

Abstract
We have shown that KRAS-TP53 genomic co-alteration is associated with immune-excluded microenvironments, chemoresistance, and poor survival in pancreatic ductal adenocarcinoma (PDAC) patients. By treating KRAS-TP53 cooperativity as a model for high-risk biology, we now identify cell-autonomous Cxcl1 as a key mediator of spatial T-cell restriction via interactions with CXCR2+ neutrophilic myeloid-derived suppressor cells in human PDAC using imaging mass cytometry. Silencing of cell-intrinsic Cxcl1 in LSL-K-rasG12D/+;Trp53R172H/+;Pdx-1Cre/+(KPC) cells reprograms trafficking and functional dynamics of neutrophils to overcome T-cell exclusion, and controls tumor growth in a T-cell-dependent manner. Mechanistically, neutrophil-derived TNF is a central regulator of this immunologic rewiring, instigating feed-forward Cxcl1 overproduction from tumor-cells and cancer-associated fibroblasts (CAF), T-cell dysfunction, and inflammatory CAF polarization via transmembraneTNF-TNFR2 interactions. TNFR2 inhibition disrupts this circuitry and improves sensitivity to chemotherapy in vivo. Our results uncover cancer cell-neutrophil crosstalk in which context-dependent TNF signaling amplifies stromal inflammation and immune tolerance to promote therapeutic resistance in PDAC.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a lethal malignancy characterized by extreme therapeutic resistance (1,2). The dominant contributors to therapeutic resistance in PDAC are an undruggable genomic landscape typified by co-occurring KRAS and TP53 mutations(3), tolerogenic signaling from myeloid cells(4,5)—particularly neutrophilic/polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs)—which promotes T-cell dysfunction and exclusion (6,7), and pro-inflammatory polarization of cancer-associated fibroblasts (iCAF) (8) that instigates stromal inflammation by elaborating soluble factors (e.g., IL-6) that further accelerate myeloid chemotaxis and induce chemoresistant CAF-tumor cell IL-6/STAT3 signaling (9). However, how these complex circuitries converge in the tumor microenvironment (TME) to mediate therapeutic resistance in PDAC is incompletely understood.
We have recently shown that KRAS-TP53 co-alteration is associated with worse survival in patients with advanced PDAC (6). Moreover, KRAS-TP53 cooperativity encodes for cell and non-cell autonomous transcriptional networks that drive innate immune cell-enriched and T-cell excluded immune microenvironments (6). Using an integrative molecular approach, we identified a KRAS-TP53 cooperative “immunoregulatory” program—encompassing cancer-cell intrinsic Cxcl1 and myeloid-derived TNF, among others—associated with chemotherapy resistance and worse overall survival in PDAC patients enrolled in the COMPASS trial (10).
Here, through the lens of KRAS-TP53 cooperativity as a model for high-risk biology, we uncover how cancer cell-autonomous programs dictate PMN-MDSC activation and functional plasticity, which in turn regulates T-cell dysfunction and stromal inflammation in the PDAC TME. Specifically, cancer cell-autonomous Cxcl1—which is transcriptionally regulated by a Creb-dependent mechanism—imposes T-cell restriction via interactions with CXCR2+ PMN-MDSCs in spatially annotated human tumors. Silencing of tumor cell-intrinsic Cxcl1 overcomes CD8+ T-cell exclusion by inducing a fundamental reprogramming of PMN-MDSC function in-vivo. Neutrophil-restricted TNF—via Cxcr2-Map3k8-Tnf signaling—emerges as a central driver of this MDSC reprogramming and TME remodeling, instigating feed-forward tumor cell/CAF-Cxcl1 overproduction, immune tolerance, and iCAF polarization via transmembrane TNF (tmTNF)-TNFR2 signaling. TNFR2 inhibition disrupts this circuitry to improve sensitivity to chemotherapy in-vivo, implicating context-dependent tmTNF-TNFR2 signaling as a promising therapeutic target in this deadly disease.
RESULTS
Imaging mass cytometry reveals tumor cell-intrinsic overexpression of CXCL1 in KRAS-TP53 co-altered human PDAC
We have previously shown that KRAS-TP53 genomic co-alteration encodes cancer cell-autonomous transcriptional programs which orchestrate innate immune enrichment and T-cell exclusion in PDAC (6,11). To identify key tumor cell-intrinsic factors mediating innate immune recruitment, we examined differentially expressed bulk transcriptomes from KRAS-TP53 co-altered (n=23) vs. KRAS-altered/TP53WT (n=5) human PDAC cell lines from the Cancer Cell Line Encyclopedia (Fig. 1A) (12). Gene set enrichment analysis (GSEA) highlighted overexpression of several pathways related to granulocyte/neutrophil function in KRAS-TP53 co-altered PDAC (Fig. 1B; Supplementary Fig. S1A; Supplementary Table S1). Among transcripts conserved across these pathways, we observed significant upregulation of CXCL1—a secreted neutrophilic chemoattractant ligand—in KRAS-TP53 co-altered transcriptomes (Fig. 1B; Supplementary Fig. S1B). We corroborated these findings in preclinical genetic models, where Cxcl1 mRNA expression via RNA in-situ hybridization (P=0.0002; Fig. 1C; Supplementary Fig. S1C) and Cxcl1 immunostaining via IHC (Supplementary Fig. S1D) was substantially increased in the epithelial/ductal compartment of LSL-K-rasG12D/+;Trp53R172H/+;Pdx-1Cre/+(KPC) compared to LSL-K-rasG12D/+;Pdx-1Cre/+(KC) tumors.
Figure 1. Cxcl1 is overexpressed in Ras-p53 cooperative PDAC and governs spatial exclusion of T-cells in human tumors.
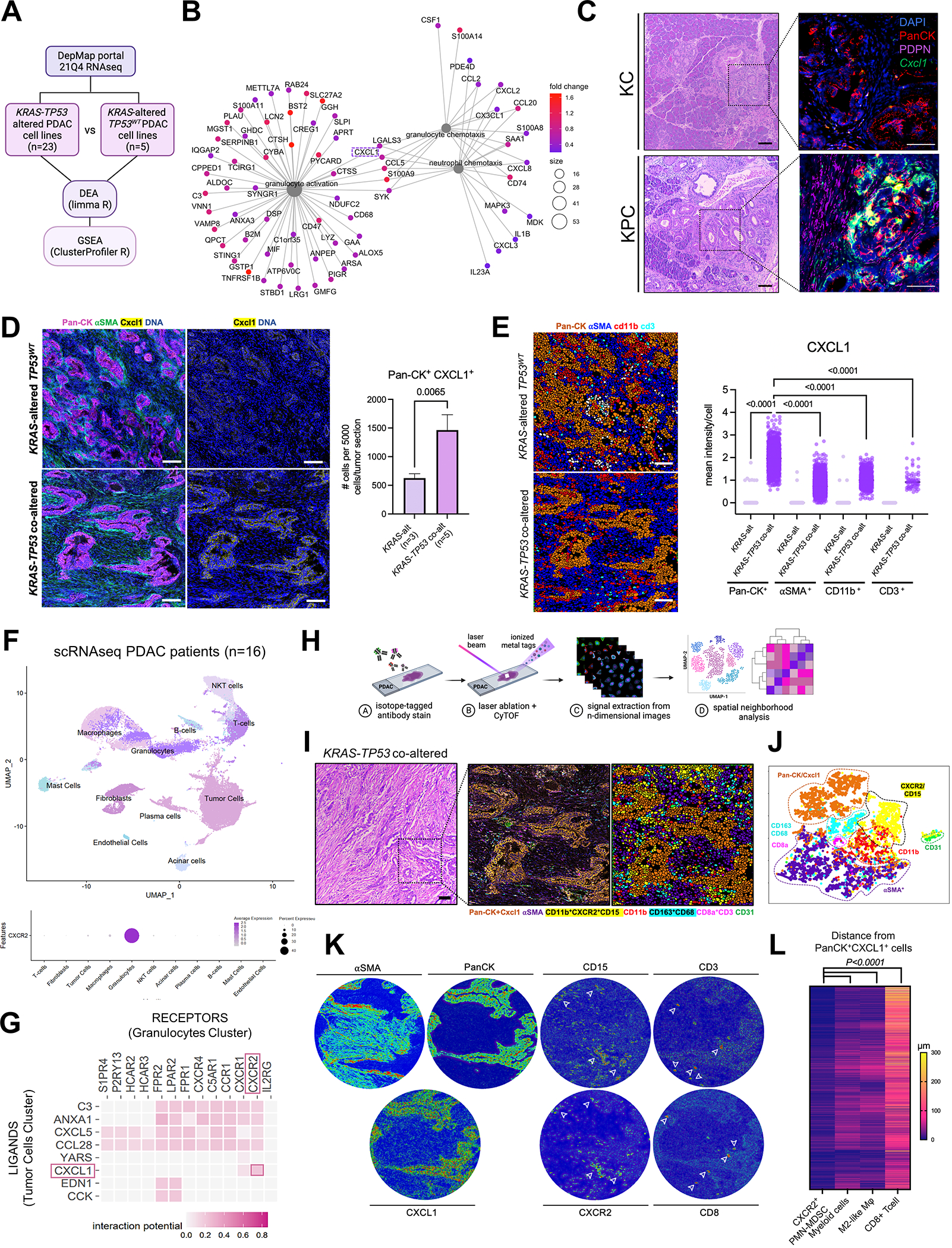
A, Schematic of transcriptomic comparison between KRAS-TP53 co-altered (n=23) and KRAS-altered/TP53WT(n=5) human PDAC cell lines from the Cancer Cell Line Encyclopedia (CCLE), with subsequent differential expression analysis (DEA) and gene set enrichment analysis (GSEA); B, Net plot visualizing the top 3 gene-sets related to granulocyte/neutrophil function overexpressed in KRAS-TP53 co-altered PDAC tumor-cell transcriptomes, with 5 transcripts conserved between these pathways (CXCL1 highlighted in dashed box) shown; C, H&E sections paired with immunostaining for pancytokeratin (PanCK), podoplanin (PDPN), and RNA-in situ hybridization to detect Cxcl1 mRNA in representative sections from volume-matched tumors in genetic models LSL-K-rasG12D/+; Trp53R172H/+; Pdx-1Cre/+ (KPC; 6-months old) and LSL-K-rasG12D/+;Pdx-1Cre/+ (KC; 12-months old). Quantification of relative Cxcl1 mRNA expression in PanCK+ cells provided in Supplementary Fig. S1C; D, Imaging Mass Cytometry (IMC) comparing epithelial expression of CXCL1 in human KRAS-TP53 co-altered (n=5) compared to KRAS-altered/TP53WT (n=3) tumors at single-cell resolution, with adjacent histogram showing quantification of average number of PanCK+CXCL1+ cells per 5000 single cells in each tumor section across groups; E, IMC image segmentation into PanCK+ tumor cells, αSMA+ fibroblasts, CD11b+ myeloid cells, CD3+ T cells in KRAS-TP53 co-altered (KRAS+TP53) vs. KRAS-altered/TP53WT (KRAS) human PDAC sections, with adjacent quantification of mean intensity of CXCL1 expression in each cell type across groups. Cell populations were grouped according to any positive pixel intensity for the respective marker, with discrepancies and overlap between phenotypes reconciled manually; F, Uniform Manifold Approximation and Projection (UMAP) showing annotated clusters from single-cell RNA sequencing (scRNAseq) data in human PDAC patient samples (n=16; top), with bubble plot showing relative CXCR2 expression between different clusters (bottom); G, Heatmap showing tumor cell (donor) to granulocyte (recipient) ligand-receptor interactome in human scRNAseq dataset using NicheNet algorithm, with CXCL1-CXCR2 interaction highlighted (red box); H, Schematic representation of IMC workflow to provide spatially resolved single-cell phenotypes of human PDAC tumors; I-J, H&E, spatial phenotype map, and image segmentation of representative KRAS-TP53 co-altered human PDAC tumor section (I), with single-cell clustering in T-distributed Stochastic Neighborhood Embedding (tSNE) maps (J) of 72,880 single-cells in 8 pre-defined ROIs each from a unique patient sample, distributed into epithelial/tumor cell (PanCK+CXCL1+), stromal/fibroblast (αSMA+), endothelial (CD31+), myeloid (CD11b+), PMN-MDSC (CD15+CXCR2+), M2-like macrophage (CD68+CD163+), and CD8+ T cell (CD3+CD8+) populations; K, Tissue heatmaps showing expression of αSMA, PanCK, CXCL1, CD15, CXCR2, CD3 and CD8 in representative KRAS-TP53 co-altered human PDAC tumor section; L, Heatmap depicting spatially resolved distances of single CXCR2+CD11b+CD15+ PMN-MDSC, CD11b+ myeloid cell, CD68+CD163+ M2-like macrophage, and CD3+CD8+ T-cells from PanCK+ tumor-cells in 8 ROIs from human PDAC sections.
To validate and extend these findings in patient-derived PDAC tumors, we utilized imaging mass cytometry (IMC) to examine tissue-level CXCL1 expression at single-cell resolution in KRAS-TP53 co-altered (n=5) vs. KRAS-altered/TP53WT (n=3) human tumors derived from patients with resectable or borderline resectable PDAC who underwent upfront surgical resection at our institution (Fig. 1D; Supplementary Table S2). Pathologist-selected regions of interest (ROI) from each tumor section probed with metal ion-conjugated antibodies for pan-cytokeratin (PanCK:epithelial), α-smooth muscle actin (α-SMA:fibroblast), CD11b (myeloid), CD3 (T-cell), and CXCL1 were laser-ablated, and atomized ions were acquired using time-of-flight mass cytometry (cyTOF). Image segmentation and quantification revealed significantly higher proportion of PanCK+CXCL1+ epithelial/tumor-cell islands in KRAS-TP53 co-altered vs. KRAS-altered/TP53WT tumors (P=0.0065; Fig. 1D). Mean CXCL1 expression was disproportionately higher in single PanCK+ tumor-cells compared with αSMA+ fibroblasts, CD11b+ myeloid cells, or CD3+ T-cells in KRAS-TP53 co-altered tumors (all P<0.001; Fig. 1E). These findings were corroborated in two independent single-cell datasets, in which highest CXCL1 gene expression was observed in tumor/malignant cells compared with all other subclusters in single-nuclear RNA sequencing (snRNAseq) data from 43 patients (13) and in single-cell RNA sequencing (scRNAseq) data from 16 human PDAC tumors (6) (Supplementary Fig. S1E–F). In IMC analysis, mean CXCL1 expression was also significantly increased in all other cellular compartments in KRAS-TP53 co-altered vs. KRAS-altered/TP53WT tumors (Fig. 1E). We also observed >10-fold higher Cxcl1 secretion by human KRAS-TP53 co-altered (Panc02.03, Capan1, Mia-Paca2) vs. KRAS-altered/TP53WT (Hs766t) cell lines in vitro (P<0.0001; Supplementary Fig. S1G).
CD8+ T-cells are spatially excluded from cellular neighborhoods comprising tumor-cell Cxcl1 and CXCR2+ PMN-MDSCs in human PDAC
CXCL1 has been implicated as a critical regulator of T-cell exclusion in congenic KPC models (14), and is also a key component of the innate immunoregulatory transcriptional program overexpressed in KPC tumor cells via scRNAseq in our recent study (6). To interrogate how tumor cell-intrinsic CXCL1 governs spatial relationships with innate and adaptive immune populations in human PDAC, we first sought to determine the localization of its cognate receptor CXCR2 within the TME. Via scRNAseq and snRNAseq, we identified that CXCR2 was near-exclusively expressed in intratumoral PMN-MDSCs from human PDAC samples (n=16 (15), Fig. 1F; Supplementary Fig. S2A–B; and n=43 (13), Supplementary Fig. S2C), as well as in KPC (Supplementary Fig. S2D) and Ptf1aCre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT (16); Supplementary Fig. S2E) genetic mouse models. Computational modeling of intercellular ligand-receptor interactomes (17) in human PDAC scRNAseq data revealed strong predicted interaction between tumor cell-CXCL1 and neutrophil-CXCR2 (Fig. 1G). Single-cell deconvolution in TCGA-PDAC dataset (n=178) confirmed robust correlations between CXCL1 expression and enrichment in computationally inferred neutrophil (P<0.0001), MDSC (P<0.001), and macrophage (P=0.002) populations (Supplementary Fig. S2F).
We next interrogated tissue-level spatial relationships between CXCL1-expressing tumor islands and immune populations in the human PDAC TME. Using IMC, we examined 72,880 cells from 8 predefined ROIs (each from a unique patient) in multicellular neighborhoods to quantify expression of 11 phenotypic markers and spatial features of each cell (Fig. 1H–I). Image segmentation into single cells generated a phenotype map, and subsequent clustering revealed distinct epithelial/tumor-cell (PanCK+), stromal (αSMA+), endothelial (CD31+), myeloid (CD11b+), PMN-MDSC (CD15+CXCR2+), M2-like macrophage (CD68+CD163+), and CD8+ T-cell (CD3+CD8+) populations (Fig. 1J). Tissue heatmaps confirmed expression of CXCL1 predominantly in PanCK+ tumor cells compared with αSMA+ CAFs or CD11b+ myeloid cells, CXCR2 near-exclusively in CD11b+CD15+ PMN-MDSCs, and CD8 in CD3+ T-cells (Fig. 1K). Neighborhood analysis to quantify statistically significant interaction or avoidance between pairs of cellular phenotypes revealed strong spatial contiguity between PanCK+CXCL1+ tumor-islands and CD11b+CD15+CXCR2+ PMN-MDSCs (median distance 7.3±11.1μm) and CD68+CD163+ M2-like macrophages (23.3±20.0μm). Importantly, CD3+CD8+ T-cells were spatially excluded from contiguous PanCK+CXCL1+ and CXCR2+CD11b+CD15+ cellular communities (95.4±63.2μm, P<0.0001; Fig. 1L).
KRAS and TP53 mutations cooperate to transcriptionally regulate CXCL1 via CREB activation in pancreatic cancer cells
To deconstruct individual elements of signaling networks driving the association between tumor cell-intrinsic Cxcl1 and T-cell exclusion in PDAC, we first investigated how KRAS and TP53 alterations cooperate to regulate cell-autonomous CXCL1 in tumor cells. To recapitulate KRAS-TP53 co-alteration in isogenic human pancreatic epithelial systems, we transiently overexpressed mutant-TP53R175H or TP53WT cDNA constructs in KRAS-wildtype hPNE-KRASWT and KRASG12D-mutant hPNE-KRASG12D cells (18), and examined dependence of Cxcl1 production on mutational status. Overexpression of KRASG12D, but not TP53R175H alone in hPNE-KRASWT cells, increased Cxcl1 secretion (P<0.001). Furthermore, compared with TP53WT overexpression in hPNE-KRASG12D cells, KRASG12D and TP53R175H mutational cooperativity (Supplementary Fig. S3A) further augmented Cxcl1 production (P<0.001; Fig. 2A).
Figure 2. KRAS and TP53 mutations cooperate to transcriptionally regulate CXCL1 via CREB activation in pancreatic cancer cells.
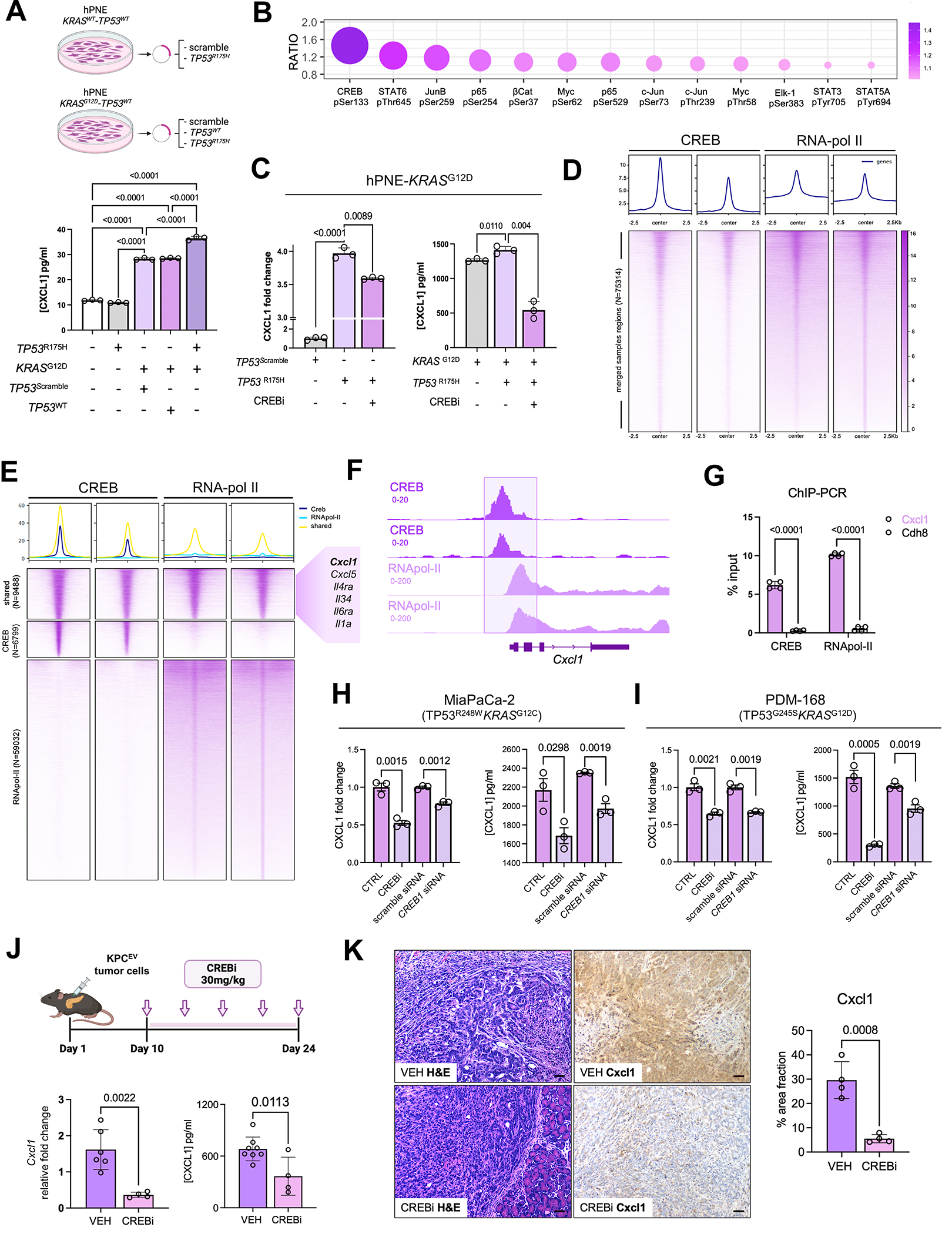
A, Schematic representing experimental constructs utilized to overexpress TP53WT or TP53R175H in isogenic hPNE-KRASWT or hPNE-KRASG12D pancreatic epithelial cells (top). Histogram showing Cxcl1 secretion from each hPNE cell system annotated by respective KRAS and/or TP53 mutational status (bottom); B, Bubble plot representing the top 10 transcription factors hyperphosphorylated in hPNE-KRASG12DTP53R175H compared with hPNE-KRASG12DTP53WT cells, with relative ratio of expression indicated on y-axis; C, Histograms representing relative fold change in CXCL1 gene expression (left) and secretion (in pg/mL; right) from hPNE-KRASG12DTP53WT and hPNE-KRASG12DTP53R175H in absence or presence of Creb inhibitor 666-15 (CREBi 0.5 μM for 24h, n=3); D, Chromatin immunoprecipitation and sequencing (ChIP-seq) peak signals and heat maps of CREB regions in CREB and RNApol-II ChIP material (n=2 biologic replicates each) in Kras-Trp53 cooperative KPC 6694c2 cells; E, ChIP-seq heatmaps showing co-occupied CREB and RNApol-II peaks (N=9488), CREB-unique peaks (N=6799), RNApol-II unique peaks (N=59032), with adjacent callout box showing curated gene module implicated in inflammatory signaling and innate immune regulation; F, Integrative Genome Viewer (IGV) plot visualizing co-occupancy of peaks in CREB and RNApol-II ChIP-seq data at the transcriptional start site of Cxcl1 promoter; G, ChIP-qPCR of Cxcl1 and Cdh8 (negative control) from CREB and RNApol-II immunoprecipitated purified DNA in KPC 6694c2 cells; H-I, CXCL1 gene expression (left) and secretion (right) each in human MiaPaCa-2 cells (H) and human PDM-168 patient-derived organoids (I) in absence or presence of CREBi 666-15 (0.5 μM for 24h) or absence or presence of Creb siRNA (n=3 each); J, Schematic of Creb inhibitor treatment of KPC orthotopic mice in vivo (top). Bar plots showing Cxcl1 gene expression via qPCR and protein levels via ELISA (in pg/mL) in whole tumor lysates from vehicle-treated vs. Crebi-treated mice (n=4–7 mice); K, Cxcl1 immunostaining with corresponding H&E staining in representative tumor sections from vehicle- vs. CREBi-treated mice (n=4–5/group; scale bar=50μm).
To determine signaling mechanisms regulating CXCL1 transcription downstream of KRAS-TP53 cooperativity, multiplex phospho-kinome arrays identified CREBSer133 as the top hyperphosphorylated transcription factor (P-adj<0.01; Fig. 2B)—and top 10 hyperphosphorylated proteins overall (Supplementary Fig. S3B)—in hPNE-KRASG12D-TP53R175H compared with hPNE-KRASG12D-TP53WT cells. Interestingly, CREB1pSer133 has been recently implicated in hyperactivating pro-metastatic transcriptional networks in KRAS-TP53 cooperative PDAC via FOXA1-Wnt/β-catenin-mediated mechanism (19). Pharmacologic CREB inhibition with 666-15 (Supplementary Fig. S3C–E) in hPNE-KRASG12D-TP53R175H cells significantly reduced CXCL1 transcription (P=0.009) and Cxcl1 secretion (P=0.004) (Fig. 2C). Moreover, 666-15-mediated reduction in Cxcl1 expression was validated in murine KPC tumor-cells (Supplementary Fig. S3F).
To interrogate if Cxcl1 is a CREB target site, we next performed chromatin immunoprecipitation and sequencing (ChIP-seq) for CREB and RNApol-II—a mark associated with transcriptional start sites—in KrasG12D-Trp53R172H KPC6694c2 tumor cells. We observed 75,314 merged peaks between CREB and RNApol-II (Fig. 2D), with the majority of peaks present in genic/promoter regions close to transcription start sites (Supplementary Fig. S4A). A robust number of co-occupied and unique targets (~9,488)—particularly genes encoding for pro-inflammatory mediators—were shared between CREB and RNApol-II (Fig. 2E; Supplementary Table S3). Notably, we observed significant co-binding of CREB and RNApol-II at the Cxcl1 promoter across biological replicates (Fig. 2E–F). Consistent with ChIP-seq findings, validation by ChIP-qPCR was performed in (i) KPC 6694c2 cells, which showed co-occupancy of CREB and RNApol-II at Cxcl1 promoter while no binding was observed for the negative control Cdh8 gene (Fig. 2G; Supplementary Fig. S4B), and in (ii) KPC K-8484 cells (20,21) (Supplementary Fig. S4C).
To corroborate the role of CREB in transcriptional regulation of CXCL1 in human KRAS-TP53 cooperative PDAC systems, we performed pharmacological inhibition and RNA interference of CREB in: (i) KRASG12D-TP53G245S PDM-168 patient-derived organoid cells; and (ii) KRASG12C-TP53R248W co-altered MiaPaCa-2 cells. In both models, Cxcl1 expression and secretion were significantly reduced with CREB1-siRNA and 666-15 treatment (Fig. 2H–I;). Finally, in vivo CREB inhibition using 666-15 in orthotopic KPC tumor-bearing mice resulted in significant diminution in Cxcl1 expression and protein levels in whole tumor lysates (P=0.011; Fig. 2J), as well as reduction in Cxcl1 immunostaining via IHC (Fig. 2K) and epithelial/ductal-specific Cxcl1 mRNA expression via RNA in-situ hybridization (Supplementary Fig. S4D) in tumor sections. Altogether, these data highlight that cell-autonomous Ras and p53 mutations cooperate to regulate Cxcl1 via a CREB-dependent mechanism.
Genetic silencing of tumor cell-intrinsic Cxcl1 overcomes T-cell exclusion in-vivo
We next investigated how cell-intrinsic Cxcl1 mechanistically governs T-cell exclusion in vivo. We utilized KPC tumor cells (KPC 6694c2 backbone) in which Cxcl1 has been genetically silenced using CRISPR-Cas9 editing (14) (Supplementary Fig. S5A) to generate syngeneic transplantation models with either control KPCEV (transduced with non-targeting sgRNA) or KPC-Cxcl1KO tumor cells in C57/BL6 mice. Coupled with reduced tumor growth kinetics in subcutaneous KPC-Cxcl1KO tumors (n=5, Supplementary Fig. S5B), there was dramatic reduction in primary tumor weights (n=20, P<0.0001; Fig. 3A) and metastatic outgrowth (Supplementary Fig. S5C–D), as well as significant improvement in overall survival (n=15, median 48 vs. 21 days; P<0.0001, Fig. 3B), in orthotopically-injected KPC-Cxcl1KO compared with KPCEV tumor-bearing mice.
Figure 3. Genetic silencing of tumor cell-intrinsic Cxcl1 overcomes T-cell exclusion and controls tumor growth in a CD8+ T-cell dependent manner in-vivo.
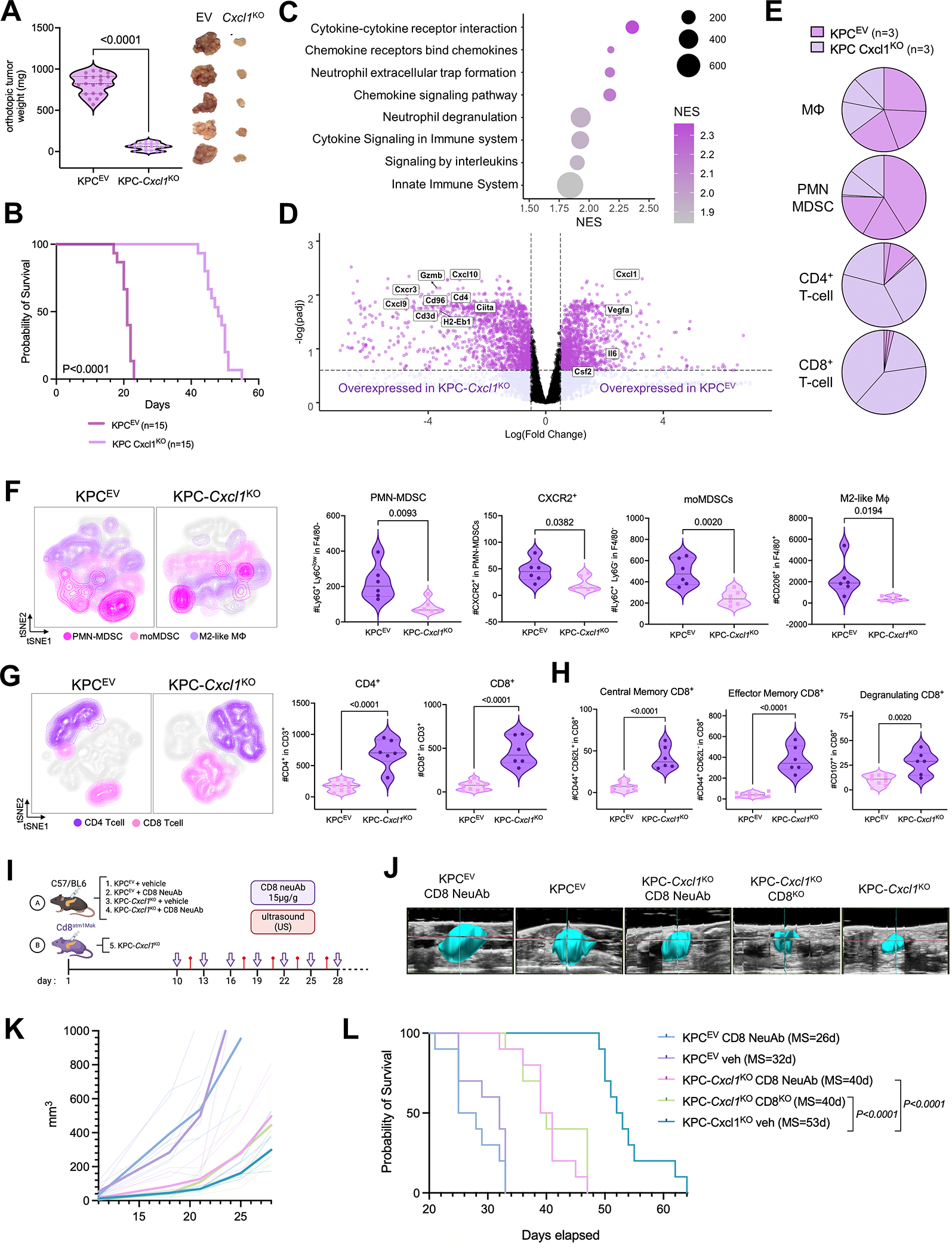
A, Violin plot representing difference in primary tumor weights in C57/BL6 mice orthotopically-injected with KPCEV or KPC-Cxcl1KO tumor-cells (n=20/group; left), with representative images of tumors from each group showing phenotypic reproducibility (n=5 each; right); B, Kaplan-Meier curves showing overall survival of KPC-Cxcl1KO and KPCEV orthotopically-injected mice (n=15; median 48 vs. 21 days); C, Bubble plot visualizing differentially upregulated pathways (using KEGG and Reactome knowledgebases) in 3-week whole-tumor transcriptomes from KPCEV compared with KPC-Cxcl1KO orthotopic tumors via RNA-sequencing (n=3 biologic replicates); D, Volcano plot depicting significantly enriched genes related to immune regulation in KPCEV (Cxcl1, Vegfa, Il6, Csf2), and KPC-Cxcl1KO (Cxcl10, Gzmb, Cxcr3, Cxcl9, Cd96, Cd3d, Cd4, Ciita, H2-Eb1); E, Pie charts showing relative proportions of immune-cell fractions of macrophages (Mϕ), PMN-MDSC, CD4+ T-cells, and CD8+ T-cells using CIBERSORT immune deconvolution from transcriptomes in KPCEV vs. KPC-Cxcl1KO tumors (n=3 biologic replicates per group); F-G, viSNE contour plots of flow cytometric immunophenotyping in concatenated single-cell suspensions from KPCEV or KPC-Cxcl1KO orthotopic tumors (left), with adjacent violin plots (right) representing absolute cell counts of PMN-MDSC, CXCR2+ PMN-MDSC, monocytic MDSC (moMDSC), M2-like macrophage (F), CD4+ T-cells and CD8+ T-cells (G), and central memory T-cells, effector memory T-cells, degranulating CD8+ T-cells (H) from each biologic replicate (n=6–8/group); I, Schematic of experimental design utilizing CD8+ T-cell neutralizing antibodies (CD8neuAb) in C57Bl/6 mice or CD8α−/− transgenic mice (B6.Cd8atm1Mak; CD8KO) for orthotopic injections; J, Representative ultrasound images from mice in each treatment group showing tumor growth dynamics in vivo; K-L, tumor growth curves (K) and Kaplan-Meier survival estimates (L) from mice across 5 groups in T-cell depletion experiments (n=10 mice/group), with median survival (MS) of each cohort indicated in parentheses.
To better understand the molecular basis for reduced tumor outgrowth in the Cxcl1-silenced model, we performed RNA sequencing on 3-weeks KPCEV and KPC-Cxcl1KO orthotopic tumors (n=3/group), and subsequent GSEA revealed effects predominantly related to immunoregulatory signaling (Fig. 3C; Supplementary Fig. S6A). KPCEV transcriptomes were expectedly enriched in genes and pathways related to innate immune recruitment/function (i.e., Il6, Csf2, neutrophil degranulation, etc.; Fig. 3C–D), while several pathways (Supplementary Fig. S6B) and genes regulating T-cell trafficking and effector activity (i.e., Cxcr3, Cxcl9, Cd96, Gzmb) were upregulated in KPC-Cxcl1KO tumors (Fig. 3D). Additionally, we observed dysregulation of multiple oncogenic signaling pathways (e.g., TNF via NF-κB, JAK/STAT3, etc.) in KPC-Cxcl1KO tumors (Supplementary Fig. S6C).
Consistent with these gene/pathway-level results, quantification of immune-cell fractions from deconvoluted RNA-seq data revealed attenuation of macrophages and PMN-MDSCs, and significant enrichment in CD4+ and CD8+ T-cells in KPC-Cxcl1KO tumors (Fig. 3E). Flow cytometric immunophenotyping confirmed significant reduction in tumor-infiltrating CD11b+F4/80−LyGhiLy6Cdim PMN-MDSCs—specifically CXCR2+ PMN-MDSC—as well as F4/80+CD206+ M2-like macrophages and F4/80−Ly6C+Ly6G− monocytic-MDSCs in KPC-Cxcl1KO tumors (Fig. 3F). Notably, we observed a dramatic increase in intratumoral CD4+ and CD8+ T-cells (Fig. 3G), as well as expansion in central memory, effector memory, and CD107+ degranulating CD8+ T-cells, in KPC-Cxcl1KO tumors (Fig. 3H). This immunologic remodeling was recapitulated by pharmacologic inhibition using anti-Cxcl1 neutralizing antibodies in Ptf1acre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) mice, confirming significant reductions in PMN-MDSC and M2-like macrophage trafficking as well as increased CD4+/CD8+ T-cell infiltration (Supplementary Fig. S5E).
Cell-intrinsic Cxcl1 controls tumor growth in a CD8+ T-cell dependent manner
To determine if anti-tumor effects of Cxcl1-silencing are CD8+ T-cell dependent, we performed KPC-Cxcl1KO orthotopic injections in transgenic CD8α−/− (B6.Cd8atm1Mak) mice or in C57/BL6 mice treated with CD8+ T-cell-depleting antibodies (n=10/group, Fig. 3I). Monitoring of in vivo tumor growth dynamics using real-time ultrasound (Fig. 3J) revealed significantly reduced latency of KPC-Cxcl1KO-mediated tumor control in transgenic CD8α−/− and anti-CD8-treated mice compared with vehicle-treated KPC-Cxcl1KO mice; notably, CD8+ T-cell depletion in KPCEV tumor-bearing mice had little impact on the aggressive tumor growth kinetics compared with vehicle-treated KPCEV mice (Fig. 3K). CD8+ T-cell depletion or CD8α silencing significantly shortened survival of KPC-Cxcl1KO mice compared with vehicle-treated KPC-Cxcl1KO controls (median 40 vs. 40 vs. 53 days, respectively; P<0.0001, Fig. 3L). Therefore, silencing of cell-intrinsic Cxcl1 overcomes T-cell exclusion and restrains tumor growth in a CD8+ T-cell-dependent manner in vivo.
Silencing of tumor-intrinsic Cxcl1 reprograms trafficking dynamics and immunosuppressive potential of PMN-MDSCs
Given the striking reduction in myeloid infiltration into KPC-Cxcl1KO tumors, we interrogated how Cxcl1 silencing regulates real-time MDSC trafficking dynamics using a novel in vivo adoptive transfer system in which splenic PMN-MDSCs from KPCEV or KPC-Cxcl1KO tumor-bearing mice are labeled ex vivo with a sulfo-Cy5.5 maleimide probe—which has nanomolar affinity for surface thiol esters on PMN-MDSCs—and intravenously injected into KPCEV or KPC-Cxcl1KO subcutaneous tumor-bearing mice, with ensuing PMN-MDSC trafficking visualized using IVIS (n=4/group, Fig. 4A). Following adoptive transfer, labeled PMN-MDSCs derived from KPCEV donor mice exhibited rapid migration to flank tumors in KPCEV recipients, but not to tumor sites in KPC-Cxcl1KO recipients (P=0.002). Interestingly, PMN-MDSCs derived from KPC-Cxcl1KO donor mice did not traffic to recipient mice harboring Cxcl1-competent KPCEV tumors (P=0.028; Fig. 4A; Supplementary Fig. S7A), suggesting reprogramming of PMN-MDSCs generated in Cxcl1-silenced tumor-bearing hosts that disrupts their migratory potential despite intact cognate chemokine gradients.
Figure 4. Silencing of cancer cell-intrinsic Cxcl1 reprograms trafficking dynamics and immunosuppressive potential of tumor-infiltrating PMN-MDSCs.
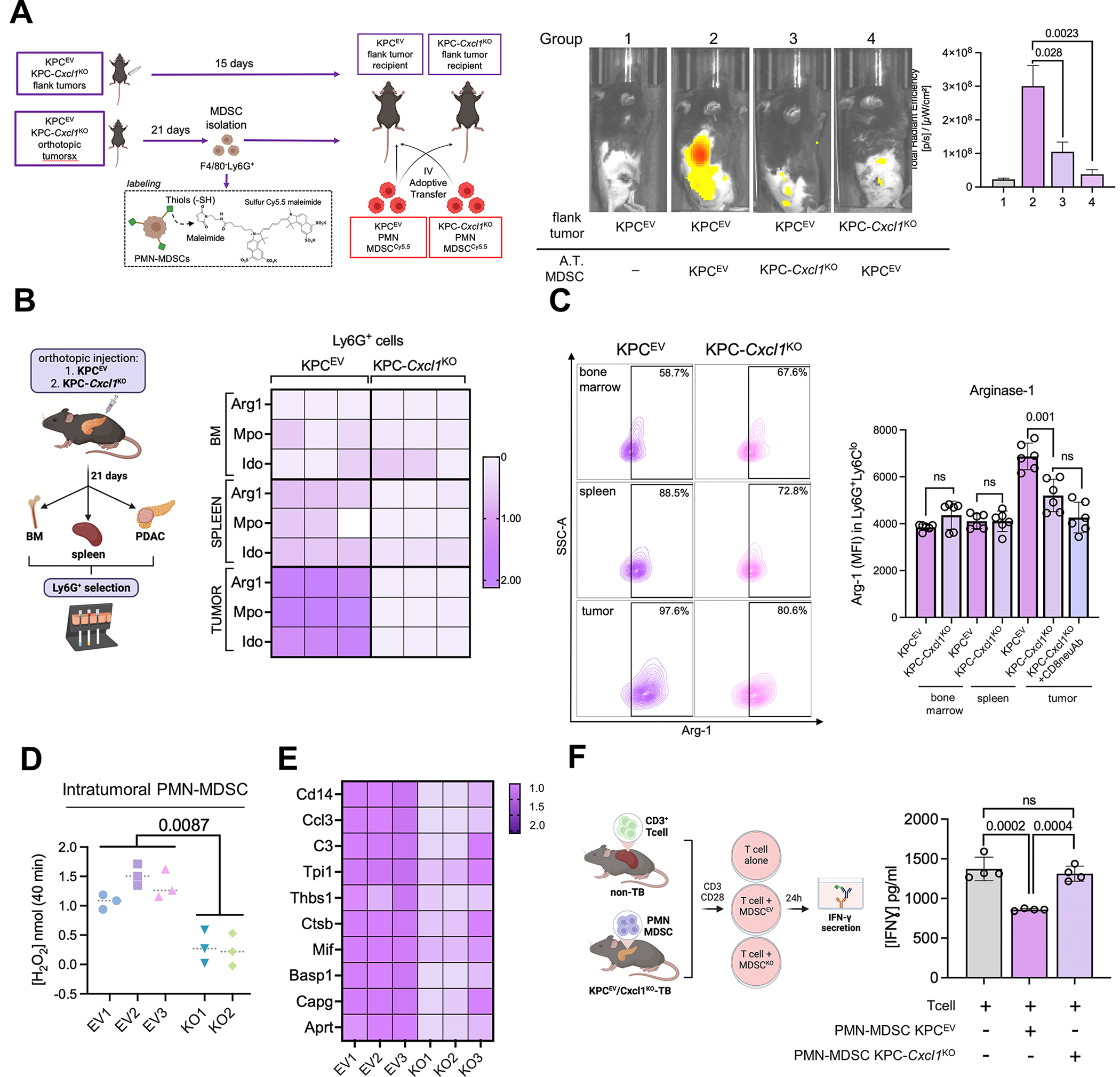
A, Experimental design of adoptive transfer experiments in which splenic neutrophils from donor mice are labeled with sulfur Cy5.5 maleimide, adoptively transferred into recipient flank tumor-bearing mice, and PMN-MDSC trafficking visualized using IVIS (left). Representative IVIS images visualizing trafficked adoptively transferred (A.T.) splenic MDSCs from KPCEV or KPC-Cxcl1KO tumor-bearing mice to subcutaneous KPCEV or KPC-Cxcl1KO tumor-bearing mice, as indicated in the legend (center), and adjacent quantification of total radiant efficiency (TRE) of trafficked Cy5.5-labeled PMN-MDSCs in each group via IVIS (n=4/group, right); B, Schematic of experimental design (left) with adjacent heatmap depicting relative fold change of Arg1, Mpo, Ido gene expressions via qPCR in Ly6G+ cells isolated from bone marrow (BM), spleen, or tumor sites in KPCEV and KPC-Cxcl1KO orthotopic tumor-bearing mice (right). Gene expression in all other groups are relative to reference expression of genes in intratumoral Ly6G+ cells from KPCEV mice; C, Representative contour plots of arginase-1 (Arg-1) expression via flow cytometry in gated F4/80−Ly6GhiLy6Cdim cells from BM, spleen, or tumor sites in KPCEV and KPC-Cxcl1KO orthotopic tumor-bearing mice (left), with adjacent histogram showing arginase-1 mean fluorescence intensity (MFI) at respective sites in designated mice (n=6/group). Arg-1 expression in intratumoral PMN-MDSCs is also quantified from KPC-Cxcl1KO mice treated with anti-CD8 neutralizing antibody (CD8neuAb); D, Arginase-1 enzymatic activity via colorimetric assay in intratumoral PMN-MDSCs from KPCEV and KPC-Cxcl1KO mice (n=2–3/group); E, Heatmap depicting relative fold change of curated gene module from activated-PMN signature (22) via qPCR from RNA extracted from intratumoral Ly6G+ cells in KPCEV vs. KPC-Cxcl1KO tumors; F, Schematic of experimental design (left), with adjacent histogram showing IFN-γ release (in pg/mL) from CD3/CD28-stimulated T-cells alone, or when co-cultured (1:3 T-cell:MDSC ratio) in combination with intratumoral PMN-MDSCs from KPCEV and/or KPC-Cxcl1KO tumor-bearing mice (n=4/group).
To interrogate if MDSC-induced T-cell suppression is another core function impacted by this reprogramming of PMN-MDSCs, we isolated F4/80−Ly6Ghi neutrophils from bone marrow, spleens, and orthotopic tumors in KPCEV or KPC-Cxcl1KO mice (n=3/group), and examined relative expression of immunosuppressive markers. Tumor-derived PMN-MDSCs from KPC-Cxcl1KO mice demonstrated the most pronounced reduction in Arg1, Ido and Mpo expression via qPCR (Fig. 4B) and Arg-1 expression via flow cytometry (n=6/group, Fig. 4C), compared with their tumor-derived counterparts in KPCEV mice, which correlated strongly with differential Cxcl1 secretion gradients (i.e., ΔCxcl1) from lysates in corresponding tumors (P<0.001; Supplementary Fig. S7B). Notably, reduction of Arg-1 expression in KPC-Cxcl1KO-derived PMN-MDSCs was unaffected by neutralization of CD8+ T-cells in these mice, which mitigates the discrepancy in tumor burden between KPCEV and KPC-Cxcl1KO tumors (refer to Fig. 3J). Colorimetry-based functional assays in intratumoral MDSCs confirmed significant reduction in arginase-1 activity in KPC-Cxcl1KO-infiltrating PMN-MDSCs (P=0.0087; Fig. 4D; Supplementary Fig. S7C). Finally, compared with KPCEV-infiltrating PMN-MDSCs, transcriptomes of KPC-Cxcl1KO-infiltrating PMN-MDSCs demonstrated striking attenuation in a recently described Cd14high “activated” gene module (22) associated with potent immunosuppressive activity (Fig. 4E).
In keeping with these findings, we observed rescue of IFN-γ release from T-cells co-cultured with KPC-Cxcl1KO-derived PMN-MDSCs compared with expected T-cell-suppressive effects of KPCEV-derived MDSCs (n=4/group, Fig. 4F). These data indicate that cell-intrinsic Cxcl1 not only modulates trafficking dynamics of PMN-MDSCs, but also governs their T-cell suppressive behavior upon arrival to the TME. This reprogramming of MDSC function may underlie the amelioration of T-cell exclusion and antitumor immunity in Cxcl1-silenced tumors.
Neutrophil-intrinsic TNF is a central regulator of MDSC function via Cxcl1-CXCR2-MAPK signaling
We next performed RNA-sequencing of sorted intratumoral PMN-MDSCs retrieved from KPCEV and KPC-Cxcl1KO orthotopic tumors to identify dominant neutrophil-intrinsic mechanisms that underlie the tumor-restraining and immune-potentiating effects of Cxcl1 silencing (Fig 5A, Supplementary Fig. S8A). Differential gene expression (Supplementary Fig. S8B) and GSEA revealed strong enrichment in mitogen-activated protein kinase (MAPK) pathway signaling in MDSC-KPCEV relative to MDSC-KPC-Cxcl1KO transcriptomes (Fig. 5A), and Ingenuity Pathway Analysis nominated Tnf as the top upstream regulator of differentially expressed MDSC transcriptomes (Fig. 5B). As such, Tnf was upregulated among canonical MAPK signaling constituents differentially overexpressed in MDSC-KPCEV transcriptomes (Fig. 5C; Supplementary Fig. S8C–D).
Figure 5. Neutrophil-intrinsic TNF is a central regulator of MDSC function via Cxcl1-CXCR2-MAPK signaling.
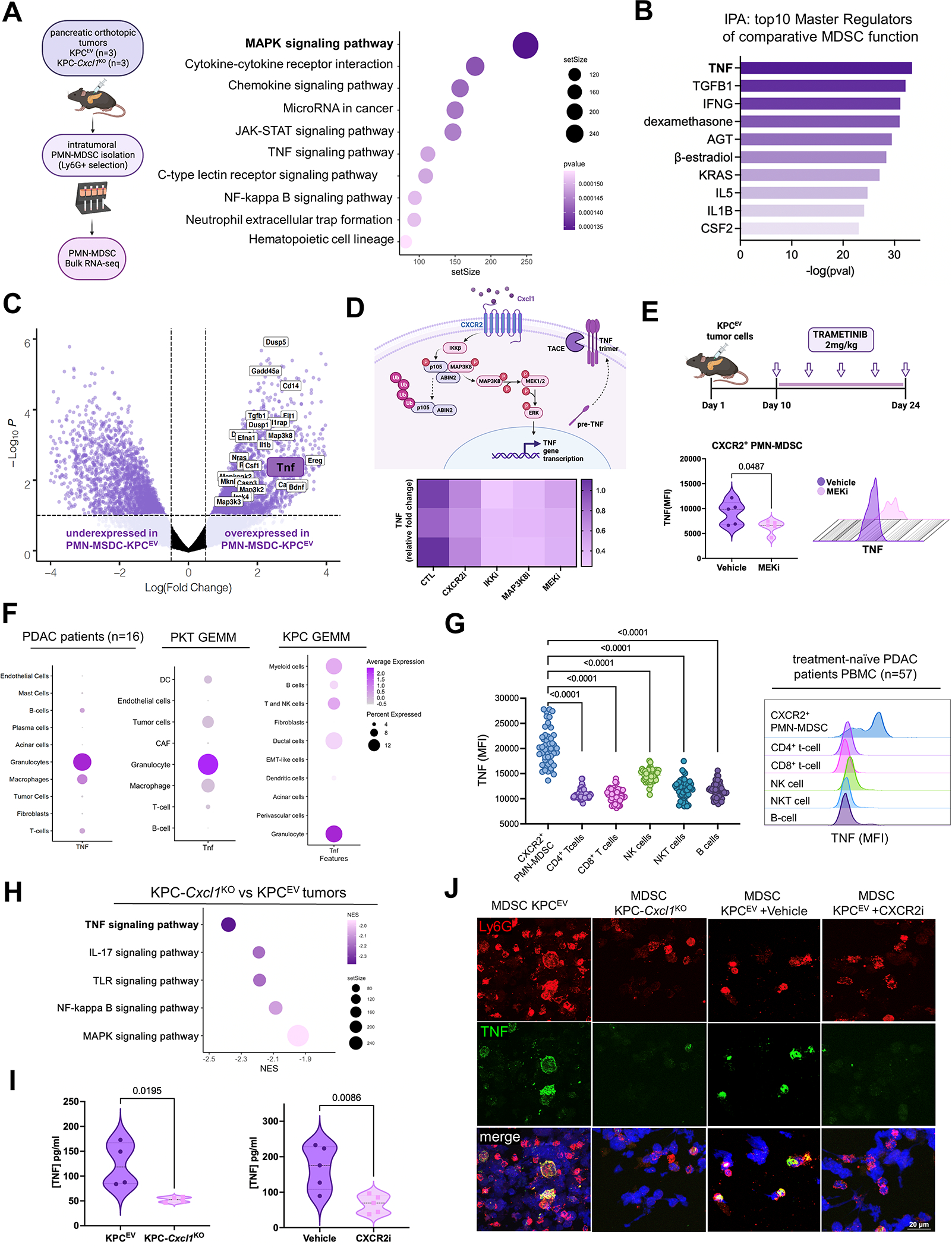
A, Schematic of intratumoral-PMN-MDSC isolation and subsequent RNA-sequencing from KPCEV and KPC-Cxcl1KO orthotopic tumors 3-weeks post-injection (left). Bubble plot depicts strongest differentially upregulated signaling pathways (using KEGG and Reactome knowledgebases) in PMN-MDSC infiltrating KPCEV relative to KPC-Cxcl1KO tumors (right), with adjusted P-value indicated in legend; B, Histogram representing top 10 predicted upstream regulators of MDSC function comparing KPCEV vs. KPC-Cxcl1KO-derived PMN-MDSCs via Ingenuity Pathway Analysis (IPA), with -log(P-value) indicated on x-axis; C, Volcano plot showing all genes from curated MAPK signaling pathways (using KEGG database) relatively enriched in MDSC-KPCEV vs. MDSC-KPC-Cxcl1KO tumors, with Tnf highlighted. Data are plotted as log(Fold Change) against -Log10P-value; D, Putative mechanism of CXCR2-MAPK-TNF signaling cascade in PMN-MDSCs (top), with heatmap visualizing relative reduction in Tnf expression upon treatment of J774M PMN-MDSCs with CXCR2 inhibitor AZD5069 (1 μM), IKK inhibitor BAY-110782 (0.1 μM), MAP3K8 inhibitor #871307–18-5 (250 nM), and MEK inhibitor trametinib (250 nM) (bottom), with relative fold change indicated in legend; E, In vivo dosing scheme of trametinib treatment in orthotopic KPC mice (top), with violin plot and representative histogram plot from flow cytometry experiments showing TNF mean fluorescence intensity (MFI) in intratumoral CXCR2+ PMN-MDSCs compared between vehicle and MEKi-treated groups (n=5/group); F, Dot plot showing relative TNF gene expression across cell clusters in single-cell RNA sequencing data from human PDAC patients (n=16)(15), PKT genetically engineered mouse model (GEMM) (16), and KPC GEMM (29); G, Plot and adjacent histogram plots showing TNF mean fluorescence intensity (MFI) via flow cytometry in peripheral blood mononuclear cells (PBMC) retrieved from treatment-naïve PDAC patients at UMiami (n=57); H, Bubble plot highlighting top 5 downregulated oncogenic signaling pathways (KEGG) in KPC-Cxcl1KO compared with KPCEV whole-tumor transcriptomes, with TNF signaling bolded; I, Violin plots showing TNF levels (in pg/mL) using ELISA in whole tumor lysates from KPCEV vs. KPC-Cxcl1KO (left), and vehicle-treated or CXCR2i AZD5069-treated orthotopic KPC mice (right) (n=5/group); J, Immunofluorescence using confocal microscopy showing Ly6G (red) and TNF (green) expression in polylysine coated cover slip-mounted intratumoral Ly6G+ cells from KPCEV, KPC-Cxcl1KO, KPCEV vehicle-treated, and KPCEV CXCR2i-treated tumor-bearing orthotopic mice.
Next, we explored the signaling link between CXCR2 ligation, MAPK pathway activation, and Tnf production in PMN-MDSCs. ScRNAseq data from Kras-Trp53 Panc02 (23) and PKT (16) models revealed robust co-expression of Cxcr2 and Tnf in single-cell PMN-MDSC transcriptomes (Supplementary Fig. S9A–B), as well as differential enrichment of MAPK signaling in Tnfhi compared with Tnflo PMN-MDSC single-cell transcriptomes (Supplementary Fig. S9C–D). Constructing STRING knowledgebase-annotated signaling interactomes within Cxcr2-Tnf co-expressing transcriptomes revealed a putative Cxclx-CXCR2-Map3k8-Tnf signaling node (Supplementary Fig. S9E). Indeed, the IKK-Map3k8 complex has been previously shown to regulate innate inflammatory signaling via MEK/ERK activation downstream of G-protein coupled receptor (e.g., PAR1) ligation (24). Pre-conditioning of murine CXCR2hi neutrophilic MDSC-like J774M cells (25) with recombinant Cxcl1, and treatment with CXCR2i AZD5069, IKKi BAY-110782(26), MAP3K8i(27), and MEKi trametinib significantly reduced Tnf gene expression (>50% in all, P<0.05; Fig. 5D). To validate the dependency of MDSC-intrinsic TNF on CXCR2-MAPK signaling in-vivo, we observed disproportionate reduction in TNF expression in CXCR2hi (P=0.04; Fig. 5E), but not CXCR2lo (Supplementary Fig. S9F), intratumoral PMN-MDSCs via flow cytometry in orthotopic KPC mice treated with MEKi trametinib (n=5/group).
PMN-MDSCs are the dominant source of TNF in PDAC
Given the unexpected emergence of MDSC-intrinsic TNF as a putative regulator of tumor-permissive MDSC function, we asked if PMN-MDSC-derived TNF was biologically relevant in human and murine PDAC. Examination of scRNAseq data from PDAC patients (15), PKT (16), or KPC GEMM mice (28) revealed that, compared with all other cellular constituents, PMN-MDSC transcriptomes exhibited highest TNF/Tnf expression (Fig. 5F). Moreover, using peripheral blood mononuclear cells retrieved from treatment-naïve PDAC patients at UMiami (n=57), we observed highest TNF expression in Lin−CD11b+CD14−CD15+ PMN-MDSCs compared with all other circulating immune populations (i.e., CD4+/CD8+ T-cells, NK, NKT, or B-cells; Fig. 5G).
Disruption of Cxcl1-CXCR2 engagement abolishes MDSC-restricted TNF production to attenuate global TNF signaling in the TME
Given these findings, we hypothesized that disrupting Cxcl1-CXCR2 axis would attenuate tumor-wide TNF signaling despite augmenting antitumor T-cell immunity. KPC-Cxcl1KO tumor transcriptomes revealed robust downregulation of TNF signaling pathways compared with KPCEV transcriptomes (P-adj<0.01; Fig. 5H; Supplementary Fig. S6C). Moreover, we observed significant reduction in TNF production from whole tumor lysates in KPC-Cxcl1KO (P=0.02, n=4) and CXCR2i AZD5069-treated (P=0.009, n=5; Fig. 5I) orthotopic KPC mice. In KPC-Cxcl1KO and CXCR2i-treated mice, abolition of intratumoral neutrophil-restricted TNF expression via confocal microscopy (Fig. 5J) is associated with global reduction in intratumoral TNF signaling. Altogether, these data reveal a previously unrecognized paradox in which inhibiting Cxcl1-CXCR2 engagement invigorates T-cell activation despite dampening tumor-wide TNF signaling, predominantly via disruption of MDSC-restricted TNF.
PMN-MDSC-derived TNF sustains tolerogenic circuitries via tmTNF-TNFR2 signaling
We have recently reported TNF as a regulator of innate immunoregulatory transcriptional networks in Ras-p53 PDAC (6). To build on these and current findings, we asked if such effects are driven by MDSC-derived TNF. Co-culture of J774M PMN-MDSCs (Supplementary Fig. S10A) with KPC tumor-cells or CAFs (Supplementary Fig. S10B) resulted in further >5-fold increase in Cxcl1 gene expression/secretion from same-well, but not transwell, co-cultures (Supplementary Fig. S11A–B). This dependence of pro-inflammatory signaling on cell-cell contact between PMN-MDSCs and tumor-cell/CAFs suggested a putative juxtacrine role for tmTNF-TNFR2—as opposed to paracrine soluble-TNF (sTNF)-TNFR1—signaling via in driving these effects. Indeed, although not constitutively expressed on CAF/tumor cells (Supplementary Fig. S11C), TNFR2 expression in CAFs was reciprocally upregulated while TNFR1 downregulated in a time-dependent manner upon co-culture with J774M cells (Supplementary Fig. S11D).
Next, compared with baseline expression, cell-intrinsic Cxcl1 expression was upregulated >4-fold when co-culturing KPC tumor cells (Fig. 6A) or CAFs (Fig. 6B) with intratumoral PMN-MDSCs derived from orthotopic KPC mice, but significantly reduced when intratumoral PMN-MDSCs were pre-conditioned with either etanercept (>70%) or CXCR2i AZD5069 (>60%) and utilized in ex vivo co-cultures (P<0.001). Notably, tumor cell/CAF-intrinsic Cxcl1 overexpression was unaffected by incubating MDSC-tumor cell or MDSC-CAF co-cultures with selective soluble-TNF inhibitor infliximab (29) (Fig. 6A–B, respectively).
Figure 6. PMN-MDSC-derived TNF sustains innate immunoregulatory and tolerogenic circuitry in the pancreatic tumor microenvironment.
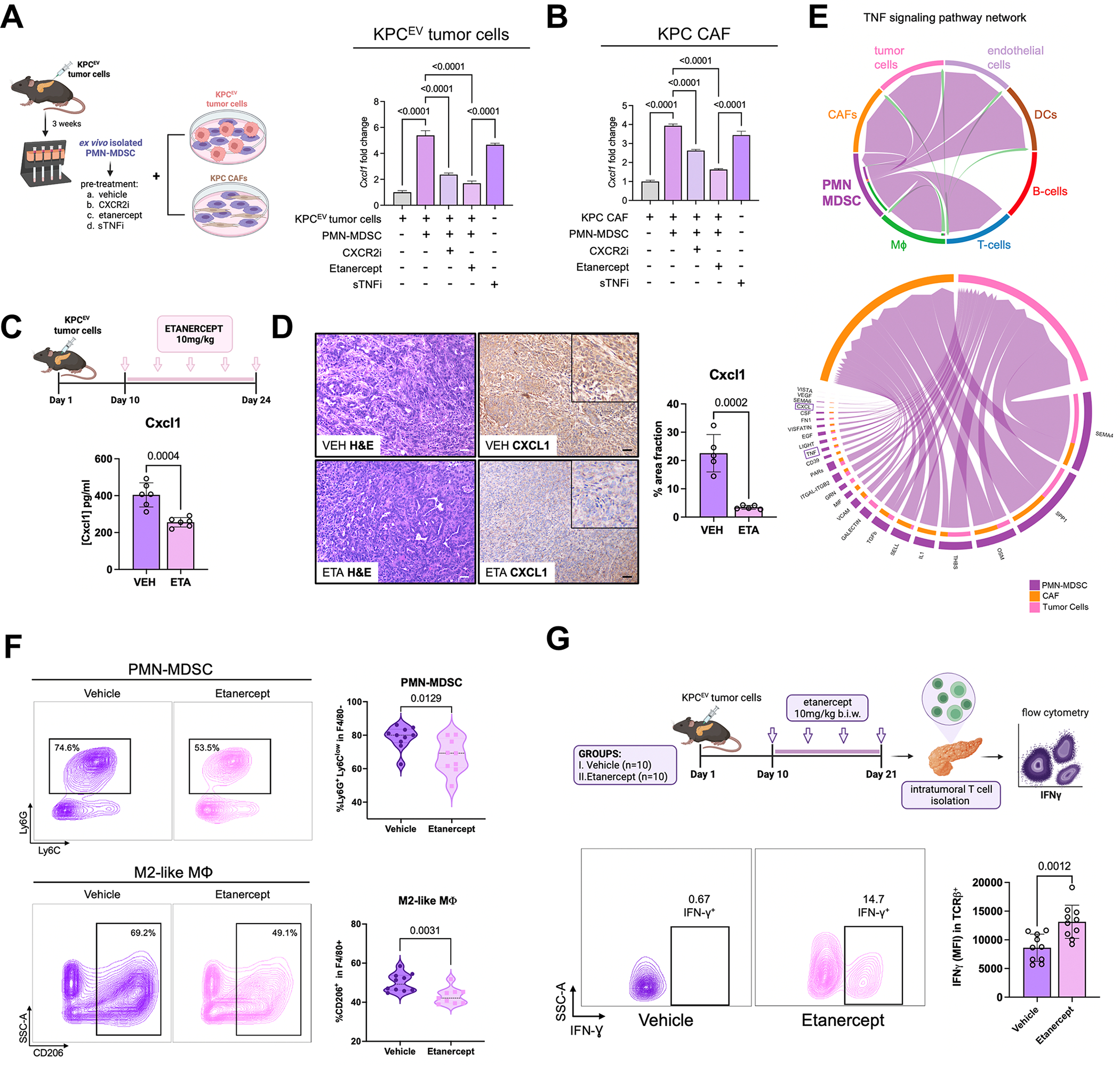
A-B, Experimental design of ex vivo co-cultures of intratumoral PMN-MDSCs with KPC tumor cells or KPC cancer-associated fibroblasts (CAF) (left). Histograms depicting relative Cxcl1 expression in KPC tumor cells (A) or KPC CAFs (B) alone, or co-cultured with PMN-MDSCs with or without preconditioning with CXCR2 inhibitor AZD5069 (1 μM), TNFR2 inhibitor etanercept (20 μg/ml), or soluble TNF inhibitor infliximab (sTNFi; 20 μg/ml) (right, n=4/group); C, In vivo dosing scheme of etanercept in orthotopic KPC mice (top), with bar plot showing Cxcl1 production (in pg/mL) by ELISA in whole tumor lysates from vehicle or etanercept (ETA)-treated mice; D, H&E and Cxcl1 immunostaining in matched tumor sections from vehicle or ETA-treated mice (both 20X; scale bar=50μm), with inset showing magnified region depicting epithelial-specific staining pattern. Adjacent bar plot shows quantification of %area Cxcl1 staining across biologic replicates (n=5 mice/group, 1 ROI/mouse); E, In single cell RNA-sequencing dataset from PKT genetically engineered mice (15), Circos plot visualizing directionality of TNF signaling pathway network from donor PMN-MDSC cluster to various cellular clusters (top), with adjoining Circos plot showing top donor ligands from PMN-MDSC (TNF highlighted in purple box) predicted to induce pro-inflammatory signaling genes/pathways (CXCL highlighted) in recipient tumor cell and CAF clusters; F, Representative contour plots and adjacent violin plots from flow cytometry experiments showing proportions (%of CD45+CD11b+) of PMN-MDSC and (%of CD45+CD11b+F4/80+) M2-like macrophages infiltrating vehicle or etanercept-treated KPC tumors (n=9–10 mice/group); G, Experimental schematic (top), with representative contour plots showing proportion of intratumoral IFN-γ+ CD3+ T-cells, and adjacent bar plot showing quantification of IFN-γ mean fluorescence intensity (MFI) within TCR-β+ T-cells across biologic replicates in vehicle- and etanercept-treated orthotopic KPC mice (n=10 mice/group).
Etanercept treatment—compared with vehicle—in orthotopic KPC mice significantly decreased tissue-level Cxcl1 production via ELISA in tumor lysates (Fig. 6C) as well as in epithelial/acinar and stromal compartments by IHC (Fig. 6D). Single-cell network analysis nominated TNF among dominant donor ligands from PMN-MDSCs which induce pro-inflammatory signaling in tumor-cell and CAF clusters—particularly expression of CXCLx genes—in PKT (Fig. 6E) and KPC (28); Supplementary Fig. S11E) tumor-derived scRNAseq datasets.
Systemic TNFR2 inhibition remodels an innate immune-enriched TME to augment T-cell activation
TNFR2i etanercept treatment in KPC tumor-bearing mice—compared with vehicle—significantly reduced neutrophil trafficking dynamics in adoptive transfer experiments (n=4, Supplementary Fig. S11F) as well as intratumoral abundance of F4/80−Ly6GhiLy6Cdim PMN-MDSCs and F4/80+MHCII−CD163+ M2-like macrophages via flow cytometry (Fig. 6F). Etanercept treatment concurrently augmented intratumoral trafficking of CD3+TCRβ+ T-cells (Supplementary Fig. S11G).
We next sought to investigate effects of tmTNF-TNFR2 inhibition on T-cell activation. Suppression of T-cell IFN-γ release following co-culture of CD3+ T-cells with untreated intratumoral PMN-MDSCs was significantly rescued following co-culture with etanercept-preconditioned PMN-MDSCs (n=4/group, P<0.001; Supplementary Fig. S11H). To corroborate these findings in vivo, intratumoral CD3+ T-cells isolated from etanercept-treated KPC mice demonstrated significantly increased IFN-γ+ expression compared with vehicle-treated mice (P=0.0012; Fig. 6G). Taken together, these data uncover a role for PMN-MDSC-derived tmTNF-TNFR2 signaling in perpetuating feed-forward immunoregulatory circuitries which promote immune tolerance in the PDAC TME.
PMN-MDSC-derived TNF promotes inflammatory CAF polarization and IL-6/STAT3 signaling
We have recently described the contribution of tumor-associated neutrophils in instigating iCAF polarization and chemoresistant IL-6/STAT3 crosstalk in PDAC (30). As such, intratumoral PMN-MDSCs derived from orthotopic KPC tumors induced expression of Il6 ~40-fold in KPC CAFs (Fig. 7A) as well as other canonical iCAF markers C3, Clec3b, and Lif (Supplementary Fig. S11I), in ex vivo co-cultures. This MDSC-induced iCAF gene expression was corroborated in human co-culture systems using HL-60 neutrophils and Patient-derived CAF PC-13 cells (31) (Supplementary Fig. S11J). In interrogating if MDSC-derived TNF mediates such iCAF polarization, CAF-Il6 expression was significantly reduced when PMN-MDSCs were pre-conditioned with etanercept (67% Il6 reduction; P<0.001) or CXCR2i (40% Il6 reduction; P<0.001). Interestingly, CAF-intrinsic Il6 was induced further when MDSC-CAF co-cultures were incubated with soluble-TNFi (Fig. 7A). Etanercept treatment of orthotopic KPC mice confirmed significant reduction in IL-6 levels in whole tumor lysates (Fig. 7B), and flow cytometric analysis demonstrated significant attenuation in iCAF (CD45−CD31−PDPN+Ly6C+MHCII−):myofibroblastic myCAF (CD45−CD31−PDPN+Ly6C−MHCII−) ratios (P=0.008; Fig. 7C). Etanercept treatment also generated striking remodeling of stromal organization—as evidenced by reduced acidic mucins (Alcian Blue) and fibrotic collagen deposition (Trichrome/Sirius Red) (Fig. 7D)—as well as substantial reduction in myofibroblast (PDPN+αSMA+) populations (Supplementary Fig. S11K).
Figure 7. Systemic inhibition of TNFR2 mitigates stromal inflammation and sensitizes PDAC to chemotherapy.
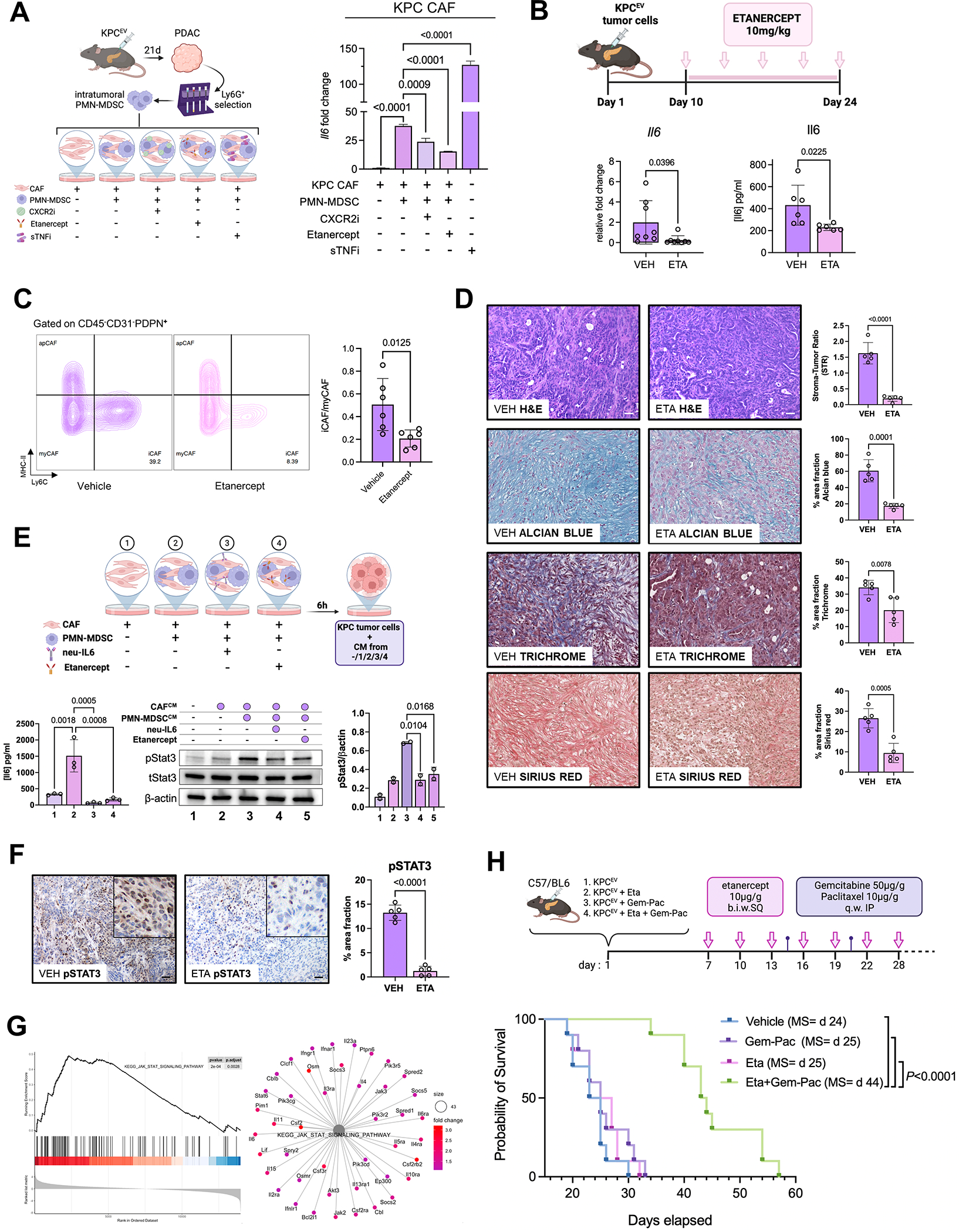
A, Experimental design of ex vivo co-cultures of intratumoral PMN-MDSCs with KPC cancer-associated fibroblasts (CAF) (left), with histograms depicting relative fold change of Il6 gene expression via qPCR in KPC CAFs alone, or CAFs co-cultured with PMN-MDSC with or without preconditioning with CXCR2i AZD5069 (1 μM), TNFR2i etanercept (20 μg/ml), or soluble TNF inhibitor infliximab (sTNFi; 20 μg/ml) (n=4/group); B, In vivo dosing scheme of etanercept in orthotopic KPC mice (top), with bar plot showing Il6 transcription (left) or protein levels by ELISA in whole tumor lysates from vehicle (VEH) or etanercept (ETA)-treated mice (n=6–8 mice/group); C, Representative contour plots showing inflammatory CAF (iCAF; Ly6C+MHC-II−), myofibroblastic CAF (myCAF; Ly6C−MHC-II−) and antigen-presenting CAF (apCAF; Ly6C−MHC-II+) populations gated on CD45−CD31−PDPN+ cells via flow cytometry in vehicle and etanercept-treated mice, and adjacent bar plot visualizing iCAF/myCAF ratio quantification across biologic replicates (n=6 mice/group); D, Representative H&E showing stromal-tumor ratio via H&E, Alcian Blue, Trichrome, and Sirius Red staining in tumor sections from vehicle or ETA-treated mice (scale bar=50μm), with adjacent bar plot visualizing respective quantifications across biologic replicates (n=5 mice/group, 1 ROI/mouse); E, Experimental design showing MDSC:CAF co-culture groups—labeled 1 through 4—from which conditioned media was generated and incubated with KPC6694c2 tumor cells (top). Bar plot showing IL-6 secretion via ELISA in KPC CAFs from experimental conditions 1–4 (bottom left, n=3). Western blot for pStat3 (tyr-705), total Stat3, and β-actin protein levels in KPC6694c2 tumor cell lysates upon conditioning with media from co-culture groups 1–4 (bottom center), with adjacent bar plot showing respective quantification of pStat3/β-actin ratio (bottom right); F, pSTAT3 immunostaining in tumor sections from vehicle or ETA-treated mice (both 20x; scale bar=50μm), with inset showing magnified region depicting epithelial-specific staining pattern. Adjacent bar plot shows quantification of %area staining for pSTAT3 across biologic replicates (n=5 mice/group); G, Enrichment plot (left) and net plot (right) showing disproportionate downregulation of KEGG_JAK_STAT_SIGNALING_PATHWAY in KPC-Cxcl1KO compared with KPCEV tumor transcriptomes; H, In vivo treatment schedules (top), and Kaplan-Meier survival curves for each of the treatment groups (n=10 mice/group): vehicle, etanercept (Eta) alone, gemcitabine+paclitaxel (Gem-Pac) alone, and Eta+Gem-Pac. Median survival (MS) of each group is indicated in parentheses.
We have previously demonstrated that paracrine activation of tumor cell-STAT3 via CAF-IL-6 is a key chemoresistant signaling node (32,33). Induction of pSTAT3 in KPC6694c2 tumor cells when incubated with conditioned media (CM) from untreated MDSC:CAF co-cultures was significantly reduced when incubated with CM from MDSC:CAF co-cultures treated with either anti-IL-6 neutralizing antibodies (P=0.017) or when using PMN-MDSCs pre-conditioned with etanercept (P=0.01; Fig. 7E). We also observed significant reduction in epithelial pSTAT3 expression in etanercept-treated KPC tumors via IHC (P<0.001; Fig. 7F). We then corroborated the relevance of Cxcl1-CXCR2-TNF axis in regulating tumor-wide STAT3 signaling by showing downregulation of KEGG_IL6_JAK_STAT3_SIGNALING pathway in transcriptomes from KPC-Cxcl1KO—with its incident paucity of TNF-licensed PMN-MDSCs (see Fig. 5J)—compared with KPCEV tumors (Fig. 7G).
Systemic TNFR2 inhibition improves sensitivity to chemotherapy
Since TNFR2i favorably remodels the immune microenvironment and mitigates stromal inflammation, we hypothesized that etanercept would improve chemosensitivity in vivo. In orthotopic KPC models, we observed no improvement in survival with etanercept monotherapy or gemcitabine+paclitaxel chemotherapy alone compared with vehicle treatment (P=ns). However, combining etanercept with gemcitabine+paclitaxel chemotherapy nearly doubled median survival (median 44 vs. 25 vs. 25 days, P<0.0001; Fig. 7H) and reduced metastatic outgrowth (Supplementary Fig. S12A) compared with single treatments. There was no additional toxicity when TNFR2 inhibition was co-administered with chemotherapy, as determined by mouse weights (Supplementary Fig. S12B). Together, these results uncover a role for TNFR2 inhibition in mitigating stromal inflammation and CAF:tumor cell IL-6/STAT3 signaling to sensitize PDAC to chemotherapy.
DISCUSSION
The present study illustrates a previously unrecognized cancer cell-neutrophil circuit that underlies the intersection between high-risk cancer genotypes (e.g., KRAS-TP53 cooperativity), tolerogenic immune contextures, and stromal inflammation that defines therapeutic resistance in PDAC. To first unravel the genotype-immunophenotype chasm, we employed integrative molecular analysis and single-cell mass cytometry in spatially annotated human tumors to show for the first time that disproportionate overexpression of Cxcl1 in epithelial/tumor cell islands exemplifying such high-risk genomic features dictates spatial exclusion of CD8+ T-cells in PDAC. These data not only provide in-human validation of the strong association between tumor cell-intrinsic Cxcl1 expression and T-cell exclusion in Kras-Trp53 cooperative KPC murine models(6), but also contribute Cxcl1 to a growing compendium of secreted cancer-cell autonomous factors (e.g., Cxcl5 (34,35), IL-8 (35), etc.) which predominantly interact with innate immune populations to thwart antitumor adaptive immunity in solid cancers. In addition, building on recent descriptions of the role of CREB in hyperactivating pro-metastatic networks in KRAS-TP53 cooperative PDAC (19) as well as the conflation of KRAS-TP53 cooperative transcriptomes, p63-mediated squamous trans-differentiation, and inflammatory gene modules in PDAC tumor cells (6,36), our data highlight CREB activation as a putative master transcriptional regulator of such inflammatory programming from cancer cells and raise the possibility that unifying cell-autonomous mechanisms may drive disparate hallmarks of its aggressive biology.
While cancer cell-autonomous inflammatory programming is undoubtedly the incipient event in establishing a hostile TME, our data suggest that these tumor cell-derived signals galvanize neutrophils into “activated” functional states, in turn rendering these PMN-MDSCs a dominant hub of extracellular signaling which perpetuate tumor-permissive circuitry in PDAC. As such, these data establish a causal link between cancer cell-autonomous factors and phenotypic plasticity in PMN-MDSCs. The pronounced dampening of T-cell suppressive function as well as “activated” CD14high signatures in intratumoral PMN-MDSCs derived from Cxcl1-silenced tumors lend support to the hypothesis that CD14high tumor-resident PMN-MDSCs are likely sculpted by tumor-derived signaling gradients in-situ and may not necessarily differentiate from obligate bone marrow-derived precursors(22). Moreover, the shared developmental ontogeny of T-cell suppressive Cd14 high and Tnf high neutrophilic states in our recently reported study employing intratumoral MDSC single-cell transcriptomes (6) may explain why disrupting Cxcl1-CXCR2 engagement invigorates adaptive antitumor immunity despite mitigating tumor-wide and neutrophil-restricted TNF signaling in PDAC. This intriguing paradox between heightened antitumor immunity and reduction in TNF signaling also offers contextual clues to help interpret recent data revealing reduced TNFα signaling as the only tumor-specific biomarker of response to gemcitabine, nab-paclitaxel, and nivolumab chemoimmunotherapy in advanced PDAC patients enrolled in the PRINCE trial (37). As such, our findings impel further investigations into understanding how chemo(immuno)therapy treatment trajectories impact the chronologic evolution of pro-inflammatory transcriptional programming (e.g., senescence-associated secretory phenotype (38)) in cancer cells and sculpt progressively tolerogenic TNFhigh myeloid ecosystems in PDAC. These efforts may inform discovery of temporally-sensitive biomarkers of response/resistance and therapeutically relevant targets at various stages of tumor progression.
The present data not only build on a growing body of evidence implicating tolerogenic and tumor-permissive TNF signaling in solid cancers (39–41), but also reveal novel insights into multiple aspects of TNF immunobiology. First, we were surprised to find that neutrophils are the major source of TNF in human—both in tumor tissue via scRNAseq, and circulation via flow cytometry in treatment-naïve patients’ PBMCs—and murine PDAC. While previous cancer-related studies investigating tolerogenic TNF functions have ascribed its cellular source to the stromal microenvironment rather than malignant cells (42), our data identify the precise cellular source of stromal TNF to recapitulate lessons learned from the infectious disease literature (43), where neutrophil-derived TNF is a dominant mediator of tissue injury and chronic inflammatory signaling. Second, we uncover direct and indirect effects of neutrophil-derived TNF on T-cell function/activation, leveraging findings from chronic inflammatory models where TNF has been vilified as a master regulator of T-cell dysfunction and exhaustion (44). While the majority of effects shown here—e.g., as observed in ex vivo co-culture experiments with etanercept-primed PMN-MDSCs and T-cells—are antigen-agnostic and may be related to activation-induced cell death and/or CD8+ regulatory polarization (CD8reg) (45,46), further investigation is warranted into whether neutrophil-derived tmTNF mediates antigen-restricted CD8+ T-cell tolerance indirectly via TNFR2high CD4+ Treg interactions (47). Third, although TNFR2 is constitutively expressed on immune cells, we discover that neutrophilic cellular contact can induce reciprocal TNFR1-to-TNFR2 expression dynamics in PDAC cancer cells and CAFs. Ensuing neutrophil-derived tmTNF-TNFR2 juxtacrine interactions amplify feed-forward pro-inflammatory cytokine production—particularly Cxcl1—in tumor cells/CAFs (48). These data expose novel redundancies in TNF-driven cell and non-cell autonomous signaling cascades that sustain immune dysfunction in the PDAC TME, and emphasize the need for thoughtful tmTNF-TNFR2 targeting strategies (49) to optimally unleash antitumor immunity in PDAC patients.
These data also establish—for the first time to our knowledge—an instructive role for neutrophil-derived tmTNF-TNFR2 signaling in regulating stromal inflammation, pro-inflammatory CAF-derived Cxcl1 and IL-6 secretion, and chemoresistant CAF:tumor cell IL-6/STAT3 signaling in PDAC. As such, TNFR2 signaling is enriched in tumor transcriptomes of advanced PDAC patients enrolled in the COMPASS trial (10) demonstrating chemo-resistant (i.e., progressive disease) compared with chemo-sensitive (i.e., stable disease and/or partial response) disease (Supplementary Fig. S12C). Fascinatingly, the relationship between tmTNF-TNFR2 signaling and CAF-derived IL-6 may offer a mechanistic explanation for the striking reduction in circulating IL-6 levels observed in metastatic breast cancer patients treated with etanercept in a non-randomized Phase II trial nearly 2 decades ago (50). Our findings can also be reconciled with the canonical model of iCAF polarization in which CAF-derived IL-6 (and Cxcl1, LIF, etc.) was predominantly driven by tumor cell-secreted IL-1α using patient-derived tumor cell/CAF organoid models(51). In such experimental models, the lack of immune cell reconstitution underestimates the contribution of innate immune cells in dictating fibroblast reprogramming. While acknowledging the undoubted contribution of tumor cell-derived IL-1α-IL1R1 signaling to iCAF skewness, the role of myeloid cell TNF-mediated effects on CAF plasticity described herein may explain the observation that silencing Il1a in tumor cells was insufficient to abolish iCAF polarization in vivo. Moreover, while recombinant TNF was sufficient to polarize iCAFs in ex vivo conditioning experiments, neutralization of soluble TNF did not impair induction of iCAF markers in pancreatic stellate cells co-cultured with organoid-conditioned media (51). These latter observations can now also be explained by our data: (i) tumor cells are not the dominant source of TNF in human or murine PDAC; and (ii) the failure to rescue inflammatory CAF polarization in experiments where soluble TNF is neutralized, underestimates the preferential dependence of iCAF determination on tmTNF-TNFR2 versus sTNF-TNFR1 signaling—findings reflected in the dramatically divergent Il6 expression in CAFs co-cultured with intratumoral PMN-MDSCs incubated with either TNFR2i or soluble-TNFi. Notwithstanding, the incomplete suppression of CAF-Il6 expression following tmTNF-TNFR2 inhibition in these MDSC-CAF co-cultures suggests additional mechanisms by which neutrophil-derived signaling governs iCAF polarization (e.g., PMN-MDSC-intrinsic IL-1β (30)). While we cannot yet estimate the relative “dosages” of neutrophilic or tumor cell contributions to inflammatory programming and transcriptional heterogeneity in PDAC CAFs, our data emphasize the need for a multi-pronged strategy to restrain both pro-inflammatory TNFR2 and IL-1 signaling to optimally subvert stromal inflammation and its tumor-permissive consequences in PDAC.
In summary, the present study “connects the dots” from high-risk cancer genotypes to unifying cell-autonomous mechanisms (i.e., CREB activation) regulating pro-inflammatory chemokines (i.e., Cxcl1) that dictate functional neutrophil plasticity to enforce T-cell exclusion in the PDAC TME, ultimately implicating an interwoven cancer cell-neutrophil Cxcl1-TNF circuit as a central regulator of stromal inflammation and T-cell dysfunction. While we utilized KRAS-TP53 cooperativity—emblematic of canonical “high-risk” PDAC genomes (6)—as a springboard to uncover this Cxcl1-tmTNF-TNFR2 circuitry, the applicability of the proposed signaling axis in PKT mice and in heterogeneous patient-derived datasets underscore its broader relevance in PDAC biology. Moreover, our findings conceptually challenge the dated model of exclusively antitumor TNF signaling in which global TNF silencing incapacitates T-cell-mediated immunologic control of tumors (52), and illustrates a more nuanced context-dependent influence of tmTNF-TNR2 signaling in mediating pro-tumorigenic effects in PDAC. Indeed, ongoing studies are examining if conditional amplification of neutrophil-restricted tmTNF recapitulates stromal and T-cell dysfunction in the PDAC TME. Moreover, a shift in our heuristic framework towards a contextually sensitive tumor-permissive TNF signaling model in cancer may also pave the way for novel bioengineered TNF-targeting immunotherapy design—e.g., strategies to intercept the competitive advantage of tolerogenic CD14highTNFhigh PMN-MDSC developmental states in the TME, or neutrophil-directed nano-targeting approaches to interrupt the transmembrane quarantining of TNF. Leveraging such novel strategies to reprogram the dynamic immunoregulatory control exerted by MDSC-derived TNF in the PDAC TME is of paramount importance since they can mitigate inflammatory stromal signaling and T-cell dysfunction to overcome therapeutic resistance in patients with PDAC.
METHODS
Transcriptomic analysis in CCLE dataset.
Genomic/transcriptomic sequencing data normalized in log2(RSEM+1),and clinical information (ver. 21Q4) were retrieved from CCLE (https://depmap.org/portal/ccle), and stratified into KRAS-TP53 co-altered (n=23) and KRAS-altered/TP53WT (n=5), as described previously(6). Comparative GSEA was performed using R package limma(53) (Supplementary Table S1). P-values was adjusted for multiple hypothesis testing based on Benjamini-Hochberg procedure(54).
Spatial annotation of human PDAC tumors via IMC.
Eight patients with resectable or borderline resectable PDAC who underwent upfront surgery at our institution were identified, archived FFPE blocks from resection specimens were retrieved, and post-hoc correlation with next-generation sequencing data from the medical record allowed genomic annotation into KRAS-TP53 co-altered (n=5) vs. KRAS-altered/TP53WT (n=3). For detailed clinicogenomic data, see Supplementary Table S2. One section was stained with hematoxylin-eosin (H&E) to enable a board-certified GI pathologist (E.M.) to select an ROI comprising tumor cells, fibroblasts, and immune cells from each unique tumor sample. A second section was stained with an IMC panel of 10 metal-conjugated antibodies and a cell intercalator (Supplementary Table S4), and the corresponding ROI was marked, and laser ablated using the Hyperion system (Fluidigm Inc.). Data were exported as MCD files and analyzed for single-cell segmentation, t-SNE clustering, and cellular neighborhood analysis using Visiopharm software. For details, refer to Supplementary Methods.
Human and mouse single-cell RNA sequencing and differential gene expression/pathway analysis.
Single nuclear RNAseq data from 43 patients were obtained from DUOS repository (ID 000139) (13). ScRNAseq data from 16 human PDAC patients and from 4 KPC GEMM mice were retrieved from the NIH Gene Expression Omnibus database (GSE155698 and GSE129455 respectively), and computational inference of cluster-specific ligand-receptor interactions was performed using NicheNet algorithm (17) and CellChat (55), as described in Supplementary Methods. scRNAseq of treatment-naïve tumors from 6.5-week-old PKT mice was performed as described previously (16), and detailed in Supplementary Methods.
Cell Lines, CRISPR/Cas9 genetic editing, and plasmid transfections.
Parent LSL-KrasG12D/+;Trp53R172H/+;Pdx1Cre/+ (KPC-6694c2) cells were transduced with CRISPR vector lentiCRISPR v2 with stable Cas9 expression (Addgene plasmid #52961) (56); non-targeting sgRNA infected cells designated as KPCEV), and deletion efficiency of Cxcl1 (KPC-Cxcl1KO) was confirmed as described previously(14). In orthotopic models, immunogenicity of parental KPC-6694c2 and KPCEV cells did not differ significantly.
Tissue-resident cancer-associated fibroblasts (CAF) from 6-month-old LSL-KrasG12D/+;Trp53R172H/+;Pdx-1Cre/+ (KPC) mice were isolated as previously described (9), and limited characterization with qPCR confirmed exclusive expression of fibroblast lineage markers Pdgfra and Col1a1 compared with neutrophil-like J774M and KPC-6694c2 cells (Supplementary Fig. S10B). Human PDAC cell lines Mia-PaCa2 (CRL-1420), Panc02 (CRL-2553), Capan-1 (HTB-79), Hs766T (HTB-134), PDM-168 (HCM-BROD-0229-C25), HL-60 (CCL-240) were purchased from American Type Culture Collection (ATCC). J774M cells were kindly gifted by Dr. Evanthia Torres/University of Southern California. Human hPNE-KRASWT and hPNE-KRASG12D cell lines were obtained from ATCC and maintained according to established guidelines. Mutant TP53R175H and/or TP53WT cDNA constructs were transiently overexpressed in hPNE-KRASWT or hPNE-KRASG12D cells as described previously (57). All cell lines were regularly tested using MycoAlert Mycoplasma Detection Kit (Lonza); for details, refer to Supplementary Methods.
Heterocellular co-culture experiments.
KPC CAFs or KPCEV tumor cells were co-cultured with either ex-vivo isolated Ly6G+ cells and/or J774M cells and conditioning media (CM) and RNA from co-cultured KPC CAFs or KPCEV tumor cells was collected. For details, refer to Supplementary Methods.
ChIP-seq and ChIP-qPCR.
Preparation of cross-linked RNA-free chromatin, sonication, and immunoprecipitation protocols using Creb (Cell Signaling, #9197S) and RNApol-II (Cell Signaling, #14958) antibodies in KPC 6694c2 tumor cells are described in Supplementary Methods. DNA was quantified with Qubit HS kit (Invitrogen), library preparation was performed using NEB Next Ultra DNA Library Prep Kit for Illumina (New England Biolabs, #E7370) following manufacturer instructions, and sequenced on a NextSeq6000 (Illumina). Fastq files were processed and analyzed as described previously (58), and detailed in Supplementary Methods. ChIP-qPCR was performed in KPC 6694c2 and KPC K-8484 tumor cells. For details on ChIP-qPCR for validation of ChIP-seq data and specific primers used, refer to Supplementary Methods.
Animal Models.
All animal experiments were performed in accordance with the NIH animal use guideline and protocol (21–057) approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Miami. C57BL/6 and Cd8atm1Mak (CD8α−/−) 6–8-week-old female mice were purchased from Jackson Laboratory. For orthotopic injection models using KPCEV or KPC-Cxcl1KO cells in these mice, refer to Supplementary Methods. PKT mice were generated as previously described (59).
RNA-sequencing from whole tumors and intratumoral PMN-MDSCs with downstream analysis.
Purified RNA was obtained from orthotopically injected KPCEV or KPC-Cxcl1KO whole tumors (following 21-day growth in-vivo), as well as intratumoral F4/80−Ly6Ghi PMN-MDSC cells (obtained after magnetic column separation from single-cell suspensions of whole tumors per manufacturer protocol (Miltenyi Biotec, #130-094-538) in biologic triplicates using RNeasy Kit (Qiagen). RNA quality was assessed on a bioanalyzer using the RNA6000 Nano kit (5067-1511; Agilent Technologies); 0.2–1 μg of RNA with RIN>7 proceeded for library preparation using Illumina TruSeq kit. Ensuing libraries were sequenced using Illumina NovaSeq SP300. Reads from RNA-Seq were mapped to Mus musculus genome (GRCm38) using STAR (v.2.5.0) aligner. Raw counts were generated based on Ensemble genes (GENCODE M13) with feature Counts (v.1.5.0). DE genes were identified using ESeq2 and determined by a threshold of FDR-corrected P-value<0.05. GSEA/pathway analysis was performed using Canonical Pathway, KEGG, REACTOME, Hallmark, Gene Ontology, and Immunologic signature gene sets retrieved from MSigDB/ImmuneSigDB databases.
Isolation of Ly6G+ cells from tumors and spleens.
Single-cell suspensions from whole pancreas or spleens obtained from KPCEV or KPC-Cxcl1KO orthotopic tumor-bearing mice were subjected to magnetic column separation, as described previously(6).
Flow Cytometry.
Single-cell suspensions from mouse tumors, spleens, and bone marrows from animals (as indicated), or PBMCs from chemotherapy-naïve PDAC patients (n=57) were thawed, washed, incubated with FcR-blocking reagent (Miltenyi Biotec), and subsequently stained with fluorescently conjugated antibodies (Supplementary Table S5). Flow cytometry data acquisition was performed on Cytek Aurora and analyzed using FlowJo v.10 software. For details of all tissue processing and flow cytometry procedures, refer to Supplementary Methods.
MDSC labeling with sulfo-Cy5.5-maleimide dye and in vivo adoptive transfer.
Cell surface thiol moieties on splenic Ly6G+ cells from KPCEV or KPC-Cxcl1KO mice were quantified using Ellman’s reagent (ThermoFisher #22582) per manufacturer protocol. Cells were washed with PBS and then resuspended at 1×106 cells/ml, incubated with 10μg of sulfo-Cy5.5-maleimide dye (Lumiprobe, #17380) at 4°C for 30 minutes. To remove unreacted dye content, cells were washed 3x times with PBS, maintained at 4°C, and used for adoptive transfer through tail vein injections in flank subcutaneous tumor-bearing animals. 24h following injection, mice were temporarily anesthetized using isoflurane and imaged using IVIS Spectrum optical imaging system (PerkinElmer).
T-cell suppression assays.
Ly6G+ cells isolated from tumor-bearing mice were co-cultured with stimulated T-cells, and T-cell activity was measured via IFN-γ secretion. T-cells were isolated from spleens of tumor-naïve C57BL/6 mice using magnetic column PanT cell isolation kit as per manufacturer’s recommendation (Miltenyi Biotec, #130-095-130). T-cells were co-cultured with Ly6G+ cells at varying ratios (1:1-1:8 T-cell:Ly6G) and anti-CD3/CD28 beads (Thermo Fisher Scientific) at a bead-to-T-cell ratio of 1:1. After 48h, condition media was collected for IFN-γ measurement by ELISA.
Confocal microscopy in intratumoral PMN-MDSCs.
Ly6G+ cells were isolated from orthotopic tumor as previously described (6,16,28). 5×105–1×106 cells were then seeded in a 24-well plate on Poly-L-lysine coated 12mm coverslips (#354085, Corning) for 2 hours. Ly6G+ cells were then co-stained as per immunofluorescence protocol described in Supplementary Methods.
RNA-in situ hybridization (ISH).
Pancreatic tissues were firstly processed for immunofluorescence analysis as previously described (6,16,28). Cxcl1 mRNA detection and amplification was performed on the same pancreatic tissue slides using HCR IHC + HCR RNA-ISH protocol (Molecular Instruments). For details, refer to Supplementary Methods.
Histopathologic analysis, western blotting, qPCR, ELISA, and cytokine array.
Histologic analysis, qPCR analysis, and western blotting procedures were performed as described previously (60). Antibodies used for western blotting are tabulated in Supplementary Table S6. Primers used for qPCR analysis are tabulated in Supplementary Table S7. For details, refer to Supplementary Methods.
Ethics Reporting.
Archival tissue acquisition was performed in accordance with protocols approved by the Institutional Review Board of the University of Miami (considered exempt). All studies were conducted in accordance with ethical guidelines outlined in the Declaration of Helsinki.
Supplementary Material
SIGNIFICANCE.
By decoding connections between high-risk tumor genotypes, cell-autonomous inflammatory programs, and myeloid-enriched/T-cell-excluded contexts, we identify a novel role for neutrophil-derived TNF in sustaining immunosuppression and stromal inflammation in pancreatic tumor microenvironments. This work offers a conceptual framework by which targeting context-dependent TNF signaling may overcome hallmarks of chemoresistance in pancreatic cancer.
ACKNOWLEDGEMENTS
We wish to thank Dr. Stephen Nimer for his mentorship and critical review of the manuscript, Dr. Wael El-Rifai for his generous mentorship and advice, Dr. Emiliano Cocco for assisting with research networking/collaborations, and Mr. Andrew Adams for technical assistance with ChIP-seq procedures. We wish to acknowledge the following Shared Resources at Sylvester Comprehensive Cancer Center for their able assistance of this project: Biospecimen (BSSR)—in particular Melinda Boone, Elena Cortizas, and Daniel Tran; Flow Cytometry (FSCR); Analytic Imaging (AISR); and Oncogenomics (OGSR)—in particular Dr. Sion Williams.
Financial Support:
This work was supported by KL2 career development grant by the Miami Clinical and Translational Science Institute (CTSI) under NIH Award UL1TR002736, American College of Surgeons Franklin H. Martin Research Fellowship, Association for Academic Surgery Joel J. Roslyn Faculty Award, Society of Surgical Oncology Young Investigator Award, Elsa U. Pardee Foundation Award, and Pancreatic Cancer Action Network Career Development Award (to J. Datta); NIH R01 CA161976 (to N. B. Merchant). Research reported in this publication was supported by the NCI/NIH Award P30CA240139.
Footnotes
Disclosure: The authors declare no potential conflicts of interest
Data Availability.
Bulk RNA sequencing data from: (1) KPCEV and KPC-Cxcl1KO whole tumors, and (2) sorted Ly6G+ intratumoral PMN-MDSCs isolated from KPCEV and KPC-Cxcl1KO tumors are available at BioProject #PRJNA926682 and #PRJNA926681, respectively. All other data generated in this study are available upon request from the corresponding author J.D. (jash.datta@med.miami.edu) and/or first author A.B. (axb2137@miami.edu).
REFERENCES
- 1.Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, et al. Pancreatic cancer. Nat Rev Dis Primers 2016;2:16022 doi 10.1038/nrdp.2016.22. [DOI] [PubMed] [Google Scholar]
- 2.Yu S, Zhang C, Xie KP. Therapeutic resistance of pancreatic cancer: Roadmap to its reversal. Biochim Biophys Acta Rev Cancer 2021;1875(1):188461 doi 10.1016/j.bbcan.2020.188461. [DOI] [PubMed] [Google Scholar]
- 3.Maddalena M, Mallel G, Nataraj NB, Shreberk-Shaked M, Hassin O, Mukherjee S, et al. TP53 missense mutations in PDAC are associated with enhanced fibrosis and an immunosuppressive microenvironment. Proc Natl Acad Sci U S A 2021;118(23) doi 10.1073/pnas.2025631118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trovato R, Fiore A, Sartori S, Cane S, Giugno R, Cascione L, et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J Immunother Cancer 2019;7(1):255 doi 10.1186/s40425-019-0734-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee K, Kumar S, Ross KA, Gautam S, Poelaert B, Nasser MW, et al. Emerging trends in the immunotherapy of pancreatic cancer. Cancer Lett 2018;417:35–46 doi 10.1016/j.canlet.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Datta J, Bianchi A, De Castro Silva I, Deshpande NU, Cao LL, Mehra S, et al. Distinct mechanisms of innate and adaptive immune regulation underlie poor oncologic outcomes associated with KRAS-TP53 co-alteration in pancreatic cancer. Oncogene 2022;41(28):3640–54 doi 10.1038/s41388-022-02368-w. [DOI] [PubMed] [Google Scholar]
- 7.Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extratumoral Macrophages. Gastroenterology 2015;149(1):201–10 doi 10.1053/j.gastro.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214(3):579–96 doi 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dosch AR, Singh S, Dai X, Mehra S, Silva IC, Bianchi A, et al. Targeting Tumor-Stromal IL6/STAT3 Signaling through IL1 Receptor Inhibition in Pancreatic Cancer. Mol Cancer Ther 2021;20(11):2280–90 doi 10.1158/1535-7163.MCT-21-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aung KL, Fischer SE, Denroche RE, Jang GH, Dodd A, Creighton S, et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin Cancer Res 2018;24(6):1344–54 doi 10.1158/1078-0432.CCR-17-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dosch AR, Chatila WK, Ban Y, Bianchi A, Deshpande NU, Silva IC, et al. Ras-p53 genomic cooperativity as a model to investigate mechanisms of innate immune regulation in gastrointestinal cancers. Oncotarget 2021;12(20):2104–10 doi 10.18632/oncotarget.27983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. Addendum: The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2019;565(7738):E5–E6 doi 10.1038/s41586-018-0722-x. [DOI] [PubMed] [Google Scholar]
- 13.Hwang WL, Jagadeesh KA, Guo JA, Hoffman HI, Yadollahpour P, Reeves JW, et al. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat Genet 2022;54(8):1178–91 doi 10.1038/s41588-022-01134-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018;49(1):178–93 e7 doi 10.1016/j.immuni.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steele NG, Carpenter ES, Kemp SB, Sirihorachai V, The S, Delrosario L, et al. Multimodal Mapping of the Tumor and Peripheral Blood Immune Landscape in Human Pancreatic Cancer. Nat Cancer 2020;1(11):1097–112 doi 10.1038/s43018-020-00121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Datta J, Dai X, Bianchi A, De Castro Silva I, Mehra S, Garrido VT, et al. Combined MEK and STAT3 Inhibition Uncovers Stromal Plasticity by Enriching for Cancer-Associated Fibroblasts With Mesenchymal Stem Cell-Like Features to Overcome Immunotherapy Resistance in Pancreatic Cancer. Gastroenterology 2022;163(6):1593–612 doi 10.1053/j.gastro.2022.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Browaeys R, Saelens W, Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods 2020;17(2):159–62 doi 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- 18.Lee KM, Nguyen C, Ulrich AB, Pour PM, Ouellette MM. Immortalization with telomerase of the Nestin-positive cells of the human pancreas. Biochem Biophys Res Commun 2003;301(4):1038–44 doi 10.1016/s0006-291x(03)00086-x. [DOI] [PubMed] [Google Scholar]
- 19.Kim MP, Li X, Deng J, Zhang Y, Dai B, Allton KL, et al. Oncogenic KRAS Recruits an Expansive Transcriptional Network through Mutant p53 to Drive Pancreatic Cancer Metastasis. Cancer Discov 2021;11(8):2094–111 doi 10.1158/2159-8290.CD-20-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Totiger TM, Srinivasan S, Jala VR, Lamichhane P, Dosch AR, Gaidarski AA 3rd, et al. Urolithin A, a Novel Natural Compound to Target PI3K/AKT/mTOR Pathway in Pancreatic Cancer. Mol Cancer Ther 2019;18(2):301–11 doi 10.1158/1535-7163.MCT-18-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Messaggio F, Mendonsa AM, Castellanos J, Nagathihalli NS, Gorden L, Merchant NB, et al. Adiponectin receptor agonists inhibit leptin induced pSTAT3 and in vivo pancreatic tumor growth. Oncotarget 2017;8(49):85378–91 doi 10.18632/oncotarget.19905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veglia F, Hashimoto A, Dweep H, Sanseviero E, De Leo A, Tcyganov E, et al. Analysis of classical neutrophils and polymorphonuclear myeloid-derived suppressor cells in cancer patients and tumor-bearing mice. J Exp Med 2021;218(4) doi 10.1084/jem.20201803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan Y, Lu F, Fei Q, Yu X, Xiong P, Yu X, et al. Single-cell RNA sequencing reveals compartmental remodeling of tumor-infiltrating immune cells induced by anti-CD47 targeting in pancreatic cancer. J Hematol Oncol 2019;12(1):124 doi 10.1186/s13045-019-0822-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hatziapostolou M, Koukos G, Polytarchou C, Kottakis F, Serebrennikova O, Kuliopulos A, et al. Tumor progression locus 2 mediates signal-induced increases in cytoplasmic calcium and cell migration. Sci Signal 2011;4(187):ra55 doi 10.1126/scisignal.2002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sidiropoulos DN, Rafie CI, Jang JK, Castanon S, Baugh AG, Gonzalez E, et al. Entinostat Decreases Immune Suppression to Promote Antitumor Responses in a HER2+ Breast Tumor Microenvironment. Cancer Immunol Res 2022;10(5):656–69 doi 10.1158/2326-6066.CIR-21-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee J, Rhee MH, Kim E, Cho JY. BAY 11–7082 is a broad-spectrum inhibitor with anti-inflammatory activity against multiple targets. Mediators Inflamm 2012;2012:416036 doi 10.1155/2012/416036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gavrin LK, Green N, Hu Y, Janz K, Kaila N, Li HQ, et al. Inhibition of Tpl2 kinase and TNF-alpha production with 1,7-naphthyridine-3-carbonitriles: synthesis and structure-activity relationships. Bioorg Med Chem Lett 2005;15(23):5288–92 doi 10.1016/j.bmcl.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 28.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 2019;9(8):1102–23 doi 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mpofu S, Fatima F, Moots RJ. Anti-TNF-alpha therapies: they are all the same (aren’t they?). Rheumatology (Oxford) 2005;44(3):271–3 doi 10.1093/rheumatology/keh483. [DOI] [PubMed] [Google Scholar]
- 30.de Castro Silva I, Bianchi A, Deshpande NU, Sharma P, Mehra S, Garrido VT, et al. Neutrophil-mediated fibroblast-tumor cell il-6/stat-3 signaling underlies the association between neutrophil-to-lymphocyte ratio dynamics and chemotherapy response in localized pancreatic cancer: A hybrid clinical-preclinical study. Elife 2022;11 doi 10.7554/eLife.78921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagathihalli NS, Castellanos JA, Lamichhane P, Messaggio F, Shi C, Dai X, et al. Inverse Correlation of STAT3 and MEK Signaling Mediates Resistance to RAS Pathway Inhibition in Pancreatic Cancer. Cancer Res 2018;78(21):6235–46 doi 10.1158/0008-5472.CAN-18-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagaraj NS, Smith JJ, Revetta F, Washington MK, Merchant NB. Targeted inhibition of SRC kinase signaling attenuates pancreatic tumorigenesis. Mol Cancer Ther 2010;9(8):2322–32 doi 10.1158/1535-7163.MCT-09-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagaraj NS, Washington MK, Merchant NB. Combined Blockade of Src Kinase and Epidermal Growth Factor Receptor with Gemcitabine Overcomes STAT3-Mediated Resistance of Inhibition of Pancreatic Tumor Growth. Clin Cancer Res 2011;17(3):483–93 doi 1078–0432.CCR-10–1670 [pii] 10.1158/1078-0432.CCR-10-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018;67(6):1112–23 doi 10.1136/gutjnl-2017-313738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lopez-Bujanda ZA, Haffner MC, Chaimowitz MG, Chowdhury N, Venturini NJ, Patel RA, et al. Castration-mediated IL-8 promotes myeloid infiltration and prostate cancer progression. Nat Cancer 2021;2(8):803–18 doi 10.1038/s43018-021-00227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Somerville TD, Biffi G, Dassler-Plenker J, Hur SK, He XY, Vance KE, et al. Squamous trans-differentiation of pancreatic cancer cells promotes stromal inflammation. Elife 2020;9 doi 10.7554/eLife.53381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Padron LJ, Maurer DM, O’Hara MH, O’Reilly EM, Wolff RA, Wainberg ZA, et al. Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nat Med 2022;28(6):1167–77 doi 10.1038/s41591-022-01829-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu X, Ghisolfi L, Keates AC, Zhang J, Xiang S, Lee DK, et al. Induction of cancer cell stemness by chemotherapy. Cell Cycle 2012;11(14):2691–8 doi 10.4161/cc.21021. [DOI] [PubMed] [Google Scholar]
- 39.Arnott CH, Scott KA, Moore RJ, Robinson SC, Thompson RG, Balkwill FR. Expression of both TNF-alpha receptor subtypes is essential for optimal skin tumour development. Oncogene 2004;23(10):1902–10 doi 10.1038/sj.onc.1207317. [DOI] [PubMed] [Google Scholar]
- 40.Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med 1999;5(7):828–31 doi 10.1038/10552. [DOI] [PubMed] [Google Scholar]
- 41.Torrey H, Butterworth J, Mera T, Okubo Y, Wang L, Baum D, et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci Signal 2017;10(462) doi 10.1126/scisignal.aaf8608. [DOI] [PubMed] [Google Scholar]
- 42.Montfort A, Colacios C, Levade T, Andrieu-Abadie N, Meyer N, Segui B. The TNF Paradox in Cancer Progression and Immunotherapy. Front Immunol 2019;10:1818 doi 10.3389/fimmu.2019.01818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cagnina RE, Michels KR, Bettina AM, Burdick MD, Scindia Y, Zhang Z, et al. Neutrophil-Derived Tumor Necrosis Factor Drives Fungal Acute Lung Injury in Chronic Granulomatous Disease. J Infect Dis 2021;224(7):1225–35 doi 10.1093/infdis/jiab188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baxter AE, Kaufmann DE. Tumor-necrosis factor is a master of T cell exhaustion. Nat Immunol 2016;17(5):476–8 doi 10.1038/ni.3436. [DOI] [PubMed] [Google Scholar]
- 45.Ban L, Zhang J, Wang L, Kuhtreiber W, Burger D, Faustman DL. Selective death of autoreactive T cells in human diabetes by TNF or TNF receptor 2 agonism. Proc Natl Acad Sci U S A 2008;105(36):13644–9 doi 10.1073/pnas.0803429105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bertrand F, Montfort A, Marcheteau E, Imbert C, Gilhodes J, Filleron T, et al. TNFalpha blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat Commun 2017;8(1):2256 doi 10.1038/s41467-017-02358-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Govindaraj C, Scalzo-Inguanti K, Madondo M, Hallo J, Flanagan K, Quinn M, et al. Impaired Th1 immunity in ovarian cancer patients is mediated by TNFR2+ Tregs within the tumor microenvironment. Clin Immunol 2013;149(1):97–110 doi 10.1016/j.clim.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 48.Ben-Baruch A Tumor Necrosis Factor alpha: Taking a Personalized Road in Cancer Therapy. Front Immunol 2022;13:903679 doi 10.3389/fimmu.2022.903679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Case K, Tran L, Yang M, Zheng H, Kuhtreiber WM, Faustman DL. TNFR2 blockade alone or in combination with PD-1 blockade shows therapeutic efficacy in murine cancer models. J Leukoc Biol 2020;107(6):981–91 doi 10.1002/JLB.5MA0420-375RRRRR. [DOI] [PubMed] [Google Scholar]
- 50.Madhusudan S, Foster M, Muthuramalingam SR, Braybrooke JP, Wilner S, Kaur K, et al. A phase II study of etanercept (Enbrel), a tumor necrosis factor alpha inhibitor in patients with metastatic breast cancer. Clin Cancer Res 2004;10(19):6528–34 doi 10.1158/1078-0432.CCR-04-0730. [DOI] [PubMed] [Google Scholar]
- 51.Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov 2019;9(2):282–301 doi 10.1158/2159-8290.CD-18-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baxevanis CN, Voutsas IF, Tsitsilonis OE, Tsiatas ML, Gritzapis AD, Papamichail M. Compromised anti-tumor responses in tumor necrosis factor-alpha knockout mice. Eur J Immunol 2000;30(7):1957–66 doi . [DOI] [PubMed] [Google Scholar]
- 53.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43(7):e47 doi 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met 1995;57(1):289–300. [Google Scholar]
- 55.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun 2021;12(1):1088 doi 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11(8):783–4 doi 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang Z, Ju H, Ling J, Zhuang Z, Li Z, Wang H, et al. Cooperativity of oncogenic K-ras and downregulated p16/INK4A in human pancreatic tumorigenesis. PLoS One 2014;9(7):e101452 doi 10.1371/journal.pone.0101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 2017;355(6331):1324–30 doi 10.1126/science.aah6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chytil A, Magnuson MA, Wright CV, Moses HL. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis 2002;32(2):73–5 doi 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
- 60.Nagathihalli NS, Castellanos JA, Shi C, Beesetty Y, Reyzer ML, Caprioli R, et al. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology 2015;149(7):1932–43 e9 doi 10.1053/j.gastro.2015.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNA sequencing data from: (1) KPCEV and KPC-Cxcl1KO whole tumors, and (2) sorted Ly6G+ intratumoral PMN-MDSCs isolated from KPCEV and KPC-Cxcl1KO tumors are available at BioProject #PRJNA926682 and #PRJNA926681, respectively. All other data generated in this study are available upon request from the corresponding author J.D. (jash.datta@med.miami.edu) and/or first author A.B. (axb2137@miami.edu).