


Abstract
Tetracycline (tet)-responsive expression vectors allow controlled inducible expression of proteins in mammalian cells. This system is widely used for experimental research both in vivo and in vitro. In our attempts to use this system to study the antiviral effect of IFNα on hepatitis B virus, we discovered an unexpected feature of the tet-responsive promoter (tet promoter) of the currently available expression vectors. IFNα was found to stimulate tet promoter activity after transient transfection in a dose- and cell type-dependent manner. By sequence inspection, an IFNα-stimulated response element (ISRE)-like sequence was identified in the linker regions located between the heptameric tet operator sequences. Gel shift assays revealed binding of IFN-stimulated gene factors to these sequences, indicating that they mediate the IFNα-mediated promoter stimulation. These data demonstrate an unexpected feature of the tet-responsive expression system which needs to be taken into acount when using this system for analysis of cytokine functions in vitro and in vivo. The data also imply that the tet promoter-based expression system can be rendered non-responsive to IFNα by mutagenesis of the ISREs and this may be essential when considering gene therapy in vivo.
INTRODUCTION
The tetracycline (tet)-responsive expression system generated by Gossen and Bujard (1) provides a genetic tool for controlled gene expression in eukaryotic cells that is highly appreciated in the scientific community, as illustrated by a correspondingly huge number of research articles. The inducibility of this system is based on regulatory elements of the Tn10 encoded tetracycline resistance operon of Escherichia coli (2). For generation of an inducible tet-responsive transactivator (tTA) the tet repressor of Tn10 of E.coli was fused to the transactivating domain of virion protein 16 (VP16) of herpes simplex virus (1). This fusion protein binds to the tetracycline-responsive operator (tet operator) region inserted as a heptamer separated by linker sequences and placed immediately upstream of a minimal CMV promoter. Mediated by the VP16 domain, the binding of tTA to the tet operators results in activation of the promoter (designated from here on as the tet promoter). In the presence of tet, tTA does not bind to the tet operator and therefore the promoter is inactive, whereas withdrawal of tet allows its activation. Thus, the promoter activity can be reversibly switched on and off, which allows controlled expression of an individual gene in vitro and in vivo (3–6). Besides exploitation of this expression system for the study of gene function in complex genetic environments, many attempts have been initiated to apply this system to gene therapy (7–9).
Inducible viral RNA synthesis, which can be switched off at specific times when using the tet system, offers an excellent opportunity to study post-transcriptional mechanisms in virus–host interactions. We have recently demonstrated that IFNα inhibits hepatitis B virus (HBV) gene expression at two levels at least, one of which involves post-transcriptional degradation of RNA and the other leads to a reduction in replicative intermediates and/or core protein (10). The aim of our current study was to characterize the corresponding mechanisms using the tet system. HBV is a small (3.2 kb) enveloped DNA virus, the replication of which involves reverse transcription of a pregenomic RNA (11). Reverse transcription of the pregenomic RNA takes place within nucleocapsids in the cytoplasm. Nucleocapsids containing replicative DNA intermediates are either shuttled to the endoplasmic reticulum and there converted to mature virions for transport out of the cell or to the nucleus for the establishment of a pool of covalently closed circular DNA (12). The circular DNA in the nucleus serves as template for transcription of the pregenomic and subgenomic RNAs, which initiate at different promoters on the circular genome but share a common 3′-end.
World wide, there are more than 300 000 000 people suffering from chronic HBV infection. Treatment of chronic HBV carriers with IFNα is one of the few effective interventions for patients with hepatitis B. It results in an efficient reduction in the viral load in only 10–20% of treated patients and rarely, if at all, in complete virus elimination. So far, the mechanisms induced by IFNα responsible for the reduction in viremia in responding patients are still elusive. IFNα is known to induce a so-called antiviral state in treated cells that interferes with viral infection (13). The antiviral state is principly mediated by the induction of IFN-inducible genes (14). Gene induction involves binding of IFNα to a specific cell surface receptor followed by tyrosine phosphorylation of the receptor, of cytoplasmic protein tyrosine kinases Tyk2 and Jak1 and of latent cytoplasmic transcription factor subunits called STATs (signal transducers and activators of transcription) (15–20). The STATs then assemble to form functional IFN-stimulated gene factors (ISGF) which translocate into the nucleus. Within the nucleus the ISGFs specifically interact with IFN-stimulated response elements (ISRE) located within the promoter region of IFNα-inducible genes. So far, three major ISGF complexes, ISGF1, ISGF2 and ISFG3, have been characterized which bind ISRE and regulate the expression of IFNα-inducible genes. Binding of ISGF2 and ISGF3 to ISRE mediates positive transcriptional regulation, and ISGF1 is currently believed to act negatively on ISGF2 (20). Binding of ISGFs can be detected by gel mobility shift assays, for example with a labeled ISRE oligonucleotide derived from the promoter of IFN-stimulated gene 15 (ISG15) (21).
Here we describe the identification of functional ISREs located within the tet promoter region which renders the promoter IFNα responsive in transiently transfected cells. The data reveal a novel feature of the tet promoter, which needs to be taken into account when using this inducible expression system and limits its use in studies on cytokine functions.
MATERIALS AND METHODS
Plasmids
Plasmid ptetHBV (22) was kindly provided by Dr Christoph Seeger. This plasmid contains a more than full-length HBV genome in which synthesis of the pregenomic RNA is under control of the tet promoter. For our experiments this plasmid was modified by converting the start codon of the HBV preS1 envelope protein encoding gene from ATG to ACG. This was achieved by PCR-mediated oligo-directed mutagenesis. Correct mutagenesis was verified by sequencing of the amplified region. This plasmid was designated ptetHBV-M1T.
Plasmid pHBV-dimer contains a HBV head-to-tail dimer DNA of subtype ayw cloned via the EcoRI site (23). All HBV RNAs derived from this plasmid are transcribed under control of the authentic viral promoters. Plasmids pUHD15-1, which expresses the tTA, and pUHC13-3, which codes for the luciferase gene of Photinus pyralis, expression of which is under control of the tet promoter, were kindly provided by Dr M. Gossen (1).
Cell culture and transfection procedure
Human HuH7-, U2-OS and 293 cells were maintained as monolayers in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum. All plasmid DNAs used for transfection were purified by ion exchange chromatography and were transfected using the transfection reagent Fugene 6 (Roche Diagnostics, Mannheim, Germany) according to the supplied protocol. Cells (5 × 105 cells/2.5 cm well) were transfected with 1 µg of ptetHBV-M1T, pHBV-dimer or pUHC13-3 and 0.2 µg pUHD15-1. At different times after transfection tet-regulated expression was turned off by addition of 2 µg/ml tet (Sigma, Deisenhofen, Germany) to the medium. Twenty-four hours after turn off by addition of tet, IFNα-2b (1.000 IU/ml, Intron A; Essex Pharma, Munich, Germany) was added to the medium and cells were harvested for analysis at the times indicated.
Purification of HBV DNA from intracellular core particles and HBV RNA
Total RNA was prepared by use of an anion exchange procedure (RNeasy; Qiagen, Hilden, Germany) according to the supplied protocol. For isolation of intracellular HBV DNA, cells were washed with ice-cold PBS and lysed in 0.5 ml lysis buffer (50 mM Tris–HCl, pH 7.4, 1 mM EDTA, 1% Nonidet P-40) per 2.5 cm well. Nuclei were pelleted by centrifugation for 1 min at 14 000 r.p.m. in a table top centrifuge. The supernatant was adjusted to 10 mM MgCl2 and treated with 100 mg/ml DNase I for 30 min at 37°C for digestion of non-encapsidated viral DNA. DNase I digestion was stopped by adding EDTA to a final concentration of 25 mM. Proteins were digested with 0.5 mg/ml proteinase K in 1% SDS for 2 h at 37°C. Nucleic acids were purified by phenol/chloroform (1:1) extraction and ethanol precipitation after adding 20 µg tRNA.
Southern and northern blot analysis
DNA isolated from cytoplasmic core particles was separated on a 1.5% agarose gel. Twenty micrograms of total RNA were separated on a 1.5% agarose–formaldehyde gel. DNAs and RNAs were blotted onto Hybond N nylon membranes (Amersham International, Little Chalfont, UK) and hybridized with a 32P-labeled gel-purified full-length HBV fragment. Probes were generated using a random primed labeling kit (Amersham International). Blots were exposed to Fuji imaging screens and quantified with a Fujix BAS 2000 Bio-imaging Analyzer (Fuji, Tokyo, Japan) and by TINA software (Raytest, Straubenhardt, Germany). Signal intensities were quantified within the linear range.
Protein extracts and gel mobility shift assay
Nuclear protein extracts were prepared as described previously (24). Briefly, after cell lysis on ice for 10 min, nuclei were collected by centifugation in a table top centrifuge for 1 min at 4°C (14 000 r.p.m.), resuspended in high salt buffer and incubated on ice for 20 min. After centrifugation for 10 min at 4°C (14 000 r.p.m.) the supernatant containing the soluble nuclear proteins was recovered. Protein content was determined according to Bradford (25). For gel mobility shift assays 5 µg of nuclear proteins were preincubated for 5 min at room temperature with 4 µg of poly(dI-dC) and 100 ng of unrelated single-strand oligonucleotide (5′-GGT TGT TGA TTA TCA TTG AGA TTC CCG AG-3′) in 25 µl of 20 mM HEPES (pH 7.9), 50 mM NaCl, 1 mM MgCl2, 0.1 mM EGTA, 0.5 mM dithiothreitol, 0.1% Nonidet P-40 and 10% glycerol. After addition of 32P-labeled double-stranded ISG15 oligonucleotide (20 000 c.p.m., corresponding to ~2 fmol) and incubation for 20 min at room temperature, DNA–protein complexes were analyzed on 6% acrylamide (80:1) gels run in 0.5× TBE. For competition unlabeled double-stranded oligonucleotides were used at 5-, 50- and 500-fold molar excess.
Double-stranded oligonucleotides used as probe and/or as competitor in gel mobility shift assays (upper strand): ISG15, 5′-GGG AAA GGG AAA CCG AAA CTG AAG CC-3′; Tet-ISRE, 5′-TGA TAG AGA AAA GTG AAA GTC GAG TTT ACC AC-3′; random oligo, 5′-GAT ATA GAT TCT GAT TTT GGA GAA GAG TCT CTC TTT GAT CTG TTC CTC TCA GA-3′.
Luciferase assay
Quantification of luciferase activity in transfected cells was performed by use of the Promega luciferase assay system (Promega, Madison, WI) and a Microlumat LB96P (Berthold, Wildbad, Germany) according to the manuals supplied.
RESULTS AND DISCUSSION
Tetracycline-mediated control of synthesis of HBV replicative intermediates in a transient transfection system
In order to study whether IFNα leads to a direct reduction in mature replicative intermediates, we needed a system which allows controlled expression of these molecules. After accumulation of mature replicative intermediates further synthesis should be prevented before addition of IFNα. We reasoned that this aim could be achieved by using the tet-controlled expression system and by preventing envelopment of the core particles and their secretion (26). Therefore, we used plasmid ptetHBV-M1T which allows tet-controlled expression of the pregenomic HBV RNA from which no preS1 protein can be synthesized (see Materials and Methods).
First, we tested whether synthesis of replicative intermediates can be regulated tightly by tet after transient transfection of human hepatoma HuH7 cells, which are known to replicate the virus. Therefore, the amount of intracellular replicative DNA intermediates produced in HuH7 cells treated with and without tet before co-transfection of the plasmids ptetHBV-M1T and pUHD15-1 was determined by Southern blotting. There were large amounts of replicative DNA intermediates in HuH7 cells when grown in the absence of tet and hardly detectable levels when tet was present (Fig. 1). Quantitative analysis of the signals with a bioimager indicated an induction factor of about 100. These data demonstrate that production of replicative intermediates can be well, though not 100% tightly, controlled by tet in this transient transfection system.
Figure 1.

Tet-regulated expression of replicative DNA intermediates after transient transfection of HuH7 cells. Cells were transfected with ptetHBV-M1T and pUHD15-1 and analyzed after 43 h in the absence or presence of tet. The amount of replicative intermediates was determined by Southern blot analysis hybridized with a 32P-labeled HBV full-length probe. The relationship between the signal and the image intensity is sigmoidal.
IFNα enhances synthesis of replicative intermediates and pregenomic RNA after tet promoter switch off by tet
Next we tested whether IFNα treatment of the transfected cells leads to a reduction in mature replicative DNA intermediates, similar to as described recently for HuH7 cells transiently transfected with a dimer of HBV DNA without foreign inducible promoter control. In order to analyze this, HuH7 cells were transfected with the tet promoter-controlled HBV construct and synthesis of pregenomic RNA was allowed for 3 h in the absence of tet. Synthesis of the pregenomic RNA was then switched off by addition of tet for the remaining time of the experiment. Twenty-four hours after switch off of pregenomic RNA synthesis, most of the pregenomic RNA was reverse transcribed, as was evident by northern blotting (data not shown), and synthesis of new intracellular intermediates was minimal. Therefore, IFNα was added at this time point and then the amount of replicative intermediates was analyzed by Southern blotting 48 h later, in both IFNα-treated and non-treated cells. Surprisingly, in cells treated with IFNα the levels of replicative intermediates were significantly higher than in non-treated cells (Fig. 2). This was observed reproducibly in three independent experiments (data not shown). In contrast, HuH7 cells transfected with the HBV dimer DNA had significantly reduced levels of intracellular replicative intermediates after IFNα treatment, as expected (10). In order to test whether the IFNα-mediated increase in intracellular replicative intermediates in the tet system is due to enhanced levels of pregenomic RNA, we analyzed the viral transcripts by northern blotting 24 and 48 h after IFNα addition. At both time points the ratio of pre-genomic RNA to envelope RNA was clearly enhanced in cells treated with IFNα (Fig. 3). As the envelope mRNA is constitutively expressed whereas the pregenomic RNA is under control of the tet promoter, these data suggest that the tet promoter is either activated by IFNα or the half-life of the pregenomic RNA is selectively increased by IFNα.
Figure 2.
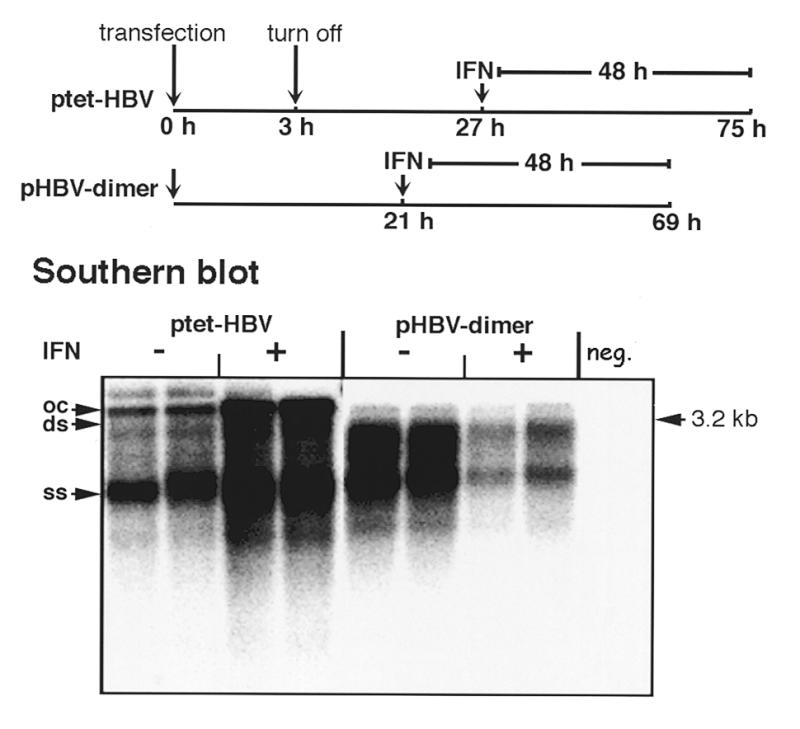
Effect of IFNα on production of replicative DNA intermediates after transfection of ptetHBV or pHBV-dimer DNA. A scheme of the time scale of the experimental protocol is shown at the top of the figure. HuH7 cells were transfected with ptetHBV-M1T and pUHD15-1. Three hours after transfection tet-controlled expression was abrogated by addition of tet and 24 h later cells were treated with 1.000 IU/ml IFNα for 2 days. In parallel cells were transfected with pHBV-dimer DNA and IFNα was added 21 h after transfection for 2 days. The amount of replicative intermediates was determined by Southern blot analysis as described in Figure 1.
Figure 3.
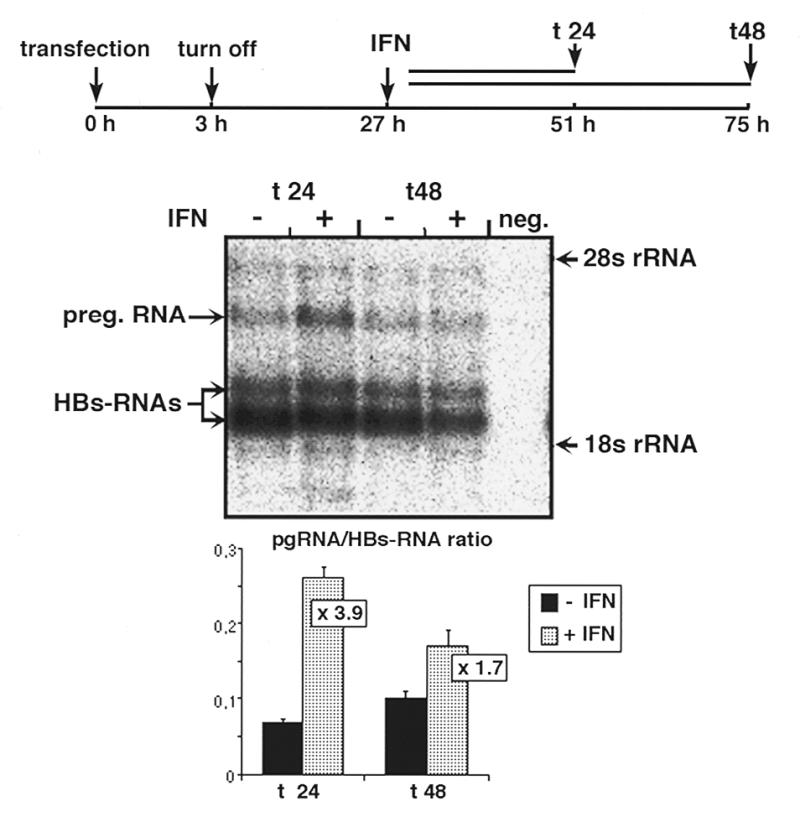
IFNα stimulates expression of the pregenomic HBV RNA controlled by the tet-responsive promoter. Cells were transfected with ptetHBV-M1T and pUHD15-1 and treated thereafter as described in the legend to Figure 2. In the sequence context of ptetHBV-M1T the pregenomic RNA is controlled by the inducible tet promoter and the envelope RNAs are constitutively expressed under control of the homologous HBV promoters. Samples were harvested 24 and 48 h after addition of IFNα. For northern analysis 20 µg of total RNA was separated by gel electrophoresis, transferred onto a nylon membrane and hybridized with a 32P-labeled HBV full-length probe. Signal intensities of the bands were measured within the linear range of the phosphorimager and the values are depicted as the ratio between pregenomic RNA and envelope RNAs.
The tet promoter contains a functional ISRE
In order to investigate whether the IFNα-mediated enhanced pregenomic RNA levels in cells transfected with the tet promoter-controlled HBV construct may be due to activation of the tet promoter, we first searched for a potential ISRE element in the tet vector. Computer-based sequence analysis revealed sequences with striking homology to the consensus sequence of ISRE in the tet promoter, diverging only at the very 5′- and 3′-ends of the region (Fig. 4). This ISRE-like sequence does not overlap with the tTA binding site in the operator sequences but is exclusively part of the linker sequence separating the tet heptameric operators (1).
Figure 4.
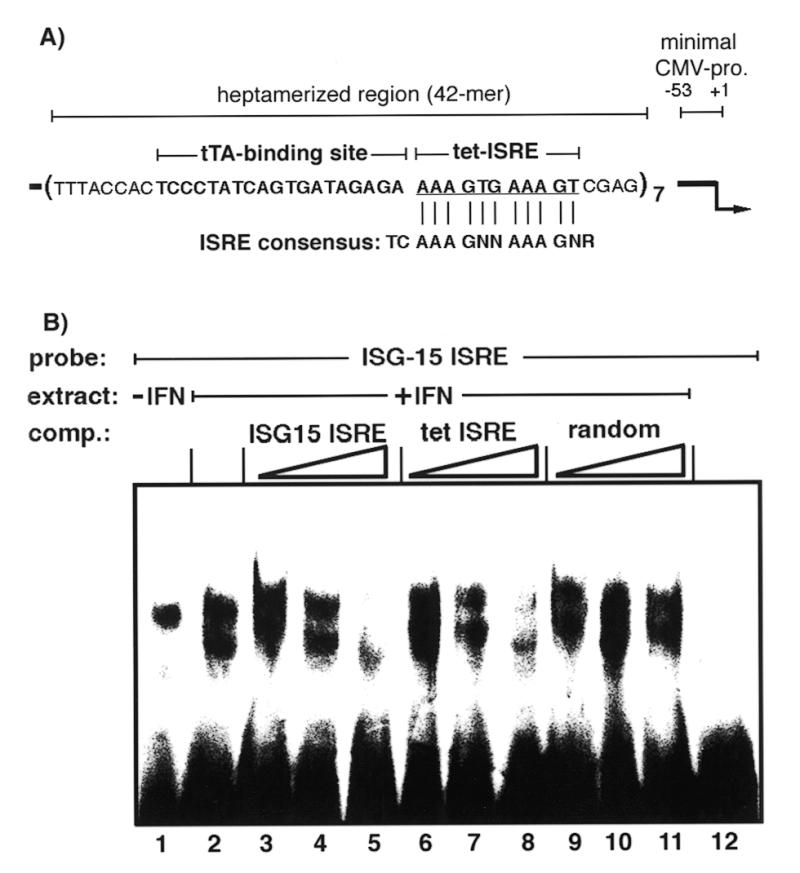
The tet ISRE specifically binds ISGFs. (A) Scheme of the tet promoter region. The region that was inserted as a heptamer upstream of a minimal CMV promoter, the tTA binding site (bold) and tet ISRE (bold underlined) is indicated. Nucleotide postitions are given in relation to the start site of transcription indicated by an arrow (+1). The ISRE consensus sequence is shown below (N, any nucleotide; R, purine). Bars between the ISRE consensus sequence and the tet ISRE show a perfect match with the consensus sequence. (B) Nuclear extracts were prepared from HuH7 cells treated with 1.000 IU/ml IFNα for 4 h or untreated cells. Extracts of IFNα-treated cells or untreated cells were incubated with a 32P-end-labeled double-stranded ISG15 oligonucleotide probe and DNA–protein complexes were separated on a 6% polyacryamide gel (lanes 1 and 2, respectively). For competition 5-, 50- or 500-fold excesses of unlabeled double-stranded ISG15 (lanes 3–5), tet ISRE (lanes 6–8) and a random oligonucleotide (lanes 9–11) were added to nuclear extracts of IFNα-treated cells in addition to the labeled ISG15 oligonucleotide probe. Lane 12 represents a sample without nuclear extract.
To determine whether the ISRE-like region represents a functional ISRE, gel mobility shift assays were performed. Nuclei of HuH7 cells treated or not for 4 h with IFNα were isolated and incubated with a 32P-labeled double-stranded ISG15 ISRE oligonucleotide as bait for IFN-stimulated gene transcription factors. Thereafter, DNA–protein complexes were analyzed on an acrylamide gel. As expected, an increase in DNA–protein complexes was detected in nuclei of IFNα-treated cells compared to untreated cells (Fig. 4, lanes 1 and 2). Specificity of the DNA–protein complexes was tested by addition of increasing amounts of unlabeled ISG15 oligonucleotide to the nuclear extract from IFNα-treated cells. Addition of unlabeled ISG15 oligonucleotide competed in a dose-dependent manner with the labeled ISG15 oligonucleotide (Fig. 4, lanes 3–5). The highest ratio of unlabeled to labeled ISG15 oligonucleotide (500-fold) led to almost complete competition and disappearance of the DNA–protein complex (Fig. 4, lane 6). Similar efficient competition was observed with increasing amounts of the unlabeled tet ISRE (Fig. 4, lanes 6–8), while an unlabeled random oligonucleotide did not compete with the labeled ISG15 ISRE (Fig. 4, lanes 9 and 10). These data clearly demonstrate that the ISRE-like sequence of the tet promoter can bind IFN-stimulated transcription factors and suggest that it confers IFNα-stimulation of the tet promoter.
IFNα-stimulated tet promoter activity is cell line specific and independent of the tet transactivator
To further characterize IFNα-induced stimulation of the tet-responsive vector system, its dependence on IFNα dosage, tTA and type of cell line was determined. As a reporter gene we used the luciferase gene under control of the tet promoter (plasmid pUHC13-3) to exclude any influence of HBV DNA sequences. First, we tested whether the reporter gene construct is tet responsive in HuH7 cells. Therefore, pUHC13-3 and pUHD15-1 were co-transfected into HuH7 cells. This resulted in an ~800-fold induction of luciferase activity in untreated cells compared to cells treated with tet as determined 24 h after transfection (data not shown). The effect of IFNα dose (0–1000 U/ml) on tet promoter stimulation was analyzed in HuH7 experiments using the same tet promoter switch on and off parameters as in Figure 2. Approximately 2-fold increased tet promoter activity was observed with an IFNα dose as low as 50 U/ml, approximately 4-fold stimulation at a dose of 200 U/ml and a 3-fold stimulation with 1000 U/ml (Fig. 5A). These data indicate that the tet promoter is increasingly stimulated by IFNα at low dosage and stimulation levels off or even decreases slightly when using higher IFNα concentrations.
Figure 5.

Stimulation of the tet promoter depends on the cell type and dosage of IFNα added and is independent of tTA. (A) HuH7 cells were transfected with pUHC13-3 and pUHD15-1 and thereafter were treated as described in the legend to Figure 2, with the exception that the cells were treated with different doses of IFNα, as indicated. Luciferase activity was determined 24 h after addition of IFNα. (B) Cells transfected with pUHC13-3 and pUHD15-1 or with pUHC13-3 alone were treated as described above and luciferase activity was determined 24 h after addition of 1.000 IU/ml IFNα. (C) IFNα-mediated stimulation of the tet promoter was determined in HuH7, 293 and U2-OS cells. Cells were treated as described in the legend to Figure 5A (1.000 IU/ml IFNα or untreated).
To determine whether IFNα-induced stimulation depends on the presence of the tTA, transfection experiments were performed with and without co-transfection of plasmid pUHD15-1, constitutively expressing tTA. Irrespective of the expression of tTA, the tet promoter was stimulated approximately 4-fold (Fig. 5B). Therefore, stimulation does not depend on the tTA. As the levels of luciferase activity measured in cells lacking tTA is even slightly higher than that in cells constitutively expressing tTA, all luciferase activity must be independent of tet promoter activation by tTA. The findings also imply that the tet promoter has a rather high activity in transiently transfected HuH7 cells which cannot be switched off by addition of tet. Note that the initial expression pulse of 3 h cannot account for the luciferase activity because the half-life of luciferase is ~3 h (27,28) and its expression was measured 2 days after addition of tet.
When the same type of experiment was performed in human embryonal kidney cells (293 cells) we also found rather high tet promoter activity which could not be switched off by addition of tet (Fig. 5C). However, in this cell line the tet promoter could not be significantly activated further by IFNα (Fig. 5C). This is most likely due to adenovirus 5-derived E1A protein, which is constitutively expressed in 293 cells (29) and is a known inhibitor of IFN-induced activation of ISGF3 (30,31). These data further confirm our conclusion that the stimulation of tet promoter activity by IFNα is mediated by ISGF3 and the ISREs of the tet promoter. Previously reported transfection experiments in HuH7 cells with an IFN-non-responsive CMV-SEAP expression vector (10), which has virtually the same vector sequences as the tet vectors except for the heptameric tet operator region, excludes the theoretical possibility that other vector sequences mediate IFN responsiveness as well.
In order to investigate whether tet promoter activity can be stimulated by IFNα in other cell lines than in HuH7 cells we performed the same experiment in human osteosarcoma U2-OS cells, which are IFNα responsive (32). Unexpectedly, IFNα addition did not increase tet promoter activity but led to a slight decrease compared to basal tet promoter activity in the presence of tet (Fig. 5C). This may be due to the fact that we are dealing with an ISRE-like sequence which is not completely identical to the consensus sequence and therefore its function may depend on cell type-specific factors binding close to or at the ISRE-like sequence. Whatever the reason may be, our data imply that the tet promoter is not IFNα responsive in all cell lines. Therefore, analysis of cytokine-mediated effects of proteins expressed under tet promoter control warrants prior testing of the IFN responsiveness of the tet promoter in the cell line to be used. This is also recommended for experiments with cell lines stably transfected with this vector system, which may or may not show the same effect. Alternatively, the ISRE-like sequences in the currently available tet promoter-containing vectors should be functionally inactivated by mutagenesis. Changing a single nucleotide in the ISRE core sequence which is known to be essential for its function should not affect tet-mediated regulation of the promoter activity since the ISREs do not overlap with the tTA binding regions.
Acknowledgments
ACKNOWLEDGEMENTS
We appreciate critical reading of the manuscript by Carol Stocking. This work was supported by grants from the Bundesministerium für Forschung und Technologie. The Heinrich-Pette-Institut für Experimentelle Virologie und Immunologie is supported by the Bundesministerium für Gesundheit and the Freie und Hansestadt Hamburg.
REFERENCES
- 1.Gossen M. and Bujard,H. (1992) Proc. Natl Acad. Sci. USA, 89, 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hillen W. and Wissmann,A. (1989) In Saenger,W. and Heinemann,U. (eds), Topics in Molecular and Structural Biology, Protein–Nucleic Acid Interaction. Macmillan, London, UK, Vol. 10, pp. 143–162.
- 3.Gossen M., Bonin,A.L., Freundlieb,S. and Bujard,H. (1994) Curr. Opin. Biotechnol., 5, 516–520. [DOI] [PubMed] [Google Scholar]
- 4.Saez E., No,D., West,A. and Evans,R.M. (1997) Curr. Opin. Biotechnol., 8, 608–616. [DOI] [PubMed] [Google Scholar]
- 5.Shockett P.E. and Schatz,D.G. (1996) Proc. Natl Acad. Sci. USA, 93, 5173–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossi F.M. and Blau,H.M. (1998) Curr. Opin. Biotechnol., 9, 451–456. [DOI] [PubMed] [Google Scholar]
- 7.Bohl D., Naffakh,N. and Heard,J.M. (1997) Nature Med., 3, 299–305. [DOI] [PubMed] [Google Scholar]
- 8.Saitoh Y., Eguchi,Y., Hagihara,Y., Arita,N., Watahiki,M., Tsujimoto,Y. and Hayakawa,T. (1998) Hum. Gene Ther., 9, 997–1002. [DOI] [PubMed] [Google Scholar]
- 9.Hagihara Y., Saitoh,Y., Arita,N., Eguchi,Y., Tsujimoto,Y., Yoshimine,T. and Hayakawa,T. (1999) Cell. Transplant., 8, 431–434. [DOI] [PubMed] [Google Scholar]
- 10.Rang A., Günther,S. and Will,H. (1999) J. Hepatol., 31, 791–799. [DOI] [PubMed] [Google Scholar]
- 11.Summers J. and Mason,W.S. (1982) Cell, 29, 403–415. [DOI] [PubMed] [Google Scholar]
- 12.Tuttleman J.S., Pourcel,C. and Summers,J. (1986) Cell, 47, 451–460. [DOI] [PubMed] [Google Scholar]
- 13.Isaacs A. and Lindenmann,J. (1987) J. Interferon Res., 7, 429–438. [DOI] [PubMed] [Google Scholar]
- 14.Staeheli P. (1990) Adv. Virus Res., 38, 147–200. [DOI] [PubMed] [Google Scholar]
- 15.Pellegrini S. and Schindler,C. (1993) Trends Biochem. Sci., 18, 338–342. [DOI] [PubMed] [Google Scholar]
- 16.Darnell J.J., Kerr,I.M. and Stark,G.R. (1994) Science, 264, 1415–1421. [DOI] [PubMed] [Google Scholar]
- 17.Schindler C., Shuai,K., Prezioso,V.R. and Darnell,J.J. (1992) Science, 257, 809–813. [DOI] [PubMed] [Google Scholar]
- 18.Starr R., Willson,T.A., Viney,E.M., Murray,L.J., Rayner,J.R., Jenkins,B.J., Gonda,T.J., Alexander,W.S., Metcalf,D., Nicola,N.A. and Hilton,D.J. (1997) Nature, 387, 917–921. [DOI] [PubMed] [Google Scholar]
- 19.Decker T., Kovarik,P. and Meinke,A. (1997) J. Interferon Cytokine Res., 17, 121–134. [DOI] [PubMed] [Google Scholar]
- 20.Stark G.R., Kerr,I.M., Williams,B.R., Silverman,R.H. and Schreiber,R.D. (1998) Annu. Rev. Biochem., 67, 227–264. [DOI] [PubMed] [Google Scholar]
- 21.Reich N., Evans,B., Levy,D., Fahey,D., Knight,E.J. and Darnell,J.J. (1987) Proc. Natl Acad. Sci. USA, 84, 6394–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ladner S.K., Otto,M.J., Barker,C.S., Zaifert,K., Wang,G.H., Guo,J.T., Seeger,C. and King,R.W. (1997) Antimicrob. Agents Chemother., 41, 1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Günther S., Li,B.C., Miska,S., Krüger,D.H., Meisel,H. and Will,H. (1995) J. Virol., 69, 5437–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grotzinger T., Jensen,K. and Will,H. (1996) J. Biol. Chem., 271, 25253–25260. [DOI] [PubMed] [Google Scholar]
- 25.Bradford M.M. (1976) Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 26.Bruss V. and Ganem,D. (1991) J. Virol., 65, 3813–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen V.T., Morange,M. and Bensaude,O. (1989) J. Biol. Chem., 264, 10487–10492. [PubMed] [Google Scholar]
- 28.Thompson J.F., Hayes,L.S. and Lloyd,D.B. (1991) Gene, 103, 171–177. [DOI] [PubMed] [Google Scholar]
- 29.Spindler K.R. and Berk,A.J. (1984) J. Virol., 52, 706–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leonard G.T. and Sen,G.C. (1997) J. Virol., 71, 5095–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalvakolanu D.V., Bandyopadhyay,S.K., Harter,M.L. and Sen,G.C. (1991) Proc. Natl Acad. Sci. USA, 88, 7459–7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heim M.H., Moradpour,D. and Blum,H.E. (1999) J. Virol., 73, 8469–8475. [DOI] [PMC free article] [PubMed] [Google Scholar]