

Abstract
The reaction of phosphorylation and phosphonylation of an oligodeoxynucleotide 3′-terminal hydroxyl (oligodeoxynucleotidyl kinase activity) catalyzed by calf thymus terminal deoxynucleotidyl transferase (TDT) was found. Triphosphates modified at Pα-, Pα,γ- or Pα,β,γ-residues served as low-molecular weight substrates. The reaction was TDT specific; human DNA polymerases α and β, as well as AMV reverse transcriptase did not catalyze it. The donor activity of modified triphosphates or triphosphonates depended on their structure and was increased with an increase in their hydrophobicity. The substrate activity of some modified triphosphates was up to one order of magnitude higher than that of ddTTP.
INTRODUCTION
The unique DNA polymerase of primitive lymphocytes, terminal deoxynucleotidyl transferase (TDT; EC 2.7.7.31), was discovered as an end-addition, template-independent enzyme of calf thymus extracts and its function has not been elucidated yet. Its strict limitation in normal animals to primitive lymphoid cells suggests a role for it in generating the functional properties of T and B cells. Some experiments strongly support a role of TDT in insertion of non-germline nucleotides (N segments) at VDJ joining sites during heavy-chain immunoglobulin gene rearrangements (1). A similar role for TDT N-segment addition in T-cell receptor gene rearrangements may also exist (2,3). It has been shown that the level of TDT in leukocytes of leukemia patients is very high (4).
In 1994–1995 a new group of very selective inhibitors of TDT in the cell-free systems was found (5,6). They belong to a group of modified α-nucleoside 5′-triphosphates of d- and l-series (I and II, Scheme 1) and their affinity to TDT was comparable with that of corresponding β-anomers. Based on these results, we suggested that a dNTP nucleic base did not participate in the genetically specific interaction with the TDT active center, and the main contribution to this interaction was made by dNTP triphosphate residues (7).

Here we describe substrate properties of modified triphosphates of types III–VI (Fig. 1) in the reaction catalyzed by calf thymus TDT in cell-free solutions.
Figure 1.

Structure of the tested compounds.
The structure of potential phosphate donors was selected by us according to the following chemical criteria: (i) triphosphates with an α-phosphate or both α- and γ-phosphates esterified by bulky hydrophobic groups sterically imitating nucleosides, but lacking chemical functional groups inherent to nucleic bases; (ii) triphosphates with one or three phosphate residues substituted by phosphonates; and (iii) dTTP with γ-phosphate substituted by a phosphonate. The basis for all substitutions will be discussed below.
Compounds III and VI phosphorylated (phosphonylated) the 3′-terminus of the oligodeoxynucleotide primers by their α- or γ-termini, whereas compounds Va–c phosphorylated the 3′-terminus only by their α-phosphonate residues. The intensity of the incorporation depended both on the structure of the substrate (Fig. 1) and the TDT preparation. Compounds IVa,b were completely inactive as phosphorylating agents. The structure of the reaction products was confirmed by direct comparison with authentic samples, or by methods of chemical modification.
MATERIALS AND METHODS
The synthesis of compounds III–V will be published elsewhere; compounds VIa–d were prepared as according toAlexandrova et al. (8).
The [5′-32P]d(CCCAGTCACGACGTOH) tetradecadeoxynucleotide primer was used. The control oligonucleotide d(CCCAGTCACGACGTOP) (VII) containing a phosphate group at 3′-position was synthesized from 5′-O-dimethoxytrityluridine coupled to the glass solid phase as described by Atkinson and Smith (9). After splitting off the support, the 5′-substituted oligonucleotide was oxidized with 100 µl of 0.1 M NaIO4 (1 h), hydrolyzed with 100 µl of 0.1 M NaOH as according to Krynetskaya et al. (10) and purified by reverse-phase HPLC. After the removal of the 5′-dimethoxytrityl protecting group (80% acetic acid, 30 min, 20°C), resulting VII was precipitated with 5% LiClO4 in acetone and purified by gel filtration on a Toyopearl HW-40 column eluting with water.
The other control oligodeoxynucleotidyl 3′-methylphosphonate d(CCCAGTCACGACGTOPMe) (VIII) was synthesized on the same uridine-modified support by coupling with the 3′-O-imidazolyl(methyl)phosphonyl 5′-O-dimethoxytritylthymidine according to Miller et al. (11). The product was treated and purified as described for VII with an additional purification by gel electrophoresis.
Enzymological procedures
Different calf thymus TDT lots were used: A401-1, A1601-2, A1801-3 (USB Amersham Life Science), M187A, M187B (Promega) and HF3403 (Gibco). TDTP+ (lots A401-1, M187A and M187B) could incorporate an unmodified γ-phosphate residue of the substrate into oligodeoxynucleotide 3′-termini. TDTP– (lots A1601-2, A1801-3 and HF3403) lacked this property. AMV reverse transcriptase was from Amersham; DNA polymerases α and β were isolated from human placenta in accordance with the methods of Mozzherin et al. (12) and Kolocheva and Nevinsky (13), respectively. Tetradecadeoxynucleotide primer as well as oligonucleotides VII–VIII were labeled at the 5′-termini using [γ-32P]ATP (Radioisotop, Moscow, Russia) and T4 polynucleotide kinase (Amersham) and purified on an Ultragel AcA54 (LKB) column in the standard TE buffer (10 mM Tris–HCl pH 7.6, 1 mM EDTA). For the experiments with DNA polymerases α and β and reverse transcriptase, the [5′-32P]primer was annealed with M13mp10 phage DNA, and the primer–template complex was isolated on a BioGel A-1.5M (Bio-Rad) column in the TE buffer.
Primer extension assays
For TDT, the assay mixture (6 µl) contained 0.02 µM [5′-32P]primer, the compounds under study or dNTP, two activity units of TDT from Amersham or Gibco, 100 mM sodium cacodylate pH 7.2, 2 mM CoCl2 and 0.05 mM DTT; or six activity units of the TDT from Promega, 100 mM sodium cacodylate pH 6.8, 1 mM CoCl2 and 0.05 mM DTT. For AMV reverse transcriptase, the assay mixture (6 µl) contained 0.01 µM DNA–[5′-32P]primer complex, the compounds under study or dNTP, two activity units of the enzyme, 10 mM Tris–HCl buffer pH 8.2, 5 mM MgCl2, 40 mM KCl and 1 mM DTT. In the case of DNA polymerase α, the assay mixture (6 µl) contained 0.01 µM template–primer complex, the compounds under study or dNTP, one activity unit of the enzyme, 10 mM Tris–HCl buffer pH 7.4, 6 mM MgCl2 and 0.4 mM DTT. For DNA polymerase β, the assay mixture (6 µl) contained 0.01 µM template–primer complex, the compounds under study or dNTP, two activity units of the enzyme, 10 mM Tris–HCl buffer pH 8.6, 6 mM MgCl2 and 0.4 mM DTT. The reactions were carried out for 30 min at 37°C and terminated by adding 3 µl of deionized formamide containing 0.5 mM EDTA, 0.1% bromophenol blue and 0.1% xylene cyanol. Reaction products were separated by electrophoresis in 20% polyacrylamide/7 M urea sequencing gel, and the gels obtained were autoradiographed. Protein electrophoreses of the TDT samples were carried out in the separating 0.1% SDS/8% polyacrylamide gel in 25 mM Tris–HCl, 250 mM glycine, 0.1% SDS buffer pH 8.3, according to Sambrook et al. (14).
The N3→NH2 reduction of 3′-phosphonylated oligonucleotides by DTT
After the reaction of compounds VIb,c with the oligonucleotide primer catalyzed by TDT (30 min, 37°C), the samples (6 µl) were heated at 100°C for 3 min. After adding H2O (2 µl), 25% NH4OH (1 µl) and 0.5 or 1 M DTT (1 µl), the assays were incubated for 60 min at 30°C. The reactions were stopped by adding 5 µl of deionized formamide containing 0.5 M EDTA, 0.1% bromophenol blue and 0.1% xylene cyanol, and reaction products were separated by electrophoresis in a 20% polyacrylamide/7 M urea sequencing gel.
Kinetic constants
For kinetic assays, the reaction mixture (6 µl) contained 0.02 µM [5′-32P]primer, two activity units of TDT (lot A1601-2), ddTTP or compounds Va,c in the 100 mM sodium cacodylate buffer pH 7.2, 2 mM CoCl2 and 0.05 mM DTT. The reaction was carried out for 2 min at 37°C and stopped by adding 3 µl of formamide containing 0.5 M EDTA, 0.1% of bromophenol blue and xylene cyanol. The products were separated by 20% PAGE, and the autoradiographs were scanned on a Molecular Dynamics 300A Computing densitometer. The apparent Km and Vmax were determined from the rate of the product formation as a function of substrate concentration.
RESULTS AND DISCUSSION
Introduction of a phosphate or phosphate ester group into the oligodeoxynucleotide primer
The unique ability of TDT to polymerize dNTPs in a template-independent manner, as well as to incorporate highly selectively unusual α-anomers of d- or l-series of carbocyclic nucleotides I and II allowed us to suggest that the triphosphate moiety of dNTP is crucial for the interaction with enzyme active center, and synthetic triphosphates esterified at the α-position by alkyl or aryl groups can be substrates in TDT-catalyzed extension of the oligodeoxynucleotide primer. Therefore, we synthesized a series of α-esterified triphosphates bearing aryl groups differing in size (phenyl, β-naphtyl) or groups capable of hydrogen bond formation (p-nitrophenyl), as well as aryl groups attached to a phosphate through a flexible ethyl arm (p-nitrophenylethyl). In addition, we synthesized α,γ-disubstituted triphosphates IV to elucidate the role of the γ-substitution in nucleoside-lacking triphosphates. This series could be useful for studying some sterical aspects of the substrate–enzyme interaction.
The pattern of the primer extension by III is given in Figure 2A and B. When the primer extension is catalyzed by TDTP–, new bands of products (i), whose mobility was lower than that of the primer were observed. In the case of TDTP+, additional bands (ii) with higher mobility were found. Bands (ii) for all compounds under study were located at the same level, whereas locations of bands (i) were different for different substrates. The bands (i) belong to the primers extended by the corresponding phosphate esters, namely, phenyl (IIIa, lanes 3–6), p-nitrophenyl (IIIb, lanes 7–10), p-nitrophenylethyl (IIId, lanes 11–14 and 15–17) and β-naphtyl (IIIc, lanes 18–20) phosphates, respectively (Fig. 2A). β-Naphtyl triphosphate IIIc showed weak substrate properties in this experiment. We assign the bands (ii) as belonging to the primer phosphorylated at the 3′-position, which was confirmed by direct comparison with the authentic sample VII (Fig. 2B). As seen in Figure 2B, lane 2, a small amount of product (ii) was also formed when ddTTP was used as substrate. Evidently, the band intensity was increased and mobility was decreased with an increase in hydrophobicity of the incorporated residues. Thus, the mobility of both modified primers upon gel electrophoresis corresponded to their structures.
Figure 2.
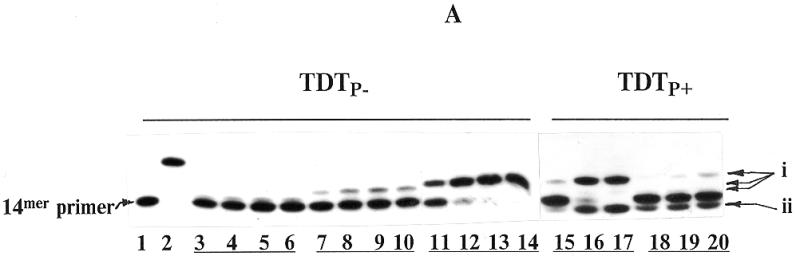

Primer extension by compounds IIIa–d catalyzed by TDTP– (Series A, lanes 1–14) and TDTP+ (Series A, lanes 15–20, Series B, lanes 1–4). (A) Lane 1, primer + enzyme (control); lane 2, as in lane 1 + 0.5 µM ddTTP; lanes 3–6, as in lane 1 + 2.5, 10, 25 and 100 µM IIIa, respectively; lanes 7–10, as in lane 1 + 2.5, 10, 25 and 100 µM IIIb, respectively; lanes 11–14, as in lane 1 + 0.025, 0.1, 0.25 and 1 µM IIId, respectively; lanes 15–17, as in lane 1 + 0.01, 0.1 and 1 µM IIId, respectively; lanes 18–20, as in lane 1 + 1, 10 and 100 µM IIIc, respectively. (B) Lane 1, primer + enzyme (control); lane 2, as in lane 1 + 0.2 µM ddTTP; lane 3, as in lane 1 + 5 µM IIId; lane 4, 3′-phosphorylated 14mer oligodeoxynucleotide VII.
The TDTP+-catalyzed primer extension reaction in the presence of dTTP and triphosphate derivatives IIIb, IIIc, IVa and IVb is illustrated in Figure 3. As seen in lanes 3–5 for IIIb and lanes 9–11 for IIIc, p-nitrophenyl and β-naphtyl phosphate residues were incorporated both into the initial primer and into the dTMP-extended oligodeoxynucleotides, which resulted in the electrophoretic upper- and lower-located bands relative to those corresponding to the incorporation of the thymidylate residue (Fig. 3, lanes 3–5, 9–11 and 8 or 14). The locations of these bands reflected hydrophobicity of the incorporated residues. Compounds IVa (lanes 6–8) and IVb (lanes 12–14) did not phosphorylate oligonucleotides, although the dose-dependent inhibition of the primer extension by dTTP was evident (compare lanes 6–8 and 12–14 with lane 2).
Figure 3.
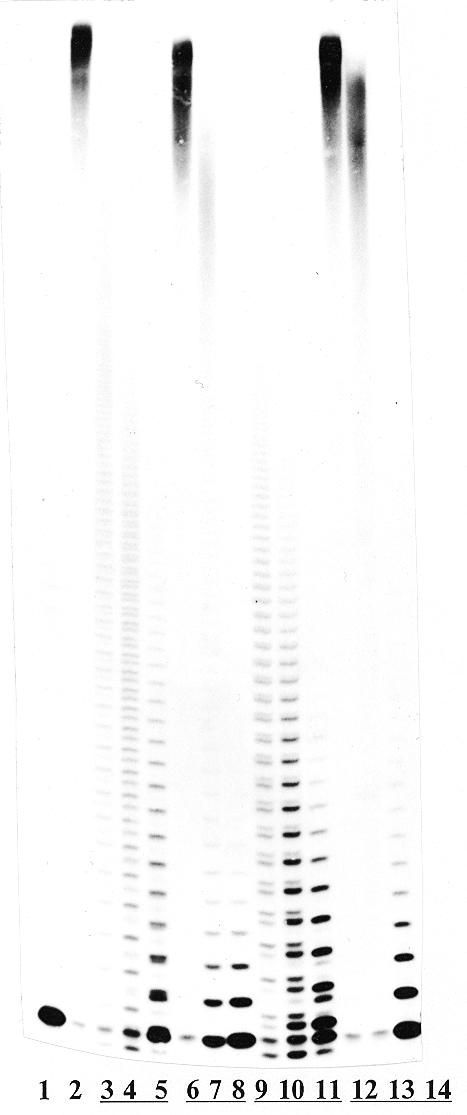
Primer extension by compounds III–IV. Lane 1, primer + TDTP+ (control); lane 2, as in lane 1 + 10 µM dTTP; lanes 3–5, as in lane 2 + 50, 100 and 300 µM IIIb, respectively; lanes 6–8, as in lane 2 + 50, 100 and 300 µM IVa, respectively; lanes 9–11, as in lane 2 + 50, 100 and 300 µM IIIc, respectively; lanes 12–14, as in lane 2 + 50, 100 and 300 µM IVb, respectively.
The incorporation of phosphate and ester phosphate residues into the oligodeoxynucleotide occur in a different concentration-dependent manner. The formation of the ester phosphorylated product (i) is parallel to the concentration of IIIb or IIIc, whereas the amount of phosphorylated product (ii) is changed in a bell-like mode (compare lanes 3–5 and 9–11). It is worth noting that formation of product (ii) was not observed with an increase of the concentration of IIIb,c when the primer was elongated at least once by a dTMP residue (lanes 5 and 11).
Thus, among the modified triphosphates tested, compound IIId containing hydrophobic substituent linked through a flexible ethyl chain showed the best substrate properties. Probably, this structure best supports the enzyme tuning in substrate binding to the active center.
Incorporation of a phosphonate group into the oligodeoxynucleotide primer
The ability of TDT to introduce a phosphate or ester phosphate group into DNA may be used for non-radioactive 3′-labeling of DNA, but this approach is limited due to instability of phosphate esters in biological media. It is well known that phosphonate groups are good mimics of phosphates, the stability of the P–C bond being much higher than that of the P–O–C one (15,16). In addition, 3′-labeling of DNA needs the presence of a reactive group. With this in mind, we synthesized compounds V containing an Fmoc-aminoethyl residue, which is bulky and highly hydrophobic.
As seen in Figure 4A, the compounds Va–c can phosphonylate the primer at the TDT catalysis. The dose-dependent primer extension in the presence of Va is observed in lanes 2–6. As a phosphonate donor, compound Vc (lanes 7–9) was more active than Va, which is supported by the kinetic data (Table 1). Compound Vb, differing from Vc by the bulky dibromomethylene fragment, was by two orders of magnitude less active (lanes 10–14).
Figure 4.
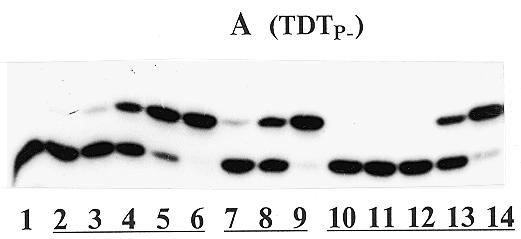

Primer extension by compounds V catalyzed by TDTP– (Series A) and TDTP+ (Series B). (A) Lane 1, primer + enzyme (control); lanes 2–6, as in lane 1 + 0.0005, 0.001, 0.01, 0.1 and 1 µM Va, respectively; lanes 7–9, as in lane 1 + 0.001, 0.01 and 0.1 µM Vc, respectively; lanes 10–14, as in lane 1 + 0.001, 0.01, 0.1, 1 and 10 µM Vb, respectively. (B) Lane 1, primer + enzyme (control); lane 2, as in lane 1 + 5 µM dTTP; lanes 3–5, as in lane 2 + 5, 10 and 20 µM IIId, respectively; lanes 6–8, as in lane 2 + 0.5, 2 and 10 µM Va, respectively; lane 9, as in lane 1 + 2 µM ddTTP.
Table 1. Kinetic parameters of the TDT-catalyzed primer extension by ddTTP and compounds Va and Vc.
| Compound | Km (µM) | Vmax/Vmax for ddTTP |
|---|---|---|
| ddTTP | 0.032 ± 0.001 | 1.00 |
| Va | 0.033 ± 0.003 | 0.56 |
| Vc | 0.009 ± 0.001 | 0.56 |
When the reaction was catalyzed by TDTP+ in the presence of dTTP (Fig. 4B), phosphonate Va, unlike compound IIId (lanes 3–5), extended the primer only by its α-phosphonate residue (lanes 6–8). Because of the high hydrophobicity of Fmoc-aminoethyl phosphonate attached to the oligodeoxynucleotide, the electrophoretic mobilities of the products were smaller than those of the respective oligodeoxynucleotides elongated with dTMP residues. The high substrate efficacy of Va can be confirmed by the absence of bands corresponding to unmodified oligodeoxynucleotides (lanes 6–8). An increase in the concentration of Va from 0.5 to 10 µM results in the considerable shortening of oligo sets (compare lanes 6–8 and 2).
Kinetic monitoring of the reaction (Table 1) showed that Km and Vmax of Va,c were similar to or lower than that of ddTTP. It implies that compounds Va,c have higher affinity to the enzyme and form the productive substrate–enzyme complex faster than ddTTP. This is in coincidence with the results obtained for IIId when a compound bearing a flexible and hydrophobic substituent was the best TDT substrate.
Phosphate incorporation into the DNA primer in the presence of ddTTP observed in Figure 2B (lane 2) led us to reassess substrate properties of previously synthesized thymidine 5′-γ-phosphonyl α,β-diphosphates VI. These nucleotides were studied in the primer elongation reaction catalyzed by AMV RT and DNA polymerases α and β (8,17). Figure 5A illustrates the results of the primer extension by VIa catalyzed by TDTP+ (lanes 4–6). Sets of doublets can easily be seen in each lane, and the intensities of these doublets are dose-dependent. The upper bands of the doublets correspond to the incorporation of a nucleotide residue, whereas lower ones correspond to the incorporation of a methyl phosphonate fragment. The incorporation of the methyl phosphonate fragment into the 14mer oligodeoxynucleotide was confirmed by direct comparison with the synthetic oligodeoxynucleotide VIII (lane 7). It is worth mentioning that the 3′-phosphorylated primer obtained in the presence of ddTTP (lane 2), moved somewhat faster than the methyl phosphonate-bearing primer because of an additional negative charge (compare the corresponding bands in lane 2 and lanes 4–7). With TDTP–, compound VIa showed similar results (data not shown).
Figure 5.

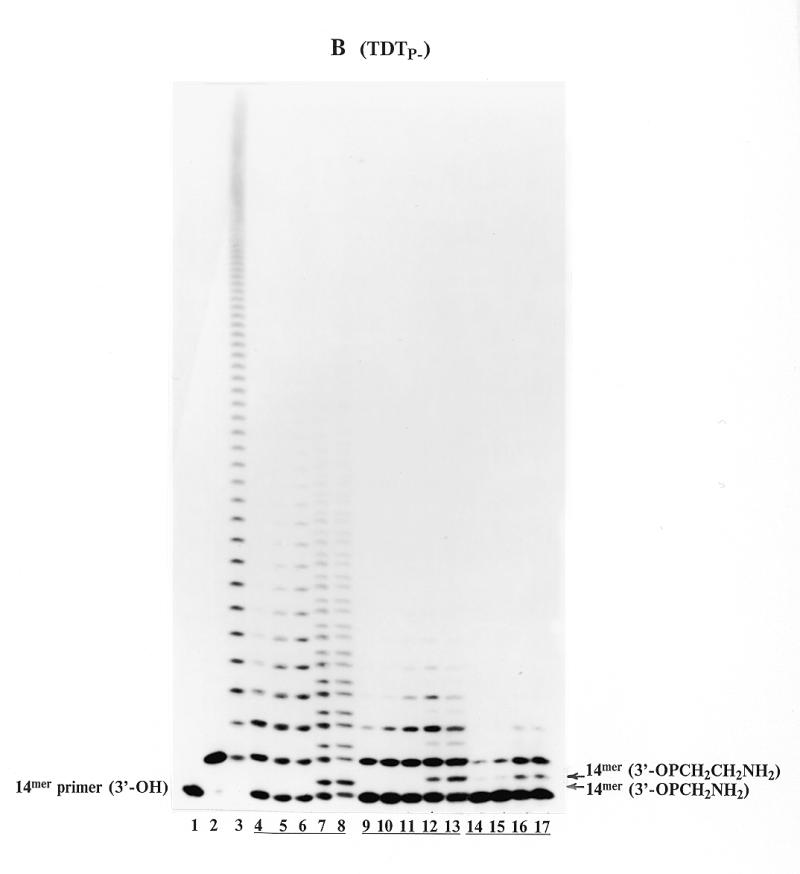
Primer extension by compounds VI catalyzed by TDTP+ (Series A) and TDTP– (Series B). (A) Lane 1, primer+ enzyme (control); lane 2, as in lane 1 + 0.5 µM ddTTP; lane 3, as in lane 1 + 5 µM dTTP; lanes 4–6, as in lane 1 + 5, 50 and 500 µM VIa, respectively; lane 7, synthetic 3′-methylphosphonylated oligodeoxynucleotide VIII. (B) Lane 1, primer + enzyme (control); lane 2, as in lane 1 + 0.1 µM ddTTP; lane 3, as in lane 1 + 1 µM ddTTP and 10 µM dTTP; lanes 4–6, as in lane 1 + 100, 300 and 600 µM VIb, respectively; lanes 7 and 8, as in lane 6, primer extension reaction followed by addition of 0.05 and 0.1 M DTT, respectively; lanes 9–11, as in lane 1 + 100, 300 and 600 µM VIc, respectively; lanes 12 and 13, as in lane 11, primer extension reaction followed by addition of 0.05 and 0.1 M DTT, respectively; lanes 14–17, as in lane 1 + 100, 300, 600 and 1000 µM VId, respectively.
Figure 5B presents the TDTP– catalyzed primer extension in the presence of thymidine 5′-(γ-alkylphosphonyl)-α,β-diphosphates (VIb–d). These compounds were involved in the reaction by both thymidylate and alkylphosphonate residues, although in lanes 4–6 (for VIb) and lanes 9–11 (for VIc) one can see the only set of bands. The incorporation of the azidomethyl phosphonate fragment in the primer was not observed because of the similarity of mobilities of the extended and the starting primers (lanes 4–6). However, this incorporation became evident after the azido group of the azidomethyl phosphonate-extended primer was reduced with DTT to an amino group (lanes 7–8, upper bands). Owing to an additional NH3+ charge, the mobility of the reduced oligodeoxynucleotide was lowered. The same result was obtained for VIc (lanes 9–11), when the reduced oligodeoxynucleotide was clearly discriminated at the electrophoresis pattern (lanes 12–13, upper bands). The mobility of the primer extended with aminoethyl phosphonate of VId (lanes 14–17) coincided with those of the products resulted from the reaction with VIc followed by reduction with DTT (lanes 12–13, upper bands). Similar results were obtained with compounds VIa–d at the catalysis of the reaction by TDTP+ (data not shown).
Compounds VIa–d also showed weaker substrate properties than that of Va,c.
Analysis of TDT
We analyzed TDT preparations of six lots available from Amersham, Promega and Gibco companies in the reactions of primer phosphorylation and phosphonylation. Although all of them catalyzed the transfer reaction of substituted phosphates and phosphonates approximately to the same degree, they revealed different properties when transferring unmodified γ-phosphates. TDT from Promega (lots M187A, M187B) and Amersham (lot A401-1) catalyzed both reactions, whereas TDT from Amersham (lots A1601-2, A1801-3) and Gibco (lot HF3403) could not extend the primer with an unmodified phosphate under various conditions. Figure 6 demonstrates these properties for TDTP– (lot A1801-3) and TDTP+ (lot A401-1) either in the presence of 2 mM Co2+ (standard conditions) or 10 mM Mn2+. In the case of 10 mM Mg2+ or 10 mM Zn2+ buffers both enzymes were inactive in this reaction (data not shown). As substrates, we used dTTP, ddTTP, Vb and IIId. Evidently, for both enzymes, the Mn2+ buffer (Series B and D) was somewhat less effective than the Co2+ buffer (Series A and C). Under the standard conditions, as well as in the Mn2+ buffer (Series A and B, respectively), TDTP– incorporates into the primer only nucleotide residues from dTTP (lane 2) and ddTTP (lane 3), a phosphonate fragment from Va (lane 4) and an esterified phosphate residue from IIId (lane 5). In contrast, in the Co2+ buffer (Series C), TDTP+ incorporates into the primer nucleotide and phosphate residues of ddTTP (lane 3, upper and lower bands, respectively), modified α-phosphonate residue of Va (lane 4), or p-nitrophenylethyl phosphate and unmodified γ-phosphate residues of IIId (lane 5, upper and lower bands, respectively). In the Mn2+ buffer no phosphorylation occurs in the presence of ddTTP and IIId (Series D, lanes 3 and 5). TDTP+ transfer an unmodified phosphate residue into the primer 3′-terminus only under standard buffer conditions. The cause of the difference in the properties of the enzyme lots tested is unclear. The protein electrophoresis of the lots used demonstrated that TDTP– contains a major band with a molecular mass of ∼58 kDa [which correlates with the earlier data (18)], whereas TDTP+ contains two major proteins with molecular masses of ∼58 and 62 kDa [previously published by Beach et al. (19)]. Thus, the difference in the phosphorylating activity can be accounted for by various enzyme composition. However, we cannot exclude that incorporation of the non-modified γ-phosphate residue of the substrate analogues (ddTTP, IIIa–d) is induced by a contaminant presenting in certain batches of TDT.
Figure 6.

Primer extension by dTTP, ddTTP, IIId and Va, catalyzed by TDTP– (Series A, B) and TDTP+ (Series C, D) in the presence of 2 mM Co2+ buffer (Series A, C) or 10 mM Mn2+ buffer (Series B, D). Lane 1, primer + enzyme (control); lanes 2–5, as in lane 1 + 5 µM dTTP, 0.1 µM ddTTP, 0.1 µM Va and 0.1 µM IIId, respectively.
From the results obtained, it is evident that all TDT samples catalyzed the transfer of phosphate ester and phosphonate residues from the corresponding triphosphate derivatives into the 3′-terminus of the DNA primer. These reactions are specific towards triphosphates (triphosphonates); neither corresponding monophosphates nor diphosphates are active in it. Moreover, these reactions are specific for TDT. Template-dependent DNA polymerases α and β as well as AMV reverse transcriptase cannot catalyze such transfer (data not shown).
To conclude, the higher the hydrophobicity of the substituent (at least among the tested compounds), the higher the efficiency of transfer of a substituted phosphate (or phosphonate) residue. On the other hand, the higher the substrate properties, the more pronounced the incorporation of a ‘standard’ α-phosphate (or phosphonate) fragment into the DNA chain; and the weaker the substrate, the more probable is the unusual reaction of incorporation of both α- and γ-residues.
Since the compounds with increased hydrophobicity and increased stability may be active phosphonate donors in the cellular media and in cells, they may be used for the investigation of TDT cellular functions. Modified triphosphates (especially those containing reporter and ligand groups) may be useful tools in biotechnological processes.
One can assign the reaction under discussion to the artifactual activity of this template-independent enzyme largely because of the use as substrates of so unnatural compounds. It may be a relevant explanation, indeed. However, it is also possible that according to the life cycle of primitive lymphoid or leukemic cells, any triphosphate (dNTP or rNTP) can serve as a phosphate donor for a molecular target currently unknown, and this reaction is catalyzed by TDT (20). Moreover, such a fact was recently found in the process of the TDT-dependent cell apoptosis (20).
In any case, the results obtained could be interesting for studying the TDT enzymology as well as the cellular biology of TDT-dependent cells. The compounds of this type with an increased stability in human blood may be useful as competitors of the natural substrates of the enzyme in the treatment of TDT-accompanied leukemia. In addition, to our knowledge, it is the only known biochemical method for the incorporation of a phosphate residue (including phosphates bearing reporter or ligand groups) to a 3′-hydroxyl of oligodeoxynucleotides.
Acknowledgments
ACKNOWLEDGEMENTS
The authors are grateful to Dr Elena Shirokova and Dr Marina Kukhanova for valuable discussion. This work was supported by the Russian Foundation for Basic Research, projects 98-03-32930a and 99-04-48315; by the Program ‘Leading Scientific Schools’, grant 96-15-97646; and by the ISTC, grant 1244.
REFERENCES
- 1.Alt F. and Baltimore,D. (1982) Proc. Natl Acad. Sci. USA, 79, 4118–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komori T., Okada,A., Stewart,V. and Alt,F.W. (1993) Science, 261, 1171–1175. [DOI] [PubMed] [Google Scholar]
- 3.Gilfillan S., Dierich,A., Lemeuur,M., Beniost,C. and Mathis,D. (1993) Science, 261, 1175–1178. [DOI] [PubMed] [Google Scholar]
- 4.McCaffrey R.P., Harrison,T.A., Parkman,R. and Baltimore,D. (1975) N. Engl. J. Med., 292, 775–781. [DOI] [PubMed] [Google Scholar]
- 5.Semizarov D.G., Victorova,L.S., Dyatkina,N.B., von Janta Lipinsky,M. and Krayevsky,A.A. (1994) FEBS Lett., 354, 23–26. [DOI] [PubMed] [Google Scholar]
- 6.Dyatkina N., Semizarov,D., Victorova,L., Krayevsky,A., Theil,F. and von Janta-Lipinski,M. (1995) Nucl. Nucl., 14, 723–726. [Google Scholar]
- 7.Krayevsky A.A. and Chernov,D.N. (1996) J. Biomol. Struct. Dyn., 14, 225–230. [DOI] [PubMed] [Google Scholar]
- 8.Alexandrova L.A., Skoblov,A.Yu., Jasko,M.V., Victorova,L.S. and Krayevsky,A.A. (1998) Nucleic Acids Res., 26, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atkinson T. and Smith,M. (1984) In Gait M.J. (ed.), Oligonucleotide Synthesis. A Practical Approach. IRL Press Limited, Oxford, UK, pp. 31–85.
- 10.Krynetskaya N.F., Zayakina,G.V., Oretskaya,T.S., Volkov,E.M. and Shabarova,Z.A. (1986) Nucl. Nucl., 5, 33–43. [Google Scholar]
- 11.Miller P., Reddy,M., Muracami,A., Blake,K., Lin,S. and Agris,C. (1986) Biochemistry, 25, 5092–5097. [DOI] [PubMed] [Google Scholar]
- 12.Mozzherin D.Yu., Atrazhev,A.M. and Kukhanova,M.K. (1992) Mol. Biol. Russian, 26, 999–1010. [PubMed] [Google Scholar]
- 13.Kolocheva T.A. and Nevinsky,G.A. (1993) Mol. Biol. Russian, 27, 1368–1379. [PubMed] [Google Scholar]
- 14.Sambrook J., Fritsch,E.E. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 15.Blackburn G.M., Taylor,G.E., Tattershall,R.U., Thathcher,G.R.J. and McLennan,M.G. (1986) In Bruzik,K.S. and Stec,W.I. (eds), Biophosphates and their Analogues—Synthesis, Structure, Metal and Activity. Elsevier, Amsterdam, The Netherlands, pp. 451–464.
- 16.Hamilton C.J., Roberts,S.M. and Shipitsin,A.V. (1998) Chem. Commun., 1087–1088. [Google Scholar]
- 17.Arzumanov A.A., Semizarov,D.G., Victorova,L.S., Dyatkina,N.B. and Krayevsky,A.A. (1996) J. Biol. Chem., 271, 24389–24394. [DOI] [PubMed] [Google Scholar]
- 18.Johnson D. and Morgan,A.R. (1976) Biochem. Biophys. Res. Commun., 72, 840–849. [DOI] [PubMed] [Google Scholar]
- 19.Beach C.M., Chan,S.K., Vanaman,T.C. and Coleman,M.S. (1985) Biochem. J., 227, 1003–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koc Y., Urbano,A.G., Sweeney,E.B. and McCaffrey,R. (1996) Leukemia, 10, 1019–1024. [PubMed] [Google Scholar]