


Abstract
Central to understanding the process of V(D)J recombination is appreciation of the protein–DNA complex which assembles on the recombination signal sequences (RSS). In addition to RAG1 and RAG2, the protein HMG1 is known to stimulate the efficiency of the cleavage reaction. Using electrophoretic mobility shift analysis we show that HMG1 stimulates the in vitro assembly of a stable complex with the RAG proteins on each RSS. We use UV crosslinking studies of this complex with azido-phenacyl derivatized probes to map the contact sites between the RAG proteins, HMG1 derivatives and the RSS. We find that the RAG proteins make contacts at the nonamer, heptamer and adjacent coding region. The HMG1 protein by itself appears to localize at the 3′ side of the nonamer, but a cooperative complex with the RAG proteins is positioned at the 3′ side of the heptamer and adjacent spacer in the 12RSS. In the complex with RAG proteins, HMG1 is positioned primarily in the spacer of the 23RSS. We suggest that bends introduced into these DNA substrates at specific locations by the RAG proteins and HMG1 may help distinguish the 12RSS from the 23RSS and may therefore play an important role in the coordinated reaction.
INTRODUCTION
V(D)J recombination is the site-specific rearrangement of DNA in the developing immune system which assembles functional T-cell receptor or immunoglobulin genes from the arrays of inactive segments inherited in the germline (reviewed in 1,2). Each of the various segments, named V, D and J, are targeted for rearrangement by an adjacent recombination signal sequence (RSS). During the recombination reaction, a double-strand break is introduced into the DNA precisely between the RSS and its associated coding region. Since this process occurs at two segments at a time, there is an intermediate stage where two cleavage events yield four broken DNA ends. In the subsequent normal reaction, pairs of these ends are joined in a directed manner. The two DNA ends belonging to coding regions are joined to each other to form the ‘coding junction’. The two RSS-containing ends are also joined to each other to form the ‘signal junction’.
The RSS is composed of a conserved heptamer (CACAGTG) and nonamer (ACAAAAACC) motif, separated by a spacer of either 12 or 23 bp in length. The two sequences of differing length are called the 12RSS and 23RSS respectively. Within a chromosomal locus, similar segments generally carry RSS of the same length. A productive rearrangement in cells always occurs between pairs of DNA segments bordered by RSS elements of the two different spacer lengths (the 12/23 rule; 3). Owing to the 12/23 rule this organization permits a V segment, for example, to join to a D segment but not a second V segment.
The proteins RAG1 and RAG2 (4,5) have been shown to bind cooperatively to individual DNA molecules containing a single RSS (6–8) and bind simultaneously to DNA molecules in a manner that obeys the 12/23 rule (9–13). The cleavage reaction is aided by DNA bending proteins. Either HMG1 (14,15) or HMG2 (16) function in this capacity and a direct role for HMG1 in the binding step is one subject of this paper.
The HMG proteins are abundant and ubiquitous nuclear proteins found throughout eukaryotic evolution (for review see 17). HMG1 is present in chromatin at a ratio of 0.2–0.3 molecules per nucleosome. HMG1 and the closely related HMG2 contain two DNA binding domains known as HMG boxes which, in this case, do not exhibit any sequence specificity for binding but rather show a clear preference for distortions of the DNA helix such as occur at cruciforms or bulges. In addition, HMG1 has been shown to enhance the sequence-specific binding of several transcription factors in ways that include direct protein–protein interactions.
The individual roles for the proteins in each step of the recombination reaction will be needed to understand the complexity of sequence recognition, the restriction of the 12/23 rule, the coordinated cleavage at two sites, the continued binding to the liberated ends and subsequent reorganization of the ends to promote the directed ligation of certain pairs over others. In this study, we use electrophoretic gel mobility shift (EMSA) assays and UV crosslinking to localize the position of these proteins on RSS-containing DNA during the earliest binding step of the reaction. We show that HMG1 stimulates the assembly of a cleavage complex on individual oligonucleotides containing 12RSS or 23RSS and may be acting through protein–protein interactions with the RAG proteins in addition to contacts with DNA. The crosslinking studies confirm our previous observation that RAG1 contacts DNA at both the heptamer and nonamer (8) and extend it to include contacts in the proximal coding DNA as well. We find that RAG2 is closely associated with the DNA near each site of interaction with RAG1. Finally, crosslinking to derivatives of HMG1 indicate that this protein positions itself at the nonamer in the absence of the RAG proteins, and additionally localizes to the heptamer and spacer region at the 12RSS and predominantly in the spacer of the 23RSS in the presence of the RAG proteins, suggesting a bend induced in these regions of DNA upon interaction with the proteins.
MATERIALS AND METHODS
Proteins
Baculovirus stocks for MR1 and MR2 (18) were obtained from Martin Gellert (NIH). MR1 and MR2 are fusion proteins, each containing an N-terminal maltose binding protein followed by the functional core region of mouse RAG1 (residues 384–1008) or RAG2 (residues 1–387) respectively. The C-termini carry a poly-histidine tag followed by three tandem copies of the c-myc epitope tag as used previously (19). MR1 and MR2 proteins were expressed simultaneously, when used together, by coinfection of the SF9 insect cell line. Proteins were harvested after 66 h of infection and purified on NTA-agarose (Qiagen) charged with Ni2+ as described (20). Fractions containing the fusion proteins were pooled, and loaded onto amylose resin (New England Biolabs). The column was washed extensively with buffer A (20 mM Tris–HCl pH 7.4, 500 mM NaCl, 10 mM β-mercaptoethanol, 1 mM EDTA) containing 0.2% Tween-20, followed by elution in buffer A plus 10 mM maltose. Protein-containing fractions were pooled and dialyzed against buffer R (25 mM Tris–HCl pH 8.0, 150 mM KCl, 2 mM DTT, 10% glycerol) for 3 h. Aliquots were stored at –80°C.
Plasmids pDVG83 and pDVG84 were constructed by Dik van Gent (Erasmus University, The Netherlands) and used with permission though they have not been published previously. They each encode human HMG1 residues 1–163 which spans the two HMG boxes but does not include the acidic C-terminal tail. Plasmid pDVG83 carries an additional 13 amino acid residues at the C-terminus from the vector. Plasmid pDVG84 has an N-terminal GST fusion plus five C-terminal residues from the vector. Additional details are available upon request. Both proteins were expressed in Escherichia coli by induction of strain DH5α with IPTG (1 mM) for 3 h at 37°C. A cleared lysate in 20 mM Tris–HCl pH 7.5, 100 mM NaCl, 0.1% Triton X-100, 2 mM β-mercaptoethanol, 1 mM EDTA and 1 mM PMSF was prepared following freeze–thaw and sonication. The soluble fraction following precipitation to 40% saturation with ammonium sulfate was precipitated with trichloroacetic acid (4% final). The pellet was neutralized with NaOH and solubilized in 0.5 M NaCl and MOPS pH 7.7. The sample was diluted to 250 mM NaCl and subjected to mono-S chromatography with a gradient of NaCl. The protein elutes at ∼700 mM NaCl. Peak fractions were pooled and dialyzed into 20 mM Tris pH 7.5, 100 mM KCl, 10% glycerol, 2 mM β-mercaptoethanol, and stored at –80°C.
Oligonucleotides and probe preparation
Oligonucleotides were synthesized using a Perceptive Biosystems 8909 synthesizer. Oligonucleotides for EMSA probes were 5′-end labeled with [γ-32P]ATP (NEN) using T4 polynucleotide kinase (Amersham Pharmacia) as described by the provider. Probes were labeled on the sense strand (shown below), annealed to the complementary strand and gel purified.
The sequences of the oligonucleotide substrates used in Figures 1 and 2 are double stranded with only one shown below (significant changes are in bold).
Figure 1.
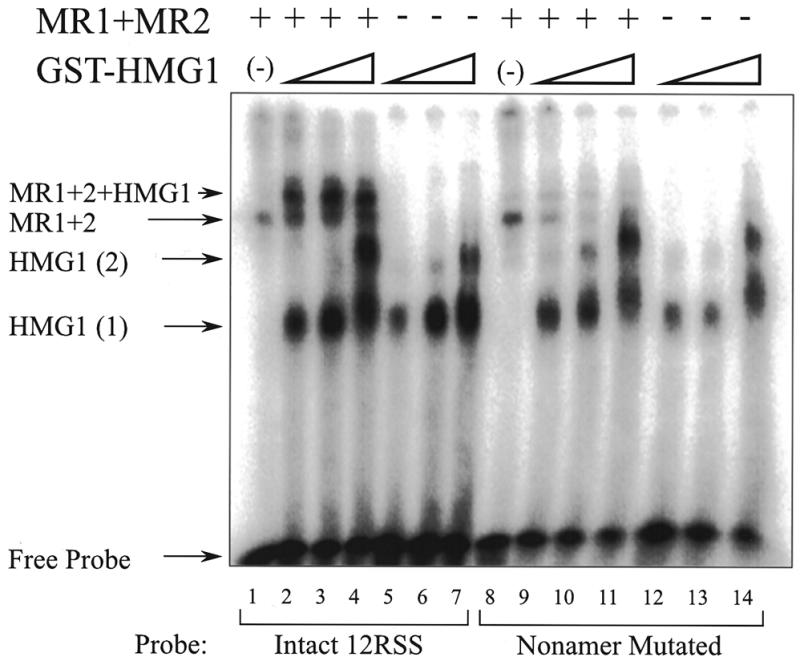
GST–HMG1 forms a complex with RAG1 and RAG2 on 12RSS DNA. This EMSA shows protein–DNA complexes resolved under native conditions in the absence of competitor DNA. Two probes are used: the 12RSS probe (lanes 1–7) and an equivalent probe in which the nonamer element has been mutated (lanes 8–14). The RAG proteins MR1 and MR2 are present in lanes marked with a plus sign at the top of the figure. GST–HMG1 is present in varying concentrations except in lanes 1 and 8. Complexes consisting of a single GST–HMG1 molecule per probe DNA or two molecules per DNA are indicated as HMG1 (1) and HMG1 (2) respectively. The complex formed by the RAG proteins without HMG1 is indicated as MR1+2. The complex containing all three proteins is marked as MR1+2+HMG1. The figure is a composite from two parallel gels.
Figure 2.
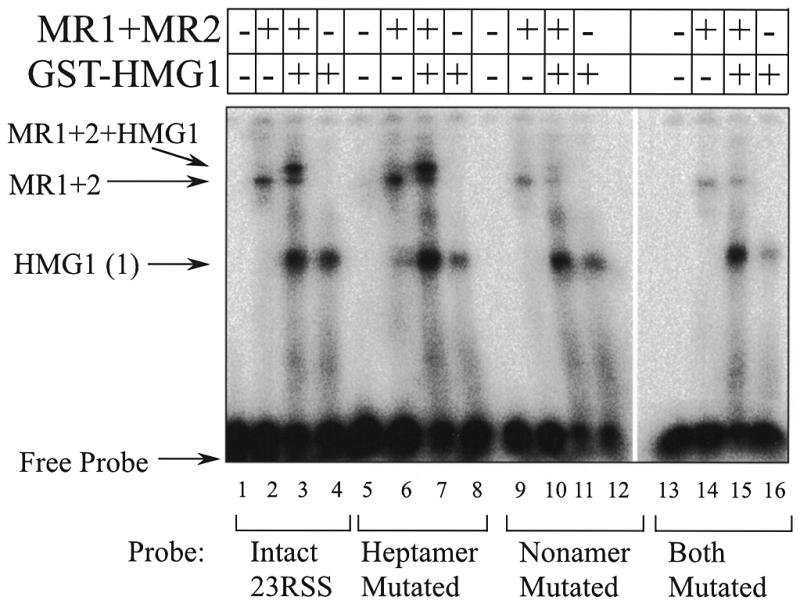
GST–HMG1 forms a complex with RAG1 and RAG2 on 23RSS DNA. This EMSA gel shows complexes formed on four related DNA probes with various combinations of RAG1 and RAG2 and GST–HMG1 proteins at a single concentration. The probes differ in the indicated regions. No competitor DNA was used. Identities of the complexes are marked as in the previous figure. The figure is a composite from two parallel gels.
12RSS substrates and mutants (sense strand shown):
intact 12RSS #245, 5′-CGGTCGACGTA CACAGTG CTTCCGGCTGGT ACAAAAACC CTCGTG-3′;
nonamer mutated 12RSS #32, 5′-CGGTCGACGTA CACAGTG CTTCCGGCTGGT TAGCTAGCT CTCGTG-3′.
23RSS and mutant substrates (sense strand shown):
intact 23RSS #36, 5′-GGGGATCCACTTA CACAGTG GTAGTACTCTGCTGTCTGGCTGT ACAAAAACC ACGCGT-3′;
mutated heptamer 23RSS #38, 5′-GGGGATCCACTTA TGGTTCC GTAGTACTCTGCTGTCTGGCTGT ACAAAA-ACC ACGCGT-3′;
mutated nonamer 23RSS #40, 5′-GGGGATCCACTTA CACAGTG GTAGTACTCTGCTGTCTGGCTGT TAGCT-AGCT ACGCGT-3′;
both motifs mutated 23RSS, 5′-GGGGATCCACTTA TGGT-TCC GTAGTACTCTGCTGTCTGGCTGT TAGCTAGCT ACGCGT-3′.
Oligonucleotides used for crosslinking were synthesized by replacing the oxidizing reagent with a sulfurizing reagent (Glen Research) at the one position where a phosphorothioate linkage was desired during synthesis of that strand. Positions are shown in Figure 3. Since synthesis proceeds from 3′ to 5′, probe number 18 would be sulfurized following the addition of the T base indicated by the number 18, creating the phosphorothioate linkage between that base and the next added T. Each probe is generated by annealing three oligonucleotides. The bottom oligonucleotide for the 12RSS is 5′-CTCGAGGG-TTTTTGTTCCAGTCTGTAGCACTGTGTAAGTCGACCTGCAGC-3′.
Figure 3.

UV crosslinking substrates. The top panel shows the 12RSS sequence. Probes are formed by annealing three oligonucleotides leaving a nick at the 5′ border of the heptamer. Individual probes are modified at a single position per probe at the locations shown by the numbers. The modified single phosphorothioate is 5′ to the indicated base pair. The bottom panel shows the equivalent 23RSS. Note that the same short oligonucleotides that form the coding region top strand (left of the heptamer) are used in both 12SS and 23RSS.
The bottom oligonucleotide for the 23RSS is 5′-CTC-GAGGGTTTTTGTACAGCCAGACAGTGGAGTAGTACC-ACTGTGTAAGTCGACCTGCAGC-3′. The common short oligonucleotide used for coding DNA top strand sequence is 5′-GCTGCAGGTCGACTTA-3′. The 12RSS sense top strand sequence is 5′-CACAGTGCTACAGACTGGAACAAAAACCC-TCGAG-3′. The 23RSS sense top strand sequence is 5′-CA-CAGTGGTACTACTCCACTGTCTGGCTGTACAAAAACCCTCGAG-3′.
Derivatized probes were prepared by 5′-end labeling the phosphorothioate-containing strand and annealing it to the remaining two. The resulting nicked double-stranded DNA was gel purified, eluted, precipitated and resuspended in 50 µl buffer TN (10 mM Tris–HCl pH 7.0, 30 mM NaCl). To this was added 4 µl of 1 M Tris–HCl pH 7.0, 36 µl methanol and 12 µl azido-phenacyl bromide (Sigma) (10 mM in methanol). The reaction was incubated in the dark for 3 h at room temperature, then the probe was purified from the reactants by passing it through a G-50 Sepharose spin column (5′–3′ Corp.) pre-equilibrated with buffer TN in the dark.
DNA binding assays
Binding mixture (typically 10 µl) contained 0.02–0.1 pmol 32P-labeled oligonucleotide substrate DNA in 25 mM MOPS pH 7.0, 5 mM MgCl2, 1 mM dithiothreitol, 50 µg/ml of bovine serum albumin and 50 mM KCl. Non-specific DNA pdIdC·pdIdC (Amersham Pharmacia) was added to 50 µg/ml final concentration when it was used. The coexpressed RAG protein was added in the range of 50 ng each per reaction. When used, HMG1 protein was added at 50 ng per reaction unless indicated otherwise. Typically, binding reactions were assembled on ice, with protein always the last added component and incubated at 37°C for 10 min. To each reaction, 4 µl of gel loading buffer dye (25% glycerol, 0.01% bromphenol blue and 0.01% xylene cyanol) was added, and samples were analyzed by electrophoresis through a polyacrylamide gel using a Tris–borate buffer system. Probes were detected by autoradiography and quantified using a Molecular Dynamics Phosphorimager and ImageQuant software (v2.1).
UV crosslinking
Binding reactions were assembled as above containing 106 c.p.m. of probe and incubated in the binding buffer for 10 min on ice. UV exposure for 1 min was performed using a 6 W UV lamp equipped with a 302 nm filter at a distance of 3 cm from the sample. The sample was supplemented with 1 µl denaturation cocktail (1% SDS, 1 M DTT, 10% glycerol) and boiled. SDS–PAGE was performed on continuous gels ranging from 6 to 10% acrylamide.
RESULTS
HMG1 increases the specificity of the RSS–protein complex
We have previously studied the binding of the RAG proteins to DNA oligonucleotides using truncated but functional forms of RAG1 and RAG2 (MR1 and MR2 respectively) (8,21). Each protein alone showed non-specific DNA binding, but these two proteins together can form a more specific complex containing both proteins. This was best observed by including non-specific DNA in the binding reaction. Others have shown that the DNA bending protein HMG1 can increase the yield of product in the cleavage reaction (14,16) and is necessary to assemble a paired complex containing both 12RSS and 23RSS (12). We first wished to determine whether HMG1 could participate in the DNA binding step prior to cleavage, as demonstrated by EMSA. Experiments testing the contributions of HMG1 and the DNA sequence of the 12RSS to the complexes that form with MR1 plus MR2 are shown in Figure 1. Lane 1 demonstrates DNA binding to the 12RSS probe by the copurified MR1 and MR2 proteins (∼50 ng of each per reaction). A single band, designated MR1+2, is obtained under these binding and gel conditions, which we have shown previously by excision and western blotting to contain both MR1 and MR2 proteins (8). A band of identical mobility is also obtained using the probe in which the nonamer motif has been fully mutated (lane 8). We have shown previously that this mutated probe forms a less specific association with the RAG proteins and can be competed more easily than the intact 12RSS probe. Lanes 5–7 and 12–14 show binding by a GST–HMG1 fusion protein at three concentrations (50, 100 and 150 ng) to the same two probes. A similar pattern is obtained on both probes. We see a single band at lower concentrations of GST–HMG1 and a second band at higher concentrations which we interpret as binding by one or two molecules of HMG1. No strong sequence preference is observed in comparing the binding between the two probes. Valid comparison is best obtained within a series assembled on one probe since the intensity difference seen between the two probes must be normalized for the difference in relative labeling, as reflected in the intensities of the free probe.
A difference in binding behavior using these two probes is obtained, however, when binding is studied with both RAG proteins and GST–HMG1 simultaneously. Lanes 2–4 show a new upper band that we interpret as a supershift of the MR1 plus MR2 band by the additional binding of GST–HMG1. The band is at maximal intensity in lane 2, and is not altered by further increasing the concentration of GST–HMG1. We have excised this new band, denatured the complex and have demonstrated that all three proteins are present (data not shown).
Notice that the intensity of the new supershifted band in lanes 2–5 is several fold higher than that seen in lane 1 in the absence of GST–HMG1. In contrast, the probe lacking the nonamer developed much less of the equivalent supershifted band. Moreover, the intensity of the MR1 plus MR2 band formed on this probe actually decreased with increasing GST–HMG1 (lanes 8–11). One possible explanation of this behavior is a cooperative interaction between HMG1 and the RAG proteins when the RAG complex is assembled on an intact RSS. This interaction would serve to stabilize the correctly formed specific complex. In contrast, the probe lacking the nonamer may not permit the assembly of the same complex, so that now the binding of HMG1 by itself competed with and displaced the non-specifically bound RAG1 plus RAG2 proteins. Effectively, the specificity of the RAG complex on the 12RSS is increased by the presence of HMG1, widening the difference in binding obtained between the two probes. Quantitation of the complexes seen with the intact 12RSS probe indicates an additional 3-fold yield of the specific supershifted complex in the presence of HMG1 protein. We employ the enhancement of binding in the crosslinking studies that follow.
We confirm that a similar behavior is obtained on the 23RSS. Figure 2 shows a series of EMSA experiments using four different DNA probes. For each probe, the four lanes show the probe alone, then the probe bound to combinations of the RAG proteins and GST–HMG1 (at 50 ng per reaction). Just as with the 12RSS probes above, we see that GST–HMG1 augments the binding of the RAG proteins to produce a band of slower mobility. The increased intensity of this band as seen with the intact 23RSS probe and the probe with fully mutated heptamer (Fig. 2, lanes 3 and 7 respectively), suggests that this complex is more stable than the complex formed with the RAG proteins alone. Also, as seen in Figure 1, probes lacking the nonamer or both the heptamer and nonamer (lanes 11 and 15 respectively) form this upper complex much more poorly. It therefore appears that the EMSA complex containing both RAG proteins and GST–HMG1 is more specific than the binding obtained with the RAG proteins alone. Previous binding studies using the RAG proteins alone (6–8) required competitor DNA to demonstrate reduced binding to probes bearing mutated RSS. Here, even in the absence of competitor, the upper supershifted complex does not form on non-specific probes. These results have also been repeated with HMG1 protein expressed without the GST fusion partner. The same behavior was obtained (not shown).
We also note a curious behavior. The band representing GST–HMG1 bound alone seems increased in all lanes containing the RAG proteins (lanes 3, 7, 11 and 15) compared to the parallel lanes containing the same probes and buffers but lacking the RAG proteins (lanes 4, 8, 12 and 16 respectively). Similar behavior was also seen in Figure 1 (most apparent comparing lane 2 with 5, and comparing lanes 9–11 with 12–14). One explanation could be that the RAG proteins cooperate in the loading of GST–HMG1 onto the DNA. If a transient complex were to form containing all the proteins but were to subsequently dissociate leaving GST–HMG1 on the DNA, the observed pattern would be obtained. The same pattern would be obtained whether the dissociation of the RAG proteins from the complex occurred in solution or during the EMSA analysis.
We have looked for direct evidence of protein–protein interaction between the GST–HMG1 protein and the RAG proteins by means of chromatography using GST–HMG1 immobilized on glutathione–Sepharose. MR1, and to a weaker extent MR2, were each retained indicating that direct interaction in the absence of DNA is likely (data not shown).
Mapping RAG protein contacts on RSS substrates
A key requirement for appreciating the three-dimensional organization of the recombination complex is a simpler two- dimensional map of the contacts that the proteins engaged in the reaction form on their target DNA. We have previously demonstrated that DNA probes containing single iodine-containing base analogs would form covalent linkages to RAG1 at positions in both the heptamer and nonamer (8). We did not detect contacts with RAG2 using those probes and we wished to continue mapping the DNA–protein contacts using a strategy originally developed to characterize restriction enzyme active sites (22). Rather than introducing modified DNA bases, here we create oligonucleotide substrates with a flexible crosslinking side chain introduced at individual positions on the phosphate backbone of the DNA. The side chain terminates in the azido (N3) moiety which can be photoactivated into a short lived reactive nitrene by exposure to UV light. The reactivity of this crosslinker is broad, and the flexible spacer allows contacts over an 11 Å radius complementing the properties of the previous study. A set of oligonucleotides was generated in which individual phosphates on one DNA strand were substituted with phosphorothioate linkages. The single sulfur atoms were subsequently derivatized in a separate step. The family of oligonucleotides is shown as Figure 3. A dot is shown at each position where a crosslinker was introduced. The phosphorothioate is actually positioned 5′ to each indicated base. Since the length of the crosslinker spacer arm is sufficient to extend well beyond the 3.4 Å that separate adjacent base pairs (in B-form DNA), this set provides sufficient coverage to adequately sample the space surrounding the entire 12RSS and into the adjacent coding DNA shown to the left of the heptamer. A subset of the most interesting crosslinking sites was similarly constructed for the 23RSS. Individual probes were assembled from three oligonucleotides. The longer bottom strand (as indicated in Fig. 3) was not radioactively labeled, while one of the two top strands was 5′-end labeled with 32P. The structure leaves a nick at the position preceding the heptamer. These nicked substrates have previously been shown to represent reaction intermediates in V(D)J recombination, which makes them ideal for studies of the reaction mechanism. This scheme also allowed us to utilize the same shorter top strand for both 12RSS and 23RSS probes.
It is important to determine whether these modified probes behave equivalently or, conversely, whether the modification at certain positions interferes with the formation of certain complexes. Binding of the RAG proteins using conditions that allow non-specific binding (in the absence of HMG1) showed that the different probes formed complexes (as judged by EMSA) and crosslinked equally well to the RAG proteins regardless of the location of the active group. MR1 consistently gave stronger signals than MR2 (data not shown). Similarly, GST–HMG1 alone also formed EMSA complexes comparably among each of the probes. A representative sampling of probes across the 12RSS in EMSA with GST–HMG1 is presented in Figure 4A. Most of the probes shown are bound similarly although probe 15 yielded slightly weaker bands which may reflect some interference in binding when the crosslinking adduct is positioned at that phosphate. This is consistent with additional observations presented later.
Figure 4.
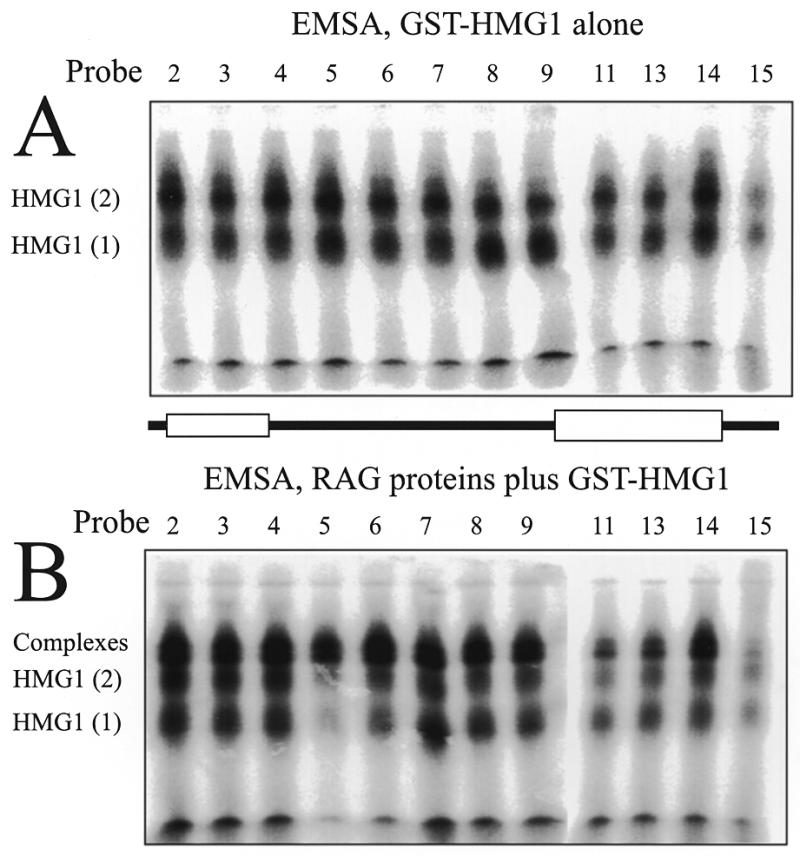
EMSA shows interference by the crosslinking adduct at certain positions in formation of specific complexes. Probes numbered as in Figure 3 are compared by EMSA in ability to form the complexes with GST–HMG1 (A) or GST–HMG1 plus the RAG proteins MR1 and MR2 (B). Each image was assembled from two wet gel autoradiograms. See text for analysis.
In contrast, when the RAG proteins and GST–HMG1 were bound to the probes to form the specific complexes, the EMSA complex containing all three proteins no longer behaves uniformly over the 12RSS. Figure 4B shows the EMSA complexes obtained with the set of indicated probes. The differences obtained in this analysis compared to Figure 4A appear to represent a degree of interference in the assembly of the specific complex by the crosslinking adduct at certain positions. Consideration of the data in Figure 4B as well as additional data (not shown) shows that positioning the crosslinker at the 5′ side of the nonamer provoked the strongest interference. Probes 10–13 exhibited specific binding in EMSA at levels of 35–70% of the spacer region probes. Intermediate levels of interference were also seen with probes 2–5 derivatized at positions in the heptamer. These data confirm ethylation interference data that showed similar effects (6). The interference demonstrates the tradeoff in the requirements for placing a crosslinker on the DNA. One desires the crosslinker to be placed as closely as possible to the position of protein contact without actually disturbing the interaction.
Crosslinking within the specific complex assembled with MR1, MR2 and the GST–HMG1 protein was explored with the set of 12RSS probes (Fig. 5A). This figure shows the SDS–PAGE analysis of crosslinked products obtained with complexes assembled in solution, exposed to UV light and denatured. Under these experimental conditions, the majority of complexes containing the RAG proteins are specifically bound whereas the GST–HMG1 protein is present in both specific and non-specific complexes. Consequently, in this panel only the positions of the RAG proteins can be considered meaningful. Within the RSS, strong crosslinking of MR1 was obtained in the heptamer (probes 2 and 3) and nonamer (probes 10–13). Additional crosslinking was also seen with the probes adjacent to those motifs; probe 4, located just 3′ to the heptamer, and probe 9, located 5′ to the nonamer with respect to the top strand (Fig. 3). Equally striking is the strong crosslinking obtained by probe 18, located two bases 5′ to the cleavage site in the coding DNA. It is evident that MR2 also crosslinks to these probes at most of the positions where MR1 is detected. A clear MR2 band is seen in the coding DNA (probe 18) in and near the heptamer (probes 2–4) and similarly in or near the nonamer (probes 9–13). Contacts with the RAG proteins are largely excluded from the spacer region.
Figure 5.

UV crosslinking of the RAG proteins to 12RSS and 23RSS probes under specific binding conditions. (A) 12RSS probes were assembled into complexes in the presence of MR1, MR2 and GST–HMG1 and crosslinked in solution. Complexes were then analyzed by SDS–PAGE. No correction for interference in complex assembly was applied to this image. (B) Selected 23RSS probes were used as above. Probe 18 is located in the 23RSS coding DNA, 21 in the heptamer and 24 in the nonamer.
We next repeated crosslinking experiments using key probes built into the context of the 23RSS. The major findings obtained with the 12RSS probes were repeated here (Fig. 5B). Contacts with MR1 and MR2 were found in the coding DNA and in the heptamer (probes 18 and 21 respectively). Contacts primarily with MR1 were also found in the nonamer (probe 24). As witnessed with the 12RSS, no contacts were found in the spacer region (not shown).
A more technically demanding procedure was required to address the position of the GST–HMG1 protein within the specific complex. As above, binding reactions containing the RAG proteins and GST–HMG1 were assembled on various probes and irradiated with UV light in solution. The products were separated by EMSA under native conditions and the positions of the various complexes were determined by autoradiography of the wet gel (presented as Fig. 4). Two slices were excised representing the gel-shift of the GST–HMG1 alone (monomer band of Fig. 4A) or the specific complex containing MR1, MR2 and GST–HMG1 (uppermost complex in Fig. 4B). These slices were soaked in SDS and heated to denature the complex. Each slice was finally incorporated into the stacking portion of an SDS–PAGE gel and the location of the GST–HMG1 protein determined after running the second gel. Figure 6A shows the result of this analysis applied to the GST–HMG1 (alone) protein complex. A clear positioning of the GST–HMG1 protein at the 3′ side of the nonamer is observed (probe 15).
Figure 6.
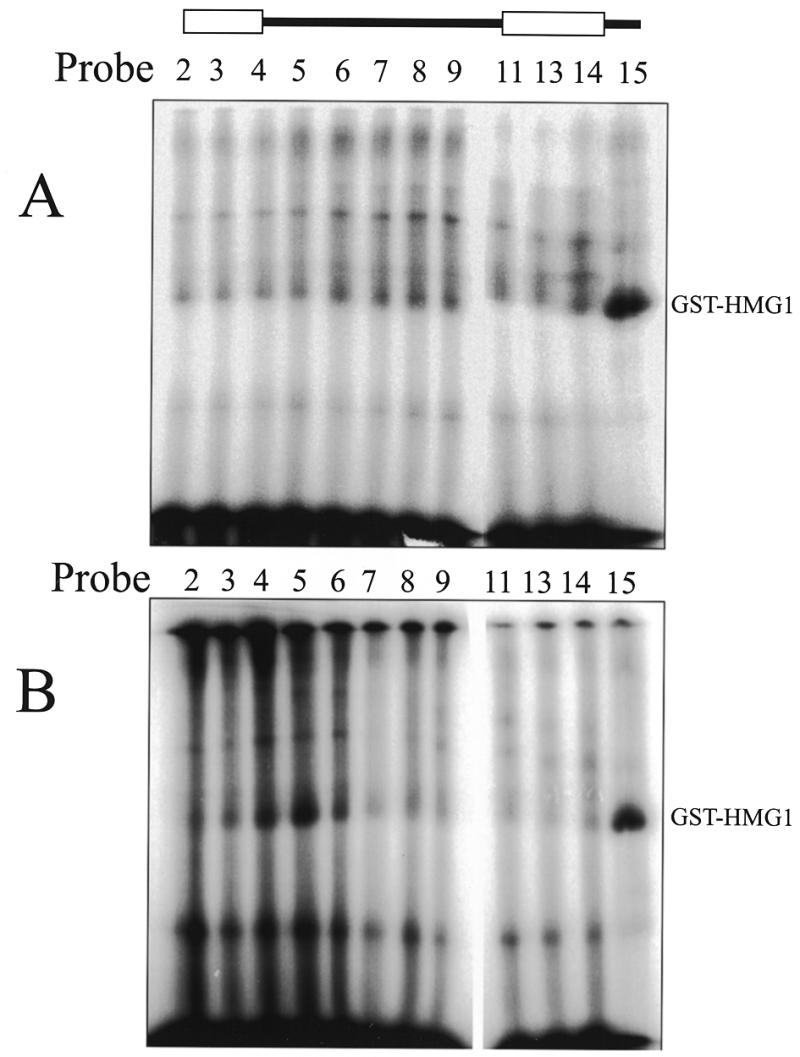
UV crosslinking of GST–HMG1 to 12RSS probes. Binding reactions containing RAG proteins and GST–HMG1 were crosslinked then separated by EMSA. (A) Native complexes containing only GST–HMG1 were excised from the wet gel of Figure 4A, denatured and analyzed by SDS–PAGE. Preferred binding is found using probe 15. (B) Native complexes containing RAG proteins and GST–HMG1 were excised from the highest complex of the wet gel presented as Figure 4B, denatured and analyzed by SDS–PAGE. New contacts with GST–HMG1 appear with probes 4–6. Both panels were assembled from parallel gels. The drawing at the top shows the heptamer and nonamer as open boxes.
When the equivalent analysis was performed on the complex containing the RAG proteins with GST–HMG1 (Fig. 6B) additional positioning was found just 3′ to the heptamer (probes 4 and 5). We note that unfortunately, the RAG proteins themselves did not enter the gel, but rather remained trapped at the interface with the resolving gel perhaps indicating that the samples were not fully denatured.
We were alerted to the disturbing fact that GST protein as a fusion partner possesses an unfortunate intrinsic ability to dimerize (see Discussion). It is not certain whether this behavior would alter the localization of HMG1 in the complex but we repeated the crosslinking experiments using HMG1 protein expressed without the GST fusion partner. The results of crosslinking the HMG1 to 12RSS or 23RSS probes within the cleavage complex containing both RAG proteins and the HMG1 protein are presented in Figure 7. As previously, 12RSS probes show localization of HMG1 to the 3′ side of the nonamer (probe 14 maximally in this case) as well as toward the 3′ side of the heptamer and the adjacent spacer (probes 3–6). The 23RSS probes showed significant enhancement of HMG1 localization only within the spacer region (probes 33, 34 and 22).
Figure 7.
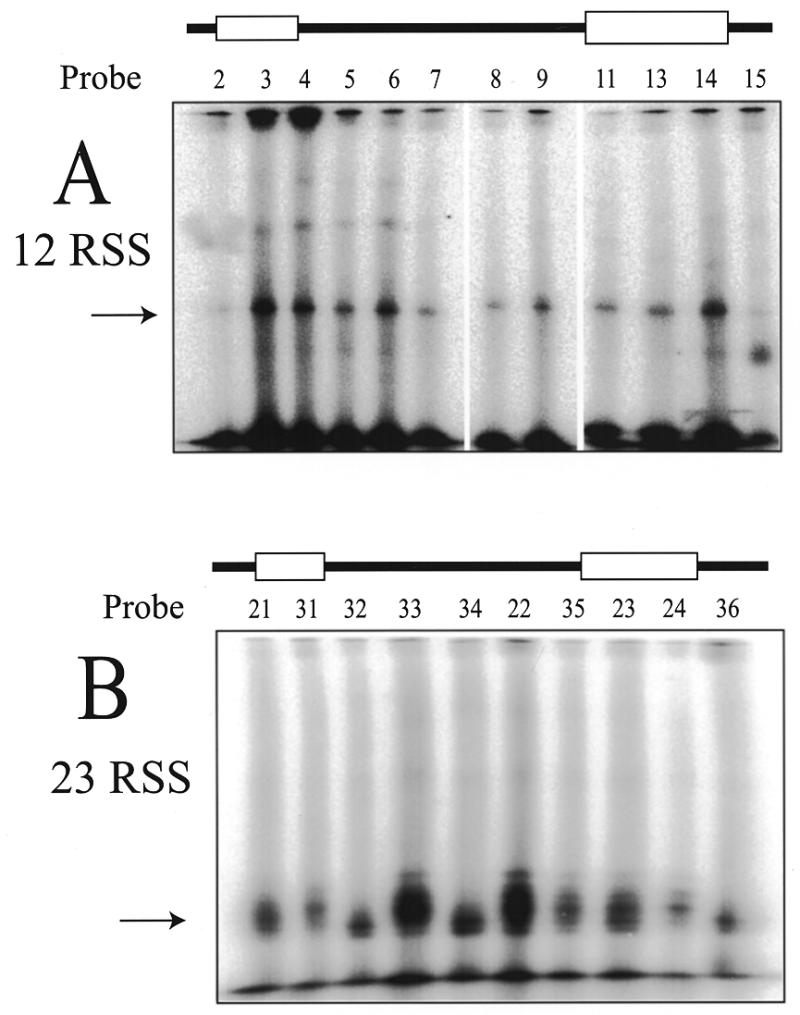
UV crosslinking of HMG1 (lacking the GST fusion partner) in complexes containing the RAG proteins. The native complexes containing the RAG proteins and HMG1 were prepared as in Figure 6 using 12RSS probes (A) or 23RSS probes (B). Subsequent SDS–PAGE demonstrates which probes make preferred contacts with HMG1 (arrow). 12RSS (A) results are similar to Figure 6B. 23RSS (B) results show contacts primarily in the spacer. The drawing at the top of each panel shows the heptamer and nonamer as open boxes.
DISCUSSION
HMG1 stabilizes the RAG protein complex on DNA
We and others have shown previously that the core regions of RAG1 and RAG2 (here expressed as maltose binding protein fusions) are together capable of binding DNA with specificity for the RSS (6–8). In our previous work we have demonstrated that specificity by competition with non-specific DNA or DNA containing mutated RSS sequences. The competition was essential because in its absence we have shown previously that the RAG proteins have a significant ability to bind DNA non-specifically. This observation is repeated here in Figure 1. An intact 12RSS probe (lane 1) and a probe lacking the nonamer (lane 8) are each capable of forming a complex with the RAG proteins in the absence of competition. Since it is known that the presence of HMG1 (or the closely related HMG2) stimulates the cleavage reaction, we wished to explore the role HMG1 could play in the initial assembly of the protein–DNA complexes prior to cleavage. In Figures 1 and 2 we show that HMG1 (in this case as a GST fusion protein) increases the specificity of the RAG proteins for both 12RSS and 23RSS substrates over that which they exhibit without HMG1. These binding experiments are conducted in the absence of competitor DNA. Not only does a new complex of slower mobility form that appears to contain all three proteins, but this complex shows a greater sequence preference for the intact RSS elements than RAG1 plus RAG2 show without HMG1. Furthermore the stability of this complex appears to be increased, judging from the increase in intensity of the signal representing the triple protein complex over that of the RAG1 plus RAG2 species. In fact, the binding of the RAG1 plus RAG2 protein to a DNA substrate mutated in the nonamer sequence actually decreases as HMG1 is titrated into the binding reaction (Fig. 1, lanes 8–11). The effect of HMG1 in this case is similar to the addition of non-specific competitor DNA. We interpret the result to indicate that here, HMG1 is binding to the substrate and is displacing the RAG proteins which are bound through non-specific interactions. In the presence of the intact nonamer, the RAG proteins and HMG1 appear to form a more specific and more stable cooperative complex. This increase in specificity may simply imply an energetically favorable conformation of the proteins on DNA without direct protein–protein interactions. However direct protein interactions involving HMG1 have been noted with other proteins.
Crosslinking suggests particular bends in the RSS DNA
HMG1 is a member of a set of highly conserved, ubiquitous nuclear proteins (17 and references therein). HMG1 and the closely related HMG2 are non-specific with regard to sequence in binding DNA but show a clear preference for binding angled structures (23,24). We anticipate that the nonamer element in each RSS would induce a DNA bend through its internal poly-adenylate tract. Such sequences are known to create a sequence-directed curvature of DNA (25), and as such may represent preferred sites of interactions with DNA-bending proteins. In fact, consistent with this expectation, preferred positioning of the GST–HMG1 protein at the 3′ end of the 12RSS nonamer was demonstrated (Fig. 6A) in the absence of the RAG proteins. In the complex containing RAG proteins and GST–HMG1 on 12RSS probes, the latter protein made close contacts with the DNA both at the previous site 3′ to the nonamer and within the heptamer and spacer (Fig. 6B). This may reflect the bending already proposed to occur at the heptamer (6,7). We interpret the bends at the heptamer and nonamer along with the reduced contacts obtained to the RAG proteins in the spacer region to reflect a looping of the spacer region away from the protein complex containing the heptamer and nonamer. The 23RSS probes showed a different pattern. The strongest sites of crosslinking to HMG1 were found in the spacer region. We find it interesting that the strongest sites of interaction (probes 33 and 22) are located 10 bp apart. The intervening probe 34 as well as additional flanking probes in the spacer gave much weaker signals. This is consistent with the suggestion that the spacer region is looped out of the protein complex with a particular sequence phasing. In this interpretation, probes 33 and 22 would orient their crosslinking adduct on the outer face of the loop while probe 34 would be half a helical turn away with crosslinker oriented toward the inside of the loop where it would be less likely to interact with HMG1. A recent study indicates that HMG1 or HMG2 binding at the RSS seems to bend associated DNA as measured by enhancement of ligation of DNA to form circular products (26).
Possible direct interaction between RAG proteins, HMG1 and chromatin
While the HMG1/2 class of proteins is commonly considered as acting through structural effects on chromatin, specific effects are obtained in some systems that imply a direct interaction with certain proteins. A HMG1 homolog was recently isolated in Caenorhabditis elegans, which is necessary for normal signaling by the Wnt pathway (27). Furthermore, direct protein–protein interactions have been reported between HMG1 or HMG2 proteins and other DNA binding proteins, usually transcription factors, resulting in an increase in the stability or specificity of the resulting complex on DNA. HMG1 has been shown to interact directly with TBP (28,29), the POU domains of Oct2 and HOXD9 (30,31), with p53 (32) and with steroid hormone receptors (33). A pull-down assay detected the HMG1 interactions with several of these proteins. The interaction with p53 was detected by a nitrocellulose membrane blotting technique. Direct protein–protein interaction between RAG1 and HMG1 or HMG2 can be detected (26). However, interaction with HMG1 itself may be less important than the structural consequences imposed on the complex by any of the HMG1-like family since knock-out of HMG1 is still compatible with V(D)J recombination (34).
HMG1 can enhance the cleavage reaction mediated by the RAG proteins in vitro (14–16). These investigators have reported that isolated cleavage at the 23RSS as well as coupled cleavage using DNA substrates containing both 12RSS and 23RSS is stimulated by the DNA bending protein, and suggest that a complex containing HMG1 may form on the 23RSS. Using EMSA we have shown that complexes containing both RAG proteins and HMG1 assemble on both 12RSS and 23RSS. Recent experiments have explored the efficiency of the RAG-mediated cleavage reaction of DNA assembled into nucleosomal structures (35,36). These studies differ in the details of their conclusions but both demonstrate that a positioned nucleosome can inhibit the cleavage reaction. If the loops that we propose to form in the spacer regions are important to the mechanism, they may require that nucleosomal structure over the RSS be dismantled.
Crosslinking to the RAG proteins confirms and extends contact information
We mapped the contact sites between DNA, the RAG proteins and HMG1 in these complexes by using a set of 12RSS probes that sample the space surrounding the entire RSS and at least 7 bp into the coding DNA adjacent to the heptamer (Fig. 3). Our probes are made by substituting the azido-phenacyl moiety at individual positions along the phosphate backbone of one strand of DNA. In principle, the rather bulky adduct may interfere with DNA binding that makes contacts at the substituted phosphate. This effect was observed in Figure 4B which shows that equivalent probes were not equal in their ability to form EMSA bands under conditions that support specific binding. When the crosslinker was placed in the spacer region, the strongest EMSA bands were detected, equivalent to unsubstituted probes. Placing the crosslinker in the nonamer had the strongest interfering effect but interference was also seen with probes substituted in the heptamer region. As such, these studies confirm the patterns obtained by ethylation interference by others (6). Despite this partial interference at certain positions, crosslinks were obtained upon UV illumination of the azido-modified probes bound to protein. Figure 5A and B shows the RAG protein–DNA crosslinked products obtained from 12RSS and 23RSS probes analyzed by SDS–PAGE without any correction for the interference effects. If such correction was applied it would only further enhance the clear difference in crosslinking obtained between the conserved motifs, which crosslinked well, and the spacer region where essentially no crosslinking was found. It is clear that RAG1 makes contacts at both the heptamer and nonamer, as we have already reported using a complementary crosslinking strategy (8). It is also clear that RAG2 is generally found to crosslink where RAG1 is detected, albeit with varying intensity. It is fair to conclude that RAG2 must approach the DNA within the radius of the crosslinker (11 Å) in the heptamer, the adjacent coding DNA, and also perhaps to a lesser degree, near the nonamer. This may not be a surprising conclusion based on our previous report that RAG1 forms a stable complex with RAG2 even in the absence of DNA. A strong crosslink of both RAG1 and RAG2 within the coding DNA at the border with the heptamer confirms our observation that the RAG complex makes contacts in the coding region and was capable of retaining the coding DNA in the complex following cleavage (21). This establishes at the biochemical level an observation made in cells that implicated RAG1 in forming contacts with the 2 bp of coding DNA adjacent to the heptamer (37). It is also consistent with the role of RAG2 in the initial cleavage reaction and in opening the hairpins later in the reaction pathway (38).
Obtaining the equivalent localization for the HMG1 protein in the complex required a more complicated experimental protocol. The EMSA band corresponding to the complex of interest was first separated under native conditions, disassembled and analyzed in a second gel. We find that the GST–HMG1 protein alone localizes 3′ to the nonamer in the 12RSS (Fig. 6A), but that the complex containing both the RAG proteins and GST–HMG1 now additionally positions GST–HMG1 in the heptamer and adjacent spacer region (Fig. 6B).
During the course of these experiments we became concerned that the use of the GST-fusion partner could alter the DNA binding properties of HMG1. This consequence has been seen in other systems (39). We therefore repeated the DNA binding experiments (not shown) and the crosslinking experiments (Fig. 7) using HMG1 expressed without the fusion partner. We found that the behaviors originally observed remained true. The non-fusion HMG1 still supershifted and enhanced the stability of the EMSA complex with the RAG proteins. Positioning of the HMG1 protein on the 12RSS crosslinking probes yielded similar results to those obtained with GST–HMG1. Crosslinking to the 23RSS probes showed predominant localization within the spacer region. The difference in behavior between the 12RSS and 23RSS with regard to HMG1 localization may reflect spatial differences important to the structure of the reaction.
We suspect that DNA bending plays a role in the assembly of complexes on both types of RSS, and that differential bending helps distinguish the 12RSS complex from the 23RSS.
Recently, results using the same chemical crosslinking approach to map contacts with the RAG proteins on DNA have shown similar contacts to those we have obtained (40). The positioning of the RAG proteins in the DNA complex has also been explored by UV crosslinking DNA substrates containing iodinated base analogs (41). Our experiments differ in a few ways. Our complexes were formed in the presence of HMG1 or GST–HMG1 which could have changed the positioning of the RAG proteins. Our DNA substrates also differ slightly in containing a nick at the 5′ position of the heptamer. As such, our pre-nicked substrates mimic a reaction intermediate slightly later in the reaction course than fully double-stranded DNA. The results for positioning of the RAG proteins turn out to be quite similar.
In this report we focus on the DNA–protein complex that assembles prior to cleavage in V(D)J recombination. We find a direct role for the HMG1 protein in the DNA binding step that appears to reflect a cooperative interaction with the RAG proteins. We also map the contact sites between the RAG proteins, HMG1 derivatives and the RSS. We find that the RAG proteins make contacts at the nonamer, heptamer and adjacent coding region. The HMG1 protein by itself appears to localize at the 3′ side of the nonamer, but in a cooperative complex with the RAG proteins, is positioned at the 3′ side of the heptamer and adjacent spacer in the 12RSS. In the complex with RAG protein, HMG1 is positioned primarily in the spacer of the 23RSS. The bends introduced into these DNA substrates at specific locations may help distinguish the 12RSS from the 23RSS and may therefore play an important role in the coordinated reaction.
Acknowledgments
ACKNOWLEDGEMENTS
The oligonucleotides were prepared in our Molecular Biology Core Facility. We would like to thank John Nechtman. We are grateful to Dik van Gent for sharing with us the HMG1 expressing plasmids. The phasing of the 23RSS loop was suggested to us by Stephen Desiderio. This work was funded by NIH grant AI41711. M.S. is a Scholar of the Leukemia Society of America.
REFERENCES
- 1.Lewis S.M. (1994) Adv. Immunol., 56, 27–150. [DOI] [PubMed] [Google Scholar]
- 2.Gellert M. (1997) Adv. Immunol., 64, 39–64. [DOI] [PubMed] [Google Scholar]
- 3.Tonegawa S. (1983) Nature, 302, 575–581. [DOI] [PubMed] [Google Scholar]
- 4.Schatz D.G., Oettinger,M.A. and Baltimore,D. (1989) Cell, 59, 1035–1048. [DOI] [PubMed] [Google Scholar]
- 5.Oettinger M.A., Schatz,D.G., Gorka,C. and Baltimore,D. (1990) Science, 248, 1517–1523. [DOI] [PubMed] [Google Scholar]
- 6.Swanson P.C. and Desiderio,S. (1998) Immunity, 9, 115–125. [DOI] [PubMed] [Google Scholar]
- 7.Akamatsu Y. and Oettinger,M.A. (1998) Mol. Cell. Biol., 18, 4670–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mo X., Bailin,T. and Sadofsky,M.J. (1999) J. Biol. Chem., 274, 7025–7032. [DOI] [PubMed] [Google Scholar]
- 9.van Gent D.C., Ramsden,D.A. and Gellert,M. (1996) Cell, 85, 107–113. [DOI] [PubMed] [Google Scholar]
- 10.Eastman Q.M., Leu,T.M.J. and Schatz,D.G. (1996) Nature, 380, 85–88. [DOI] [PubMed] [Google Scholar]
- 11.Hiom K. and Gellert,M. (1997) Cell, 88, 65–72. [DOI] [PubMed] [Google Scholar]
- 12.Hiom K. and Gellert,M. (1998) Mol. Cell, 1, 1011–1019. [DOI] [PubMed] [Google Scholar]
- 13.West R.B. and Lieber,M.R. (1998) Mol. Cell. Biol., 18, 6408–6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Gent D.C., Hiom,K., Paull,T.T. and Gellert,M. (1997) EMBO J., 16, 2665–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim D.R. and Oettinger,M.A. (1998) Mol. Cell. Biol., 18, 4679–4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawchuk D.J., Weis-Garcia,F., Malik,S., Besmer,E., Bustin,M., Nussenzweig,M.C. and Cortes,P. (1997) J. Exp. Med., 185, 2025–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bustin M. and Reeves,R. (1996) Prog. Nucleic Acid Res. Mol. Biol., 54, 35–100. [DOI] [PubMed] [Google Scholar]
- 18.McBlane J.F., van Gent,D.C., Ramsden,D.A., Romeo,C., Cuomo,C.A., Gellert,M. and Oettinger,M.A. (1995) Cell, 83, 387–395. [DOI] [PubMed] [Google Scholar]
- 19.Sadofsky M.J., Hesse,J.E., McBlane,J.F. and Gellert,M. (1993) Nucleic Acids Res., 21, 5644–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Gent D.C., McBlane,J.F., Ramsden,D.A., Sadofsky,M.J., Hesse,J.E. and Gellert,M. (1995) Cell, 81, 925–934. [DOI] [PubMed] [Google Scholar]
- 21.Bailin T., Mo,X. and Sadofsky,M.J. (1999) Mol. Cell. Biol., 19, 4664–4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayer A.N. and Barany,F. (1995) Gene, 153, 1–8. [DOI] [PubMed] [Google Scholar]
- 23.Bianchi M.E., Beltrame,M. and Paonessa,G. (1989) Science, 243, 1056–1059. [DOI] [PubMed] [Google Scholar]
- 24.Locker D., Decoville,M., Maurizot,J.C., Bianchi,M.E. and Leng,M. (1995) J. Mol. Biol., 246, 243–247. [DOI] [PubMed] [Google Scholar]
- 25.Hagerman P.J. (1990) Annu. Rev. Biochem., 59, 755–781. [DOI] [PubMed] [Google Scholar]
- 26.Aidinis V., Bonaldi,T., Beltrame,M., Santagata,S., Bianchi,M.E. and Spanopoulou,E. (1999) Mol. Cell. Biol., 19, 6532–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang L.I. and Sternberg,P.W. (1999) Genes Dev., 13, 877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ge H. and Roeder,R.G. (1994) J. Biol. Chem., 269, 17136–17140. [PubMed] [Google Scholar]
- 29.Sutrias-Grau M., Bianchi,M.E. and Bernues,J. (1999) J. Biol. Chem., 274, 1628–1634. [DOI] [PubMed] [Google Scholar]
- 30.Zwilling S., Konig,H. and Wirth,T. (1995) EMBO J., 14, 1198–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zappavigna V., Falciola,L., Citterich,M.H., Mavilio,F. and Bianchi,M.E. (1996) EMBO J., 15, 4981–4991. [PMC free article] [PubMed] [Google Scholar]
- 32.Jayaraman L., Moorthy,N.C., Murthy,K.G., Manley,J.L., Bustin,M. and Prives,C. (1998) Genes Dev., 12, 462–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boonyaratanakornkit V., Melvin,V., Prendergast,P., Altmann,M., Ronfani,L., Bianchi,M.E., Taraseviciene,L., Nordeen,S.K., Allegretto,E.A. and Edwards,D.P. (1998) Mol. Cell. Biol., 18, 4471–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calogero S., Grassi,F., Aguzzi,A., Voigtlander,T., Ferrier,P., Ferrari,S. and Bianchi,M.E. (1999) Nature Genet., 22, 276–280. [DOI] [PubMed] [Google Scholar]
- 35.Golding A., Chandler,S., Ballestar,E., Wolffe,A.P. and Schlissel,M.S. (1999) EMBO J., 18, 3712–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwon J., Imbalzano,A.N., Matthews,A. and Oettinger,M.A. (1998) Mol. Cell, 2, 829–839. [DOI] [PubMed] [Google Scholar]
- 37.Sadofsky M.J., Hesse,J.E., van Gent,D.C. and Gellert,M. (1995) Genes Dev., 9, 2193–2199. [DOI] [PubMed] [Google Scholar]
- 38.Besmer E., Mansilla-Soto,J., Cassard,S., Sawchuk,D.J., Brown,G., Sadofsky,M., Lewis,S.M., Nussenzweig,M.C. and Cortes,P. (1998) Mol. Cell, 2, 817–828. [DOI] [PubMed] [Google Scholar]
- 39.Niedziela-Majka A., Rymarczyk,G., Kochman,M. and Ozyhar,A. (1998) Prot. Expr. Purif., 14, 208–220. [DOI] [PubMed] [Google Scholar]
- 40.Swanson P.C. and Desiderio,S. (1999) Mol. Cell. Biol., 19, 3674–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eastman Q.M., Villey,I.J. and Schatz,D.G. (1999) Mol. Cell. Biol., 19, 3788–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]