


Abstract
Previous studies have shown that the small subunit of Xenopus DNA polymerase γ (pol γB) acts as a processivity factor to stimulate the 140 kDa catalytic subunit of human DNA polymerase γ. A putative human pol γB initially identified by analysis of DNA sequence had not been shown to be functional, and appeared to be an incomplete clone. In this paper, we report the cloning of full-length human and mouse pol γB. Both human and mouse pol γB proteins were expressed in their mature forms, without their apparent mitochondrial localization signals, and shown to stimulate processivity of the recombinant catalytic subunit of human pol γA. Deletion analysis of human pol γB indicated that blocks of sequence conserved with prokaryotic class II aminoacyl-tRNA synthetases are necessary for activity and interaction with human pol γA. Purification of DNA pol γ from HeLa cells indicated that both proteins are associated in vivo.
INTRODUCTION
The enzyme responsible for mitochondrial DNA replication and repair, DNA polymerase γ (pol γ) has been characterized in several higher eukaryotes as a heterodimeric enzyme (1–4). The larger catalytic subunit (pol γA) has been cloned in Drosophila (5), Xenopus (6) and humans (7,8). It has a size ranging from 125 to 140 kDa and a sequence relationship to the family A of DNA polymerases (6,9). The smaller subunits (pol γB) from Drosophila (10) and Xenopus (11) have been cloned and expressed in recombinant form. We recently reported that recombinant Xenopus pol γB can function as a processivity factor for human pol γA in in vitro replication assays (11).
Human pol γB has been difficult to study. Although Gray and Wong (3) reported co-purification of a smaller protein with the large subunit of human DNA pol γ, this protein was not obviously present in the enzyme purified by Longley et al. (12). Putative cDNA clones for human and mouse pol γB have been identified in sequence databases (10). These clones lacked N-terminal mitochondrial localization signals and appeared to be truncated at their N-termini in comparison with the active Xenopus pol γB. Although the mouse clone (GenBank accession number AF006072) was clearly an incomplete cDNA, the reported cDNA sequence of human pol γB (GenBank accession number U94703) did contain sequences preceding the putative translation start site. Analysis of the human clone revealed sequences related to the N-terminus of Xenopus pol γB in a small open reading frame (ORF) separated from the remainder of the protein by two frame shift mutations. We have confirmed that this reflects an error in the original sequence. This was not evident upon comparison of the previously available human sequence with its Drosophila homolog, both of which were inactive in recombinant form, but became clear upon availability of the Xenopus homolog. We report the cloning of intact versions of both the human and mouse cDNAs encoding pol γB. The complete cDNA sequences extend the human and mouse ORFs further upstream to include sequences conserved in the N-terminal region of the Xenopus protein. The N-termini of all three vertebrate DNA pol γB proteins appear to encode mitochondrial localization signals. Both human and mouse pol γB were expressed and shown to stimulate the processivity of recombinant human pol γA in in vitro replication assays. The recombinant human pol γB protein has the same size as the protein purified from HeLa cell mitochondria. Purification of human pol γ from HeLa cell mitochondria and western blot analyses indicated that pol γA and γB subunits are associated in vivo.
Alignment of human, mouse and Xenopus pol γB revealed a clear conservation of several sequence blocks also present in some class II aminoacyl-tRNA synthetases. Deletion analyses of human pol γB indicated that the conserved sequences are required for interaction with the catalytic subunit.
MATERIALS AND METHODS
Cloning and expression of human pol γB
Primers HGBNDE (5′-ACTTCGGAGACATATGCGCTCT-CGTGTAGCCG-3′) and HGBNOT (5′-CAAATATAGCGG-CCGCTACATTCTTAGCTGATGAT-3′) were used to amplify a 1.5 kb PCR product from a HeLa Marathon-ready cDNA (Clontech), using Pfu Turbo DNA polymerase (Stratagene). This product was cloned as an NdeI–NotI fragment in the vector pET22b(+) (Novagen) to generate clone pJAC39. Proteins were expressed in Escherichia coli strain BL21(DE3) after induction with 1 mM IPTG. Sequencing of this clone revealed the presence of several differences with the previously published sequence, including two frame shifts. Primer HGB1 (5′-TCATATGGATGCGGGGCAGCCGGAGC-3′), corresponding to sequence DAGQPE preceded by a methionine codon, was used in combination with the HGBNOT primer to clone by PCR a cDNA encoding the putative mature protein (clone pJAC44). For expression of proteins in insect cells, the NdeI–NotI insert was directly transferred into a pFastBacI baculovirus expression vector (Life Technologies) modified as previously described (11) to allow expression in insect cells of C-terminal his-tagged proteins. The human sequence has been deposited in GenBank under accession number AF177201.
Cloning and expression of mouse pol γB
Primer PMGB1 (5′-CTGCAGGTGCTGGCTGTCCG-3′) was designed from EST clone AA499566 and used in PCR together with primer MGBNOT (5′-GGCGGCCGCCACATTGCTAGCTGACGCTAG-3′) using a mouse Marathon-ready cDNA (Clontech) as template and Pfu DNA polymerase (Stratagene). We amplified two fragments of 1.2 and 1.3 kb that were cloned into the EcoRV site of plasmid pBluescript II KS(+) to generate clones pJAC52 (short product) and pJAC54 (large product). Sequencing revealed a 125 bp fragment missing in pJAC52 with respect to pJAC54. For bacterial expression, PCR on pJAC54 DNA with primer MGBNDE (5′-CGCAGGGC-CTGCCATATGTGGCTGTCCGGGTACGCG-3′) and primer MGBNOT was used to produce an NdeI–NotI fragment to express the putative mature protein in bacteria using the pET22b(+) vector. The mouse sequence has been deposited in GenBank under accession number AF177202.
Purification of his-tagged proteins
Bacteria expressing the recombinant protein were washed in a buffer containing 50 mM HEPES (pH 7.5), 0.2 M sucrose, 1 mM EDTA, 1 mM PMSF, 1 µM pepstatin and 7 mM β-mercaptoethanol, and sonicated in lysis buffer (50 mM sodium phosphate, pH 8, 300 mM NaCl, 20% glycerol, 1 mM β-mercaptoethanol, 0.5 mM benzamidine–HCl, 1 µM pepstatin, 5 µg/ml leupeptin and 0.2 mM PMSF). Insoluble proteins were solubilized in the same buffer containing 8 M urea without glycerol. Insect cells expressing recombinant protein were lysed by sonication in binding buffer (50 mM sodium phosphate, pH 8, 300 mM NaCl, 10% glycerol, 3 mM β-mercaptoethanol and 0.2 mM PMSF).
For affinity purification on Ni-NTA columns, imidazole was added to 10 mM final concentration to the cell lysate containing soluble proteins. Both bacterial and insect cell lysates were precleared by centrifugation at 23 500 g for 10 min. The cleared lysates were incubated for 1 h with Ni-NTA beads (Qiagen) pre-washed in binding buffer containing 20 mM imidazole (W20). Proteins from insect cells were then washed three times with W20 and proteins from bacteria were washed in W60 (binding buffer containing 60 mM imidazole). Elution was carried out with EB250 (binding buffer containing 250 mM imidazole). Purified protein was dialyzed against a buffer containing 50 mM sodium phosphate, pH 8, 300 mM NaCl, 50% glycerol, 2 mM β-mercaptoethanol and 0.2 mM PMSF, and stored at –80°C in aliquots after freezing in liquid nitrogen. To prepare protein to be used as antigen, bacterially expressed protein was purified from the urea-soluble fraction as described above but using buffers containing 8 M urea. Purified protein was dialyzed against PBS and used to inoculate a rabbit.
Partial purification of DNA pol γ from HeLa cells
HeLa cells were grown in 12 l of DMEM to a cell density of 7 × 105 cells/ml. Cells were harvested by centrifugation and washed in 240 ml TD buffer (135 mM NaCl, 5 mM KCl, 25 mM Tris–Cl, pH 7.6). After centrifugation as above, cells were resuspended in 210 ml of CaRSB buffer (10 mM NaCl, 1.5 mM CaCl2, 10 mM Tris, pH 7.5) and allowed to swell for 10 min on ice. Cells were then homogenized with a glass Dounce homogenizer and two-thirds volume of 2.5× MS buffer was added (1× MS is 210 mM mannitol, 70 mM sucrose, 5 mM EDTA, 5 mM Tris, pH 7.6). Nuclei were pelleted by centrifugation at 1500 g for 5 min, twice. A mitochondrial pellet was obtained by centrifugation at 27 000 g for 15 min, mitochondria were resuspended in MS buffer and then centrifuged again. Mitochondrial lysis was achieved by homogenization in MS buffer after addition of KCl to 250 mM and Triton X-100 to 0.5%. The mitochondrial lysate was clarified by centrifugation at 50 000 r.p.m. in a Beckman 70Ti rotor for 1 h and stored at –80°C.
Purification of pol γ was carried out by chromatography of the lysate through DEAE-Sephacel at 250 mM KCl in buffer Z (20 mM Tris pH 8.4, 1 mM DTT, 1 mM EDTA, 10% glycerol, 0.2 mM PMSF, 10 mM benzamidine–HCl, 1 µM pepstatin, 1 µg/ml aprotinin, 1 µg/ml leupeptin). The flow through was dialyzed against buffer Z to a KCl concentration below 50 mM, loaded onto a SP-Sepharose column and proteins were eluted with a KCl gradient from 50 to 750 mM in the same buffer. The peak fractions were collected, pooled, diluted to a KCl concentration of 200 mM and loaded onto a Heparin Hi Trap column. Proteins were eluted with a KCl gradient from 200 mM to 1 M. Pol γ was detected by assay for reverse transcriptase activity on poly(rA):oligo(dT), as described previously (2).
Generation of a rabbit antiserum against human pol γA
The polymerase domain of human pol γA was expressed in bacteria as a C-terminal his-tagged protein from an expression vector constructed as follows. An NdeI-containing primer was made in such a way that the ATG in the NdeI site matched the ATG in the sequence MDQEDL (between the exonuclease and the polymerase domains in the protein). Another primer was made containing a NotI site corresponding to the stop codon of the cDNA. PCR with these two primers on a HeLa Marathon-ready cDNA (Clontech) amplified a 2180 bp fragment that was then digested with NdeI and NotI and cloned into pET22B(+) (a three piece ligation was done due to the existence of an internal NdeI site in the amplified DNA). The 81.7 kDa protein was expressed in E.coli and purified from urea-solubilized inclusion bodies by affinity chromatography on Ni-NTA under denaturing conditions (8 M urea). A rabbit was immunized using standard protocols.
Human pol γB deletion mutants
To generate the different deletion mutants described in this work, NdeI–NotI fragments produced by PCR were cloned into pET22b(+) to produce C-terminal his-tagged proteins as described for the wild-type protein. N-terminal deletions were made by PCR with Pfu turbo DNA polymerase on pJAC44 DNA using the T7 terminator primer in combination with each of the following primers: HGBDN1 (5′-GAAGGCATTTCCTAC-ATATGAGCAAGCAGCAGCTTA-3′) and HGBDN2 (5′-GACCCTTGGGCGTACATATGCGGAAGAACCTGGC-3′). C-terminal deletions were produced using the same approach with the T7 promoter primer in combination with each of the following: HGBDC1 (5′-TCATTGTGGTGTCTGCGGCCG-CCAGATGTATTAATCCA-3′), HGBDC2 (5′-AACAAGGGT-GAAGTGCGGCCGCCTTTCTATGAAGATTT-3′), HGBDC3 (5′-CCACATTTTTTCGTGCGGCCGCGCCATGTAATTTAG-AC-3′). These PCR products were digested with NdeI and NotI and cloned into pET22b(+). Internal deletion HGBΔI1 was produced by elimination of a 240 bp SmaI fragment and religation. HGBΔI2 was generated by replacing the NdeI–HindIII fragment from pJAC 44 with an NdeI–HindIII fragment from a PCR product obtained with the T7 promoter primer and primer HGBHIND (5′-AATCTGAGAAGCTTCATAAGGTAGCC-TCTTGTT-3′). To generate HGBΔI3, a PCR product was obtained with the T7 promoter primer and primer HGBECO1 (5′-ACTGAATTCGGGTAGCAAAGGGCCTG-3′), a second PCR product was generated with primer HGBECO2 (5′-GTT-GAATTCGAAACTCTACGCGAAAT-3′) and the T7 terminator primer. NdeI–EcoRI and EcoRI–NotI fragments from these two PCR products were ligated together with NdeI–NotI digested pET22b(+). Restriction analysis and sequencing confirmed that the clones were correct. Protein expression in bacteria and purification by affinity chromatography on Ni-NTA was achieved as described above.
Pol γ activity assays
Pol γ assays and electrophoretic mobility shift assays (EMSA) were carried out as described (11,13). For the EMSA assays shown in Figure 4, we used an internally labeled [α32P]dCTP 45mer (5′-ATCCAACCTCGCGTCGTATCGAATCGGATCA-GATCGGGTCGTCAA-3′, labeled C is underlined) annealed to a 26mer (5′-TTGACGACCCGATCTGATCCGATTCG-3′), which leaves a 19-nt single-stranded region.
Figure 4.
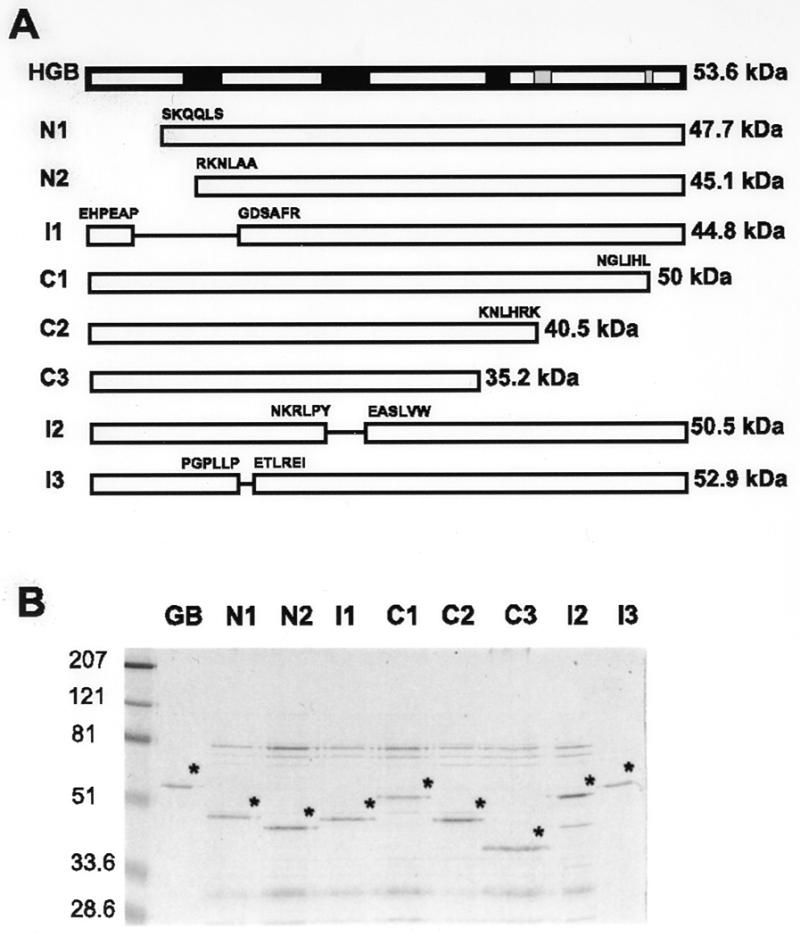
Deletion analysis of human pol γ B. (A) Diagram showing the locations of deletions within human pol γB in comparison to the full-length mature protein (HGB). Black rectangles indicate the regions that align with conserved motifs 1–3 (from left to right, respectively) of class II aminoacyl-tRNA synthetases; gray rectangles represent the two conserved blocks close to the C-terminus. Amino acid sequences at the end points of the deletion sites are indicated. ΔI1 is depicted with the two N-terminal deletions due to the proximity of the deleted region to the N-terminus. For ΔI3, two extra amino acids, E and F, were inserted in the deletion site. The size of each protein (including the his-tag) is indicated on the right. (B) Coomassie blue-stained gel showing the mutant proteins purified by Ni-NTA affinity chromatography together with recombinant wild-type pol γB (GB). The pol γB derived proteins are indicated with asterisks. Note that protein samples contained a variable contamination with bacterial proteins in the 70 to 80 kDa range. The sizes in kDa of molecular mass markers run in parallel are indicated on the left. (C) Activity of wild-type human pol γB and deletion mutants on poly(dA):oligo(dT). (D) EMSA assays to test the ability of pol γB derivatives to bind with pol γA to a primer-template oligonucleotide. 0 indicates no enzyme. A, human pol γA alone; B, human pol γA plus pol γB; N1 to I3, human pol γA plus each of the deletion mutants. Binding reactions contained 40 fmol of pol γA and 40 or 400 fmol (triangles) of pol γB derived protein with 40 fmol of a 26mer:45mer primer:template.
RESULTS
Cloning and expression of human and mouse pol γB
A putative sequence for human pol γB has been available in sequence databases for some time, originally identified based on the homology of expressed sequence tag clones to the Drosophila accessory subunit (10). It was surprising that this putative protein did not contain N-terminal sequences related to those of its functional Xenopus counterpart (11). This putative human pol γB also lacked an N-terminal mitochondrial localization signal. We therefore decided to search for human sequences in the expressed sequence tag (EST) database with homology to the N-terminus of the mature Xenopus protein, using BLAST (14). We found highly conserved sequences in the putative 5′ UTR of the available human pol γ accessory subunit cDNA sequence (GenBank accession number U94703), but in a different reading frame than that identified by Wang et al. (10). We realized that by introducing two nucleotides in the correct positions in the human sequence, the ORF could be extended further upstream to a putative translation start codon. With these insertions, this modified sequence would be slightly larger than the Xenopus one and the homology between the N-terminal regions of the proteins would be significantly increased. Furthermore, computer analysis of this sequence predicted a mitochondrial localization sequence comprising the first 25 amino acids (aa). These observations suggested that the original sequence might be in error and that the two reading frames might be continuous.
We designed primers to permit recloning of a human cDNA to determine whether the original sequence contained the expected frameshift mutations. The PCR approach described in Materials and Methods was used to clone a 1458 cDNA that contains an ORF for a putative protein of 485 aa and 54.9 kDa (Fig. 1, HSGB). Sequencing revealed that this clone contained the two expected frame shifts with respect to the previously published sequence (GenBank U94703; a G after nucleotide position 230 and an A after position 453). Other sequence differences were also noted as described in the legend to Figure 1. The putative protein encoded by this cDNA was now similar in size to its Xenopus homolog. The mature protein, after removing the computer-predicted mitochondrial localization signal of 25 aa has 460 residues, a predicted molecular mass of 52.2 kDa, and exhibits 50.4% identity and 79.3% similarity with its Xenopus homolog (without considering the mitochondrial localization signals). The revised mature human pol γB also showed 18% identity/54.1% similarity with the complete Drosophila pol γB and 20.2% identity/54.5% similarity with the glycyl–tRNA synthetase (GRS) from Thermus thermophilus.
Figure 1.

Alignment of vertebrate pol γB subunits with T.Thermophilus GRS. The program Clustal (PCGene) was used to align the sequences of human (HSGB; GenBank accession number AF177201), mouse (MMGB; accession number AF177202) and Xenopus (XLGB; accession number AF124606) pol γB subunits with T.thermophilus GRS (TTGRS; accession number P56206). Identical amino acids are indicated with asterisks; similar ones, with dots. The first amino acid of the mature Xenopus protein and the predicted ones for the human and mouse proteins are indicated in bold. The consensus motifs in class II aaRS sequences are underlined in the TTGRS sequence (31). The DNA sequence we determined for human pol γB includes a few changes with respect to the original sequence reported in GenBank U94703, as follows: silent T to C changes at positions 371 and 998; C to G at 448, resulting in a T122 to R change in the protein sequence; A to G at 488, resulting in S136 changed to G; G to A in 587, resulting in A169 changed to T; and a short frame shift caused by an insertion of an A in position 940 and a deletion of a C in position 961, resulting in a change of sequence TNFTTI to NKLYYN.
The BLAST searches described above also identified a cDNA fragment from rat (EST H33033) that contained a putative ORF with very good homology to the N-terminal region of the Xenopus pol γB. We repeated a BLAST search as described above to find mouse EST clones with homology to the N-terminal regions of Xenopus pol γB, human pol γB and clone H33033 from rat. This strategy identified two clones, AA499566 and AI615346.1, which differed from one another due to a frame shift in the N-terminal region of the protein and due to the presence of additional sequences in clone AI165346.1 that included a potential 16 residue mitochondrial localization signal. Primers designed from these clones and from the C-terminal side of the published partial mouse pol γB sequence (10) were used to clone the coding region for the mature protein from a mouse Marathon-ready cDNA (Clontech) using the same approach described for the human gene. Figure 1 shows the composite sequence including the amplified product and its predicted mitochondrial localization signal. The predicted mature mouse pol γB contains 443 residues, with a molecular mass of 49.7 kDa. The extreme 5′ end of the cDNA is derived from the GenBank entry AI165346.1. Although there are no in-frame stop codons located upstream of the mouse cDNA, the N-terminal methionine shown in Figure 1 is the first one in frame. Figure 1 shows that the three vertebrate pol γB sequences are highly conserved throughout, including the novel N-terminal sequences. The sequence of T.thermophilus GRS is included in this alignment to show that all three pol γB proteins share extensive homology with this protein.
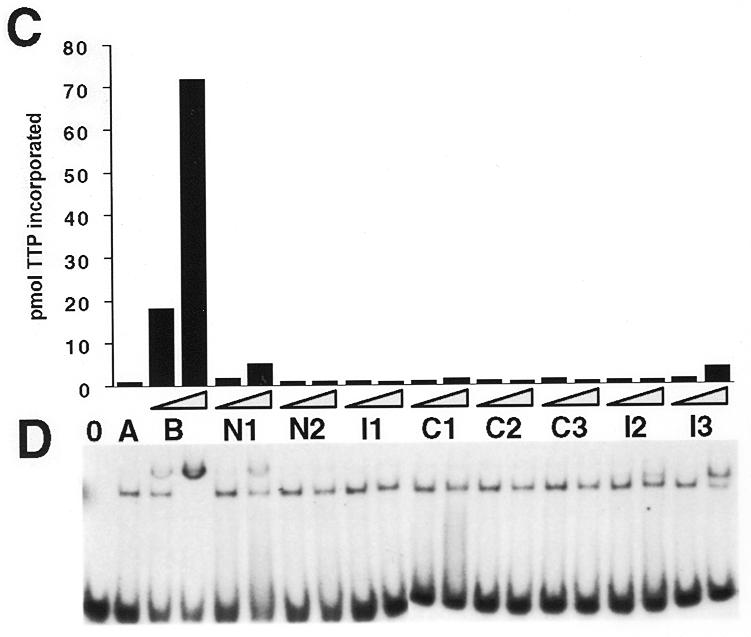
Bacterial clones expressing his-tagged versions of the putative mature human and mouse pol γB (without the mitochondrial localization signal) were constructed as described in Materials and Methods. A substantial fraction of both bacterially expressed proteins was insoluble. Nevertheless the soluble fraction from both expression systems could be purified by affinity chromatography in Ni-NTA (Fig. 2A). The insoluble human pol γB protein found in inclusion bodies was solubilized using 8 M urea. The protein regained some activity after refolding but was not as active as the original soluble fraction. The protein recovered from inclusion bodies was used to immunize a rabbit to produce an antiserum.
Figure 2.
All three vertebrate pol γB subunits stimulate processive DNA synthesis by human pol γA. (A) Coomassie blue stained SDS gel showing recombinant Xenopus (X), mouse (M) and human (H) pol γB subunits. 4.7 pmol of each protein was loaded on the gel, corresponding to ~230 ng of each polypeptide. The sizes in kDa of molecular mass markers run in parallel are indicated on the left. (B) Primer extension assay on oligo(dT)-primed poly(dA). Reactions were carried out as described in Materials and Methods using 40 fmol of pol γA and 240 fmol of pol γB per reaction. Lane 1, no protein; lanes 2–5 correspond to reactions carried out with pol γA alone, A + Xenopus B, A + mouse B and A + human B, respectively, incubated for 1 min; lanes 6–9 are as 2–5 but incubated for 2 min; lanes 10–13 correspond to reactions incubated for 5 min. The sizes in nucleotides of MspI restriction fragments of pUC 18 are indicated on the left.
Human and mouse pol γB increase processivity of human pol γA
We determined the effect of human and mouse pol γB on processivity of the human pol γA catalytic subunit using primer extension assays in vitro with singly-primed M13 DNA and oligo(dT)-primed poly(dA) as previously described for Xenopus pol γB (11). These experiments were performed with an excess of template-primer in order to permit determination of the number of nucleotides incorporated in a single binding event. Titration experiments showed that a slight excess of pol γB protein was generally required for maximal stimulation of the human pol γA. Figure 2B shows the results obtained with oligo(dT)-primed poly(dA) with recombinant pol γB from Xenopus, mouse and human. As expected, the human and mouse homologs of Xenopus pol γB functioned as processivity factors for human DNA pol γ. The specific activity of the Xenopus protein was higher than that of the mouse and human proteins, which could be due in part to the fact that the Xenopus protein used in this assay was generated in insect cells while the mouse and human proteins were of bacterial origin. The human pol γB produced in insect cells also showed a slightly higher activity than the bacterially produced protein (data not shown).
Copurification of DNA pol γA and B subunits from HeLa cell mitochondria
As noted above, Gray and Wong (3) described the co-purification of human DNA pol γ with two polypeptides of 140 and 54 kDa. The size of the 54 kDa subunit is consistent with the revised sequence reported above, but would be substantially larger than the human pol γB candidate identified by Wang et al. (10). We generated a polyclonal antiserum directed against the recombinant human pol γB to determine its relationship to the protein reported by Gray and Wong (3). We partially purified DNA pol γ from HeLa cell mitochondria by chromatography on DEAE-Sephacel, SP-Sepharose and heparin Sepharose as described in Materials and Methods. Proteins in S-Sepharose and heparin Sepharose fractions containing polymerase activity were analyzed by SDS–PAGE and immunoblotting. Membranes were cut in two at the level of the 81 kDa molecular weight marker to permit the upper part to be probed with antiserum directed against pol γA while the lower part was probed with antiserum directed against human pol γB. Figure 3 shows that the mitochondrial pol γB exhibited the same electrophoretic mobility as the recombinant protein and that both subunits of DNA pol γ precisely copurified with the polymerase activity.
Figure 3.
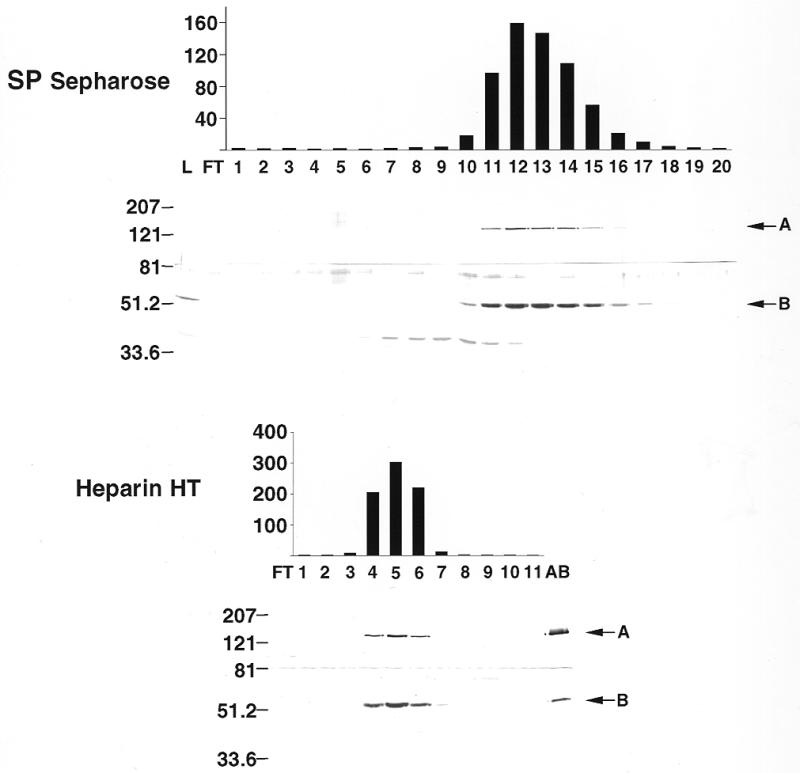
Copurification of human γA and B subunits with pol γ activity. Mitochondrial DNA polymerase was purified from HeLa cells as described in Materials and Methods. Fractions from the SP Sepharose and Heparin Hi Trap chromatography steps were assayed for reverse transcriptase activity (indicated as c.p.m. × 1000 on the right of each chart) and also analyzed by western blot. The upper part of each membrane (containing proteins above 81 kDa) was incubated with an anti-human pol γA antibody (A). The lower part was incubated with an anti-human pol γB antibody (B). Fraction numbers indicated at the bottom of the histogram also refer to the lanes in the associated western blot; L, loaded fraction; FT, flow through; AB, recombinant pol γA plus γB used as positive control. The sizes in kDa of protein molecular mass markers run in parallel are indicated on the left.
Deletion analysis of human pol γB
We constructed several deletion mutants of human pol γB in order to characterize protein domains required for binding pol γA and stimulating polymerase activity. The N-terminal deletion clones, ΔN1 and ΔN2, lacked the first 56 and 81 residues, respectively. Clone ΔN2 was designed to remove the sequence GPLGVEL conserved between all three vertebrate DNA pol γB proteins and GRS motif I. The C-terminal deletion clones, ΔC1, ΔC2 and ΔC3, lacked the final 30, 115 and 161 residues, respectively. Clones ΔC1 and ΔC2 were designed to remove the sequences RSRDT and VLKLHPCLAPIKVA that are highly conserved with GRS sequences. Clone ΔC3 was designed to remove sequences conserved with GRS motif 3. These constructs are illustrated in Figure 4A along with three internally deleted mutants ΔI1, ΔI2 and ΔI3. All proteins were expressed and purified as described for the full-length protein. In every case a sufficient fraction of the bacterially expressed protein was soluble to permit affinity purification using Ni-NTA columns. The proteins eluted from the Ni-NTA columns were contaminated with some bacterial proteins as shown in Figure 4B. This contamination was more evident with the less soluble mutants, due to the lower yield of soluble protein. These contaminants did not affect the activity of the recombinant proteins when the mutant proteins were added to reactions containing the wild-type protein (data not shown). Note that some of these proteins displayed an aberrant electrophoretic mobility. For example, ΔI1 (44.8 kDa) migrated more slowly than ΔN2 (45.1 kDa). Activity assays on poly(dA):oligo(dT) followed by quantitation of acid-insoluble products indicated that all deleted proteins were essentially inactive (Fig. 4C) when used with pol γ in equimolar amounts. ΔN1 and ΔI3 showed a low but detectable level of stimulation activity when used in a 10-fold molar excess over pol γA. These mutants were also able to interact with pol γA–DNA complexes in EMSA (Fig. 4C, lower panel). Mutant ΔI2 was also capable of some degree of interaction in the binding assay, but did not significantly stimulate pol γA activity. Migration of the supershifted protein:DNA complexes was slower for mutants ΔI2 and ΔI3. This difference in the extent of the mobility shift cannot be explained simply by a difference in charge of the mutant proteins, since the pK values of these two mutant proteins (6.67 and 7.08, respectively) is similar to the pK of the wild-type protein (7.36). A more plausible explanation for the altered mobility of the protein–DNA complexes involving these two internal deletion mutants is that these proteins may adopt a different conformation than the wild-type protein.
DISCUSSION
The complete sequences of human and mouse DNA pol γB
Gray and Wong (3) identified two polypeptides co-purifying with DNA pol γ activity from HeLa cell mitochondria, a catalytic subunit of 140 kDa and a small subunit of 54 kDa of unknown function. The 190 kDa native molecular mass of the enzyme suggested a heterodimeric structure containing one copy of each polypeptide. The size of the putative small subunit of human pol γ, which we designate pol γB, was significantly larger than the 42.4 kDa human pol γB homolog of Drosophila pol γB identified in a database search by Wang et al. (10). When we cloned Xenopus pol γB (11), we noted that the Xenopus protein contained additional N-terminal sequences lacking in the putative human pol γB. We employed the strategy described above to clone a full-length version of the human pol γB that does exhibit homology to the N-terminus of the Xenopus protein, as shown in Figure 1. The cloned sequence contains a potential mitochondrial targeting signal and predicts a molecular mass of 52.2 kDa for the mature pol γB, which agrees well with the molecular mass of 54 kDa estimated by Gray and Wong (3). The recombinant human pol γB protein co-migrates with the pol γB purified from HeLa cells (Fig. 3, lower panel, compare lanes 5 and AB).
The sequences of Xenopus and human pol γB also allowed us to clone an apparently full-length form of murine pol γB. The identification of this protein as the small subunit of mouse pol γ relies mainly on sequence homology and activity in vitro at this time, since no extensive efforts to purify mouse pol γ have been reported. However, both the human and mouse DNA pol γB stimulate the human catalytic subunit at physiological salt concentrations (Fig. 2B), as has previously been shown for Xenopus pol γB (11). All three vertebrate pol γB subunits act as processivity factors for human pol γA.
Domains of human pol γB conserved in aminoacyl-tRNA synthetases from prokaryotes are important for polymerase activity
When Carrodeguas et al. (11) first reported the homology between Xenopus pol γB and class II aminoacyl-tRNA synthetases (aaRSs), it was unclear how well this would apply to human pol γB since the sequence reported by Wang et al. (10) was significantly shorter than that of Xenopus pol γB. Our elucidation of the complete sequences of human and mouse pol γB has extended the homology between pol γB and aaRS proteins to include motif I of aaRSs. Since Xenopus and mouse pol γB stimulate the activity of human pol γA as well as human pol γB, it is virtually certain that all three proteins will share a common structure. The homology of all three proteins with aaRS sequences is so extensive that computer algorithms such as PredictProtein (15) predict that the common structure of pol γB will closely resemble that determined for T.thermophilus GRS. After the Xenopus pol γB sequence was published (11), a theoretical molecular modeling study appeared which supported this conclusion (16). Unfortunately, these authors did not employ the complete human pol γB in their analysis, so that the model lacks the N-terminal domain. The definitive analysis of the homology between DNA pol γB and tRNA synthetases will require experimental determination of the structure of the polymerase subunit.
We constructed several deletion mutants of human DNA pol γB to determine whether sequences related to aaRS proteins were essential for stimulation of pol γA activity and for binding of pol γB in complexes with pol γA and primer–template oligonucleotides. All of the mutants we generated were markedly impaired in both assays. The general lack of activity we observed for pol γB deletion mutants suggests that the protein requires a highly conserved structure in order to serve as an accessory factor for pol γA. Removal of 56 amino acid residues from the N-terminus is sufficient to essentially inactivate the protein. Since the mouse pol γB is truncated slightly (26 residues) with respect to human pol γB and is still fully active, this indicates that the remaining 30 residues in the N-terminal region are important for activity. Similarly, removal of 30 amino acid residues from the C-terminus, including the RSRDT motif conserved in GRS, inactivates human pol γB. Two mutants, ΔN1 and ΔI3, exhibited detectable activity when used in a molar excess over the catalytic subunit. These mutants are also able to supershift pol γA:DNA complexes when used in excess. This is consistent with a weaker or more unstable interaction as compared to the wild-type protein.
It is interesting that deletions that completely inactivate the protein involve regions that are conserved in aminoacyl-tRNA synthetase sequences. The two deletion mutants that retain some activity, ΔN1 and ΔI3, are missing regions that are only weakly conserved between these two groups of proteins. ΔN2 differs from ΔN1 by the deletion of an additional 25 amino acids including a very well conserved sequence block, GPLGVEL, and is completely inactive. This block is part of motif 1 in class II aaRSs, which has been implicated in dimerization and orientation of motifs 2 and 3 (17,18). Mutant ΔI2 protein is still capable of interaction with the pol γA:DNA complex but it is unable to activate pol γA. This mutant is missing a region that aligns with motif 2 of aaRSs which, together with motif 3, is believed to form the active site of the tRNA synthetase.
The functional relationship, if any, between a subunit of an eukaryotic mitochondrial DNA polymerase and a prokaryotic aminoacyl-tRNA synthetase is still a matter of speculation. Although pol γB is more closely related to GRS than to other tRNA synthetases in genetic databanks, there are several reasons to think it is not an active mitochondrial GRS. First, preliminary experiments carried out in our laboratory to detect adenylation of amino acids as a subreaction of the tRNA aminoacylation reaction have not given positive results (data not shown). Second, we did not detect a free pool of pol γB not associated with pol γA in HeLa cell or Xenopus oocyte mitochondria. Finally, a likely candidate for the functional mitochondrial GRS has been identified by Shiba et al. (19).
It appears that the pol γB subunit was acquired by the catalytic subunit during evolution to enhance the processivity of the enzyme and possibly to serve other roles in replication. What other activities of an aminoacyl-tRNA synthetase may play a role in replication? There is growing evidence that aminoacyl-tRNA synthetases are involved in other cellular processes not related to tRNA aminoacylation. Escherichia coli GRS catalyzes the synthesis of dinucleoside polyphosphates (20), with suspected regulatory functions in replication and cell proliferation (21). Aminoacyl-tRNA synthetases also play a role in intron splicing (22) and transcription termination (23,24). Phenylalanyl-tRNA synthetase from T.thermophilus binds DNA specifically (25), although the functional significance of this binding has not yet been identified. More recently, Magrath and Hyman (26) reported that GRS is involved in 3′-end formation in yeast, possibly by direct interaction with tRNA-like structures located at the 3′ end of several yeast mRNAs.
Recent studies of the mechanism of priming of mtDNA replication suggest a hypothetical role for pol γB in replication initiation. As shown in Figure 5, DNA sequences in the control regions of several vertebrate mitochondrial DNAs that may be capable of adopting tRNA-like structures have been proposed to participate in initiation of replication (27). Priming of leading strand replication is thought to require synthesis of a highly folded RNA that can be recognized and cleaved by a mitochondrial RNA processing enzyme (MRP endonuclease; 28). The specificity of primer cleavage differs for free RNA or for RNA associated with mtDNA in an R-loop structure. It is possible that binding of DNA pol γ in this region, possibly mediated by the small subunit, may influence the cleavage of the RNA by MRP endonuclease to generate primers or may facilitate primer utilization. A different priming mechanism has been proposed for the lagging strand origin of replication, OL, which is located within a cluster of tRNA genes and adopts a stem–loop conformation (29; see Fig. 5). The lagging strand primer is thought to be synthesized by a DNA primase (30). It is possible that binding of DNA pol γ to tRNA-like structures near OL may influence the efficiency of primer utilization at this origin as well. These proposed functions for the small subunit are reminiscent of the function of aminoacyl-tRNA synthetase in mRNA 3′ end formation in yeast and transcription termination in other systems noted above. The availability of an active heterodimeric pol γ will permit direct experimental testing of such models in future experiments.
Figure 5.

Model for the recognition of the origin of replication by mitochondrial DNA polymerase. (A) Structures present at the heavy strand (OH) and light strand (OL) origins of replication of human mitochondrial DNA [adapted from Brown et al. (27) and Wong and Clayton (30)]. RNA primers are indicated by the gray outline, sites of DNA synthesis are indicated by black arrows. Two alternative start sites are shown at OH. (B) mtRNA polymerase initiates transcription at the light strand promoter (LSP) and proceeds through conserved sequence block 2 (CSB2). The GC-rich sequence at CSB2 has been proposed to stabilize the RNA–DNA hybrid. (C) The RNA adopts a secondary structure that can be recognized by MRP endonuclease. The double headed arrow at the bottom of the panel indicates that single-stranded DNA in the origin region may fold in an RNA-like secondary structure. We suggest that DNA pol γB may interact with the nascent RNA, with the folded single-stranded DNA, or with both.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Kevin Pinz for technical assistance. This research was supported by grants from the NIH (GM 29681) and NIEHS (ES04068).
DDBJ/EMBL/GenBank accession nos AF177201, AF177202
REFERENCES
- 1.Wernette C.M. and Kaguni,L.S. (1986) J. Biol. Chem., 261, 14764–14770. [PubMed] [Google Scholar]
- 2.Insdorf N.F. and Bogenhagen,D.F. (1989) J. Biol. Chem., 264, 21491–21497. [PubMed] [Google Scholar]
- 3.Gray H. and Wong,T.W. (1992) J. Biol. Chem., 267, 5835–5841. [PubMed] [Google Scholar]
- 4.Olson M.W., Wang,Y., Elder,R.H. and Kaguni,L.S. (1995) J. Biol. Chem., 270, 28932–28937. [DOI] [PubMed] [Google Scholar]
- 5.Lewis D.L., Farr,C.L., Wang,Y., Lagina,A.T.,III and Kaguni,L.S. (1996) J. Biol. Chem., 271, 23389–23394. [DOI] [PubMed] [Google Scholar]
- 6.Ye F., Carrodeguas,J.A. and Bogenhagen,D.F. (1996) Nucleic Acids Res., 24, 1481–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ropp P.A. and Copeland,W.C. (1996) Genomics, 36, 449–458. [DOI] [PubMed] [Google Scholar]
- 8.Graves S.W., Johnson,A.A. and Johnson,K.A. (1998) Biochemistry, 37, 6050–6058. [DOI] [PubMed] [Google Scholar]
- 9.Lecrenier N., Van Der Bruggen,P. and Foury,F. (1997) Gene, 185, 147–152. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y., Farr,C.L. and Kaguni,L.S. (1997) J. Biol. Chem., 272, 13640–13646. [DOI] [PubMed] [Google Scholar]
- 11.Carrodeguas J.A., Kobayashi,R., Lim,S.E., Copeland,W.C. and Bogenhagen,D.F. (1999) Mol. Cell. Biol., 19, 4039–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Longley M.J., Ropp,P.A., Lim,S.E. and Copeland,W.C. (1998) Biochemistry, 37, 10529–10539. [DOI] [PubMed] [Google Scholar]
- 13.Mikhailov V.S. and Bogenhagen,D.F. (1996) J. Biol. Chem., 271, 18939–18946. [DOI] [PubMed] [Google Scholar]
- 14.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 15.Rost B. (1996) Methods Enzymol., 266, 525–539. [DOI] [PubMed] [Google Scholar]
- 16.Fan L., Sanschagrin,P.C., Kaguni,L.S. and Kuhn,L.A. (1999) Proc. Natl Acad. Sci. USA, 96, 9527–9532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eriani G., Cavarelli,J., Martin,F., Dirheimer,G., Moras,D. and Gangloff,J. (1993) Proc. Natl Acad. Sci. USA, 90, 10816–10820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cavarelli J., Eriani,G., Rees,B., Ruff,M., Boeglin,M., Mitschler,A., Martin,F., Gangloff,J., Thierry,J.-C. and Moras,D. (1994) EMBO J., 13, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shiba K., Schimmel,P., Motegi,H. and Noda,T. (1994) J. Biol. Chem., 269, 30049–30055. [PubMed] [Google Scholar]
- 20.Led J.J., Switon,W.K. and Jensen,K.F. (1983) Eur. J. Biochem., 136, 469–479. [DOI] [PubMed] [Google Scholar]
- 21.Plateau P. and Blanquet,S. (1994) Adv. Microb. Physiol., 36, 81–109. [DOI] [PubMed] [Google Scholar]
- 22.Lambowitz A.M. and Perlman,P.S. (1990) Trends Biochem. Sci., 15, 440–444. [DOI] [PubMed] [Google Scholar]
- 23.Yanofsky C., Konan,K.V. and Sarsero,J.P. (1996) Biochimie, 78, 1017–1024. [DOI] [PubMed] [Google Scholar]
- 24.Schray B. and Knippers,R. (1991) Nucleic Acids Res., 19, 5307–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lechler A. and Kreutzer,R. (1998) J. Mol. Biol., 278, 897–901. [DOI] [PubMed] [Google Scholar]
- 26.Magrath C. and Hyman,L.E. (1999) Genetics, 152, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown G.G., Gadaleta,G., Pepe,G., Saccone,C. and Sbisa,E. (1986) J. Mol. Biol., 192, 503–511. [DOI] [PubMed] [Google Scholar]
- 28.Lee D.Y. and Clayton,D.A. (1997) Genes Dev., 11, 582–592. [DOI] [PubMed] [Google Scholar]
- 29.Hixson J.E., Wong,T.W. and Clayton,D.A. (1986) J. Biol. Chem., 261, 2384–2390. [PubMed] [Google Scholar]
- 30.Wong T.W. and Clayton,D.A. (1985) Cell, 42, 951–958. [DOI] [PubMed] [Google Scholar]
- 31.Logan D.T., Mazauric,M.-H., Kern,D. and Moras,D. (1995) EMBO J., 14, 4156–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]