 V and 960 µF. After electroporation, cells were maintained at 4°C for 10 min and then cultured in pre-equilibrated RPMI supplemented with 10% fetal serum for 24 h. The following amounts of plasmids were transfected where needed: CMV-Ref-1, 5 µg; CMV-Luc, 5 µg; Δ71-BSAP, 7.5 µg; CMV-BSAP, 7.5 µg. CAT (27) and Luc (28) activities were measured as described in the cited references.
The protein levels achieved in all transfection experiments were comparable and were monitored by western blot analysis using specific antibodies raised against BSAP/Pax-5 and Ref-1, respectively. Moreover, the levels of transactivation were calculated as the CAT/Luc ratios, the luciferase gene activity being the normalizer.
Preparation of nuclear extracts and western blot analysis
Cell nuclear extracts were prepared as previously described (20). The nuclear extracts were then quantitated for protein levels according to the Bradford method (24) and used immediately for western blot or EMSA analysis or kept at –80°C.
For western blot analysis, 10 µg of nuclear extracts were electrophoresed on a 12% SDS–PAGE minigel and transferred to nitrocellulose membrane (Schleicher & Schuell, Keene, NH) as previously described (20,29). The blots were developed using the ECL chemiluminescence method (Amersham Pharmacia Biotech) using Biomax-Light films (Kodak) and bands were quantitated by densitometric scanning of the autoradiograms using an LKB densitometer. All data were confirmed by Gel Doc 2000 analysis (Bio-Rad Inc.).
Immunofluorescence and confocal laser scanning microscopy
Suspension cultures of Raji cells, untreated (control) or treated with 50 µM H2O2 for the indicated times, were centrifuged onto ethanol-cleaned slides. Cytospins were air dried and fixed with 3.7% formaldehyde in PBS for 10 min at room temperature. After washing with 0.1% BSA/PBS, cells were permeabilized with 0.1% Triton X-100 in 0.1% BSA/PBS and 4% FCS for 1 min at room temperature. The slides were extensively washed with 0.1% BSA/PBS and blocked for 30 min with 2% BSA/PBS prior to application of antibodies. Incubation with both primary rabbit polyclonal anti-Ref-1 (29) and secondary (FITC-labeled anti-rabbit IgG; Diagnostics Pasteur, Marnes-La-Coquette, France) antibodies, diluted in 0.1% BSA/PBS (1:100), was for 1 h at room temperature in a moist chamber. Nuclei were counterstained with 80 nM ethidium homo- dimer-1 (EthD-1) (Molecular Probes Inc., Eugene, OR). The slides were then mounted in Mowiol (Calbiochem, La Jolla, CA) containing 2.5% (w/v) 1,4-diazabicyclo[2,2,2]octane (DABCO) (Sigma Chemical Co., St Louis, MO) as antifading agent.
Immunofluorescence-labeled cell preparations were studied using a Bio-Rad MRC 1024 confocal laser scanning microscope (Bio-Rad Inc.) equipped with an argon laser attached to a Nikon Diaphot inverted microscope with a 60× oil immersion objective lens of numerical aperture 1.4. The excitation wavelengths used were 488 (FITC) and 568 nm (EthD-1). Filters used for collecting emission signals were 522/35 (FITC) and 605/32 nm (EthD-1). For acquisition the same conditions were used. For two color fluorescence the red and green signals were collected simultaneously. To create a two color image, the red and green images were merged. Co-localization appears as yellow because of the mixture of red and green.
Metabolic labeling and immunoprecipitation
Metabolic pulse labeling was performed as previously described (30). After chasing, cells were treated with 50 µM H2O2 and collected at the indicated times for immunoprecipitation of the endogenous Ref-1 protein from nuclear extracts. Nuclear extracts were incubated for 2 h at 4°C with gentle tumbling with 20 µl of total rabbit IgG linked to CNBr-activated Sepharose 4B beads (Amersham Pharmacia Biotech). The pre-cleared samples were then centrifuged and incubated, overnight at 4°C and with gentle tumbling, with 25 µl of polyclonal anti-Ref-1 antibody (29) linked to CNBr-activated Sepharose 4B beads at a final concentration of 50 µg antibody/ml resin. After incubation, the immunoprecipitated material was washed sequentially with 100 µl 1% Nonidet P-40 (Pierce), 100 µl 1% Triton X-100 (Pierce) and 100 µl 1% SDS. Elution was carried out with 50 µl of 2× Laemmli sample buffer. The eluted proteins were run on a 12% denaturing SDS–polyacrylamide reducing gel. Gels containing [35S]methionine-labeled material were processed for autoradiography. After fixation for 20 min with 10% acetic acid and 10% methanol, the gel was enhanced with an enlightening solution (NEN Life Science Products), dried and exposed to film (Kodak X-Omat AR) at –80°C for 3–5 days.
RESULTS
The initial event in activation of Ref-1 is its translocation into the nucleus
An increase in Ref-1 expression by treatment with ROS and ROS generators such as H2O2 and NaOCl has been demonstrated in a few cellular systems (18,31). To assess whether this phenomenon also occurs in B lymphoid cells, the ability of H2O2 to induce Ref-1 expression in the Raji cell line was investigated. A 6 h treatment with increasing amounts of H2O2, ranging from 50 nM to 250 µM, showed a clear increase (2- to 6-fold) in Ref-1 protein levels in both the nuclear and cytoplasmic compartments as evaluated by western blot analysis with a specific antibody (data not shown). A time course experiment (Fig. 1A) demonstrated a time-dependent increase in Ref-1 levels whose values reached a maximum at 9 h after treatment with 50 µM H2O2, then declined. The activation of Ref-1 is not unique to H2O2, as bleomycin, a pro-oxidant substance commonly used in anti-leukemic therapy, is able to exert similar effects (data not shown), as previously reported for other cell lines (18).
Figure 1.
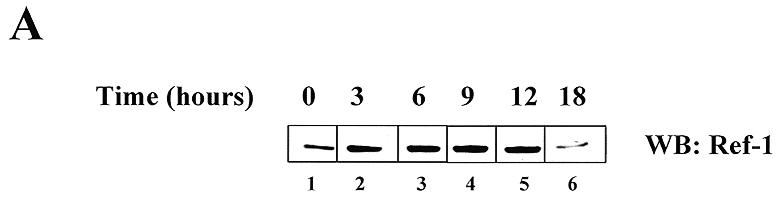

Effect of H2O2 treatment on Ref-1 expression levels in Raji cells. (A) Time course analysis of Ref-1 stimulation in Raji cells after treatment with H2O2. Cells (5 × 106) were treated with 50 µM H2O2 and then harvested for western blot analysis. Aliquots of 20 µg of nuclear extracts were separated by SDS–12% PAGE. A rabbit polyclonal specific antibody (29) detected endogenous Ref-1 protein. (B and C) Ref-1 translocation occurs in Raji cells pre-treated with CH. Raji cells were either preincubated in the presence (filled symbols) or in the absence (open symbols) of the protein synthesis inhibitor CH (10 µg/ml) for 1 h before exposure to 50 µM H2O2. The cells were then processed to obtain cytoplasmic (B) and nuclear (C) extracts. Aliquots of 20 µg of each extract were loaded onto a SDS–PAGE gel for western blot analysis with anti Ref-1 polyclonal antibody. Values obtained from densitometric scannings of films, after exposure to ECL development, are reported. (B) Cytoplasmic extracts of Raji cells untreated (circles) and treated (triangles) with 50 µM H2O2. (C) Nuclear extracts of Raji cells untreated (squares) and treated (rhombuses) with 50 µM H2O2. Bars indicate the means ± SD of at least three independent experiments.
Figure 1 also shows that the cytoplasmic form of Ref-1 increases upon H2O2 treatment (Fig. 1B, open triangles). Therefore, the increment in the total amount of Ref-1 protein necessitates new protein synthesis and, therefore, is time consuming. In order to establish whether a faster way of Ref-1 activation, cytosol to nucleus translocation, may occur, we performed a series of experiments with the protein synthesis inhibitor CH. Data obtained from densitometric scanning of Ref-1 bands obtained by western blot analysis demonstrate that CH prevents the increase in Ref-1 levels in the cytoplasmic fraction (Fig. 1B, filled triangles) but not the Ref-1 increase in the nucleus (Fig. 1C, filled diamonds). In fact, at 3 h, the increase in Ref-1 levels in the nuclear fraction was largely independent of de novo protein synthesis (Fig. 1C, filled diamonds, see arrow). At later times, 6–9 h, the nuclear increase was greatly, if not completely, due to de novo synthesis of protein, while at 12 h there was a decrease in total protein (Fig. 1C, open diamonds). Therefore, an early event occurring soon after H2O2 treatment was accelerated translocation of Ref-1 from the cytoplasmic compartment to the nucleus. Supportive evidence for this phenomenon was provided by a pulse–chase experiment. [35S]methionine-labeled Raji cells were pulsed overnight, maintained in chase medium for 1 h and then treated with 50 µM H2O2 for different lengths of time (3, 6 and 9 h). Nuclear extracts were immunoprecipitated with an anti-Ref-1 antibody and it was found that the ratio of Ref-1 levels in treated versus non-treated cells was higher at 3 h (ratio ~3.5) and progressively decreased at 6 (ratio ~2) and 9 h (ratio ~1) (Fig. 2).
Figure 2.
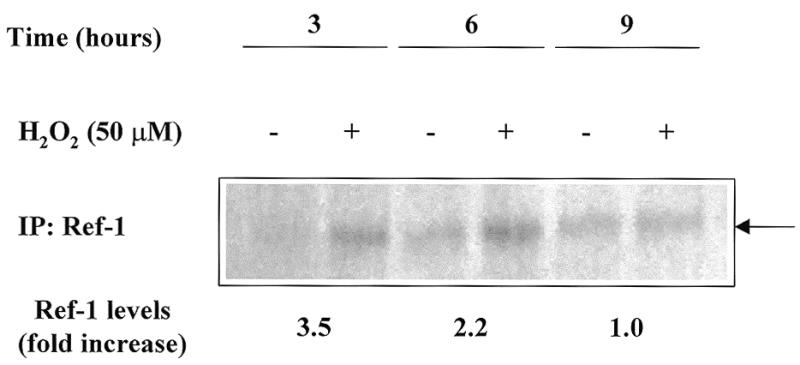
Metabolic labeling of Raji cells confirms that the first event in Ref-1 activation is its translocation into the nucleus. SDS–PAGE analysis of immunoprecipitates from Raji nuclear extracts after exposing the cells for different times to 50 µM H2O2 during a pulse–chase experiment. At the bottom the 35S-labeled Ref-1 values calculated after densitometric analysis of the immunoprecipitated bands are indicated as the fold increase in the ratio between H2O2-stimulated and unstimulated Raji cells.
To further investigate nuclear translocation at early times after H2O2 treatment the subcellular distribution of Ref-1 was determined by two color analysis and confocal microscopy (Fig. 3). In untreated cells (Fig. 3a) Ref-1 was detected mostly in the cytoplasm (green signal) and not in the nucleus, which is stained red by EthD-1. Treatment with H2O2 induced a visible Ref-1 mobilization towards the nucleus, as could be seen by the considerable increase in yellow staining indicative of co-localization at the periphery of the nucleus after 30 and 120 min of treatment (Fig. 3b and c, respectively). At these early times there was little evidence of de novo protein synthesis, since the ‘green’ signal decreased consistently in the cell cytoplasm. The signal for both cytoplasmic and nuclear localization of Ref-1 became much more intense after a 6 h treatment (Fig. 3d). These data show that Ref-1 had not only migrated ‘massively’ into the nucleus (marked yellow staining) but also that synthesis of new Ref-1 protein was increased (abundant green staining in the cytoplasm). Thus, the primary consequence of H2O2 treatment is that at early times (0–3 h) Ref-1 is translocated from the cytoplasm to nucleus.
Figure 3.
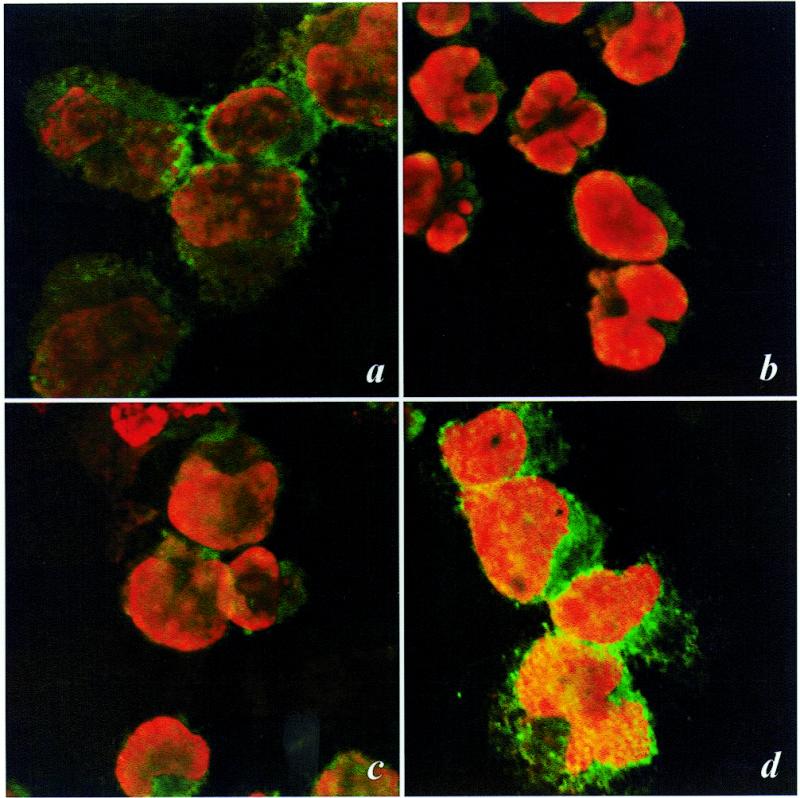
The effect of H2O2 treatment of Raji cells on Ref-1 translocation as demonstrated by confocal microscopy analysis. Raji cells were treated with H2O2 for 30 (b), 120 (c) and 360 min (d). Ref-1 localization was analyzed by indirect immunolabeling with specific antibodies, while nuclei were stained with EthD-1. Control (untreated) cells (a) showed a predominant cytoplasmic distribution of Ref-1; after a 30 (b) or 120 min (c) treatment Ref-1 was visible in the peripheral region of the nucleus (yellow), while it was less evident in the cytoplasm. A large increment in Ref-1 both in the nucleus and in the cytoplasm appeared after a 6 h incubation with H2O2, indicating both a consistent translocation of Ref-1 from the cytoplasm to the nucleus and de novo synthesis of the protein
The DNA-binding activity of BSAP/Pax-5 is affected by H2O2 in vivo
Whether the nuclear translocation of Ref-1, observed at early times, is effective in modulating the DNA-binding activity of endogenous BSAP/Pax-5 was investigated by treating Raji cells with 50 µM H2O2 for 30 min and then assaying nuclear extracts for the level of specific BSAP/Pax-5 DNA-binding activity. The nuclear extracts were made in the absence of DTT to determine only the active form of Pax-5. A 30 min treatment of Raji cells with H2O2 induced a clear increase in DNA-binding activity by endogenous BSAP/Pax-5 compared to the activity of a similar amount of nuclear extract from non-H2O2-treated cells (compare Fig. 4A, lanes 2 and 3). This increase in BSAP/Pax-5 DNA-binding activity was not due to an increase in the protein levels since they were not influenced by H2O2 treatment (Fig. 4B). Since at 30 min there was a consistent increase in Ref-1, it is very likely that the enhanced DNA-binding activity was the consequence of Ref-1 translocation. Thus, translocation of Ref-1 into the nucleus after H2O2 treatment is able to activate BSAP/Pax-5 binding, indicating the importance of Ref-1–BSAP/Pax-5 regulation in vivo and that changes in the environmental redox potential can influence BSAP/Pax-5 activity.
Figure 4.
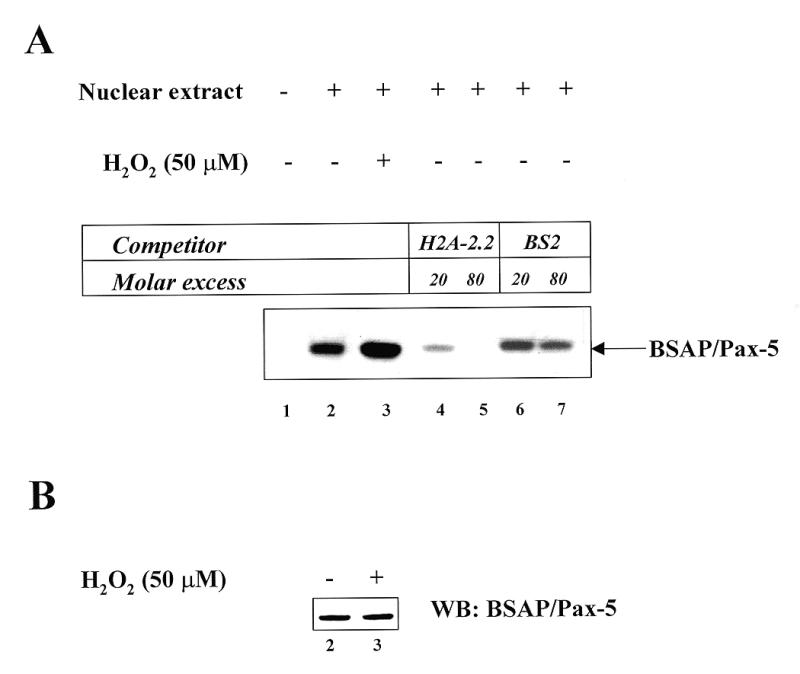
The effect of H2O2 treatment of Raji cells on BSAP/Pax5 DNA-binding activity. (A) Induction of BSAP/Pax5 DNA-binding activity by treatment of B cells with H2O2. Raji cells were left untreated (lane 2) or incubated for 30 min with 50 µM H2O2 (lane 3). Nuclear extracts (lanes 2–3) were prepared as described in Materials and Methods. An aliquot of 5 µg of protein was reacted with a 32P-labeled H2A-2.2 probe (25). Samples were analyzed on a native 7% polyacrylamide gel. An autoradiograph of the gel is shown. The arrow indicates the position of the BSAP/Pax-5–DNA specific complex. To test for the specificity of the complex, a competition assay with cold specific and non-specific oligonucleotide was performed. As expected, the specific H2A-2.2 oligonucleotide efficiently reduces the BSAP/Pax-5–DNA complex (lanes 4 and 5), whereas the non-specific BS2 oligonucleotide is almost ineffective (lanes 6 and 7). (B) The expression levels of BSAP/Pax-5 are not induced by a 30 min treatment of B cells with H2O2. The nuclear samples, as in (A) (10 µg/lane), were loaded onto a SDS–PAGE gel for western blot analysis with anti BSAP/Pax-5 monoclonal antibody. The blot was developed using an ECL chemiluminescence detection kit (Amersham Biotech).
Ref-1 protein is directly responsible for the induction of BSAP/Pax-5 DNA-binding activity
In order to support the hypothesis cited above, we tested whether Ref-1 was able to directly increase the DNA-binding activity of BSAP/Pax-5. Another set of experiments with rRef-1 and rPax-5 were performed. The oxidized form of rPax-5 protein, obtained by prolonged air exposure, and the reduced form of rRef-1 protein were used in the EMSA assay. The oxidized form of rPax-5 alone or in the presence of a low concentration (0.05 mM) of DTT (data not shown) did not display any DNA-binding activity (Fig. 5, lane 3). Instead, a strong reducing environment (5 mM DTT) was necessary to demonstrate DNA-binding activity (lane 2). The addition of reduced rRef-1 protein (final concentration 0.05 mM DTT) resulted in rPax-5 DNA-binding activity (Fig. 5, lanes 7 and 8). The oxidized form of rRef-1 was totally ineffective (data not shown). In addition, it is evident that the interaction between rPax-5 and rRef-1 is likely to be of a transient nature because there was no change in mobility of rPax-5–DNA complexes when comparing the samples stimulated with DTT (Fig. 5, lane 2) and those stimulated with Ref-1 (Fig. 5, lanes 7 and 8).
Figure 5.
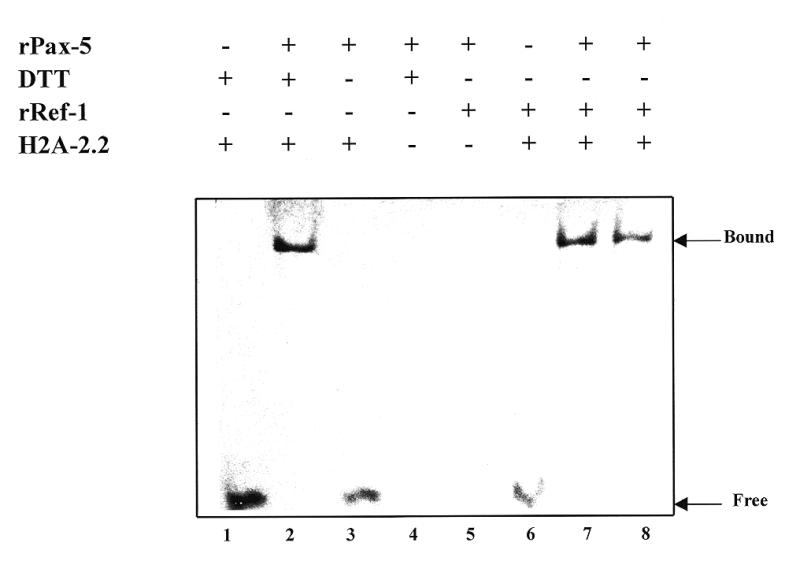
rRef-1 is a stimulator of Pax-5 DNA-binding activity. DNA-binding by 1 µg of rPax-5 protein, in the absence (lane 3) or presence of 1.5 (lane 7) or 1 µg (lane 8) of rRef-1 or of the reducing agent DTT (5 mM) (lane 2) was analyzed by EMSA. Lane 1 contains 0.5 µg of probe alone; lane 4 contains rPax-5 under reducing conditions without probe; lane 5 contains rRef-1 and rPax-5; lane 6 contains rRef-1 and the DNA probe encompassing the promoter of the sea urchin histon H2A-2.2 (25). The oxidized form of rPax-5 was obtained by prolonged air exposure. The EMSA was developed by silver nitrate staining.
Ref-1 increases Pax-5 transcriptional activity
The Δ71-BSAP promoter is not functional when transfected into HeLa cells, but its activity can be reconstituted by forced expression of BSAP/Pax-5 due to the presence of polymerized BSAP/Pax-5 binding sites in the CD19 promoter (32). Thus, in order to test whether the stimulatory effect of BSAP/Ref-1 on Pax-5 DNA-binding activity could have any relevance in vivo, co-transfection with a Ref-1 expression vector was used. As expected, the Δ71-BSAP construct was inactive in HeLa cells while co-expression of BSAP/Pax-5 was able to activate it. Co-transfection with the Ref-1 expression vector increased BSAP/Pax-5-induced activation of the Δ71-BSAP construct by 5-fold (Fig. 6A). The Ref-1 effect is specific, since the activity of the CMV promoter was not modified by co-transfection of the Ref-1 expression vector (not shown). Moreover, in the absence of BSAP/Pax-5, transfection of Ref-1 reduced the basal level of Δ71-BSAP construct activity by ~50% (data not shown), excluding the possibility that Ref-1 had any activating effect on the basal transcriptional machinery of the Δ71-BSAP promoter. To extend and confirm the relevance of these observations in a lymphoid system, we performed the same type of experiment using non-secreting myeloma cell line NS-1, in which BSAP/Pax-5 protein is not expressed (unpublished data). Only forced expression of BSAP/Pax-5 allowed activation of the Δ71-BSAP promoter. Co-transfection of a Ref-1 expression vector increased Pax-5-induced activation of the Δ71-BSAP promoter 8-fold. Although we cannot formally exclude a direct effect of Ref-1 on the transcriptional activation domain of BSAP/Pax-5 (33), these data, together with those shown in Figure 5, strongly suggest that the stimulatory effect of Ref-1 on Δ71-BSAP construct activation might be due to an increase in Pax-5 DNA-binding activity.
Figure 6.

Effect of Ref-1 on transactivation activity of the Δ71-BSAP promoter. (A) Plasmids were transfected by the calcium phosphate method in HeLa cells at the concentrations indicated in Materials and Methods. Forty-eight hours after transfection, cells were harvested and CAT and Luc activities were measured in nuclear extracts. Bars indicate the means ± SD of at least three independent experiments. (B) Plasmids were transfected by electroporation into NS-1 cells at the concentrations indicated in Materials and Methods. Twenty-four hours after transfection, cells were harvested and CAT and Luc activities were measured in nuclear extracts. Bars indicate the means ± SD of at least three independent experiments.
DISCUSSION
ROS are harmful by-products of oxidative metabolism but are also used in eukaryotic cells as second messengers in signal transduction pathways from cytokine–receptor interactions to activation of TF (10). In fact, it has been shown that IL-1β induces NF-κB activation through the production of ROS in lymphoid cells (34) and this activity is regulated through oxidation/reduction of cysteine residues in critical positions (35). Redox regulation of TF activation mostly occurs in the nuclear compartment and, in turn, requires a system of regulation. The nuclear protein Ref-1 is a clear candidate for the role of shuttle factor between the cytoplasm and the nucleus, being able to modify, after stimulation, the nuclear redox potential of the cell. Therefore, Ref-1 seems to fit with previous requirements. In fact: (i) it is able to stimulate the DNA-binding activity of TFs AP-1, NF-κB, Myb and p53 (14,15); (ii) it is an inducible enzyme, whose expression is up-regulated when cells are exposed to oxidative stresses (36–38); (iii) its expression level is chronologically strictly controlled and another redox enzyme (TRX) is a cytoplasmatic partner which seems to be involved in extracellular to nuclear environment signaling (39). We have recently demonstrated that the structure and in vitro binding activity of the Prd domain of Pax proteins is regulated through the oxidation/reduction of conserved cysteine residues (20). Here we have demonstrated that Ref-1 could act as a signaling molecule in response to oxidative stress by translocating from the cytoplasm to the nucleus of a B cell. Moreover, Ref-1 is also able, both in vitro and in vivo, to control the DNA-binding activity of BSAP/Pax-5 in lymphoid cells. These findings represent the very first examples of the regulation of activity of a lymphoid-specific TF through Ref-1.
To our knowledge, Ref-1 expression levels have never previously been investigated in a lymphoid cell system. In fact, all studies have concerned its expression in epithelial cells (skin, thyroid, liver and duodenum) (29,40), in the central nervous system (brain, hippocampus and cerebellum) (29,41) and in the myeloid cell line HL-60 (42). Here we have demonstrated that exposure of Raji cells to H2O2 induces an increment in the total amount of Ref-1 and accumulation in the nucleus. The timing of activation clearly demonstrated a two-stage process: translocation of the protein from the cytoplasmic compartment to the nucleus at very early times (<30 min), followed by de novo synthesis/translocation of Ref-1. Therefore, the B cell responds promptly to oxidative stress by the accumulation of Ref-1 in the nucleus, a quick means of activation. Since it has been reported that H2O2 is efficiently able to mimic the effect of ROS in the cell (18,38,43) and that ROS are directly involved, as second messengers, one can argue that Ref-1 could play a role during B cell activation. To this end, the use of H2O2 appears useful in the study of BSAP/Pax-5 activation mediated by Ref-1. In fact, the increase in nuclear levels of Ref-1 protein after H2O2 treatment of Raji cells has a direct role in induction of the DNA-binding activity of endogenous BSAP/Pax-5. This finding suggests that in the nucleus of the B cell there could be at least two forms of BSAP/Pax-5: the DNA-binding active form (reduced) and a non-DNA-binding active form (oxidized). Ref-1 could modulate the transcriptional activity of BSAP/Pax-5 by increasing the reduced form of this TF in the nucleus. Therefore, our model could represent a chronologically and energetically more economical means of regulating BSAP/Pax-5 activity.
Several experimental approaches have consistently suggested that BSAP/Pax-5 regulates distinct classes of genes during B cell differentiation, acting either as a positive or negative regulator of gene expression (2,44). This double nature of BSAP/Pax-5 (as positive or negative regulator of gene expression) includes, in the same TF, the possibility of accurately regulating B cell functions and suggests that this TF plays a fundamental role in activation and proliferation. Thus, mechanisms able to control the activity of BSAP/Pax-5 will enable B cell differentiation and activation. A good model to test this could be the mechanism of isotype switching in B cells. Since transcription of the Ref-1 gene is itself regulated, requiring elements such as a multiprotein complex containing c-jun, and is modulated by means of second messengers such as cAMP (17), it is possible to speculate that stimuli other than ROS could modulate the transcriptional activity of TFs activated by Ref-1. PGE2 is known to promote B lymphocyte Ig class switching from IgA to IgE through a mechanism which up-regulates intracellular cAMP levels (45). It is remarkable that the same result is produced by overexpressing BSAP/Pax-5 in the I.29µ murine B cell line (46), which inhibits switching to IgA and enhances switching to IgE. Therefore, it could be hypothesized that Ref-1 could play a role in mediating BSAP/Pax-5-regulated IgE class switching following PGE2 treatment of B cells. The switch between repression and activation of BSAP/Pax-5-regulated gene expression might be through modulation of BSAP/Pax-5 protein levels by synthesis or degradation. However, a change in nuclear levels of BSAP/Pax-5 through de novo synthesis of protein is time and energy consuming. Moreover, exposure to cytokines and chemical stimuli requires rapid modification of the transcription apparatus. This can only happen if control of TF activity is dependent on a fast ‘shuttle factor’ that transduces, at the nuclear level, signals generated in the cytoplasm by intra- or extracellular signal pathways.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr John Kehrl for helpful discussions and comments on the manuscript. We are grateful to Agostino Riva for providing the Δ71-BSAP plasmid, Markus Neurath for the pCRII-BSAP construct and Tom Curran for the CMV-Ref-1 and His-tagged Ref-1 plasmids. We also thank David Cimarosti for the help in recombinant protein expression and purification. This work was supported by the Italian National Research Council, Target Project on Biotechnology, by the Italian Association for Cancer Research and by the Ministero per l’ Università e la Ricerca Scientifica e Tecnologica (MURST). Dr Kelley was supported by NIH/NCI Program Project Grant PO1-CA75426, NIH grant ES07815 and NIH/NCI grant CA76643.
REFERENCES
- 1.Darnell J.R. (1982) Nature, 297, 365–371. [DOI] [PubMed] [Google Scholar]
- 2.Wallin J.J., Gackstetter,E.R. and Koshland,M.E. (1998) Science, 279, 1961–1964. [DOI] [PubMed] [Google Scholar]
- 3.Tjian R. and Maniatis,T. (1994) Cell, 77, 5–8. [DOI] [PubMed] [Google Scholar]
- 4.Epstein J., Glaser,T., Cai,J., Jepeal,L., Walton,D.S. and Maas,R.L. (1994) Genes Dev., 8, 2022–2034. [DOI] [PubMed] [Google Scholar]
- 5.Kozmik Z., Czerny,T. and Busslinger,M. (1997) EMBO J., 16, 6793–6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson S.P. and Tjian,R. (1988) Cell, 55, 125–133. [DOI] [PubMed] [Google Scholar]
- 7.Boyes J., Byfield,P., Nakatani,Y. and Ogryzko,V. (1998) Nature, 396, 594–598. [DOI] [PubMed] [Google Scholar]
- 8.Hunter T. and Karin,M. (1992) Cell, 70, 375–387. [DOI] [PubMed] [Google Scholar]
- 9.Xanthoudakis S. and Curran,T. (1996) Adv. Exp. Med. Biol., 387, 69–75. [PubMed] [Google Scholar]
- 10.Nakamura H., Nakamura,K. and Yodoi,J. (1997) Annu. Rev. Immunol., 15, 351–369. [DOI] [PubMed] [Google Scholar]
- 11.Schreck R., Rieber,P. and Baeuerle,P.A. (1991) EMBO J., 10, 2247–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sen C.K. and Packer,L. (1996) FASEB J., 10, 709–720. [DOI] [PubMed] [Google Scholar]
- 13.Schenk H., Klein,M., Erdbrugger,W., Droge,W. and Schulze-Osthoff,K. (1994) Proc. Natl Acad. Sci. USA, 91, 1672–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xanthoudakis S., Miao,G., Wang,F., Pan,Y.C. and Curran,T. (1992) EMBO J., 11, 3323–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaiddon C., Moorthy,N.C. and Prives,C. (1999) EMBO J., 18, 5609–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward J.F. (1994) Int. J. Radiat. Biol., 66, 427–432. [DOI] [PubMed] [Google Scholar]
- 17.Grosch S. and Kaina,B. (1999) Biochem. Biophys. Res. Commun., 261, 859–863. [DOI] [PubMed] [Google Scholar]
- 18.Ramana C.V., Boldogh,I., Izumi,T. and Mitra,S. (1998) Proc. Natl Acad. Sci. USA, 95, 5061–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mansouri A., Hallonet,M. and Gruss,P. (1996) Curr. Opin. Cell. Biol., 8, 851–857. [DOI] [PubMed] [Google Scholar]
- 20.Tell G., Scaloni,A., Pellizzari,L., Formisano,S., Pucillo,C. and Damante,G. (1998) J. Biol. Chem., 273, 25062–25072. [DOI] [PubMed] [Google Scholar]
- 21.Tell G., Pellizzari,L., Cimarosti,D., Pucillo,C. and Damante,G. (1998) Biochem. Biophys. Res. Commun., 252, 178–183. [DOI] [PubMed] [Google Scholar]
- 22.Abate C., Patel,L., Rauscher,F.J. and Curran,T. (1990) Science, 249, 1157–1161. [DOI] [PubMed] [Google Scholar]
- 23.Studier F.W., Rosenberg,A.H., Dunn,J.J. and Dubendorff,J.W. (1991) Methods Enzymol., 185, 60–89. [DOI] [PubMed] [Google Scholar]
- 24.Bradford M. (1976) Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 25.Adams B., Dorfler,P., Aguzzi,A., Kozmik,Z., Urbanek,P., Maurer-Fogy,I. and Busslinger,M. (1992) Genes Dev., 6, 1589–1607. [DOI] [PubMed] [Google Scholar]
- 26.Graham F.L. and van der Eb,A.J. (1973) Virology, 52, 456–467. [DOI] [PubMed] [Google Scholar]
- 27.Gorman C.M., Moffat,L.M. and Howard,B.H.. (1982) Mol. Cell. Biol., 2, 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Wet J.R., Wood,K.V., Deluca,M., Helinski,D.R. and Subramani,S. (1987) Mol. Cell. Biol., 7, 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duguid J.R., Eble,J.N., Wilson,T.M. and Kelley,M.R. (1995) Cancer Res., 55, 6097–6102. [PubMed] [Google Scholar]
- 30.Jost C.R., Kurucz,I., Jacobus,C.M., Titus,J.A., George,A.J.T. and Segal,D.M. (1994) J. Biol. Chem., 269, 26267–26273. [PubMed] [Google Scholar]
- 31.Grosch S., Fritz,G. and Kaina,B. (1998) Cancer Res., 58, 4410–4416. [PubMed] [Google Scholar]
- 32.Riva A., Wilson,G.L. and Kehrl,J.H. (1997) J. Immunol., 159, 1284–1292. [PubMed] [Google Scholar]
- 33.Dorfler P. and Busslinger,M. (1996) EMBO J., 15, 1971–1982. [PMC free article] [PubMed] [Google Scholar]
- 34.Bonizzi G., Piette,J., Merville M.P. and Bours,V. (1997) J. Immunol., 159, 5264–5272. [PubMed] [Google Scholar]
- 35.Matthews J.R., Wakasugi,N., Virelizier,J.L., Yodoi,J. and Hay,R.T. (1992) Nucleic Acids Res., 20, 3821–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao K.S., Xanthoudakis,S., Curran,T. and O’Dwyer,P.J. (1994) Mol. Cell. Biol., 14, 5997–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang L.E., Arany,Z., Livingston,D.M. and Bunn,H.F. (1996) J. Biol. Chem., 271, 32253–32259. [DOI] [PubMed] [Google Scholar]
- 38.Walker L.J., Craig,R.B., Harris,A.L. and Hickson,I.D. (1994) Nucleic Acids Res., 22, 4884–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirota K., Matsui,M., Hiwata,S., Nishiyama,A., Mori,K. and Yodoi,J. (1997) Proc. Natl Acad. Sci. USA, 94, 3633–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asai T., Kambe,F., Kikumori,T. and Seo,H. (1997) Biochem. Biophys. Res. Commun., 236, 71–74. [DOI] [PubMed] [Google Scholar]
- 41.Wilson T.M., Rivkees,S.A., Deutsch,W.A. and Kelley,M.R. (1996) Mutat. Res., 362, 237–248. [DOI] [PubMed] [Google Scholar]
- 42.Robertson K.A., Hill,D.P., Xu,Y., Liu,L., Van Epps,S., Hockenbery,D.M., Park,J.R., Wilson,T.M. and Kelley,M.R. (1997) Cell Growth Differ., 8, 443–449. [PubMed] [Google Scholar]
- 43.Meyer M., Schreck,R. and Baeuerle,P.A. (1993) EMBO J., 12, 2005–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nutt S.L., Morrison,A.M., Dorfler,P., Rolink,A. and Busslinger,M. (1998) EMBO J., 17, 2319–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roper R.L., Brown,D.M. and Phipps,R.P. (1995) J. Immunol., 154, 162–170. [PubMed] [Google Scholar]
- 46.Qiu G. and Stavnezer,J. (1998) J. Immunol., 161, 2906–2918. [PubMed] [Google Scholar]