

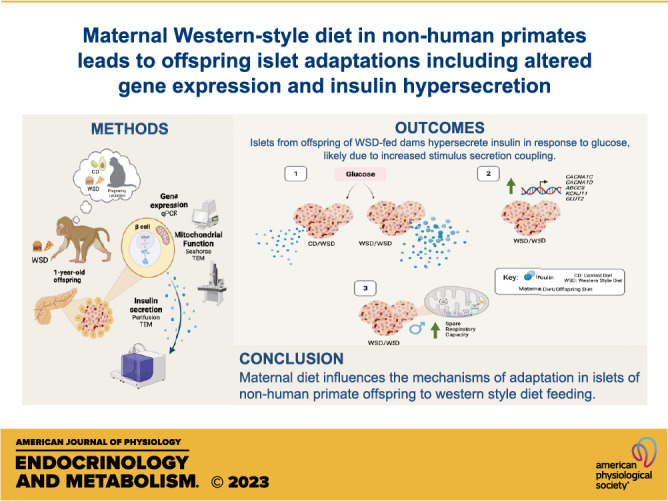
Keywords: developmental origins of health and disease, developmental programming, maternal overnutrition, mitochondrial respiration, pancreatic β cell
Abstract
Maternal overnutrition is associated with increased susceptibility to type 2 diabetes in the offspring. Rodent models have shown that maternal overnutrition influences islet function in offspring. To determine whether maternal Western-style diet (WSD) alters prejuvenile islet function in a model that approximates that of human offspring, we utilized a well-characterized Japanese macaque model. We compared islet function from offspring exposed to WSD throughout pregnancy and lactation and weaned to WSD (WSD/WSD) compared with islets from offspring exposed only to postweaning WSD (CD/WSD) at 1 yr of age. WSD/WSD offspring islets showed increased basal insulin secretion and an exaggerated increase in glucose-stimulated insulin secretion, as assessed by dynamic ex vivo perifusion assays, relative to CD/WSD-exposed offspring. We probed potential mechanisms underlying insulin hypersecretion using transmission electron microscopy to evaluate β-cell ultrastructure, qRT-PCR to quantify candidate gene expression, and Seahorse assay to assess mitochondrial function. Insulin granule density, mitochondrial density, and mitochondrial DNA ratio were similar between groups. However, islets from WSD/WSD male and female offspring had increased expression of transcripts known to facilitate stimulus-secretion coupling and changes in the expression of cell stress genes. Seahorse assay revealed increased spare respiratory capacity in islets from WSD/WSD male offspring. Overall, these results show that maternal WSD feeding confers changes to genes governing insulin secretory coupling and results in insulin hypersecretion as early as the postweaning period. The results suggest a maternal diet leads to early adaptation and developmental programming in offspring islet genes that may underlie future β-cell dysfunction.
NEW & NOTEWORTHY Programed adaptations in islets in response to maternal WSD exposure may alter β-cell response to metabolic stress in offspring. We show that islets from maternal WSD-exposed offspring hypersecrete insulin, possibly due to increased components of stimulus-secretion coupling. These findings suggest that islet hyperfunction is programed by maternal diet, and changes can be detected as early as the postweaning period in nonhuman primate offspring.
INTRODUCTION
Over 36% of pregnancies in the United States are in women classified as obese (1). A growing body of evidence demonstrates that consumption of a calorically dense, Western-style diet (WSD), rather than obesity per se, results in increased risk of cardiometabolic disease, including type 2 diabetes (T2D), in the offspring (2, 3). These findings align with the Developmental Origins of Health and Disease hypothesis, which suggests that the fetal and early postnatal environments program susceptibility to disease in offspring (4, 5). Most concerning is the increased risk of childhood obesity and its sequelae, including hypertension, nonalcoholic fatty liver disease, and T2D in youth born to mothers with obesity (3).
With the goal of understanding and, ultimately, interrupting the cycle of obesity and metabolic disease, our group has generated and utilized a Japanese macaque model of maternal WSD consumption during gestation and lactation (Fig. 1; 6–17). The importance of the nonhuman primate (NHP) model is that the developmental changes in placenta, islets, liver, and brain are similar to humans and is the only model that develops the full spectrum of metabolic disease as in humans, including complex psychosocial behaviors in the offspring (6, 18, 19). Studies in this model have identified lasting effects of maternal WSD exposure on the offspring from the fetal stage of development through adolescence in key metabolic organs. For instance, fetal and postnatal WSD-exposed offspring have increased liver triglycerides and hepatic periportal collagen deposition, supporting early signs of pediatric fatty liver disease (8, 20). In addition, in skeletal muscle, maternal WSD impairs oxidative capacity in fetuses, which is present even when obese, WSD-fed mothers are switched to a control diet (CD) before pregnancy (8, 12, 20, 21). Postnatally, offspring exposed to maternal WSD have a greater preference for high-fat, high-sugar foods (22). Maternal overnutrition also leads to gut dysbiosis in the offspring, which is only partially corrected when offspring consume a healthy diet postweaning (17). Furthermore, maternal WSD intake confers reduced insulin-stimulated glucose uptake in skeletal muscle from fetal and 1-yr-old NHP offspring. These effects are persistent, even in the absence of elevated adiposity, or elevated fasting insulin or glucose, or insulin area under the curve during an intravenous glucose tolerance test (23).
Figure 1.

Schematic of experimental design. Female Japanese macaques were assigned to a control diet (CD) or Western-style diet (WSD) for 4–7 yr before pregnancy and maintained on the same diet throughout gestation and lactation. On weaning, offspring were all maintained on WSD. One-year-old offspring were necropsied at 14 mo of age. Created with Biorender.com.
In the pancreatic islet, maternal WSD feeding similarly leads to programed changes in fetal islets that persist in 3-yr-old offspring. In the fetus, exposure to a maternal WSD results in reduced α-cell mass (24), which persists to 3 yr of age even when weaned at 7 mo onto a control diet (25). These islets from 3-yr-old offspring are functionally aberrant and hypersecrete insulin in response to glucose ex vivo, despite having been weaned onto a CD (25). These findings are in line with a large body of literature demonstrating that maternal caloric dense and high-fat diet (HFD) feeding is associated with persistent structural and functional adaptations in the islet in different animal models, ranging from zebrafish to mammals (26–31).
With the goal of ascertaining whether adaptations in islet function following postweaning WSD consumption are altered by developmental WSD exposure, we compared live, isolated islets and fixed pancreas tissue from 1-yr-old offspring (postweaning) from our NHP model of maternal WSD feeding and postnatal WSD exposure on the molecular phenotype and functional effects on islets in the offspring. These results confirm an increase in glucose-stimulated insulin secretion (GSIS) from islets of 1-yr-old offspring exposed to maternal WSD and reveal changes in islet gene expression that are predicted to facilitate insulin secretion and modulate cell stress, suggesting coordination between β-cell metabolism and dysfunction in response to the nutritional milieu experienced during development.
METHODS
Animal Care
All animal procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the Oregon National Primate Research Center (ONPRC) and Oregon Health and Sciences University (OHSU) and were approved by the ONPRC IACUC. The ONPRC abides by the Animal Welfare Act and Regulations enforced by the United States Department of Agriculture (USDA) and the Public Health Service Policy on Humane Care and Use of Laboratory Animals in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Offspring Generation and Diet Assignment
Before mating, adult female Japanese macaques (Macaca fuscata), starting at 4–7 yr of age, were placed on either CD (Fiber Balanced Diet 5000; Purina Mills, Gray Summit, MO) or WSD (TAD Diet No. 5LOP, Test Diet, Purina Mills, Gray Summit, MO) for a minimum of 2 yr. The CD is made up of 15% of calories from fat, whereas the WSD has ∼36% of calories from fat. Both diets contain sufficient vitamin, mineral, and protein content for normal growth. Animals in the WSD group also received calorically dense treats once per day as previously described (8, 19). All animals were housed in social environments comprising several females and a single male (fed the same diet as dam) to facilitate natural, optimal breeding. Females were sedated two to three times during pregnancy for fetal dating and third trimester measures. Dams gave birth naturally in their social groups, and most offspring began independently ingesting the maternal diet by 4 mo of age but were allowed to stay with their mothers until weaning, beginning at 7 mo of age. Birth weight did not differ at 1 mo of age between male and female offspring exposed to either CD or WSD (data not shown). Weaned offspring were placed in social groups with similarly aged offspring and one to two adult females. Maternal CD and WSD-exposed offspring were all fed WSD at weaning, generating two experimental groups: CD/WSD and WSD/WSD. At 14 mo of age, offspring were sedated with 15–20 mg/kg ketamine and humanely euthanized with sodium pentobarbital followed by exsanguination under the AVMA guidelines for the Euthanasia of Animals. Before the study, two offspring were excluded due to injury and death as monitored by animal care technicians during routine maintenance of colony. A total of 15 CD/WSD and 14 WSD/WSD animals were eligible for use in the current study. The number of islets and tissue samples utilized in experiments herein was based on the number of offspring of the correct age available during the study period.
Isolation of Pancreatic Islets
Pancreata were excised from surrounding tissues and placed in ice-cold phosphate-buffered saline (PBS, Sigma-Aldrich, St. Louis, MO) before islet isolations. Pancreata were then removed from PBS and inflated with collagenase P (0.5 mg/mL, 80 mL total volume, Sigma-Aldrich, St. Louis, MO) via cannulation of the pancreatic duct using 18–20-gauge catheters. Once inflated, each pancreas was divided into 12 sections and digested for 27–30 min at 37°C in a collagenase solution. Using a Histopaque gradient (Sigma-Aldrich, St. Louis, MO), islets were collected at the interphase between Histopaque and medium. Islets were cultured overnight in supplemented RMPI 1640 media (Sigma-Aldrich, St. Louis, MO) at 37°C and 5% CO2.
Glucose-Stimulated Insulin Secretion Assay
After overnight recovery, islets were transferred into prepared columns and placed in a perifusion system (PERI-4.2, Biorep Technologies, Miami Lakes, FL) maintained at 37°C. Islets were preincubated in Krebs-Ringer bicarbonate HEPES buffer (KRBH) containing 4.0 mM glucose for 1 h at a flow rate of 100 µL/min. After preincubation, islets underwent 4 × 15-min washes in 4.0 or 16.7 mM glucose-supplemented KRBH for a total of 60 min, with collections every 3 min. Collections were stored at 4°C and processed the same day. All perifusion GSIS assays were done in triplicate. Insulin was assayed using a Human Insulin ELISA kit (ALPCO, Salem, NH) and normalized to the number of islets present in each column. Technical replicates were averaged to achieve each datapoint.
Oxygen Consumption Assay
Following perifusion, islets were shipped overnight to Vanderbilt University Medical Center in plastic vials containing supplemented RPMI media (Sigma-Aldrich, St.Louis, MO) at room temperature for ex vivo oxygen consumption assays. Islet quality was assessed by picking islets into 60-mm tissue culture-treated dishes (Corning, Oneonta, NY) containing Seahorse media assembled by supplementing a minimal-DMEM Seahorse base media (Agilent Technologies, Santa Clara, CA) with 0.5 mL each of 1 mM sodium pyruvate, 2 mM glutamine, and 0.496 mL of 200 mg/mL glucose. Islets that were smaller than 50 μm, were attached to acinar tissue, or had poor integrity based on the islet quantification and ranking guidelines from the Integrated Islet Distribution Program (IIDP) were excluded from the oxygen consumption assay (32, 33). Viability dyes were not used. Oxygen consumption was assessed using the Seahorse XFe96 instrument (Agilent Technologies, Santa Clara, CA) as previously described (34). On the day before the assay, a cell adhesive solution was prepared by combining 50 μL of Cell-Tak (Corning, Oneonta, NY) with 700 μL of filter-sterilized sodium bicarbonate. Wells of a XFe96 spheroid microplate (Agilent Technologies, Santa Clara, CA) were coated with 20 μL of cell adhesive solution, incubated at 37°C for 1 h, and then washed with 400 μL of 37°C sterile water, allowed to air dry for 40 min, and then covered and stored overnight at 4°C. Also, on the day before the assay, the sensor cartridge and utility plate were removed from an XFe96 Extracellular Flux Assay Kit (Agilent Technologies, Santa Clara, CA). The sensor cartridge was hydrated overnight in a 37°C, non-CO2 incubator. On the day of the assay, the sensor cartridge was removed from the incubator and the water in the utility plate was replaced with the prewarmed XF Calibrant (Agilent Technologies, Santa Clara, CA) and incubated at 37°C. Next, the wells of the cell adhesive-coated microplate were loaded with 175 μL of assembled medium. Isolated islets, 15 per well, were added to noncorner wells of the microplate and then the microplate was covered and placed in a 37°C incubator. The first three ports of sensor cartridge were loaded with 45 μM oligomycin, 10 μM FCCP, and 25 μM rotenone/antimycin A. Concentrations of oligomycin and rotenone/antimycin A were based on the literature (35), and the concentration of FCCP was titrated to produce maximal islet respiration. A wave assay (Agilent Technologies, Santa Clara, CA) was programed for a 30-min baseline period, 42-min oligomycin period, 30-min FCCP period, and 30-min rotenone/antimycin A period as previously described (34).
Tissue Processing
The pancreas was weighed and divided into 10 pieces from head to tail, with 1 being head and 10 being tail as follows: 1–4 head, 5–8 body, and 9–10 tail. For electron microscopy, a portion of the pancreatic tail was minced in 1× PBS, incubated in fixative composed of 2.5% paraformaldehyde/2.5% glutaraldehyde/2 mM calcium chloride in 0.1 M cacodylate (Electron Microscopy Sciences, Hatfield, PA) for 1 h at room temperature, and shipped in 2.5% glutaraldehyde in 0.1 M cacodylate at 4°C overnight to Vanderbilt University Medical Center (Nashville, TN). On arrival, samples were prepared by the Vanderbilt Cell Imaging Shared Resource and washed in 0.1 M cacodylate buffer, 1% tannic acid in cacodylate buffer. After washes, samples were incubated in osmium tetroxide, washed, and stained in Enbloc with 1% uranyl acetate for 1 h. Following washes in ethanol and propylene oxide, samples were epoxy embedded.
Transmission Electron Microscopy
For ultrastructural analysis of β cells, 80 nm epoxy sections were cut with a diamond knife and stained with 2% uranyl acetate and lead citrate. Images were acquired on a Philips/FEI T-12 transmission electron microscope using Serial EM at 2.7 kx magnification. Following image acquisition, montages were stitched using Etomo blending software. At least 1,500 μm2 of β-cell area (∼20 β cells) was analyzed per sample in ImageJ. To assess mitochondrial density, mitochondria within β cells were annotated using the freehand selection tool and area was quantified using ImageJ. To measure insulin granule density and maturity, individual granules were manually counted and binned based on electron density using the ImageJ multipoint selector tool function. Granule maturity was calculated per montage based on the average gray value calculated by ImageJ for all selected granules. Granules with higher-than-average gray values were counted as mature, indicating darker granules, whereas immature granules were counted as those with lower-than-average gray values. Granule density and mitochondrial density were normalized to β-cell cytoplasmic area (after the exclusion of nuclear area).
Tissue Processing for RNA Acquisition and Quantitative RT-PCR
Islets frozen in Trizol reagent (Thermo Fisher, Waltham, MA) were thawed over ice and lysed by vortexing. RNA was isolated and eluted in RNAse-free water using the RNeasy Mini kit (Qiagen, Germantown, MD) according to the manufacturer’s instructions. Concentration and integrity (260/280 ratio) were assessed using a ND-1000 Spectrophotometer (NanoDrop). cDNA was prepared from isolated RNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA). Gene quantitation was performed using the iQ5 Real Time PCR Detection method (BioRad, Hercules, CA) with iQ5 Real QPCR SYBR mastermix (BioRad, Hercules, CA). Fold-change was calculated based on the 2−ΔΔCt method (36), and gene expression was normalized to GAPDH. Primers were validated for specificity in silico using National Institutes of Health (NIH) Primer Blast (Table 1). In addition, an incremental melt curve was run after each cycle to ensure the amplification of single products in each reaction.
Table 1.
Primer sequences
| Transcript | Gene Name | Forward (5′>3′) | Reverse (5′>3′) |
|---|---|---|---|
| ND1 | NADH dehydrogenase I | ATGCCGTAAAACTTTTCACTAAAG | GGGTTCATAGGAGGAGGGCAATGA |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | TCAGCCGCATTTTCTCTTGCA | AGGCGCCCAATACGACCAAAT |
| PPARGC1A | Peroxisome proliferatoractivated receptor gamma coactivator 1-alpha | GGAGTATCAGCACGAGAGGC | GTCCCTCAGTTCTGTCCGTG |
| GCK | Glucokinase | CTCCGTGTACAAACTGCACC | TCGATGAAGGTGATCTCGCA |
| CACNA1C | Voltage-gated channel subunit alpha1 c | GAGGTGCGCTGCTCAATTCT | CTTTCCACTACTGCCTCCTCG |
| CACNA1D | Voltage-gated channel subunit alpha1 d | CCAGTGTGAAAAGGTCCGACT | TCCTCATAGCACTCCTCGCT |
| IPF1 (PDX1) | Pancreatic and duodenal homeobox 1 | ACCTCCACCACCACCTCC | CCAGGCTTACCTGCCCAC |
| ABCC8 | ATP binding cassette subfamily c member 8 | CACCATCGCTGATCCCAAAG | CTGTCGTAGCGCACACTC |
| KCNJ11 | Potassium inwardly rectifying channel subfamily j member 11 | TGCCAACAGCCCACTCTATG | AGTAACGTCCGTCCTCCTCA |
| NQO1 | NAD(P)H quinone dehydrogenase 1 | GGATGGAAGAAACGCCTGGA | TCTGGTTGTCCGTTGGGATG |
| DDIT3 (CHOP) | Nuclear factor erythroid 2-related factor 2 | CTTGTTCCAGCCACTCCCC | CCTCTTGCAGGTCCTCATACC |
| SOD2 | Manganese superoxide dismutase | GAAGTACCAGGAGGCGTTGG | CCACCACCATTAGGGCTGAG |
| ATF6 | Activating transcription factor 6 | TGGATTTGATGCCTTGGGAGT | CTTGAGGAGGCTGGAGAAAGT |
| HSPA5 (BiP) | Heat shock protein protein family A member 5 | ACTGCTTGATGTGTGTCCCC | GTCAGGGGTCGTTCACCTTC |
| SLC2A1 (GLUT1) | Solute carrier family 2 member 1 | GCCTGGATCTCCCCACTCTA | AAGGAGACTAGAACCCGGCA |
| SLC2A2 (GLUT2) | Solute carrier family 2 member 2 | CAGGCAGGAGTTAGTCAGGT | CTGGGTTCCATTGTTAGGCAGT |
Tissue Processing for DNA Acquisition and Measurement of Mitochondrial DNA Copy Number
Islets utilized for oxygen consumption assay were washed twice with 1× PBS and frozen at −80°C after completion of the assay. DNA was isolated from thawed pellets utilizing the DNeasy Blood and Tissue kit (Qiagen, Germantown, MD) using the Cultured Cells Protocol. Genomic DNA was amplified using NADH dehydrogenase I (ND1) primers for mitochondrial DNA (mtDNA) and GAPDH primers for nuclear DNA. Ratios of mtDNA relative to nuclear DNA were calculated utilizing the 2−ΔΔCt method as previously described (37).
Statistics
Data are expressed as means ± SE. All data were analyzed utilizing GraphPad Prism Software (Graphpad Software, Inc., La Jolla, CA). For islet perifusion data, a two-way ANOVA was performed to test for effects of maternal diet, time, or interaction. Insulin perifusion area under the curve (IAUC) was calculated from a zero baseline. For direct comparisons of continuous measures between two groups, an unpaired t test was used for normally distributed measures. Data from animals that were deemed as outliers in GraphPad Prism Grubb’s statistical outlier test or animals for which there was no RNA stock due to availability during experimental timeline were excluded from analysis of qRT-PCR targets. Primates that were included in qRT-PCR and ex vivo islet perifusion assays were included in a correlation matrix of fold-change values for gene transcripts and IAUC was generated using a multivariable table in Graphpad, allowing for the generation of multiple Pearson’s Product-Moment Correlation (r) values. Statistical significance was determined as P values less than 0.05.
RESULTS
Developmental Exposure to Maternal WSD Confers Insulin Hypersecretion in Isolated Islets from Offspring
We previously reported that 3-yr-old peripubertal offspring exposed to maternal WSD, but weaned onto a CD, hypersecrete insulin in response to high glucose (25). In the current study, a separate cohort of offspring from this NHP model of maternal overnutrition was weaned onto a WSD and analyzed at 1 yr of age. In this model, we analyzed whether insulin hypersecretion in response to glucose is present at an even earlier timepoint following developmental exposure to WSD (WSD/WSD), compared with a group of animals fed only a postweaning WSD (CD/WSD). Indeed, even at 1-yr of age, islets from WSD/WSD offspring hypersecrete insulin in response to a glucose challenge compared with CD/WSD offspring, which show the expected response to elevated glucose (Fig. 2A). In addition, islets from WSD/WSD offspring exhibited increased basal insulin secretion at 4 mM glucose, before exposure to 16.7 mM glucose (Fig. 2B).
Figure 2.

Maternal Western-style diet is associated with hypersecretion of insulin in 1-yr-old offspring. A: perifusion of isolated islets demonstrates that WSD/WSD offspring secreted significantly more insulin when stimulated with 16.7 mM glucose. B: basal insulin secretion during the first 15 min of perifusion assay on isolated islets was increased in WSD/WSD offspring. Each color corresponds to a different offspring and open symbols denote female offspring in B. **P < 0.001. CD/WSD (n = 14, 9 M/5 F), WSD/WSD (n = 10, 5 M/5 F). CD, control diet; F, female; M, male; WSD, Western-style diet.
β Cells of WSD-Exposed Offspring Do Not Have Increased Insulin Granule Density
One possibility for the increase in insulin secretion in islets from WSD/WSD offspring was an increase in insulin production in β cells. To this end, insulin granule density and maturity were evaluated via transmission electron microscopy (TEM; Fig. 3A). There was no difference in the density of insulin granules or the proportion of mature insulin granules in the cytoplasm of β cells from 1-yr-old WSD/WSD offspring compared with CD/WSD (Fig. 3, B and C). Given that PDX1 is a marker of β cells and a transcriptional regulator of insulin production, we evaluated transcript levels of PDX1 via qRT-PCR and found a trend for increased mRNA expression in WSD/WSD versus CD/WSD offspring (Fig. 3D).
Figure 3.
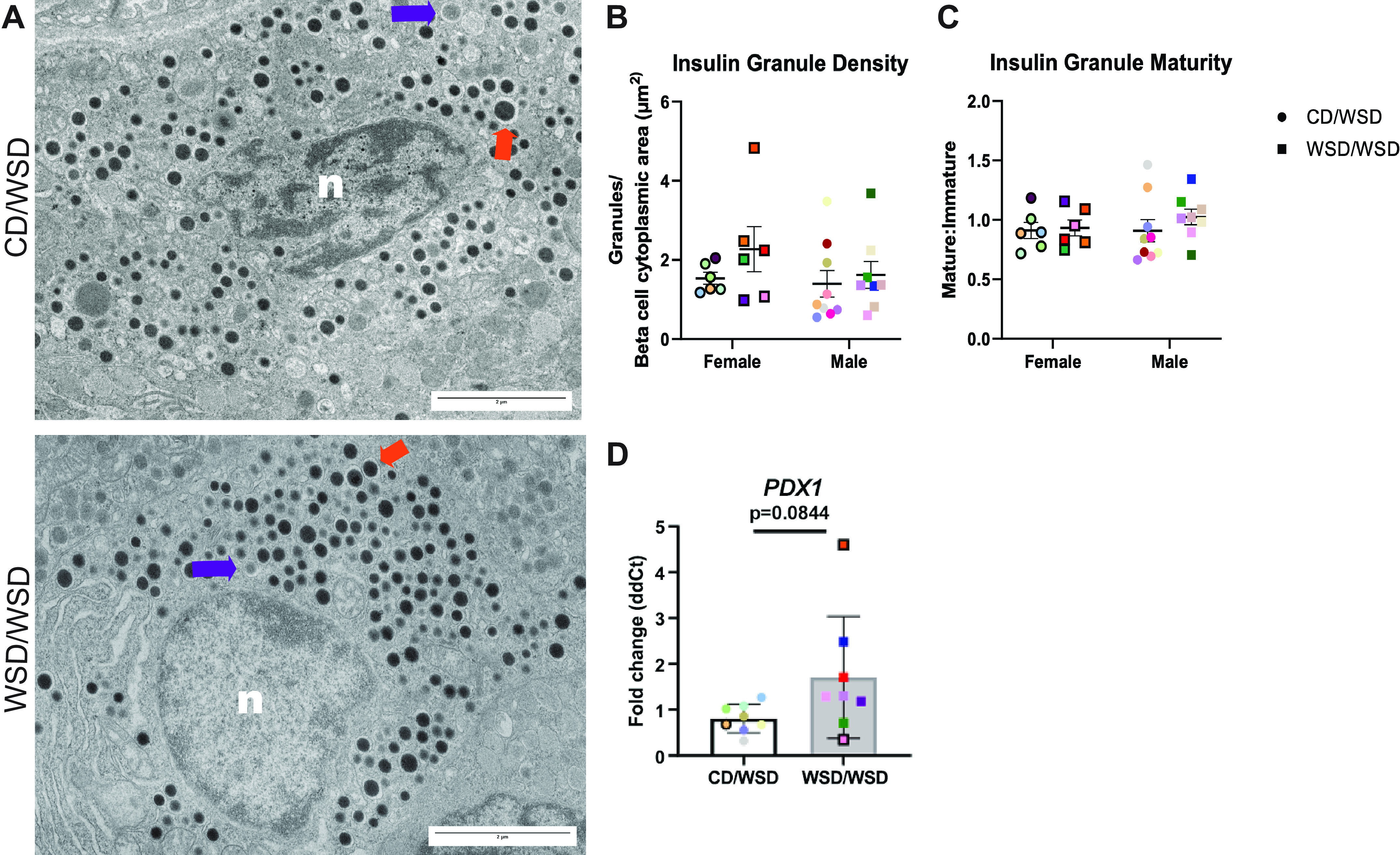
Maternal Western-style diet feeding has no effect on insulin granule density or maturity. A: representative images of β cells from 1-yr-old female offspring. In total, 20 β cells were analyzed per offspring. n: nucleus; orange arrow: mature insulin granule; purple arrow: immature insulin granule. B: insulin granule density, quantified as the number of insulin granules per β-cell cytoplasmic area, was not changed in islets of male or female WSD/WSD offspring. C: insulin granule maturity, calculated based on average gray value within an insulin granule, was not influenced by maternal WSD consumption. D: expression of PDX1 trends to be increased in WSD/WSD offspring. Each color corresponds to a different offspring and open symbols denote female offspring. TEM image magnification 2.7 kx, scale bars = 2 μm. CD, control diet; TEM, transmission electron microscopy; WSD, Western-style diet.
Components of Stimulus-Secretion Coupling Are Increased in Islets of WSD/WSD Offspring
Increased offspring insulin secretion in response to maternal obesity and high-fat diet feeding has been associated with increased calcium channel activity in rodents (27). Thus, qRT-PCR was used to evaluate transcript levels of membrane channels involved in stimulus-secretion coupling. Islets from WSD/WSD offspring have increased expression of l-type voltage-dependent calcium channel subunits, CACNA1C and CACNA1D, as well as increased expression of the ATP-sensitive potassium channel (KATP) subunits, KCNJ11 and ABCC8 (Fig. 4, A–D). Based on the fact that increased KATP activity normally inhibits insulin secretion, the observed increase in KATP channel component expression seems contradictory. Thus, we performed a correlation analysis of fold-change transcript levels of KCNJ11, ABCC8, CACNA1D, CACNA1C, and the insulin area under the curve (IAUC) in the ex vivo islet perifusion assays shown in Fig. 2. Although each of these transcripts exhibits strong correlations with IAUC during perifusion, expression of ABCC8 and CACNA1D most closely correlated with increased insulin secretion in islets of 1-yr-old WSD/WSD offspring (Fig. 4E).
Figure 4.

Expression of calcium and potassium channel components is elevated in islets from offspring of Western-style diet fed dams. CACNA1C (A), CACNA1D (B), KCNJ11 (C), and ABCC8 (D) transcripts were increased in male and female WSD/WSD offspring. E: insulin area under the curve during perifusion assay was positively correlated with the fold-change expression of KCNJ11, ABCC8, CACNA1C, and CACNA1D in offspring. n = 8 per maternal diet group, *P < 0.05, **P < 0.001. Each color corresponds to a different offspring and open symbols denote female offspring in A–D. CD, control diet; WSD, Western-style diet.
Islets from WSD/WSD Offspring Show Altered Expression of Markers of Endoplasmic Reticulum or Oxidative Stress
β-Cell hyperactivity has been shown to precede β-cell failure due to excitotoxicity and increased reactive oxygen species related to increased glucose metabolism (38). Therefore, we evaluated the expression of transcripts associated with endoplasmic reticulum (ER) stress (DDIT3, ATF6, HSPA5) or oxidative stress (NQO1, SOD2), that might signal elevated β-cell stress in the face of increased activity. There was no difference in the expression of DDIT3 (CHOP) or ATF6 (Fig. 5, A and B); however, there was a trend for increased expression of NQO1 (Fig. 5C) and decreased expression of SOD2 and HSPA5 (BiP; Fig. 5, D and E) in WSD/WSD offspring compared with CD/WSD offspring islets.
Figure 5.

Maternal Western-style diet feeding is associated with altered expression of cell stress markers in offspring islets. Offspring of dams fed a WSD showed no change in expression of DDIT3 (A) or ATF6 (B) but showed increased expression of NQO1 (C) and reduced expression of SOD2 (D) and HSPA5 (E). Each color corresponds to a different offspring and open symbols denote female offspring. CD, control diet; WSD, Western-style diet.
Mitochondrial Density Is Not Increased in β Cells of WSD-Exposed Offspring
Based on our observation that islets from 1-yr-old WSD/WSD offspring hypersecrete insulin and have increased expression of proteins associated with stimulus-secretion coupling, we predicted that β cells from these offspring would have increased mitochondrial density, and therefore increased ATP production. To assess mitochondrial phenotype, we first evaluated mitochondrial area relative to β-cell cytoplasmic area and found no difference in the density of mitochondria in β cells from CD/WSD and WSD/WSD offspring, though there was a trend for increased mitochondrial density in male WSD/WSD offspring (Fig. 6B). In addition, there was no difference in the ratio of mitochondrial DNA relative to nuclear DNA, or expression of PPARGC1A (gene for PGC1α), which is involved in mitochondrial biogenesis (Fig. 6, C and D).
Figure 6.
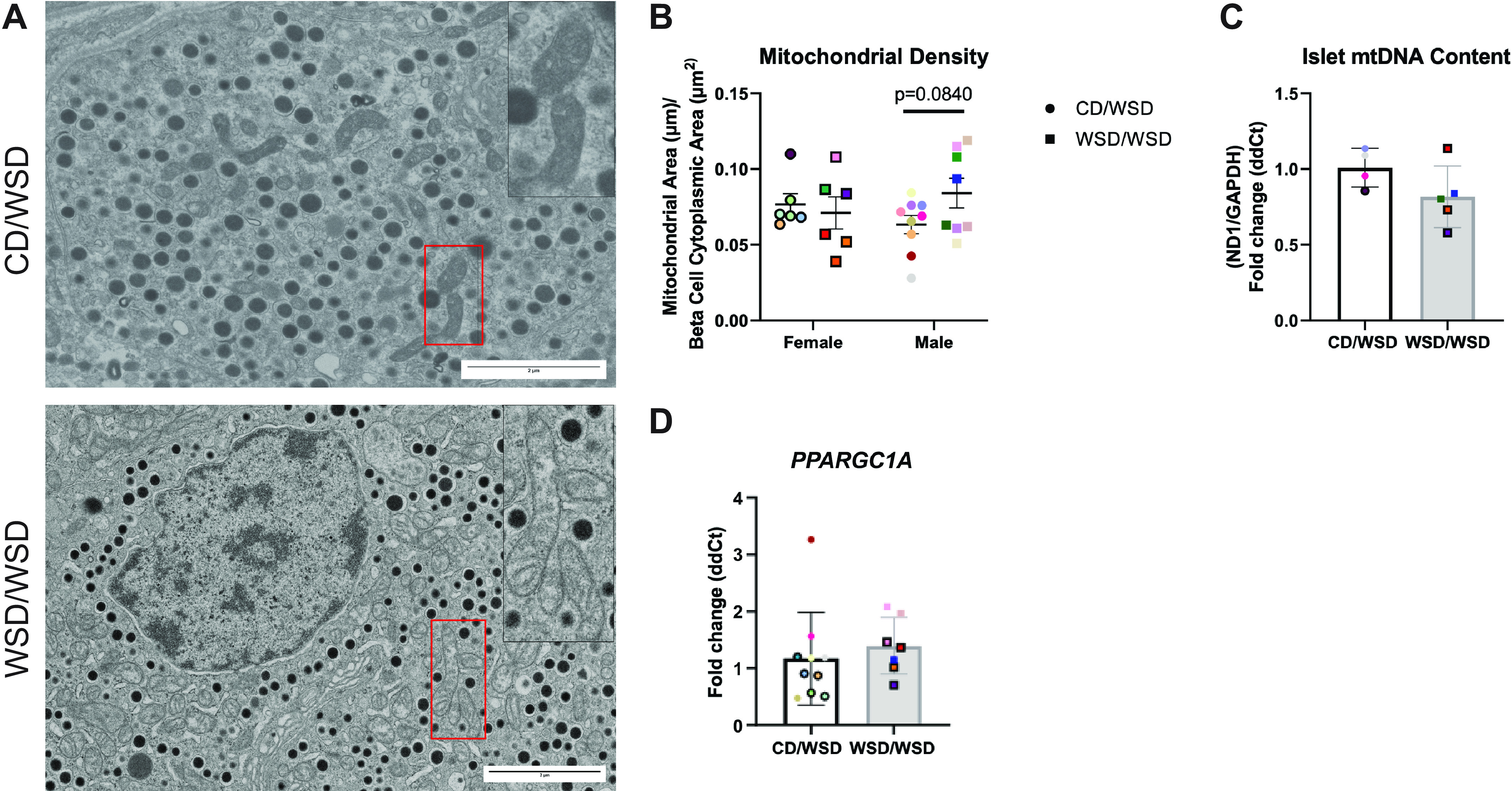
No evidence for increased mitochondrial density in β cells from offspring exposed to maternal Western-style diet. A: representative images of β cells from male offspring. 2× zoom factor insets show β-cell mitochondria. In total, 20 β cells were analyzed per offspring. n: nucleus. B: mitochondrial density relative to total β-cell area was not changed in WSD/WSD offspring. C: the ratio of expression of the mitochondrial gene ND1 relative to nuclear-encoded GAPDH was not altered in islets of WSD/WSD offspring. D: transcript expression of PPARGC1A was not changed in islets from WSD/WSD offspring. Each color corresponds to a different offspring and open symbols denote female offspring. TEM image magnification 2.7 kx, scale bars = 2 μm. CD, control diet, TEM, transmission electron microscopy; WSD, Western-style diet.
Islets from 1-Yr-Old Male WSD/WSD Offspring Exhibit Increased Spare Respiratory Capacity
Since increased glycolysis and ATP can each trigger insulin secretion (39, 40), transcript expression of the glucose transporters SLC2A1 (GLUT1), SLC2A2 (GLUT2), and GCK (glucokinase), the first enzyme in the glycolytic pathway, was next examined in WSD/WSD versus CD/WSD offspring. There was no difference in SLC2A1 expression, but expression of SLC2A2 was increased in islets of WSD/WSD offspring (Fig. 7, A and B). The expression of GCK did not differ between offspring groups (Fig. 7C). There was also no evidence of increased glycolytic rate in islets isolated from 1-yr-old WSD-exposed offspring based on the basal and total extracellular acidification rate during the Cell Mito Stress assay (41; Fig. 7, D and E).
Figure 7.

Hallmarks of glycolysis are not altered in islets from offspring exposed to maternal Western-style diet. Transcript expression of SLC2A1 (GLUT1; A) was not altered in islets of WSD/WSD offspring, whereas SLC2A2 (GLUT2; B) is increased. C: there was no difference in transcript expression of GCK. D: extracellular acidification rate (ECAR) for isolated islets in Agilent Seahorse Cell Mito Stress Test showed no difference between treatment groups. E: basal ECAR rate before the addition of oligomycin was not changed in islets from offspring exposed to maternal Western-style diet. Each color corresponds to a different offspring and open symbols denote female offspring in A and C. P < 0.05; n = 11–13/group. CD, control diet; WSD, Western-style diet.
Mitochondrial respiration is another potential source of ATP in β cells. There were no differences in the overall oxygen consumption rate in islets from WSD/WSD versus CD/WSD offspring (Fig. 8A). In addition, maternal WSD feeding did not confer increased basal respiration, maximal respiration, or ATP-linked respiration in offspring. However, islets from male WSD-exposed offspring exhibited increased spare respiratory capacity relative to male CD-exposed offspring (Fig. 8, B–E).
Figure 8.

Spare respiratory capacity is increased in male offspring of dams fed a Western-style diet. Isolated islets were subjected to Agilent Seahorse Cell Mito Stress Test. A: oxygen consumption rate (OCR) profiles of islets assessed ex vivo. Basal respiration (B), maximal respiration (C), and ATP-linked respiration (D) were not changed in male or female WSD/WSD offspring. E: spare respiratory capacity was increased in islets of male WSD/WSD offspring. *P < 0.05, n = 9–13 per maternal diet group. Each color corresponds to a different offspring and open symbols denote female offspring in B and C. CD, control diet; WSD, Western-style diet.
DISCUSSION
The key finding from the current study is that maternal WSD consumption is associated with persistent offspring islet dysfunction as early as 1 yr of age. This dysfunction occurs in both WSD/WSD (this study) and WSD/CD offspring as shown previously (25). Consistent with our previous observations from peripubertal 3-yr-old offspring in which CD and WSD-exposed offspring were weaned onto CD (25), islets from 1-yr-old WSD/WSD offspring secrete more insulin at low glucose and also when stimulated with high glucose. In human subjects, insulin hypersecretion is associated with adverse clinical outcomes of insulin resistance, including increased fat mass and T2D (42). Systemic insulin resistance may develop as an adaptive response to chronic insulin oversecretion, as seen in our animals (43–45).
The reasons for the observed insulin hypersecretion in ex vivo islets have not been completely elucidated. In the same cohort of 1-yr-old offspring, developmental WSD exposure does not confer increased fasting insulin, glucose, or insulin area under the curve during an intravenous glucose tolerance test (23). However, ex vivo insulin-stimulated glucose uptake is persistently reduced in skeletal muscle cells from WSD/WSD 1-yr-old offspring (23), suggesting some level of muscle insulin resistance early in life. A hyperinsulinemic-euglycemic clamp would be optimal for assessing insulin resistance in 1-yr-old offspring. Given the observation of reduced insulin sensitivity in the skeletal muscle of the same offspring that we here show have insulin hypersecretion, we anticipate that this could develop into an insulin-resistant phenotype as these offspring age.
Increased β-to-α cell ratio was observed in WSD-exposed 1-yr-old offspring from an earlier cohort in this model, although this was attributed to reduced α-cell mass rather than increased β-cell mass (24). Glucagon action at its receptor in β cells has been shown to increase insulin secretion; however, rodent studies suggest that near total loss of α-cell mass does not have a significant impact on glucose tolerance (46). Therefore, it is unlikely that the observed persistence of insulin hypersecretion can be attributed to an increased β-cell ratio or decreased paracrine regulation via α cells. Another possible mechanism for the observed increase in GSIS could have been increased insulin production. However, based on the analyses performed here, we have no evidence for an increase in insulin production or insulin granule content in islets from 1-yr-old WSD-exposed offspring.
In the current study, we found that islets of maternal WSD-exposed offspring weaned onto a WSD had increased expression of voltage-dependent calcium channel genes and genes encoding KATP channel components involved in stimulus-secretion coupling. Although we did not observe sex-dependent differences in insulin secretion in response to maternal WSD in this study, in a study in mice, female offspring of obese dams displayed increased GSIS, which was attributed to increased transcript expression of Cacna1d (27). Taken together, these data support a calcium-dependent mechanism of fetal programming affecting insulin secretion in response to maternal overnutrition. Our consortium previously published that levels of KCNJ11 and ABCC8 transcripts were unchanged in islets of fetal offspring exposed to maternal WSD; however, we report here that 1-yr-old offspring have elevated expression of transcripts encoding the KATP channel subunits, SUR1 and Kir6.2 (24). This may be a change that occurred on weaning. Increased KATP channel activity is normally associated with reduced β-cell excitability and poor insulin secretion, and therapeutics, such as sulfonylureas that reduce KATP channel activity enhance insulin secretion (47). Therefore, our findings seem paradoxical to the persistent hypersecretory phenotype we observed in offspring islets. As β cells transition from an adaptive, hypersecretory state, to maladaptive secretion, the mechanisms underlying regulation of KATP channel gene expression are unknown (47). To attempt to address this gap in our understanding, we examined the correlation between IAUC in the ex vivo perifusion assay of islets exposed to stimulatory glucose conditions to the fold-change expression of KCNJ11, ABCC8, CACNA1C, and CACNA1D, all of which facilitate stimulus-secretion coupling. Interestingly, a positive correlation exists for each transcript and the IAUC in the perifusion assay. This suggests that insulin secretion in offspring islets cannot solely be attributed to changes in KATP channel activity, but coordination with calcium dynamics in islets may dictate function. Our studies are limited by an insufficient number of islets available to also evaluate calcium flux dynamics and potassium chloride-stimulated insulin secretion. Future electrophysiological studies in islets of NHP offspring exposed to maternal WSD may tease out the developmental programming of the coordination of potassium, ATP, and calcium activity in the regulation and adaptation in insulin secretion.
Although there is ample literature suggesting that systemic insulin resistance precedes β-cell dysfunction in the etiology of T2D (48–50), an alternate explanation for the mechanism responsible for the onset of dysglycemia is that chronic insulin hypersecretion due to β-cell dysfunction arises before the development of systemic insulin resistance (51, 52). The offspring in this cohort do not have elevated fasting blood glucose or insulin, which supports the likelihood of this alternative mechanism. Chronic elevations in insulin production and secretion can lead to ER stress, oxidative stress, and ultimately β-cell dysfunction and failure. With the goal of ascertaining early signs of β-cell stress, we examined transcripts associated with antioxidant responses and the adaptive ER stress response and saw no evidence for increased stress in islets of WSD/WSD offspring using these chosen markers. We observed increased expression of the antioxidant gene, NQO1, which scavenges superoxide. Expression of SOD2, also involved in neutralization of superoxide, was decreased in WSD/WSD offspring relative to CD/WSD offspring. A study in which human islets were transplanted into hyperglycemic mice whose β cells were ablated by diphtheria toxin (DT-HG), found that although islets exhibited reduced transcript expression of the antioxidant transcription factor, NFE2L2 (Nrf2), there were no changes in SOD2 (53). In the same study, human islets grafts in NOD scid gamma mice fed a HFD (60% kcal from fat) (HFD + I) for 12 wk showed decreased transcript expression of both SOD2 and nuclear factor erythroid 2-related factor 2 (NFE2L2) (53). The authors concluded that the contribution of hyperglycemia (DT-HG) and lipotoxicity (HFD + I) separately exhibit different antioxidant responses in human islets. Given that NQO1 is modulated via the Nrf2 antioxidant pathway, and SOD2 is regulated via a different pathway, there may be preferential antioxidant responses in the islets of these offspring. Transcript expression of the ER chaperone, HSPA5 (BiP), was higher in offspring exposed to maternal CD compared with those exposed to maternal WSD, suggesting that weaning onto a WSD after developmental exposure to CD leads to an acute adaptation to the increased nutrient milieu and insulin demand, whereas islets from WSD/WSD offspring may have already been programed during development to account for increased nutritional demand at an earlier timepoint that was not captured in our analysis.
Given the important role of ATP production and coordination of cellular metabolism by mitochondria in β cells for insulin secretion, we evaluated whether changes in mitochondrial abundance or function could explain insulin hypersecretion in WSD-exposed offspring. Maternal WSD did not confer increased mitochondrial density, mitochondrial DNA ratio, or expression of the mitochondrial biogenesis transcript, PPARGC1A, indicating that maternal overnutrition does not elicit changes in mitochondrial mass in offspring islets. Nevertheless, we observed a trend for increased mitochondrial density in male offspring of dams fed a WSD. Although mitochondrial density was not significantly changed in response to maternal overnutrition, mitochondrial function could have still been elevated in response to maternal WSD. Glucose uptake and metabolism are important for the triggering of insulin secretion; we observed increased expression of the transcript encoding GLUT2 in maternal WSD-exposed offspring. Although GLUT2 is coexpressed with GLUT1 in NHP islets, we did not observe an increase in SLC2A1 in WSD/WSD offspring. This might suggest that increased insulin secretion in islets from WSD-exposed offspring is a result of increased glucose uptake; however, the variability in SLC2A2 expression among offspring in the WSD/WSD group suggests that there are multiple mechanisms that are causing insulin hypersecretion. Despite the possibility of increased glucose uptake, we did not observe an increase in GCK, which encodes the first enzyme of glycolysis, or in the extracellular acidification rate during Seahorse assay. We conclude that increased glucose metabolism does not contribute to insulin hypersecretion in WSD-exposed macaque offspring. Although we also did not observe increased basal respiration, maximal respiration, or ATP-linked respiration in islets of WSD/WSD offspring, we observed a sex-specific increase in spare respiratory capacity in islets of 1-yr-old male WSD/WSD offspring. This adaptation likely does not explain increased insulin secretion from offspring islets, which was observed in both sexes, and would not be expected to increase insulin secretion under nonstress conditions. However, it does suggest a functional adaptation in the mitochondria of offspring islets to developmental WSD exposure. Male islets may have an increased ability to adapt to increased metabolic demand when weaned from dams fed a WSD, and increased spare respiratory capacity in islets from these offspring could be attributed to the slight increase in mitochondrial density observed. Studies of human islets exposed to acute glucotoxic stress also exhibit augmented mitochondrial metabolism, but this change was associated with a reduction in glucose-stimulated insulin secretion (54). Future studies should evaluate the mechanisms underlying spare respiratory capacity in β cells and the relative contribution of such changes to insulin secretion.
In conclusion, the islets of offspring exposed to maternal WSD are hyperreactive to glucose, and the resultant increase in insulin secretion can be attributed to increased components of stimulus-secretion coupling. In addition to functionally adapting to a maternal WSD, 1-yr-old male offspring have increased spare respiratory capacity, suggesting that islet mitochondrial function is dependent on the nutritional milieu during development, even after weaning. These data suggest that developmental WSD exposure is more effective in programming than postweaning WSD. The total developmental diet exposure period was ∼11 mo, whereas the postweaning exposure period was 7 mo. We cannot rule out the possibility that the strong programming effect seen with maternal WSD is due to the slightly longer WSD exposure during development. We conclude that maternal diet influences the mechanisms of adaptation in offspring islets in response to WSD feeding. At this timepoint, these changes do not result in β-cell failure; however, the documented reductions in muscle insulin sensitivity by our group in these same offspring suggest an increased susceptibility to early onset metabolic syndrome and future β-cell dysfunction.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
All nonhuman primate studies were supported in part by the Oregon National Primate Research Center grant P51 OD011092 from the National Institutes of Health/Office of the Director. D.T.C. was supported by the Vanderbilt University Training Program in Molecular Endocrinology (5T32 DK563-30) and a predoctoral fellowship from the NIH/NIDDK (1F31 DK135164). J.M.E. was supported by the NIH/NIGMS under Award No. T32 GM007347. D.L.T., T.A.D., C.E.M., K.M.A., J.E.F., P.K., and M.G. were supported by NIH/NIDDK Grants R24 DK090964 and R01 DK128187. J.E.F. was also supported by R01 DK128416. M.G. was also supported by VA Merit Awards (I01 BX003744-01 and I01 BX005399). The Agilent Seahorse Extracellular Flux Analyzer is housed and managed within the Vanderbilt High-Throughput Screening Core Facility, an institutionally supported core, and was funded by NIH Shared Instrumentation Grant 1S10OD018015. TEM imaging was performed in collaboration with the Vanderbilt Cell Imaging Shared Resource and supported by NIH Grants CA68485, DK20593, DK58404, DK59637, and EY08126.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.T.C., J.M.E., S.R.L., M.K., D.L.T., T.A.D., S.R.W., C.E.M., J.E.F., K.M.A., P.K., and M.G. conceived and designed research; D.T.C., J.M.E., A.M., J.F., S.R.L., M.K., D.L.T., and T.A.D. performed experiments; D.T.C., J.M.E., A.M., S.R.L. M.K., P.K., and M.G. analyzed data; D.T.C., A.M., S.R.W., C.E.M., J.E.F., K.M.A., P.K., and M.G. interpreted results of experiments; D.T.C., J.M.E., S.R.L., and M.K. prepared figures; D.T.C. drafted manuscript; D.T.C., S.R.W., C.E.M., J.E.F., K.M.A., P.K., and M.G. edited and revised manuscript; D.T.C., J.M.E., A.M., J.F., S.R.L., M.K., D.L.T., T.A.D., S.R.W., C.E.M., J.E.F., K.M.A., P.K., and M.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank members of the Gannon lab for their input during the course of these studies. In particular, we thank Valerie Ricciardi and Matthew Shipley for technical assistance with TEM analysis. We further acknowledge Dr. Evan Krystofiak, Dr. Rafael Arrojo e Drigo, Dr. Antentor Hinton, Rachel Hart, and Maria Vinogradova for their support in preparing tissues for TEM and improvements to tissue staining protocols. Figure 1 and graphical abstract created with BioRender and published with permission.
REFERENCES
- 1. Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of obesity among adults and youth: United States, 2015-2016. NCHS Data Brief (288): 1–8, 2017. [PubMed] [Google Scholar]
- 2. O'Hara SE, Gembus KM, Nicholas LM. Understanding the long-lasting effects of fetal nutrient restriction versus exposure to an obesogenic diet on islet-cell mass and function. Metabolites 11: 514, 2021. doi: 10.3390/metabo11080514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Perng W, Oken E, Dabelea D. Developmental overnutrition and obesity and type 2 diabetes in offspring. Diabetologia 62: 1779–1788, 2019. doi: 10.1007/s00125-019-4914-1. [DOI] [PubMed] [Google Scholar]
- 4. Barker DJ, Eriksson JG, Forsén T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol 31: 1235–1239, 2002. doi: 10.1093/ije/31.6.1235. [DOI] [PubMed] [Google Scholar]
- 5. Barker DJ. The developmental origins of well-being. Philos Trans R Soc Lond B Biol Sci 359: 1359–1366, 2004. doi: 10.1098/rstb.2004.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aagaard-Tillery KM, Grove K, Bishop J, Ke X, Fu Q, McKnight R, Lane RH. Developmental origins of disease and determinants of chromatin structure: maternal diet modifies the primate fetal epigenome. J Mol Endocrinol 41: 91–102, 2008. doi: 10.1677/JME-08-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cox J, Williams S, Grove K, Lane RH, Aagaard-Tillery KM. A maternal high-fat diet is accompanied by alterations in the fetal primate metabolome. Am J Obstet Gynecol 201: 281.e1–281.e9, 2009. doi: 10.1016/j.ajog.2009.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE, Grove KL. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 119: 323–335, 2009. doi: 10.1172/JCI32661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pace RM, Chu DM, Prince AL, Ma J, Seferovic MD, Aagaard KM. Complex species and strain ecology of the vaginal microbiome from pregnancy to postpartum and association with preterm birth. Med 2: 1027–1049, 2021. doi: 10.1016/j.medj.2021.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prince AL, Pace RM, Dean T, Takahashi D, Kievit P, Friedman JE, Aagaard KM. The development and ecology of the Japanese macaque gut microbiome from weaning to early adolescence in association with diet. Am J Primatol 81: e22980, 2019. doi: 10.1002/ajp.22980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pace RM, Prince AL, Ma J, Belfort BDW, Harvey AS, Hu M, Baquero K, Blundell P, Takahashi D, Dean T, Kievit P, Sullivan EL, Friedman JE, Grove K, Aagaard KM. Modulations in the offspring gut microbiome are refractory to postnatal synbiotic supplementation among juvenile primates. BMC Microbiol 18: 28, 2018. doi: 10.1186/s12866-018-1169-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McCurdy CE, Schenk S, Hetrick B, Houck J, Drew BG, Kaye S, Lashbrook M, Bergman BC, Takahashi DL, Dean TA, Nemkov T, Gertsman I, Hansen KC, Philp A, Hevener AL, Chicco AJ, Aagaard KM, Grove KL, Friedman JE. Maternal obesity reduces oxidative capacity in fetal skeletal muscle of Japanese macaques. JCI Insight 1: e86612, 2016. doi: 10.1172/jci.insight.86612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harris RA, Alcott CE, Sullivan EL, Takahashi D, McCurdy CE, Comstock S, Baquero K, Blundell P, Frias AE, Kahr M, Suter M, Wesolowski S, Friedman JE, Grove KL, Aagaard KM. Genomic variants associated with resistance to high fat diet induced obesity in a primate model. Sci Rep 6: 36123, 2016. doi: 10.1038/srep36123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suter M, Bocock P, Showalter L, Hu M, Shope C, McKnight R, Grove K, Lane R, Aagaard-Tillery K. Epigenomics: maternal high-fat diet exposure in utero disrupts peripheral circadian gene expression in nonhuman primates. FASEB J 25: 714–726, 2011. doi: 10.1096/fj.10-172080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suter MA, Chen A, Burdine MS, Choudhury M, Harris RA, Lane RH, Friedman JE, Grove KL, Tackett AJ, Aagaard KM. A maternal high-fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. FASEB J 26: 5106–5114, 2012. doi: 10.1096/fj.12-212878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Suter MA, Sangi-Haghpeykar H, Showalter L, Shope C, Hu M, Brown K, Williams S, Harris RA, Grove KL, Lane RH, Aagaard KM. Maternal high-fat diet modulates the fetal thyroid axis and thyroid gene expression in a nonhuman primate model. Mol Endocrinol 26: 2071–2080, 2012. doi: 10.1210/me.2012-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ma J, Prince AL, Bader D, Hu M, Ganu R, Baquero K, Blundell P, Alan Harris R, Frias AE, Grove KL, Aagaard KM. High-fat maternal diet during pregnancy persistently alters the offspring microbiome in a primate model. Nat Commun 5: 3889, 2014. doi: 10.1038/ncomms4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pound LD, Kievit P, Grove KL. The nonhuman primate as a model for type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 21: 89–94, 2014. doi: 10.1097/MED.0000000000000043. [DOI] [PubMed] [Google Scholar]
- 19. Elsakr JM, Zhao SK, Ricciardi V, Dean TA, Takahashi DL, Sullivan E, Wesolowski SR, McCurdy CE, Kievit P, Friedman JE, Aagaard KM, Edwards DRV, Gannon M. Western-style diet consumption impairs maternal insulin sensitivity and glucose metabolism during pregnancy in a Japanese macaque model. Sci Rep 11: 12977, 2021. doi: 10.1038/s41598-021-92464-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nash MJ, Dobrinskikh E, Newsom SA, Messaoudi I, Janssen RC, Aagaard KM, McCurdy CE, Gannon M, Kievit P, Friedman JE, Wesolowski SR. Maternal Western diet exposure increases periportal fibrosis beginning in utero in nonhuman primate offspring. JCI Insight 6: e154093, 2021. doi: 10.1172/jci.insight.154093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nash MJ, Dobrinskikh E, Janssen RC, Lovell MA, Schady DA, Levek C, Jones KL, D'Alessandro A, Kievit P, Aagaard KM, McCurdy CE, Gannon M, Friedman JE, Wesolowski SR. Maternal Western diet is associated with distinct preclinical pediatric NAFLD phenotypes in juvenile nonhuman primate offspring. Hepatol Commun 7: e0014, 2023. doi: 10.1097/HC9.0000000000000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rivera HM, Kievit P, Kirigiti MA, Bauman LA, Baquero K, Blundell P, Dean TA, Valleau JC, Takahashi DL, Frazee T, Douville L, Majer J, Smith MS, Grove KL, Sullivan EL. Maternal high-fat diet and obesity impact palatable food intake and dopamine signaling in nonhuman primate offspring. Obesity (Silver Spring) 23: 2157–2164, 2015. doi: 10.1002/oby.21306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Campodonico-Burnett W, Hetrick B, Wesolowski SR, Schenk S, Takahashi DL, Dean TA, Sullivan EL, Kievit P, Gannon M, Aagaard K, Friedman JE, McCurdy CE. Maternal obesity and western-style diet impair fetal and juvenile offspring skeletal muscle insulin-stimulated glucose transport in nonhuman primates. Diabetes 69: 1389–1400, 2020. doi: 10.2337/db19-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Comstock SM, Pound LD, Bishop JM, Takahashi DL, Kostrba AM, Smith MS, Grove KL. High-fat diet consumption during pregnancy and the early post-natal period leads to decreased α cell plasticity in the nonhuman primate. Mol Metab 2: 10–22, 2012. doi: 10.1016/j.molmet.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elsakr JM, Dunn JC, Tennant K, Zhao SK, Kroeten K, Pasek RC, Takahashi DL, Dean TA, Velez Edwards DR, McCurdy CE, Aagaard KM, Powers AC, Friedman JE, Kievit P, Gannon M. Maternal Western-style diet affects offspring islet composition and function in a non-human primate model of maternal over-nutrition. Mol Metab 25: 73–82, 2019. doi: 10.1016/j.molmet.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cerf ME, Louw J. Islet cell response to high fat programming in neonate, weanling and adolescent Wistar rats. JOP 15: 228–236, 2014. doi: 10.6092/1590-8577/1534. [DOI] [PubMed] [Google Scholar]
- 27. Nicholas LM, Nagao M, Kusinski LC, Fernandez-Twinn DS, Eliasson L, Ozanne SE. Exposure to maternal obesity programs sex differences in pancreatic islets of the offspring in mice. Diabetologia 63: 324–337, 2020. doi: 10.1007/s00125-019-05037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ford SP, Zhang L, Zhu M, Miller MM, Smith DT, Hess BW, Moss GE, Nathanielsz PW, Nijland MJ. Maternal obesity accelerates fetal pancreatic beta-cell but not alpha-cell development in sheep: prenatal consequences. Am J Physiol Regul Integr Comp Physiol 297: R835–R843, 2009. doi: 10.1152/ajpregu.00072.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Graus-Nunes F, Dalla Corte Frantz E, Lannes WR, da Silva Menezes MC, Mandarim-de-Lacerda CA, Souza-Mello V. Pregestational maternal obesity impairs endocrine pancreas in male F1 and F2 progeny. Nutrition 31: 380–387, 2015. doi: 10.1016/j.nut.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 30. Maddison LA, Chen W. Nutrient excess stimulates β-cell neogenesis in zebrafish. Diabetes 61: 2517–2524, 2012. doi: 10.2337/db11-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang L, Long NM, Hein SM, Ma Y, Nathanielsz PW, Ford SP. Maternal obesity in ewes results in reduced fetal pancreatic β-cell numbers in late gestation and decreased circulating insulin concentration at term. Domest Anim Endocrinol 40: 30–39, 2011. doi: 10.1016/j.domaniend.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ricordi C, Gray DW, Hering BJ, Kaufman DB, Warnock GL, Kneteman NM, Lake SP, London NJ, Socci C, Alejandro R. Islet isolation assessment in man and large animals. Acta Diabetol Lat 27: 185–195, 1990. doi: 10.1007/BF02581331. [DOI] [PubMed] [Google Scholar]
- 33.NIH CIT Consortium Chemistry Manufacturing Controls Monitoring Committee; NIH CIT Consortium. Purified human pancreatic islet: qualitative and quantitative assessment of islets using dithizone (DTZ): standard operating procedure of the NIH Clinical Islet Transplantation Consortium. CellR4 Repair Replace Regen Reprogram 3: e1369, 2015. [PMC free article] [PubMed] [Google Scholar]
- 34. Elsakr JM, Deeter C, Ricciardi V, Gannon M. Analysis of non-human primate pancreatic islet oxygen consumption. J Vis Exp (154): e60696, 2019. doi: 10.3791/60696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Taddeo EP, Stiles L, Sereda S, Ritou E, Wolf DM, Abdullah M, Swanson Z, Wilhelm J, Bellin M, McDonald P, Caradonna K, Neilson A, Liesa M, Shirihai OS. Individual islet respirometry reveals functional diversity within the islet population of mice and human donors. Mol Metab 16: 150–159, 2018. doi: 10.1016/j.molmet.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 25: 402–408, 2001. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37. Quiros PM, Goyal A, Jha P, Auwerx J. Analysis of mtDNA/nDNA ratio in mice. Curr Protoc Mouse Biol 7: 47–54, 2017. doi: 10.1002/cpmo.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Osipovich AB, Stancill JS, Cartailler JP, Dudek KD, Magnuson MA. Excitotoxicity and overnutrition additively impair metabolic function and identity of pancreatic β-cells. Diabetes 69: 1476–1491, 2020. doi: 10.2337/db19-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Merrins MJ, Corkey BE, Kibbey RG, Prentki M. Metabolic cycles and signals for insulin secretion. Cell Metab 34: 947–968, 2022. doi: 10.1016/j.cmet.2022.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Foster HR, Ho T, Potapenko E, Sdao SM, Huang SM, Lewandowski SL, VanDeusen HR, Davidson SM, Cardone RL, Prentki M, Kibbey RG, Merrins MJ. β-Cell deletion of the PKm1 and PKm2 isoforms of pyruvate kinase in mice reveals their essential role as nutrient sensors for the KATP channel. eLife 11: e79422, 2022. doi: 10.7554/eLife.79422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mookerjee SA, Brand MD. Measurement and analysis of extracellular acid production to determine glycolytic rate. J Vis Exp e53464, 2015. doi: 10.3791/53464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tricò D, Natali A, Arslanian S, Mari A, Ferrannini E. Identification, pathophysiology, and clinical implications of primary insulin hypersecretion in nondiabetic adults and adolescents. JCI Insight 3: e124912, 2018. doi: 10.1172/jci.insight.124912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nolan CJ, Prentki M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: time for a conceptual framework shift. Diab Vasc Dis Res 16: 118–127, 2019. doi: 10.1177/1479164119827611. [DOI] [PubMed] [Google Scholar]
- 44. Corkey BE, Deeney JT, Merrins MJ. What regulates basal insulin secretion and causes hyperinsulinemia? Diabetes 70: 2174–2182, 2021. doi: 10.2337/dbi21-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Johnson JD. On the causal relationships between hyperinsulinaemia, insulin resistance, obesity and dysglycaemia in type 2 diabetes. Diabetologia 64: 2138–2146, 2021. doi: 10.1007/s00125-021-05505-4. [DOI] [PubMed] [Google Scholar]
- 46. Thorel F, Damond N, Chera S, Wiederkehr A, Thorens B, Meda P, Wollheim CB, Herrera PL. Normal glucagon signaling and β-cell function after near-total α-cell ablation in adult mice. Diabetes 60: 2872–2882, 2011. doi: 10.2337/db11-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nichols CG, York NW, Remedi MS. ATP-sensitive potassium channels in hyperinsulinism and type 2 diabetes: inconvenient paradox or new paradigm? Diabetes 71: 367–375, 2022. doi: 10.2337/db21-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37: 1595–1607, 1988. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 49. Reaven GM. Role of insulin resistance in human disease (syndrome X): an expanded definition. Annu Rev Med 44: 121–131, 1993. doi: 10.1146/annurev.me.44.020193.001005. [DOI] [PubMed] [Google Scholar]
- 50. Erion K, Corkey BE. β-Cell failure or β-cell abuse? Front Endocrinol (Lausanne) 9: 532, 2018. doi: 10.3389/fendo.2018.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Esser N, Utzschneider KM, Kahn SE. Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia. Diabetologia 63: 2007–2021, 2020. doi: 10.1007/s00125-020-05245-x. [DOI] [PubMed] [Google Scholar]
- 52. Shanik MH, Xu Y, Skrha J, Dankner R, Zick Y, Roth J. Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care 31, Suppl 2: S262–S268, 2008. doi: 10.2337/dc08-s264. [DOI] [PubMed] [Google Scholar]
- 53. Dai C, Kayton NS, Shostak A, Poffenberger G, Cyphert HA, Aramandla R, Thompson C, Papagiannis IG, Emfinger C, Shiota M, Stafford JM, Greiner DL, Herrera PL, Shultz LD, Stein R, Powers AC. Stress-impaired transcription factor expression and insulin secretion in transplanted human islets. J Clin Invest 126: 1857–1870, 2016. doi: 10.1172/JCI83657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chareyron I, Christen S, Moco S, Valsesia A, Lassueur S, Dayon L, Wollheim CB, Santo Domingo J, Wiederkehr A. Augmented mitochondrial energy metabolism is an early response to chronic glucose stress in human pancreatic beta cells. Diabetologia 63: 2628–2640, 2020. doi: 10.1007/s00125-020-05275-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.