

Abstract
INTRODUCTION:
Thousands of genetic variants have been associated with human diseases and traits through genome-wide association studies (GWASs). Translating these discoveries into improved therapeutics requires discerning which variants among hundreds of candidates are causally related to disease risk. To date, only a handful of causal variants have been confirmed. Here, we leverage 100 million years of mammalian evolution to address this major challenge.
RATIONALE:
We compared genomes from hundreds of mammals and identified bases with unusually few variants (evolutionarily constrained). Constraint is a measure of functional importance that is agnostic to cell type or developmental stage. It can be applied to investigate any heritable disease or trait and is complementary to resources using cell type– and time point–specific functional assays like Encyclopedia of DNA Elements (ENCODE) and Genotype-Tissue Expression (GTEx).
RESULTS:
Using constraint calculated across placental mammals, 3.3% of bases in the human genome are significantly constrained, including 57.6% of coding bases. Most constrained bases (80.7%) are noncoding. Common variants (allele frequency ≥ 5%) and low-frequency variants (0.5% ≤ allele frequency < 5%) are depleted for constrained bases (1.85 versus 3.26% expected by chance, P < 2.2 × 10−308).Pathogenic ClinVar variants are more constrained than benign variants (P < 2.2 × 10−16).
The most constrained common variants are more enriched for disease single-nucleotide polymorphism (SNP)–heritability in 63 independent GWASs. The enrichment of SNP-heritability in constrained regions is greater (7.8-fold) than previously reported in mammals and is even higher in primates (11.1-fold). It exceeds the enrichment of SNP-heritability in nonsynonymous coding variants (7.2-fold) and fine-mapped expression quantitative trait loci (eQTL)–SNPs (4.8-fold). The enrichment peaks near constrained bases, with a log-linear decrease of SNP-heritability enrichment as a function of the distance to a constrained base.
Zoonomia constraint scores improve functionally informed fine-mapping. Variants at sites constrained in mammals and primates have greater posterior inclusion probabilities and higher per-SNP contributions. In addition, using both constraint and functional annotations improves polygenic risk score accuracy across a range of traits. Finally, incorporating constraint information into the analysis of noncoding somatic variants in medulloblastomas identifies new candidate driver genes.
CONCLUSION:
Genome-wide measures of evolutionary constraint can help discern which variants are functionally important. This information may accelerate the translation of genomic discoveries into the biological, clinical, and therapeutic knowledge that is required to understand and treat human disease.
Thousands of genomic regions have been associated with heritable human diseases, but attempts to elucidate biological mechanisms are impeded by an inability to discern which genomic positions are functionally important. Evolutionary constraint is a powerful predictor of function, agnostic to cell type or disease mechanism. Single-base phyloP scores from 240 mammals identified 3.3% of the human genome as significantly constrained and likely functional. We compared phyloP scores to genome annotation, association studies, copy-number variation, clinical genetics findings, and cancer data. Constrained positions are enriched for variants that explain common disease heritability more than other functional annotations. Our results improve variant annotation but also highlight that the regulatory landscape of the human genome still needs to be further explored and linked to disease.
Graphical Abstract
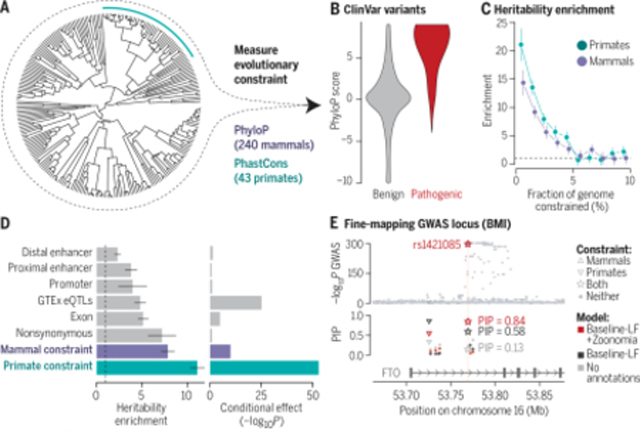
Using evolutionary constraint in genomic studies of human diseases. (A) Constraint was calculated across 240 mammal species, including 43 primates (teal line). (B) Pathogenic ClinVar variants (N = 73,885) are more constrained across mammals than benign variants (N = 231,642; P < 2.2 × 10−16). (C) More-constrained bases are more enriched for trait-associated variants (63 GWASs). (D) Enrichment of heritability is higher in constrained regions than in functional annotations (left), even in a joint model with 106 annotations (right). (E) Fine-mapping (PolyFun) using a model that includes constraint scores identifies an experimentally validated association at rs1421085. Error bars represent 95% confidence intervals. BMI, body mass index; LF, low frequency; PIP, posterior inclusion probability.
In the past 15 years, increasingly larger genomic studies have delivered many previously unknown associations for a wide array of human diseases, disorders, biomarkers, and other traits. About 400,000 genetic associations have been identified that span the allelic spectrum, from ultrarare variants in large sequencing datasets to common variants that are present in many humans, in both coding and regulatory regions [see supplementary methods (SM), section 1]. Although these associations meet rigorous standards for statistical significance and replicability, their functional importance is generally unknown. Inferring functional importance is crucial to translating the results of rare and common variant association studies into the biological, clinical, and therapeutic knowledge required to understand and treat human disease. Exceptional efforts have been made to annotate the human genome using functional genomics—e.g., Encyclopedia of DNA Elements (ENCODE) (1) and Genotype-Tissue Expression (GTEx) (2)—as well as inferring deleterious effects from allele frequencies and location in coding sequence—e.g., Genome Aggregation Database (gnomAD) (3) and Trans-Omics for Precision Medicine (TOPMed) (4). Although these seminal projects greatly expanded our knowledge base, this “central problem in biology” is unresolved and motivated the National Human Genome Research Institute (NHGRI) Impact of Genomic Variation on Function initiative.
Evolutionary constraint is complementary to these efforts. Functional importance is inferred from the signatures of evolution in the human genome: “Constraint” indicates genomic positions that have changed more slowly than expected under neutral drift because of purifying selection. A key advantage of constraint lies in its mechanistic agnosticism; a highly constrained base has an impact on some biological process, in some cell, at some life stage (discussed in SM, section 2). Constraint has been used in efforts to understand the human genome for more than 50 years, beginning with cross-species protein-sequence comparisons. More recently, at the extremes of the allelic spectrum, constraint is often used by clinical geneticists to prioritize potentially causal rare variants (5, 6), and common variants in regions under constraint are highly enriched in genome-wide association study (GWAS) results (7–9). However, evolutionary constraint is underused in the functional interpretation and prioritization of GWAS loci (10–15).
Our companion paper describes the Zoonomia reference-free alignment of 240 placental mammals spanning ~100 million years of evolution (16). The analyses showed the unprecedented informativeness of this alignment at multiple scales, from exceptionally constrained 100-kb bins (e.g., all HOX clusters) to smaller ultraconserved elements and human accelerated regions, noncoding regulatory regions, and specific base positions in binding motifs. These results strongly suggest the utility of constraint as a functional annotation that can be leveraged to deepen our understanding of heritable human diseases. Here, we demonstrate the importance of mammalian constraint for connecting genotype to phenotype for human disease.
The properties of evolutionary constraint at single-base resolution
Defining constraint
Placental mammalian constraint was estimated using phyloP scores (17) across 240 species for 2,852,623,265 bases in the human genome (chromosomes 1 to 22, X, and Y; SM, section 3). In our companion paper (16), we estimated that 10.7% of the human genome is under some degree of constraint because of purifying selection; for these disease-focused analyses, we used a subset with the strongest constraint signatures. We defined a base as constrained in mammals if its phyloP score was ≥2.27 [false discovery rate (FDR) 0.05 threshold]. At this threshold, 100,651,377 bases or 3.26% of the human genome is constrained. We defined constraint across 43 primates using a phastCons (18) threshold (≥0.961, 101,134,907 bases) selected to match the fraction of the genome annotated as constrained in the placental mammals studied here. Mammalian and primate constraint overlapped considerably but not fully (Jaccard index 0.30). In section 4 of the SM, we describe the properties of constrained genomic positions, from base-level to higher-order annotations. Briefly, we found that mammalian constrained bases had a marked tendency to cluster (median distance two bases) compared with random expectations (median distance 24 bases), that specific genomic elements were highly enriched in constrained bases [e.g., 57.6% of coding sequence (CDS) is constrained] (Fig. 1A and fig. S1), that constraint scores captured nuances of the genetic code (fig. S2), and that constrained bases mainly spanned regulatory features (e.g., 80.7% of constrained bases are within non-coding regions versus 19.3% within CDS).
Fig. 1. Overview of constraint distribution.
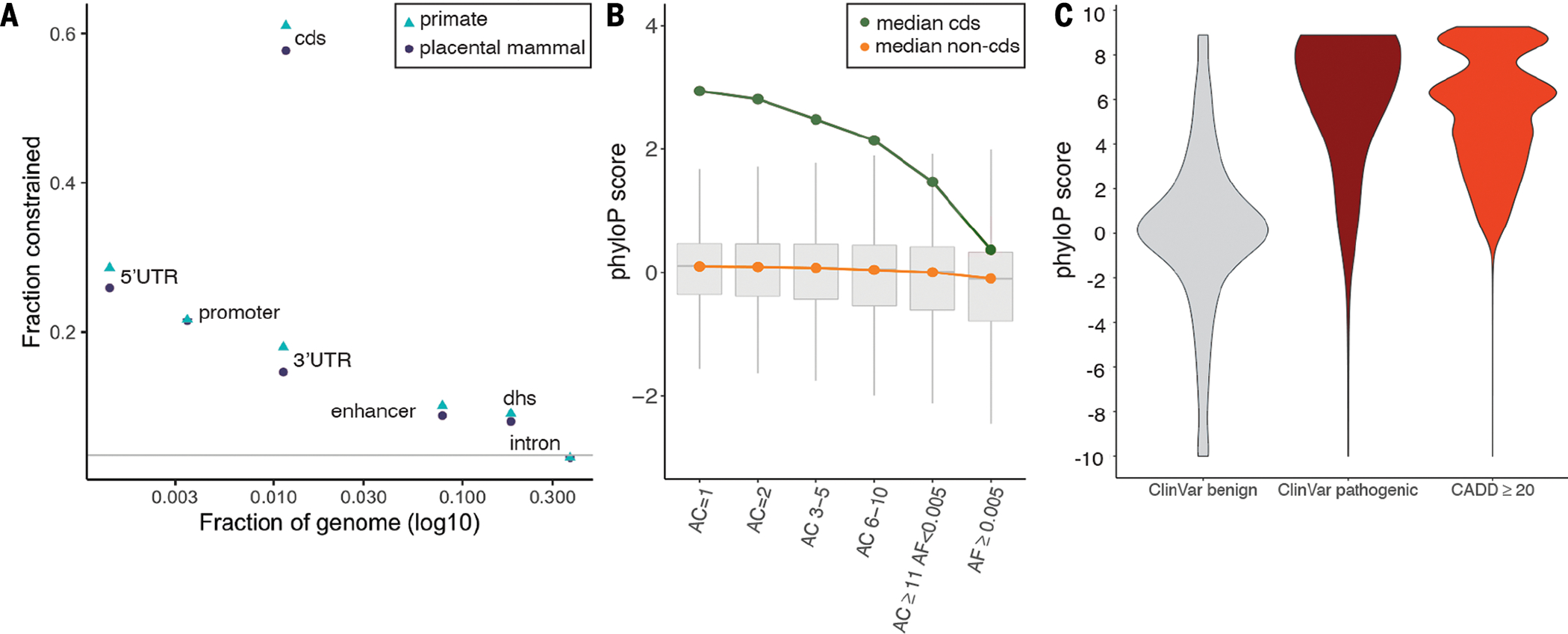
(A) Evolutionary constraint in multiple genomic partitions. The x axis is the fraction of the genome occupied by a partition, the y axis is the fraction of partition under constraint in placental mammals (purple circles) and primates (blue triangles), and the gray line is the genome mean (0.033). The greatest constraint is found in CDS and key regulatory regions (5′UTRs, ENCODE promoter-like elements, and 3′UTRs). The higher fraction constrained in primates versus mammals is due to different constraint definitions and does not necessarily reflect biology. This figure is a subset of fig. S1 and data from section 4 of the SM, which shows more biotypes, PC gene parts, and regulatory regions. dhs, DNase I hypersensitive sites. (B) Whisker plots of constraint in variants from TOPMed whole-genome sequencing (WGS), stratified by CDS (green, 6.14 million biallelic SNPs) and non-CDS variants (orange, 549.64 million biallelic SNPs). The x axis shows six AC bins, from singletons (bin AC = 1, 44.8% of total variants) to common and low-frequency variants (AF ≥ 0.5%, 1.4% of total variants). For the plots, the center line represents the median, box limits are upper and lower quartiles, and whiskers are minimum and maximum values. Outliers are hidden for clarity. (C) PhyloP score density for ClinVar benign (N = 231,642), ClinVar pathogenic (N = 73,885), and gnomAD WGS variant positions with CADD ≥ 20 (N = 3,958,488).
Constraint across the allelic spectrum
Genetic variation is fundamental to heritable human diseases, disorders, and other traits. We thus evaluated the relationship between allele frequency (AF) and constraint (Fig. 1B). Using whole-genome sequencing data from more than 140,000 humans (TOPMed, v8) (4), we observed an inverse correlation between allele count and phyloP score [Spearman’s correlation coefficient (ρ) = −0.07], with stronger correlations in CDS regions and for nonsynonymous variants (Spearman’s ρ = −0.12 and −0.18, all P < 2.2 × 10−308). As expected, owing to negative selection, common (defined as AF ≥ 5%) and low-frequency (0.5% ≤ AF < 5%) genetic variants were depleted for constrained bases (1.85 versus 3.26% expected by chance, P < 2.2 × 10−308). This relatively high fraction of constrained bases highlights the ability of mammalian constraint to predict deleterious effects across the AF spectrum. To evaluate these relations more formally, genome-wide models contrasting singletons [allele count (AC) = 1] to common and low-frequency variants (AF ≥ 0.005) found that common and low-frequency variants had lower phyloP scores and a marked increase in CG context (fig. S3 and SM, section 4). Models for CDS single-nucleotide polymorphisms (SNPs) found an inverse association of AC with constraint and that common and low-frequency SNPs had greater odds of occurring at a C or G base and tend not to occur in important CDS positions (e.g., codon position 1 or 2, or at bases that could mutate to stop).
Common and low-frequency constrained SNPs are relevant for human diseases
We conducted additional analyses of common and low-frequency SNPs (AF ≥ 0.5%) because these variants are the main focus of GWASs (SM, section 4). Of these 15,777,878 SNPs in TOPMed, 1.85% (N = 291,669) are constrained, far less than genome-wide constraint (3.26%). Our modeling showed that constrained SNPs are 22 times more likely to occur in CDS, 3 times more likely to occur in promoters, and ~2 times more likely to be a “fine-mapped” expression quantitative trait loci (eQTL)–SNP or to occur in open chromatin or an enhancer compared with outside those regions.
The strong tendency of these constrained SNPs to occur in CDS was unexpected given that (by definition) these positions are highly constrained in placental mammals and yet variable in humans. We hypothesized that this could occur if selection effects were variable across genes (some generate peptide variability whereas others are highly intolerant of CDS variation). We found that 37.8% of protein-coding (PC) genes had no constrained CDS SNPs and other genes had appreciable fractions (up to 10% of all CDS bases are common and low-frequency SNPs). A gene-set analysis of the top 5% (N = 980) of genes containing the greatest number of constrained CDS SNPs showed that this set was enriched for genes with medical relevance [an Online Mendelian Inheritance in Man (OMIM) entry including multiple neurological disorders], G protein–coupled receptor genes, “druggable” genes (19), taste receptor genes, skin development genes, and genes involved in multiple immune processes. These biological processes are at the interface of a mammal and its environment and allow adaptation to an environmental niche. We suggest that many of these genes could be prioritized for gene-environment interaction searches because constrained variants that reach high frequency in human populations may be particularly relevant for human diseases.
Base-pair resolution of deleterious effects
We contrasted constraint scores to metrics that are used to aid the interpretation of functional variation for human health. First, pathogenic ClinVar (20) variants were significantly skewed to higher phyloP in comparison to benign variants (two-tailed Wilcoxon rank sum test, P < 2.2 × 10−16; Fig. 1C), and phyloP scores were strongly associated with the improvement in annotations of variants in ClinVar from 2016 to 2021 (e.g., uncertain to benign or to pathogenic; SM, section 5). For a second metric, Combined Annotation–Dependent Depletion (CADD) (6), which incorporates evolutionary constraint, we found that variant positions with a higher likelihood of deleteriousness were also enriched for constrained phyloP scores (two-tailed Wilcoxon rank sum test, P < 2.2 × 10−16; Fig. 1C). A focused analysis of human nonsynonymous variants at constrained sites across the mammalian tree using Tool to infer Orthologs from Genome Alignments (TOGA) (16, 21) identified 1570 genes for which a nonsynonymous change resulted in a ClinVar pathogenic or likely pathogenic phenotype in humans (SM, section 5). For example, the CFTR gene that underlies cystic fibrosis (22) showed a high burden of pathogenic sites compared with benign sites (123 versus 1 out of 1585 alignment sites). A further 12,889 genes had identifiable constrained sites but lacked records of nonsynonymous pathogenic alterations (SM, section 5). Several of these constrained positions, which presently lack ClinVar pathogenic annotations, likely represent previously uncharacterized sources of deleterious variation resulting in a disease state. We tested this by leveraging functionally explored variation in two GPCRs, GPR75 (23) and ADRB2 (24), and showed that functionally important SNP or amino acid sites, respectively, were marked by higher constraint scores (SM, section 5). Species alignments at this scale also allow for the identification of potential model systems, those for which a substitution may result in a human disease state but is otherwise naturally occurring in nonhuman mammals. We found 697 such sites across 330 genes, including multiple positions in SOD1 (pathogenic sites for amyotrophic lateral sclerosis). These observations open a pathway for natural adaptive variants to inform the development of new therapies for treatment (SM, section 5).
Common and low-frequency variation and human diseases and complex traits
GWASs have found that the genetic architecture of human diseases and complex traits is highly polygenic and dominated by common variants with weak effects (10). Here, we dissected the impact of common and low-frequency variants on this architecture through polygenic analyses of disease SNP–heritability (h2) using stratified linkage disequilibrium (LD) score regression (S-LDSC) (7, 25, 26).
Constraint scores are proportional to common variant SNP-h2 enrichments
We first validated the relevance of our constraint scores to investigate the role of common variants in human diseases and complex traits using the results of 63 independent European ancestry GWASs (27) (mean N = 314,000; data S1 and SM, section 6). We found that common variants in the highest constraint score percentiles had greater enrichment for GWAS trait-associated variants (measured by SNP-h2 enrichment, or the proportion of h2 divided by the proportion of SNPs; Fig. 2A and data S2). We observed decreasing but significant enrichments (P < 0.0033, Bonferroni correction for 15 comparisons) for SNPs in the first four percentiles of mammalian constraint scores (phyloP) (in line with 3.26% of the genome bases being considered as constrained using a 5% FDR threshold) and in the first five percentiles of primate (phastCons) constraint scores. We justified the use of different scores to measure constraint in mammals and primates by the fact that phyloP scores were unable to detect single-base constraint in primates owing to lack of power and were too noisy to lead to significant h2 enrichment (fig. S4). Although both phyloP and phastCons element scores performed similarly in heritability analyses, phyloP is superior for having single-base resolution (fig. S4 and additional justification in SM, section 6).
Fig. 2. SNP-h2 analyses of variants at constrained positions in human complex traits and diseases.
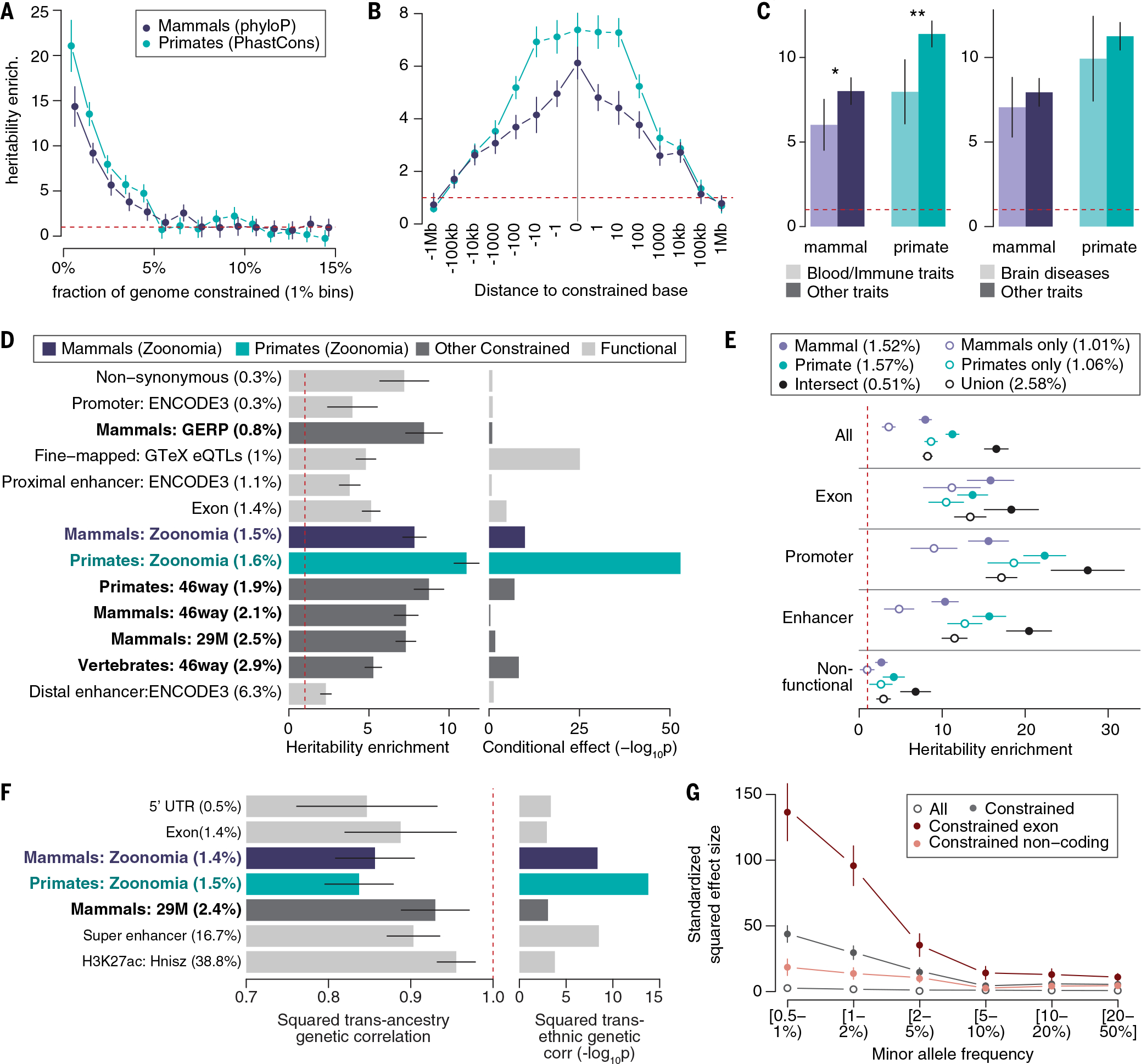
(A) Heritability enrichment of common SNPs in the top percentiles of constraint scores in placental mammals (phyloP positions) and primates (phastCons elements). (B) Heritability enrichment as a function of the distance to a constrained base. (C) Heritability enrichment of constrained annotations in 11 blood and immune traits and nine brain diseases (light color) versus other types of traits (dark color). *P < 0.05 and **P < 0.05 after Bonferroni correction. (D) Heritability enrichment of constrained and functional annotations (left) and corresponding significance of the conditional effect while considered in a joint model with 106 annotations (right). GERP, genomic evolutionary rate profiling. (E) Heritability enrichment of constrained annotations intersected together and stratified by their genomic function. (F) Squared transancestry genetic correlation enrichment (left) with corresponding significance (right) for seven annotations with significant depletion of squared transancestry genetic correlations. H3K27ac, histone H3 acetylated at lysine 27. (G) Standardized squared effect sizes as a function of AF. Results are meta-analyzed across, 63 independent GWASs [(A), (B), (D), and (E)], 31 independent traits with GWASs available in European and Japanese populations [(F)], and 27 independent UK Biobank traits [(G)]. Dashed red lines represent a null enrichment of 1 [(A) to (E)] and a null squared transancestry genetic correlation (F). Error bars are 95% confidence intervals. Numerical results are reported in data S2 to S4, S6 to S8, and S11.
Mammalian constraint scores are base pair–specific
We evaluated the resolution of constraint scores by estimating SNP-h2 with different distances to a constrained base. First, we confirmed the base-pair resolution of mammalian constraint scores by observing that SNPs ~1 base pair (bp) from a constrained variant were significantly less enriched for h2 than constrained SNPs (P ≤ 3.35 × 10−3) (Fig. 2B and data S3). We also observed a log-linear decrease of h2 enrichment as a function of the distance to a constrained base, with significant h2 enrichment up to 100 kb from constrained bases, confirming the larger-scale clustering of constrained bases. Finally, demonstrating the power of a broad mammal-wide genome sampling, constraint scores obtained only from primate species have lower resolution (10 to 100 bp; Fig. 2B) because these are based on fewer species (43), from a single mammalian order, and thus have shorter branch length.
Zoonomia constraint is distinctively informative
Annotations derived from mammal and primate constrained positions were more informative for human diseases than key functional annotations, including previously published constrained annotations (18, 28, 29) (Fig. 2D and data S4). First, their degrees of enrichment (7.84 ± 0.37–fold for mammals and 11.10 ± 0.40–fold for primates) exceeded those of previously published constraint and key functional annotations, such as nonsynonymous coding variants (7.20 ± 0.78–fold) or fine-mapped eQTL-SNPs (4.81 ± 0.31–fold) (30). We still observed high degrees of enrichment when removing exonic variants from our constraint annotations (6.15 ± 0.41–fold for mammals and 9.90 ± 0.51–fold for primates; fig. S5), confirming the informativeness of constraint to annotate noncoding common variants (see next sections). Second, in conditional analyses involving 106 annotations analyzed jointly (SM, section 6), we observed that these constrained annotations were among the most significant (P = 1.17 × 10−10 for mammals and P = 1.19 × 10−53 for primates) and were more significant than previously published constrained annotations (Fig. 2D and data S4).
Variants at constrained positions are less enriched in blood and immune trait heritability than in other complex traits
We did not observe disease-specific patterns for our constrained annotations, without any trait exhibiting higher h2 enrichment than the mean calculated for the mammal and primate constrained annotations (fig. S6 and data S5). However, we observed consistently lower h2 enrichments for constrained annotations in a meta-analysis of 11 blood and immune traits, as previously observed (7), but no differential enrichment in nine brain disorders (Fig. 2C and data S1 and S6).
Variants at positions constrained in primates are informative for noncoding common variants
SNPs constrained in primates have greater SNP-h2 enrichment than SNPs constrained in mammals (Fig. 2, A to C). To investigate, we intersected mammalian and primate constraint information and observed significantly higher h2 enrichment in SNPs constrained in both mammals and primates (16.52 ± 0.73–fold) compared with constraint only in primates (8.66 ± 0.38–fold) or only in mammals (3.56 ± 0.40–fold) (Fig. 2E and data S7). We verified that these results are mostly driven by the intersection of mammal and primate constrained bases (and are not due to the different scoring tests; fig. S7). By stratifying constrained mammalian bases by their primate constraint scores, we found that variants identified as constrained in the studied placental mammals but not in primates are not significantly enriched in h2, whereas SNPs constrained in primates were significantly enriched regardless of their constraint scores in mammals (fig. S8). These results explain the lower SNP-h2 for constraint in mammals and demonstrate increased informativeness when combining information from primates and mammals. We observed consistently higher h2 enrichment for SNPs that are constrained in both mammals and primates when stratifying by genomic function (i.e., coding regions, promoters, and enhancers), but that constraint is more informative in primates than in mammals only for noncoding variants (Fig. 2E). This confirms that the informativeness of our constraint annotations does not only reside in their high overlap with exonic bases (see also fig. S5). We observed that constrained SNPs defined as nonfunctional (see SM, section 6) were still enriched in h2 (>2.67-fold with P < 1.22 × 10−4, except for SNPs constrained only in mammals or primates; Fig. 2E), emphasizing the informativeness of our constrained annotations to annotate noncoding variants with unknown functions.
Per-allele effect sizes of common variants at constrained positions differ across human populations
Although our heritability analyses focused on European ancestry GWASs, variant per-allele effect sizes differ across human populations, especially for variants with stronger gene-environment interactions (31). To quantify how per-allele effect sizes of constrained common variants differ across populations, we applied S-LDXR (31) on 31 diseases and complex traits with GWAS data from East Asian (mean N = 90,000) and European (mean N = 267,000) populations. Here, we focused on per-allele effect sizes rather than per-SNP h2 to account for differences in allele frequencies across populations (31). Variants at constrained sites in mammals and primates were among the most significantly depleted in squared transancestry genetic correlation (P = 4.38 × 10−9 and 1.63 × 10−14, the third and most significant investigated annotations, respectively; Fig. 2F and data S8). These results highlight more population-specific causal effect sizes for variants at constrained positions, in line with stronger gene-environment interactions at these loci, and potentially explain how genetic variations at constrained bases could have become common in human populations.
Strong effect sizes for coding low-frequency variants at constrained positions
Genomic regions under purifying selection tend to have low-frequency variants (0.5% ≤ AF < 5%) with larger effect sizes, which leads to higher enrichment in low-frequency variant h2 compared with common variant h2 (8). We quantified low-frequency SNP-h2 enrichments of constrained annotations by analyzing 34 well-powered independent UK Biobank traits (mean N = 340,000; data S10). We observed that constrained annotations had consistently larger low-frequency h2 enrichment than common h2 enrichment, especially for variants at constrained sites in mammals (17.02 ± 0.89–fold versus 8.67 ± 0.71–fold; P = 1.99 × 10−13 for difference) (fig. S9 and data S10) in line with greater effect sizes as AF decreases (Fig. 2G and data S11). Similar patterns were observed for variants at constrained sites in primates (data S10). This enrichment difference was driven by exonic variants at constrained sites (50.03 ± 2.74–fold versus 19.80 ± 1.84–fold in mammals; P = 5.49 × 10−20 for difference); we note that the low-frequency h2 enrichment for these variants was similar to that of nonsynonymous variants (40.48 ± 2.37–fold), suggesting that constraint information is as informative as protein change information at the coding level. Low-frequency and common SNP h2 enrichments within regulatory constrained variants were similar (data S10), suggesting that although a very high fraction of variants within regulatory constrained elements are deleterious, their deleterious effects are moderately high (8).
In conclusion, we observed that our mammalian constraint scores have unprecedented base-pair resolution to investigate common variants in GWAS findings for human complex traits and diseases, are distinctively informative compared with known functional annotations and previously published constraint scores, are even more informative when combined with primate constraint scores, and could be used to investigate variants defined as nonfunctional.
Leveraging constraint to move from prioritization to function
Zoonomia constraint scores improve functionally informed fine-mapping analyses
Based on our heritability results, we expected that our constraint scores would improve functionally informed fine-mapping of constrained genetic variants associated with common traits. We compared PolyFun (32) fine-mapping results obtained with no annotations (nonfunctional model) with its default setting of annotations [baseline–low frequency (LF) model] and with an augmented baseline-LF annotation containing multiple Zoonomia constraint annotations (baseline-LF+Zoonomia model) on the 34 well-powered UK Biobank diseases and complex traits (data S12 and SM, section 7). We observed significantly (P < 1.00 × 10−4) greater posterior inclusion probability (PIP) for variants at constrained sites in mammals and primates when using PolyFun with the baseline-LF+Zoonomia model compared with the nonfunctional and baseline-LF models (Fig. 3, A and B). Notably, PolyFun with the baseline-LF+Zoonomia model detected 2100 variants at constrained sites fine-mapped with high confidence (PIP > 0.75) across all the UK Biobank traits (43.81% of high-confidence fine-mapped variants), against 1108 and 1840 when using the nonfunctional and baseline-LF models, respectively (33.39 and 40.92% of high-confidence fine-mapped variants, respectively) (fig. S10).
Fig. 3. Leveraging constraint to move from variation to function.
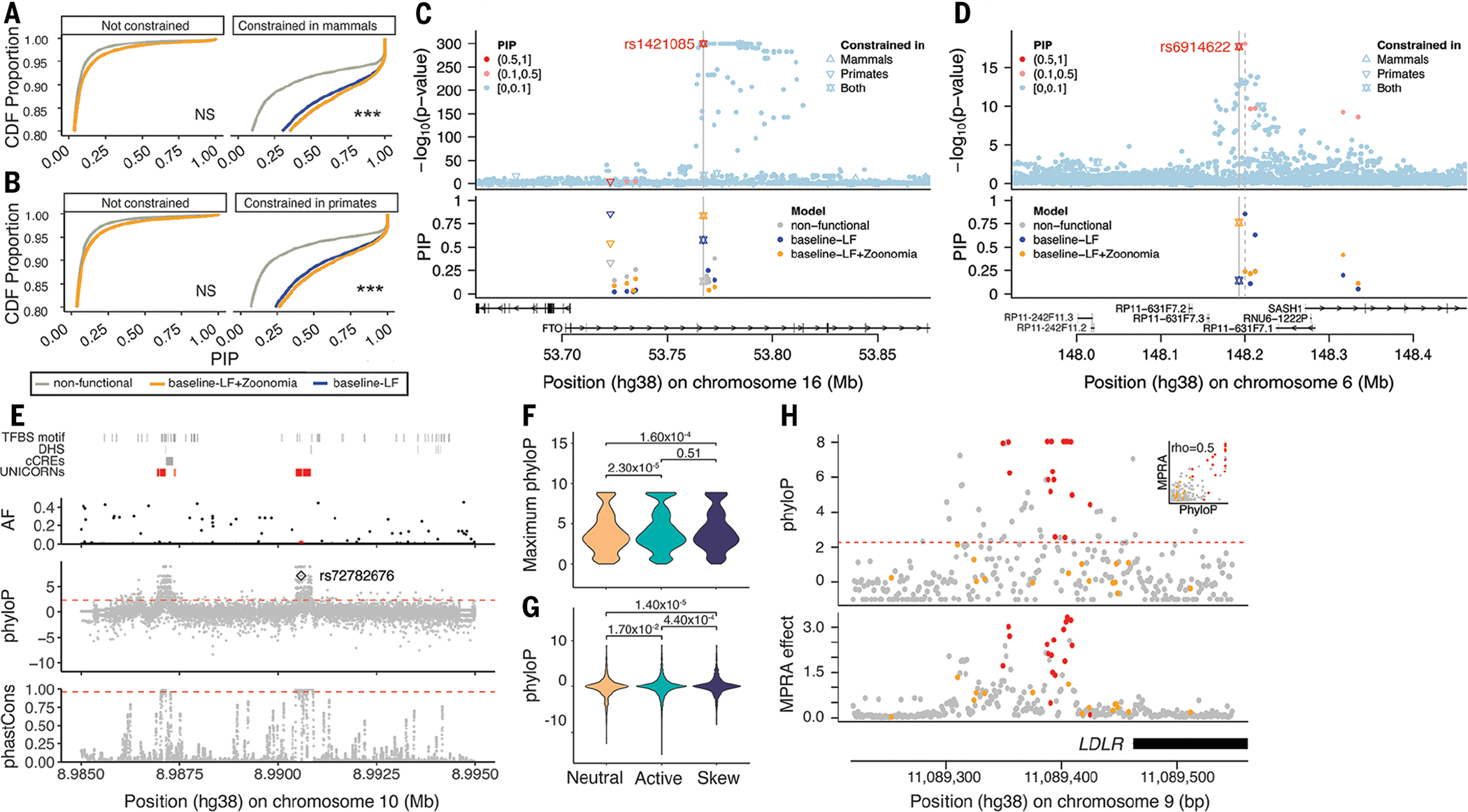
(A and B) We report the cumulative distribution function (CDF) of PIP scores using functionally informed fine-mapping with different models of functional annotations. Distribution functions are split into subpanels according to whether the fine-mapped SNP overlaps high constraint scores in mammals (A) and primates (B). One-way Kolmogorov-Smirnov tests show that CDFs for PIP scores obtained from the baseline-LF model (blue) are lower (above) than the CDFs for PIP scores obtained from the baseline-LF+Zoonomia model (orange) with Bonferroni correction for N = 4 categories across panels (***P < 0.0001; NS is not significant). (C and D) Examples of constrained fine-mapped variants. We report GWAS P values (top) and corresponding PIP scores under different functionally informed fine-mapping models (bottom). The shapes of the data points correspond to constraint information. (E) Fine-mapped variants are not limited to the annotated genome, as exemplified by rs72782676 (red dot in the AF panel) in the GATA3 UNICORN locus. TFBS, transcription factor binding site; cCREs, candidate cis-regulatory regions. (F and G) Constraint is formally linked to function through MPRAs at the regional oligo (F) and base-pair (G) level for neutral, active, and allele-specific skewed effects. (H) For the LDLR promoter locus, the MPRA effect is strongly correlated with the phyloP score. Constrained (red) and unconstrained (orange) ClinVar pathogenic variants are plotted to highlight known deleterious positions. In (E) and (H), the dashed orange lines represent the 5% FDR threshold for constraint.
Fine-mapping examples
We highlight the utility of evolutionary constraint scores in fine-mapping analyses. First, rs1421085 has a causal and experimentally validated association with body mass index (the SNP is located in FTO but has regulatory effects on IRX5 and IRX3) (33, 34); this variant is extremely constrained in mammals (phyloP = 6.31) and primates (phastCons = 1.00), leading to a higher PIP when using the baseline-LF+Zoonomia model (0.84) than when using the nonfunctional and baseline-LF models (0.13 and 0.58, respectively; Fig. 3C). The fractions of CDS and promoter bases that are constrained for IRX5 (0.79 and 0.58) and IRX3 (0.74 and 0.34) were higher than those for FTO (0.61 and 0.23), suggesting that constrained variants in regulatory regions could be more likely to target genes with constrained CDS and/or promoters (see section Evolutionary constraint, PC genes, and human disease). Second, rs6914622 is constrained in mammals and primates (phyloP = 2.37 and phastCons = 1.00) and may be causal in hypothyroidism by the baseline-LF+Zoonomia model (PIP = 0.76; Fig. 3D) but not by the nonfunctional and baseline-LF models (PIP ≤ 0.14). Conversely, the sentinel variant rs9497965 is not evolutionarily constrained but has a notable PIP in the baseline-LF model (PIP ≥ 0.85) but not in the baseline-LF+Zoonomia model (PIP = 0.24). Using epigenetic marks from four thyroid cell types (35) (functional information not in the fine-mapping models), rs6914622 was in an active enhancer in all thyroid cell types and rs9497965 was inferred as being in an enhancer in only one thyroid cell type (weak transcription and quiescent for the others), suggesting a causal role for rs6914622 over rs9497965. Although functional follow-up is necessary, these examples illustrate how Zoonomia constraint scores can affect fine-mapping. Some regulatory elements may not be conserved at the nucleotide level but lie in a cell-type regulatory element that is predicted to be conserved across mammals. Identifying associations between enhancers and phenotypes with the Tissue-Aware Conservation Inference Toolkit (TACIT) provides examples of how mammalian genomes can be leveraged to discover regulatory conservation and link variation to function (36).
Measures of constraint can reveal unannotated variants that affect human health
Because of the challenge of generating functional datasets in all cell types and all cell states, much of the genome’s regulatory space is unannotated (37). The high levels of constraint and low levels of variant diversity in unannotated intergenic constraint regions (UNICORNs) [SM, section 8; (16)] suggest that they are likely of functional importance despite lacking functional annotations (consistent with our observation that unannotated constrained SNPs are enriched in h2; Fig. 2E). Although fewer fine-mapped SNPs were located within UNICORNs (905 SNPs) compared with a matched set of random unannotated nonconstrained intergenic regions (5572 SNPs) and to SNPs located elsewhere in the genome (272,374 SNPs), those variants had higher mean PIP scores (0.14 UNICORNs versus 0.05 for the other two regions). This demonstrates that UNICORNs can reveal unannotated variants that affect human health and disease. UNICORNs contain fine-mapped SNPs with significantly higher PIP scores compared with the background sets across multiple traits (linear regression, P < 0.01 in all cases after correcting for multiple testing; data S13). For example, a 163-bp UNICORN contains rs72782676 with fine-mapping evidence for multiple traits (e.g., eosinophil count, asthma, eczema, respiratory and ear, nose, and throat diseases; AFTOPMed = 0.005; PIP > 0.99 in all GWASs) (Fig. 3E). The nearest gene, GATA3, sits 915 kb upstream, is a master transcriptional regulator for T helper 2 lineage commitment (38), and is known to play an important role in inflammatory disease (39, 40). This UNICORN highlights a strong regulatory candidate for GATA3 in a disease-relevant region that presently lacks annotation.
Predicted variant effect validated at single-base resolution
Massively parallel reporter assays (MPRAs) have been used to rapidly test thousands of genomic variants for their potential regulatory effects on gene expression. Although the functional output from these high-throughput methods is useful for localizing putative causal alleles, overlaying constraint scores may help further elucidate functional variants (SM, section 8). To investigate this, we integrated our Zoonomia-derived phyloP scores with >35,000 assayed variants from existing 3′ untranslated region (3′UTR) (41) and eQTL (42) MPRAs. Using the 3′UTR MPRA data to highlight our results, we found that phyloP scores could differentiate between sequence backgrounds with and without regulatory activity (e.g., across multiple tissues, neutral versus active: Polig = 2.32 × 10−5; Fig. 3F). PhyloP scores further highlighted variants with allele-specific regulatory effects (e.g., neutral versus skew: Pbase = 1.4 × 10−5; Fig. 3G). Additionally, we found that selection on constrained phyloP positions enriched the allele-specific regulatory effects by 1.3-fold (SM, section 8). Similar trends were observed in promoter and enhancer saturation mutagenesis MPRAs (43). For example, phyloP constraint was a strong predictor for variant effect within the LDLR promoter (Spearman’s ρ = 0.51), with five of the most constrained sites providing the strongest regulatory effects and also tagging pathogenic ClinVar positions (Fig. 3H). Further, in our companion paper (44), we use MPRAs to directly assess the regulatory impacts of bases under high constraint that have been deleted specifically in the human lineage. For many, we can precisely identify how the deletions affect transcription factor binding, which is well correlated with the observed regulatory changes, linking sequence change to mechanism. We found that these human-specific deletions were enriched to overlie psychiatric disease GWAS signals (i.e., schizophrenia or bipolar disorder) and discovered 800 deletions with significant species-specific regulatory effects, providing a set of candidate variants that may have contributed to the prevalence of human neurological disorders.
Evolutionary constraint, PC genes, and human disease
Gene-based measures of evolutionary constraint have an important role in understanding the impact of genetic variation on human disease [e.g., LOEUF (loss-of-function observed/expected upper bound fraction)] (3). As detailed in section 9 of the SM, we defined seven measures of gene constraint based on the Zoonomia alignment, including the fraction of CDS constrained, normalization against 32.13 million CDS bases, a model-based approach adjusting for 12 covariates (codon information, mutational consequences, and positional features), and cross-species amino acid constraint (normalized Shannon entropy). After evaluation, we selected the fraction of constrained CDS bases per gene (fracCdsCons) as a simple measure of gene constraint, given its continuous distribution, low missingness, high correlations with more complex measures of gene constraint, and external validation (Fig. 4A). These gene-based constraint metrics are provided in data S14.
Fig. 4. Evolutionary constraint, PC genes, and human disease.
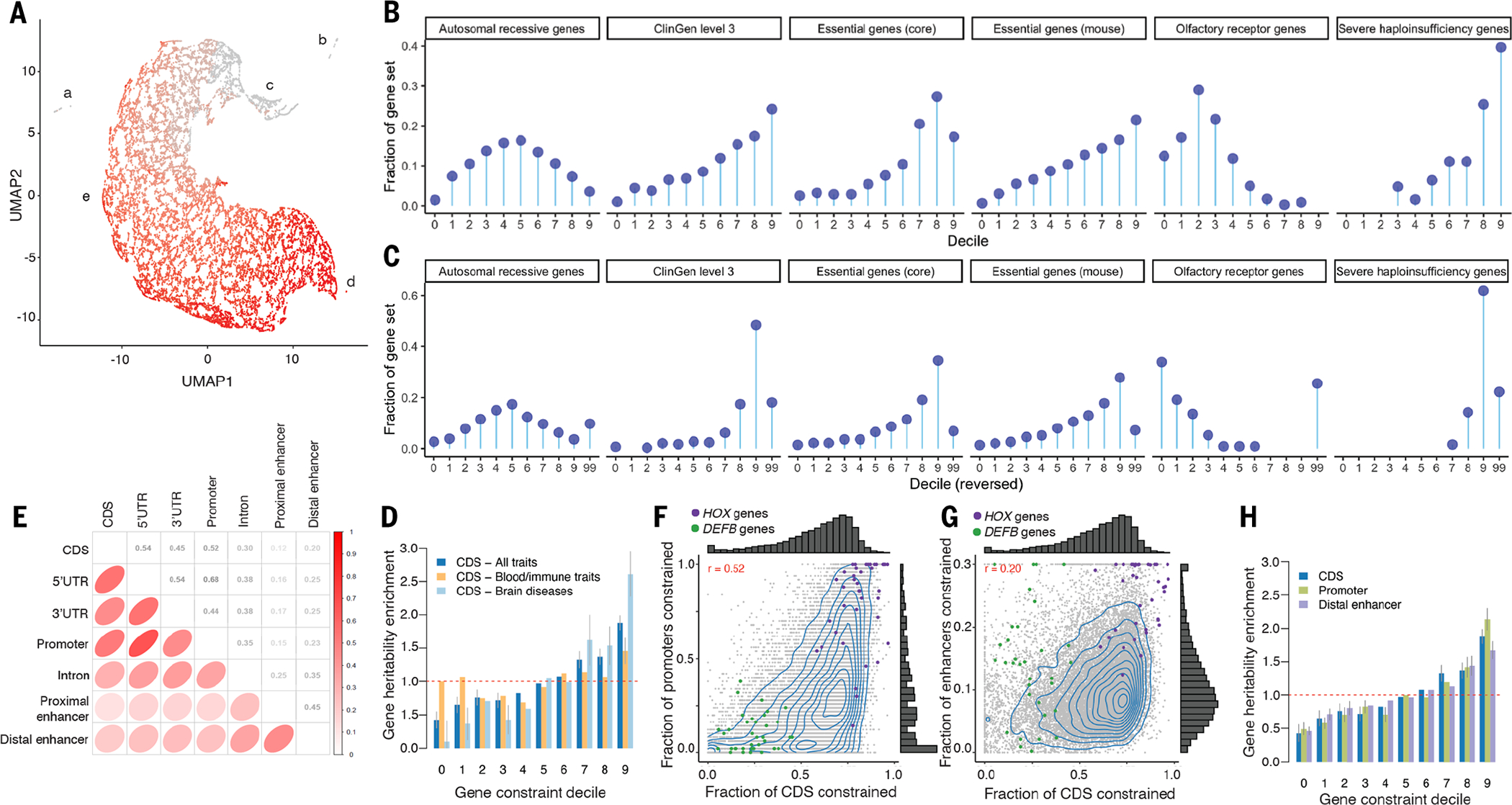
(A) Scatterplot of PC gene clustering [uniform manifold approximation and projection (UMAP) and density-based spatial clustering of applications with noise (DBSCAN)]. The x and y axes are the UMAP coordinates. Each point is a PC gene (N = 19,386). Five clusters are labeled: (a) 56 genes whose CDS bases are in complex regions that align poorly; (b) 221 genes that are apparently human- or primate-specific; (c) 669 genes with good alignment and possible human-specific functions [e.g., five human leukocyte antigen (HLA) genes and 14 interferon-α genes]; (d) 15 genes, all highly constrained; and (e) all other 18,425 PC genes. Coloring shows fracCdsCons, where gray indicates least and red indicates most constrained with an anticlockwise gradient in mammalian constraint from the upper middle to lower right. (B and C) Gene constraint deciles versus external gene sets as “lollipop plots” Zoonomia fracCdsCons are shown in (B). A recapitulation of figure 3 from (3) with the LOEUF decile reversed and missing data shown is presented in (C). Each panel has six subgraphs for autosomal-recessive genes, ClinGen level 3 genes, essential genes from Hart, essential genes in mouse, olfactory receptor genes, and severe haploinsufficiency genes. The x axis is the constraint decile (0 is least, 9 is most constrained, 99 is missing). The y axis is the fraction of the PC genes in a gene set in each decile as represented by circles. (D) Gene heritability enrichment for SNPs linked to genes of each decile of fracCdsCons. The dashed red line represents a null enrichment of 1. Error bars are 95% confidence intervals. (E) Spearman’s correlation of the constraint fraction between the parts of PC genes. (F and G) Fraction of CDS constraint (fracCdsCons) versus fraction of promoter constraint (F) and fraction of distal enhancer constraint (G) (shrunk to values <0.3). For (F) and (G), each point is a PC gene, and HOX genes (purple) and DEFB genes (green) are highlighted. (H) Gene heritability enrichment for SNPs linked to genes of decile of constraint in different gene features, plotted as per (D).
Given the complexities of human PC genes, it would be surprising if any one gene metric applies to all genes [e.g., LOEUF and pLI (probability of being loss-of-function intolerant) are missing for 10.1% of PC genes]. We used an empirical approach to identify genes behaving differently and identified 277 genes (1.43%) that are inaccessible to fracCdsCons (clusters A and B; Fig. 4A and SM, section 10). We examined fracCdsCons in several ways (SM, section 10). First, given its widespread use, we compared fracCdsCons to the inverse-scored LOEUF (3) and found Spearman’s ρ = −0.55. This is notable given the markedly different basis of each measure—constraint over ~100 million years of mammalian evolution versus statistical modeling of predicted loss of function (pLoF) counts in human whole-exome sequencing catalogs (SM, section 2): Empirical confirmation is an important validator for both measures. We next compared fracCdsCons to external gene sets with established patterns of constraint (similar to the LOEUF validation strategy) (3) and obtained similar patterns between both scores (Fig. 4, B and C).
Second, we used an empirical approach to cluster genes based on different constrained metrics (Fig. 4A, data S14, and SM, section 10). After removing 277 gene outliers inaccessible to fracCdsCons, we conducted gene set analyses for 19,109 PC genes (clusters C to E; data S15 and S16). The 5% most constrained genes (N = 955, fracCdsCons 0.811 to 0.975) were strongly enriched in the following gene sets: basic embryology (stem cell proliferation and differentiation, tube formation, anterior and posterior patterning, endoderm and mesoderm formation); organ morphogenesis (central and peripheral nervous system, connective tissue, ear, epithelium, eye, gastrointestinal tract, heart, kidney, lung, muscle, myeloid, pancreas, skeleton); cell cycle (phase transition, fate, WNT), cell signaling, positive and negative regulatory processes; and pre- and postsynaptic processes (synapse assembly, postsynaptic density, neurotransmitter regulation, synaptic vesicle cycle, modulation of transsynaptic signaling). The 5% least constrained genes (N = 956, fracCdsCons 0 to 0.150) were strongly enriched in the following gene sets: microbial defense response (adaptive immunity, bacteria and virus, cell killing, cytokine and interferon); bitter taste and olfaction; and skin development (keratinization, keratinocyte differentiation, epidermal cell differentiation, and epidermis development). The most-constrained genes captured processes fundamental to the making of a mammal, and the least-constrained genes are central to the adaptive evolution of a mammal to its environment—that is, the specific microbiota; adaptations of smell and taste to detect mates, prey, predators, and poisons; and adaptations of skin for temperature regulation, camouflage, and defense.
Finally, we evaluated the relevance of mammalian gene constraint to human disease. Figure S11A shows the relationship of fracCdsCons to multiple human disease annotations. For all comparisons, increasing constraint is correlated with increasing relevance for human disease. Figure S11B depicts the relation with GTEx gene expression, and greater gene constraint is correlated with greater expression in all tissues. “Housekeeping” genes that are uniformly expressed across tissues had greater constraint (P < 3 × 10−197) and made up 3.0% of the least-constrained decile and 30.5% of the most-constrained decile. Finally, we evaluated the impact of common SNPs linked to PC genes in each fracCdsCons decile by estimating their gene h2 enrichment (defined as h2 enrichment for the decile annotation divided by the mean h2 enrichment over all deciles) using S-LDSC on 63 independent GWAS datasets (SM, section 10). We observed significantly higher gene h2 enrichment for SNPs linked to genes in the most-constrained deciles (P = 6.96 × 10−59; Fig. 4D and data S17). We observed stronger gene h2 enrichment patterns in a meta-analysis of nine brain disorders and gene h2 enrichment patterns that were nearly independent of gene constraint in a meta-analysis of 11 blood and immune traits (Fig. 4D and data S17).
Long noncoding RNAs are depleted of constraint bases
Although less well-defined than their PC gene counterparts, long noncoding RNAs (lncRNAs) represent a genome-wide catalog of transcribed elements with broad tissue expression (SM, section 11). We found that lncRNA exons are an order of magnitude less constrained than their PC counterparts (median constraint 0.02 lncRNA versus 0.62 PC genes), and in contrast to others (45, 46), lncRNA promoters have a similar and not higher fraction of constraint compared with lncRNA exons. We found a trend of higher constraint in lncRNAs implicated in cancer or neurological disease but note that this analysis is limited by the number of lncRNAs with clear and validated biological processes. Finally, although lncRNA exons were depleted of common constrained SNPs, these positions were enriched in disease heritability (4.36 ± 2.55–fold in mammals and 9.81 ± 2.78–fold in primates), but only the primate measure was significant (P = 6 × 10−3).
Mammalian constraint is correlated between coding and regulatory elements
We further extended our approach to measure gene constraint on different regulatory features [including promoters and ENCODE3 distal enhancers linked to their genes using EpiMap (35)] because human diseases and complex traits are predominantly affected by common regulatory variants. We found substantial correlations of constraint between CDS and the regulatory parts of PC genes, with a higher correlation between CDS and promoter gene constraint (Spearman’s ρ = 0.55) than between CDS and distal enhancer gene constraint (r = 0.25) (Fig. 4, E to G; gene scores are reported in data S18). These correlations are consistent with the idea that if the function of a gene in mammals requires high conservation of protein structure, then its regulatory sequences tend to also be constrained. We observed families of genes with shared constrained patterns (such as HOX genes that have constrained exons, promoters, and enhancers) and with distinct constrained patterns [such as defensin β (DEFB) genes, which only have constrained enhancers]. Finally, we observed that common SNPs linked to genes with constrained promoters and distal enhancers are as enriched in h2 as genes with constrained CDS, suggesting that constraint in regulatory elements can be leveraged in the analyses of human diseases and complex traits (Fig. 4F and data S17).
Mammalian constraint and copy-number variation
Copy-number variants (CNVs) are genomic segments that have fewer or more copies than a reference genome. CNVs are important drivers of evolution and risk factors for multiple human diseases (47–49). However, CNVs often occur in high-repeat and low-mappability regions, meaning that detecting their presence and importance is often complex (50, 51). We thus evaluated whether mammalian constraint could help prioritize potentially disease-related CNVs. First, as a qualitative check, we evaluated a pathogenic CNV—a small distal enhancer upstream of SOX9 with a ClinVar pathogenic annotation as a cause of Pierre Robin sequence— and found that it was highly constrained (52) (SM, section 12). Second, we evaluated constraint in structural variants (SVs) identified in TOPMed (4). We found that singleton (AC = 1) SV deletions, inversions, and duplications had similar fractions of constrained bases. However, common and low-frequency (AF ≥ 0.005) SV deletions had far less constraint than SV inversions or duplications. We speculate that singletons are recent mutations that have been relatively unexposed to purifying selection, whereas common and low-frequency SV deletions are directly exposed to selection pressures because of the impacts of haploinsufficiency.
Third, these analyses suggest that constrained bases could have utility in CNV prioritization and burden calculations. Given that CNVs are known risk factors for schizophrenia (53), we obtained the CNV call set from the largest published study (21,094 cases, 20,227 controls) (54). After replicating the main analysis, we found that schizophrenia cases had greater CNV constraint burden (the total number of conserved bases affected by a CNV) compared with controls. The case-control differences were four to five logs more significant than two commonly used measures of CNV burden (total number and total bases per person). The improvements were particularly notable for CNV deletions. We suggest that the number of constrained bases affected by a CNV is a more direct assessment of functional impact—for example, a large CNV with no constrained bases is less likely to be deleterious than a far smaller CNV that deletes constrained exons, promoters, and/or enhancer elements.
Evolutionary constraint and polygenic risk scores
Polygenic risk scores (PRSs) have been widely used to summarize the inherited liability for individuals across a broad range of complex diseases, disorders, and human traits (55, 56). High PRSs can confer substantial risk of disease (57, 58). Full details are provided in section 13 of the SM, but, briefly, PRSs are calculated by selecting a subset of SNPs from a large training set (e.g., GWASs for height or diabetes) and then summarizing their impact in an independent testing set for which an estimation of inherited genetic risk in individual subjects is of interest.
Considerable prior work has compared methods of selecting the subset of genetic variants from the training set. Because of LD, a typical GWAS locus can contain hundreds of similarly strongly associated SNPs. A core challenge is to select variants that are the most likely to be causal and that yield the best performance in the testing set, and we asked whether use of constraint measures improved PRSs. Three expert groups evaluated this question using different but complementary approaches as rigorous tests of the utility of constraint scores for PRSs.
As detailed in section 13 of the SM, we found that (i) evolutionarily constrained SNPs contain a disproportionately large fraction of the PRS prediction accuracy (e.g., 3% of all common SNPs captured 88% of the PRS prediction accuracy for human height), (ii) the per-SNP contribution of evolutionarily constrained SNPs is far greater than that of nonconstrained SNPs, (iii) annotating SNPs using evolutionary constraint improves PRS across a range of quantitative and discrete traits, (iv) aggregating constraint metrics (e.g., a union set of mammalian and primate constraint) tended to perform well (but this may vary by the specific trait), and (v) generalizability is maximized by the use of different methodological approaches, traits, and samples.
Cancer driver genes identified with mammalian constraint
Moving from the germline to the somatic genomes, we demonstrated how mammalian constraint in noncoding regions of the genome can be applied to detect candidate cancer driver genes (SM, section 12). Noncoding constraint mutations [NCCMs; phyloP ≥ 1.2 (59)] were identified using whole-genome sequencing data (International Cancer Genome Consortium) (60) for two types of brain tumors that primarily affect children. Pilocytic astrocytoma is a low-grade tumor (61), and medulloblastomas are malignant brain tumors with intertumoral heterogeneity informed by subgroups determined by molecular profiling (i.e., wingless/integrated (WNT), sonic hedgehog signaling (SHH), group 3 and group 4) (62). We identified NCCMs within introns, 5′UTRs and 3′UTRs, and regions within 100 kb of each gene (59).
We found significantly different NCCM rates between the two cancers (63). In pilocytic astrocytoma, which is known to have coding and translocation mutations primarily in BRAF, high NCCM rates were restricted to the BRAF locus, in line with the low somatic mutation burden of this tumor type. Notably, for medulloblastoma, 114 genes had ≥2 NCCMs per 100 kb (Fig. 5A) and 525 genes had ≥5 NCCMs per gene. These genes were enriched for the Gene Ontology (GO) biological processes “nervous system development” (P = 1.32 × 10−26) and “generation of neurons” (P = 1.68 × 10−22). Among the top 114 genes, 15 gene loci were primarily seen in adult cases (≥18 years of age) and seven loci in pediatric cases (<18 years of age). A subset of these loci is shown in Fig. 5B. An example is ZFHX4, which was previously reported to be differentially expressed in medulloblastoma (64), where NCCMs were predominantly identified in adult patients of the SHH subgroup and found in high-constraint ZFHX4 intronic regions (Fig. 5C). For the pediatric set of medulloblastoma, potential driver genes included BMP4 and the HOXB locus (containingmultiple genes),mostly in patients diagnosed as group 3 or group 4. Multiple NCCMs in these two loci were shown to have differential DNA binding capacity in a medulloblastoma cell line (63). Further, we noted differential gene expression in medulloblastoma compared with cerebellum for multiple NCCM genes, for example, HOXB2 (65), for which expression levels correlate with patient survival (66).
Fig. 5. Cancer driver genes identified using NCCM rates.

(A) Distribution of the rates of NCCM for medulloblastoma. (B) An example set of the candidate driver genes found either in pediatric (light blue) or adult (purple) samples. Age of diagnosis (years) of the patient is indicated together with the tumor subgroup. (C) The ZFHX4 locus contains nine NCCMs drawn from eight patients.
The addition of evolutionary constraintmeasures may help advance stratification of medulloblastoma, with regard to both age and molecular subgroups. More generally, we demonstrate how NCCM analysis can be used as a tool for the identification of previously uncharacterized driver genes in cancer. We suggest that NCCM analysis should be evaluated in more cancer types for its potential to yield a better understanding of disease biology and improved diagnosis and prognosis.
Discussion
Understanding genome-wide patterns in the strength of evolutionary constraint can deepen our understanding of human diseases. Zoonomia’s alignment of 240 placental mammals, representing ~100 million years of evolution, achieves single-base resolution constraint that allows a detailed evaluation of individual mutations. This contrasts sharply with existing methodologies that offer only gene-sized resolution. Evolutionary constraint compares favorably to huge amounts of functional genomics data based on specific cell types or tissues because functionality in any tissue at any time point will be detected by constraint. The combination of constraint scores measured here, and additional empirical measures of coding and noncoding function, can only serve to refine our understanding of complex genomic processes. We demonstrate that constraint can be used to detect candidate causal mutations in both rare and common diseases, including cancer, and could be particularly leveraged for brain diseases that are more affected by constrained genes and biological processes. Finally, we note that primate constraint has a stronger heritability enrichment than mammalian constraint in noncoding regions, suggesting that sequencing more primates would complement the present efforts to validate the functions of the multitude of regulatory elements present in the human lineage.
Methods summary
The analyses in support of our study goals were organized into 14 main areas and entailed the coordinated work of more than 10 different teams. Each of these approaches is described in full length as a separate section in the SM and briefly here. The numbers below correspond to the SM section (e.g., section 4: Genomic properties of constraint scores).
-
4)
We described the properties of constrained bases, including GC content, clustering, enrichment in specific elements (gene biotypes, gene parts, regulatory elements), CDS and base-pair resolution, and constraint at variable sites in humans.
-
5)
We benchmarked constraint score against ClinVar (19) and CADD (6) with strong effects on ClinVar classification from 2016 to 2021.
-
6)
We evaluated constraint as an annotation in S-LDSC (7, 25, 26) in GWAS results for 63 independent human traits (27).
-
7)
We applied functionally informed fine-mapping, PolyFun (32), to leverage evolutionary constraint.
-
8)
We identified and evaluated UNICORNs, which are clusters of constrained bases with no known annotation.
-
9)
We created seven gene-based measures of constraint [complementary to residual variation intolerance score (RVIS), pLI, and LOEUF (3)] and selected the simplest measure, fracCdsCons, the fraction of CDS bases under significant constraint (phyloP ≥ 2.27).
-
10)
We conducted extensive evaluation of fracCdsCons, including identifying outliers, gene-set analysis of the top and bottom ventiles, and comparison to LOEUF (3).
-
11)
We developed a constraint measure for long intergenic noncoding RNA genes (lncRNA).
-
12)
We demonstrated the utility of constraint for prioritization of rare CNVs in human disease (e.g., Pierre Robin sequence and schizophrenia).
-
13)
We extensively demonstrated the utility of evolutionary constraint in the selection of SNPs in training sets for application to new data and for developing polygenic risk scores.
-
14)
Finally, we showed that mammalian constraint scores identified previously uncharacterized candidate cancer driver genes in pilocytic astrocytoma and medulloblastoma tumors.
Supplementary Material
ACKNOWLEDGMENTS
Computation and data handling were enabled by projects SNIC 2017/7-385, SNIC 2017/7-386, SNIC 2019/3-415, SNIC 2019/30-57, SNIC 2019/8-369, SNIC 2021/2-11, SNIC 2021/5-296, SNIC 2021/6-208, and SNIC 2021/5-28 provided by the Swedish National Infrastructure for Computing (SNIC) at UPPMAX, which is partially funded by the Swedish Research Council through grant agreement no. 2018-05973.
Funding:
This work was funded by the Swedish Research Council and Knut and Alice Wallenberg Foundation, Swedish Cancer Society, Swedish Childhood Cancer Fund, National Institute of Mental Health (NIMH) U01MH116438, Gladstone Institutes, National Institute on Drug Abuse (NIDA) DP1DA04658501, NIDA F30DA053020, University College Dublin (UCD) Ad Astra Fellowship, and National Human Genome Research Institute (NHGRI) R01HG008742 and U41HG002371. S.G. was supported by National Institutes of Health (NIH) grants R00 HG010160 and R35 GM147789. Y.L. was supported by NIH U01 HG011720. Additional support was provided by the Australian National Health and Medical Research Council (1113400, 1173790, and 1177268). L.M.H. was supported by NIH grants MH118278, MH124839, and ES033630. P.F.S. was supported by the Swedish Research Council (Vetenskapsrådet, award D0886501). This study makes use of data from the UK Biobank (project ID 12505).
Zoonomia Consortium
Gregory Andrews1, Joel C. Armstrong2, Matteo Bianchi3, Bruce W. Birren4, Kevin R. Bredemeyer5, Ana M. Breit6, Matthew J. Christmas3, Hiram Clawson2, Joana Damas7, Federica Di Palma8,9, Mark Diekhans2, Michael X. Dong3, Eduardo Eizirik10, Kaili Fan1, Cornelia Fanter11, Nicole M. Foley5, Karin Forsberg-Nilsson12,13, Carlos J. Garcia14, John Gatesy15, Steven Gazal16, Diane P. Genereux4, Linda Goodman17, Jenna Grimshaw14, Michaela K. Halsey14, Andrew J. Harris5, Glenn Hickey18, Michael Hiller19,20,21, Allyson G. Hindle11, Robert M. Hubley22, Graham M. Hughes23, Jeremy Johnson4, David Juan24, Irene M. Kaplow25,26, Elinor K. Karlsson1,4,27, Kathleen C. Keough17,28,29, Bogdan Kirilenko19,20,21, Klaus-Peter Koepfli30,31,32, Jennifer M. Korstian14, Amanda Kowalczyk25,26, Sergey V. Kozyrev3, Alyssa J. Lawler4,26,33, Colleen Lawless23, Thomas Lehmann34, Danielle L. Levesque6, Harris A. Lewin7,35,36, Xue Li1,4,37, Abigail Lind28,29, Kerstin Lindblad-Toh3,4, Ava Mackay-Smith38, Voichita D. Marinescu3, Tomas Marques-Bonet39,40,41,42, Victor C. Mason43, Jennifer R. S. Meadows3, Wynn K. Meyer44, Jill E. Moore1, Lucas R. Moreira1,4, Diana D. Moreno-Santillan14, Kathleen M. Morrill1,4,37, Gerard Muntané24, William J. Murphy5, Arcadi Navarro39,41,45,46, Martin Nweeia47,48,49,50, Sylvia Ortmann51, Austin Osmanski14, Benedict Paten2, Nicole S. Paulat14, Andreas R. Pfenning25,26, BaDoi N. Phan25,26,52, Katherine S. Pollard28,29,53, Henry E. Pratt1, David A. Ray14, Steven K. Reilly38, Jeb R. Rosen22, Irina Ruf54, Louise Ryan23, Oliver A. Ryder55,56, Pardis C. Sabeti4,57,58, Daniel E. Schäffer25, Aitor Serres24, Beth Shapiro59,60, Arian F. A. Smit22, Mark Springer61, Chaitanya Srinivasan25, Cynthia Steiner55, Jessica M. Storer22, Kevin A. M. Sullivan14, Patrick F. Sullivan62,63, Elisabeth Sundström3, Megan A. Supple59, Ross Swofford4, Joy-El Talbot64, Emma Teeling23, Jason Turner-Maier4, Alejandro Valenzuela24, Franziska Wagner65, Ola Wallerman3, Chao Wang3, Juehan Wang16, Zhiping Weng1, Aryn P. Wilder55, Morgan E. Wirthlin25,26,66, James R. Xue4,57, Xiaomeng Zhang4,25,26
1 Program in Bioinformatics and Integrative Biology, UMass Chan Medical School, Worcester, MA 01605, USA. 2Genomics Institute, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 3Department of Medical Biochemistry and Microbiology, Science for Life Laboratory, Uppsala University, Uppsala 751 32, Sweden. 4Broad Institute of MIT and Harvard, Cambridge, MA 02139, USA. 5Veterinary Integrative Biosciences, Texas A&M University, College Station, TX 77843, USA. 6School of Biology and Ecology, University of Maine, Orono, ME 04469, USA. 7The Genome Center, University of California Davis, Davis, CA 95616, USA. 8Genome British Columbia, Vancouver, BC, Canada. 9School of Biological Sciences, University of East Anglia, Norwich, UK. 10School of Health and Life Sciences, Pontifical Catholic University of Rio Grande do Sul, Porto Alegre 90619–900, Brazil. 11School of Life Sciences, University of Nevada Las Vegas, Las Vegas, NV 89154, USA. 12Biodiscovery Institute, University of Nottingham, Nottingham, UK. 13Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala 751 85, Sweden. 14Department of Biological Sciences, Texas Tech University, Lubbock, TX 79409, USA. 15Division of Vertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA. 16Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA. 17Fauna Bio, Inc., Emeryville, CA 94608, USA. 18Baskin School of Engineering, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 19Faculty of Biosciences, Goethe-University, 60438 Frankfurt, Germany. 20LOEWE Centre for Translational Biodiversity Genomics, 60325 Frankfurt, Germany. 21Senckenberg Research Institute, 60325 Frankfurt, Germany. 22Institute for Systems Biology, Seattle, WA 98109, USA. 23School of Biology and Environmental Science, University College Dublin, Belfield, Dublin 4, Ireland. 24Department of Experimental and Health Sciences, Institute of Evolutionary Biology (UPF-CSIC), Universitat Pompeu Fabra, Barcelona 08003, Spain. 25Department of Computational Biology, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 26Neuroscience Institute, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 27Program in Molecular Medicine, UMass Chan Medical School, Worcester, MA 01605, USA. 28Department of Epidemiology & Biostatistics, University of California San Francisco, San Francisco, CA 94158, USA. 29Gladstone Institutes, San Francisco, CA 94158, USA. 30Center for Species Survival, Smithsonian’s National Zoo and Conservation Biology Institute, Washington, DC 20008, USA. 31Computer Technologies Laboratory, ITMO University, St. Petersburg 197101, Russia. 32Smithsonian-Mason School of Conservation, George Mason University, Front Royal, VA 22630, USA. 33Department of Biological Sciences, Mellon College of Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 34Senckenberg Research Institute and Natural History Museum Frankfurt, 60325 Frankfurt am Main, Germany. 35Department of Evolution and Ecology, University of California Davis, Davis, CA 95616, USA. 36John Muir Institute for the Environment, University of California Davis, Davis, CA 95616, USA. 37Morningside Graduate School of Biomedical Sciences, UMass Chan Medical School, Worcester, MA 01605, USA. 38Department of Genetics, Yale School of Medicine, New Haven, CT 06510, USA. 39Catalan Institution of Research and Advanced Studies (ICREA), Barcelona 08010, Spain. 40CNAG-CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona 08036, Spain. 41Department of Medicine and Life Sciences, Institute of Evolutionary Biology (UPF-CSIC), Universitat Pompeu Fabra, Barcelona 08003, Spain. 42Institut Català de Paleontologia Miquel Crusafont, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Barcelona, Spain. 43Institute of Cell Biology, University of Bern, 3012 Bern, Switzerland. 44Department of Biological Sciences, Lehigh University, Bethlehem, PA 18015, USA. 45BarcelonaBeta Brain Research Center, Pasqual Maragall Foundation, Barcelona 08005, Spain. 46CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona 08003, Spain. 47Department of Comprehensive Care, School of Dental Medicine, Case Western Reserve University, Cleveland, OH 44106, USA. 48Department of Vertebrate Zoology, Canadian Museum of Nature, Ottawa, ON K2P 2R1, Canada. 49Department of Vertebrate Zoology, Smithsonian Institution, Washington, DC 20002, USA. 50Narwhal Genome Initiative, Department of Restorative Dentistry and Biomaterials Sciences, Harvard School of Dental Medicine, Boston, MA 02115, USA. 51Department of Evolutionary Ecology, Leibniz Institute for Zoo and Wildlife Research, 10315 Berlin, Germany. 52Medical Scientist Training Program, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261, USA. 53Chan Zuckerberg Biohub, San Francisco, CA 94158, USA. 54Division of Messel Research and Mammalogy, Senckenberg Research Institute and Natural History Museum Frankfurt, 60325 Frankfurt am Main, Germany. 55Conservation Genetics, San Diego Zoo Wildlife Alliance, Escondido, CA 92027, USA. 56Department of Evolution, Behavior and Ecology, School of Biological Sciences, University of California San Diego, La Jolla, CA 92039, USA. 57Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA 02138, USA. 58Howard Hughes Medical Institute, Chevy Chase, MD, USA. 59Department of Ecology and Evolutionary Biology, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 60Howard Hughes Medical Institute, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 61Department of Evolution, Ecology and Organismal Biology, University of California Riverside, Riverside, CA 92521, USA. 62Department of Genetics, University of North Carolina Medical School, Chapel Hill, NC 27599, USA. 63Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden. 64Iris Data Solutions, LLC, Orono, ME 04473, USA. 65Museum of Zoology, Senckenberg Natural History Collections Dresden, 01109 Dresden, Germany. 66Allen Institute for Brain Science, Seattle, WA 98109, USA.
Footnotes
Competing interests: P.F.S. is a consultant and shareholder for Neumora.
Diversity and inclusion: One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Data and materials availability:
Scripts for PhyloP and PhastCons constraint score calculation are available at https://github.com/michaeldong1/ZOONOMIA.git (67). Code to perform analyses benchmarking of phyloP deleteriousness and a case example are available at https://github.com/teone182/Zoonomia_Scripts (68). Scripts for analyses using TOGA are available at https://github.com/GMHughes/ZoonomiaScripts (69). LDSC software and annotations are available at https://www.github.com/bulik/ldsc (70) and https://alkesgroup.broadinstitute.org/LDSCORE/. GWAS summary statistics used in LDSC analyses are available at https://alkesgroup.broadinstitute.org/cS2G/sumstats_63/ and https://zenodo.org/record/7787039 (71). S-LDXR software is available at https://alkesgroup.broadinstitute.org/S-LDXR/. Code to perform Polyfun analyses is available at https://github.com/pfenninglab/Zoonomia_flagship2_fine-mapping (72). Polyfun annotations for fine-mapping are available at https://kilthub.cmu.edu/account/articles/19380533.
REFERENCES AND NOTES
- 1.Moore JE et al. , Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). doi: 10.1038/s41586-020-2493-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguet F et al. , The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020). doi: 10.1126/science.aaz1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karczewski KJ et al. , The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taliun D et al. , Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021). doi: 10.1038/s41586-021-03205-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper GM, Shendure J, Needles in stacks of needles: Finding disease-causal variants in a wealth of genomic data. Nat. Rev. Genet. 12, 628–640 (2011). doi: 10.1038/nrg3046 [DOI] [PubMed] [Google Scholar]
- 6.Kircher M et al. , A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014). doi: 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finucane HK et al. , Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015). doi: 10.1038/ng.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gazal S et al. , Functional architecture of low-frequency variants highlights strength of negative selection across coding and non-coding annotations. Nat. Genet. 50, 1600–1607 (2018). doi: 10.1038/s41588-018-0231-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hujoel MLA, Gazal S, Hormozdiari F, van de Geijn B, Price AL, Disease heritability enrichment of regulatory elements is concentrated in elements with ancient sequence age and conserved function across species. Am. J. Hum. Genet. 104, 611–624 (2019). doi: 10.1016/j.ajhg.2019.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Visscher PM et al. , 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 101, 5–22 (2017). doi: 10.1016/j.ajhg.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallagher MD, Chen-Plotkin AS, The post-GWAS era: From association to function. Am. J. Hum. Genet. 102, 717–730 (2018). doi: 10.1016/j.ajhg.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tam V et al. , Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484 (2019). doi: 10.1038/s41576-019-0127-1 [DOI] [PubMed] [Google Scholar]
- 13.Claussnitzer M et al. , A brief history of human disease genetics. Nature 577, 179–189 (2020). doi: 10.1038/s41586-019-1879-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uffelmann E et al. , Genome-wide association studies. Nat. Rev. Methods Primers 1, 59 (2021). doi: 10.1038/s43586-021-00056-9 [DOI] [Google Scholar]
- 15.Lappalainen T, MacArthur DG, From variant to function in human disease genetics. Science 373, 1464–1468 (2021). doi: 10.1126/science.abi8207 [DOI] [PubMed] [Google Scholar]
- 16.Christmas MJ et al. , Evolutionary constraint and innovation across hundreds of placental mammals. Science 380, eabn3943 (2023). doi: 10.1123/science.abn3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siepel A, Pollard KS, Haussler D, New methods for detecting lineage-specific selection. Lect. Notes Comput. Sci. 3909, 190–205 (2006). doi: 10.1007/11732990_17 [DOI] [Google Scholar]
- 18.Siepel A et al. , Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 (2005). doi: 10.1101/gr.3715005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finan C et al. , The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 9, eaag1166 (2017). doi: 10.1126/scitranslmed.aag1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landrum MJ et al. , ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018). doi: 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirilenko BM et al. , Integrating gene annotation with orthology inference at scale. Science 380, eabn3107 (2023). doi: 10.1126/science.abn3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lopes-Pacheco M, CFTR modulators: Shedding light on precision medicine for cystic fibrosis. Front. Pharmacol. 7, 275 (2016). doi: 10.3389/fphar.2016.00275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akbari P et al. , Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science 373, eabf8683 (2021). doi: 10.1126/science.abf8683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones EM et al. , Structural and functional characterization of G protein-coupled receptors with deep mutational scanning. eLife 9, e54895 (2020). doi: 10.7554/eLife.54895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gazal S et al. , Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat. Genet. 49, 1421–1427 (2017). doi: 10.1038/ng.3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gazal S, Marquez-Luna C, Finucane HK, Price AL, Reconciling S-LDSC and LDAK functional enrichment estimates. Nat. Genet. 51, 1202–1204 (2019). doi: 10.1038/s41588-019-0464-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gazal S et al. , Combining SNP-to-gene linking strategies to pinpoint disease genes and assess disease omnigenicity. Nat. Genet. 54, 827–836 (2022). doi: 10.1038/s41588-022-01087-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davydov EV et al. , Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLOS Comput. Biol. 6, e1001025 (2010). doi: 10.1371/journal.pcbi.1001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindblad-Toh K et al. , A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (2011). doi: 10.1038/nature10530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hormozdiari F et al. , Leveraging molecular quantitative trait loci to understand the genetic architecture of diseases and complex traits. Nat. Genet. 50, 1041–1047 (2018). doi: 10.1038/s41588-018-0148-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi H et al. , Population-specific causal disease effect sizes in functionally important regions impacted by selection. Nat. Commun. 12, 1098 (2021). doi: 10.1038/s41467-021-21286-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weissbrod O et al. , Functionally informed fine-mapping and polygenic localization of complex trait heritability. Nat. Genet. 52, 1355–1363 (2020). doi: 10.1038/s41588-020-00735-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Claussnitzer M et al. , FTO obesity variant circuitry and adipocyte browning in humans. N. Engl. J. Med. 373, 895–907 (2015). doi: 10.1056/NEJMoa1502214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Claussnitzer M, Hui C-C, Kellis M, FTO obesity variant and adipocyte browning in humans. N. Engl. J. Med. 374, 192–193 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Boix CA, James BT, Park YP, Meuleman W, Kellis M, Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature 590, 300–307 (2021). doi: 10.1038/s41586-020-03145-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaplow IM et al. , Zoonomia Consortium, Relating enhancer genetic variation across mammals to complex phenotypes using machine learning. Science 380, eabm7993 (2023). doi: 10.1126/science.abm7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Umans BD, Battle A, Gilad Y, Where are the disease-associated eQTLs? Trends Genet. 37, 109–124 (2021). doi: 10.1016/j.tig.2020.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu J, Yamane H, Cote-Sierra J, Guo L, Paul WE, GATA-3 promotes Th2 responses through three different mechanisms: Induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. 16, 3–10 (2006). doi: 10.1038/sj.cr.7310002 [DOI] [PubMed] [Google Scholar]
- 39.Mjösberg J et al. , The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity 37, 649–659 (2012). doi: 10.1016/j.immuni.2012.08.015 [DOI] [PubMed] [Google Scholar]
- 40.Wohlfert EA et al. , GATA3 controls Foxp3+ regulatory T cell fate during inflammation in mice. J. Clin. Invest. 121, 4503–4515 (2011). doi: 10.1172/JCI57456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griesemer D et al. , Genome-wide functional screen of 3′UTR variants uncovers causal variants for human disease and evolution. Cell 184, 5247–5260.e19 (2021). doi: 10.1016/j.cell.2021.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tewhey R et al. , Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 172, 1132–1134 (2018). doi: 10.1016/j.cell.2018.02.021 [DOI] [PubMed] [Google Scholar]
- 43.Kircher M et al. , Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat. Commun. 10, 3583 (2019). doi: 10.1038/s41467-019-11526-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xue JR et al. , The functional and evolutionary impacts of human-specific deletions in conserved elements. Science 380, eabn2253 (2023). doi: 10.1126/science.abn2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Necsulea A et al. , The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505, 635–640 (2014). doi: 10.1038/nature12943 [DOI] [PubMed] [Google Scholar]
- 46.Chodroff RA et al. , Long noncoding RNA genes: Conservation of sequence and brain expression among diverse amniotes. Genome Biol. 11, R72 (2010). doi: 10.1186/gb-2010-11-7-r72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Innan H, Kondrashov F, The evolution of gene duplications: Classifying and distinguishing between models. Nat. Rev. Genet. 11, 97–108 (2010). doi: 10.1038/nrg2689 [DOI] [PubMed] [Google Scholar]
- 48.Zarrei M, MacDonald JR, Merico D, Scherer SW, A copy number variation map of the human genome. Nat. Rev. Genet. 16, 172–183 (2015). doi: 10.1038/nrg3871 [DOI] [PubMed] [Google Scholar]
- 49.Mérot C, Oomen RA, Tigano A, Wellenreuther M, A roadmapfor understanding the evolutionary significance of structural genomic variation. Trends Ecol. Evol. 35, 561–572 (2020). doi: 10.1016/j.tree.2020.03.002 [DOI] [PubMed] [Google Scholar]
- 50.Lappalainen T, Scott AJ, Brandt M, Hall IM, Genomic analysis in the age of human genome sequencing. Cell 177, 70–84 (2019). doi: 10.1016/j.cell.2019.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mahmoud M et al. , Structural variant calling: The long and the short of it. Genome Biol. 20, 246 (2019). doi: 10.1186/s13059-019-1828-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Long HK et al. , Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell 27, 765–783.e14 (2020). doi: 10.1016/j.stem.2020.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sullivan PF, Geschwind DH, Defining the genetic, genomic, cellular, and diagnostic architectures of psychiatric disorders. Cell 177, 162–183 (2019). doi: 10.1016/j.cell.2019.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marshall CR et al. , Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 49, 27–35 (2017). doi: 10.1038/ng.3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.International Schizophrenia Consortium, Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752 (2009). doi: 10.1038/nature08185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wray NR et al. , From basic science to clinical application of polygenic risk scores: A primer. JAMA Psychiatry 78, 101–109 (2021). doi: 10.1001/jamapsychiatry.2020.3049 [DOI] [PubMed] [Google Scholar]
- 57.Schizophrenia Working Group of the Psychiatric Genomics Consortium, Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). doi: 10.1038/nature13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khera AV et al. , Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018). doi: 10.1038/s41588-018-0183-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sakthikumar S et al. , Whole-genome sequencing of glioblastoma reveals enrichment of non-coding constraint mutations in known and novel genes. Genome Biol. 21, 127 (2020). doi: 10.1186/s13059-020-02035-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J et al. , The International Cancer Genome Consortium Data Portal. Nat. Biotechnol. 37, 367–369 (2019). doi: 10.1038/s41587-019-0055-9 [DOI] [PubMed] [Google Scholar]
- 61.Louis DN et al. , The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 131, 803–820 (2016). doi: 10.1007/s00401-016-1545-1 [DOI] [PubMed] [Google Scholar]
- 62.Northcott PA et al. , The whole-genome landscape of medulloblastoma subtypes. Nature 547, 311–317 (2017). doi: 10.1038/nature22973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roy A et al. , Using evolutionary constraint to define novel candidate driver genes in medulloblastoma. bioRxiv 2022.11.02.514465 [Preprint] (2022); doi: 10.1101/2022.11.02.514465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smits M et al. , EZH2-regulated DAB2IP is a medulloblastoma tumor suppressor and a positive marker for survival. Clin. Cancer Res. 18, 4048–4058 (2012). doi: 10.1158/1078-0432.CCR-12-0399 [DOI] [PubMed] [Google Scholar]
- 65.Weishaupt H et al. , Batch-normalization of cerebellar and medulloblastoma gene expression datasets utilizing empirically defined negative control genes. Bioinformatics 35, 3357–3364 (2019). doi: 10.1093/bioinformatics/btz066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cavalli FMG et al. , Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 31, 737–754.e6 (2017). doi: 10.1016/j.ccell.2017.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.michaeldong1, michaeldong1/ZOONOMIA: v1.0.0. Zenodo: (2022); 10.5281/zenodo.7276319. [DOI] [Google Scholar]
- 68.Bianchi M, teone182/Zoonomia_Scripts: ZoonomiaScripts_MB. Zenodo: (2022); 10.5281/zenodo.7276329. [DOI] [Google Scholar]
- 69.HughesLab, GMHughes/ZoonomiaScripts: v1.0.0. Zenodo: (2022); 10.5281/zenodo.7276583. [DOI] [Google Scholar]
- 70.Gazal S, Zoonomia annotation files for S-LDSC. (2022); 10.5281/zenodo.7292919. [DOI]
- 71.Gazal S, Baseline-LF model. Zenodo: (2023); 10.5281/zenodo.7787039. [DOI] [Google Scholar]
- 72.Phan B, pfenninglab/Zoonomia_flagship2_fine-mapping: v1.0.0 publication. Zenodo: (2022); 10.5281/zenodo.7277007. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Scripts for PhyloP and PhastCons constraint score calculation are available at https://github.com/michaeldong1/ZOONOMIA.git (67). Code to perform analyses benchmarking of phyloP deleteriousness and a case example are available at https://github.com/teone182/Zoonomia_Scripts (68). Scripts for analyses using TOGA are available at https://github.com/GMHughes/ZoonomiaScripts (69). LDSC software and annotations are available at https://www.github.com/bulik/ldsc (70) and https://alkesgroup.broadinstitute.org/LDSCORE/. GWAS summary statistics used in LDSC analyses are available at https://alkesgroup.broadinstitute.org/cS2G/sumstats_63/ and https://zenodo.org/record/7787039 (71). S-LDXR software is available at https://alkesgroup.broadinstitute.org/S-LDXR/. Code to perform Polyfun analyses is available at https://github.com/pfenninglab/Zoonomia_flagship2_fine-mapping (72). Polyfun annotations for fine-mapping are available at https://kilthub.cmu.edu/account/articles/19380533.