


Abstract
We have previously identified a mitochondrial Y-box protein in Trypanosoma brucei that we designated RBP16. The predicted RBP16 amino acid sequence revealed the presence of a cold-shock domain at its N-terminus and a glycine- and arginine-rich C-terminus reminiscent of an RGG RNA-binding motif. Since RBP16 is capable of interacting with different guide RNAs (gRNAs) in vitro and in vivo primarily via the oligo(U) tail, as well as with ribosomal RNAs, possible functions of RBP16 may be in kinetoplastid RNA editing and/or translation. Herein, we report experiments that further define the RNA-binding properties of RBP16. RBP16 forms a single stable complex with the gRNA gA6[14] at low protein concentration, while at higher protein concentration two stable complexes that possibly represent two different conformations are observed. Both complexes are stable at relatively high salt and moderate heparin concentrations indicating that the binding of RBP16 to gA6[14] does not rely primarily on ionic interactions. Phenylglyoxal treatment of the protein indicates that arginine residues are important in RNA binding. The minimal length of RNA sequence necessary for the binding of RBP16 was assessed by gel retardation and UV cross-linking competition assays using oligo(U) ribonucleotides of varying lengths (4–40 nt). Although RBP16 can bind to oligonucleotides as small as U4, its affinity increases with the length of the oligo(U) ribonucleotide, with a dramatic increase in binding efficiency observed when the length is increased to 10 nt. Gel retardation assays employing T.brucei mRNAs demonstrated that, although it acts as a major binding determinant, a 3′ U tail is not an absolute requirement for efficient RBP16–RNA binding. Experiments with oligonucleotides containing U stretches embedded at different positions in oligo(dC) indicated that high-affinity binding requires both a uridine stretch, as well as 5′ and 3′ non-specific sequences. These results suggest a model for the molecular interactions involved in RBP16–RNA binding.
INTRODUCTION
We have previously reported the identification of a gene encoding a mitochondrial 16 kDa Y-box protein from the parasitic protozoan Trypanosoma brucei (1). We designated this protein RBP16 (RNA-binding protein of 16 kDa). Analysis of the predicted amino acid sequence of RBP16 revealed the presence of a cold-shock domain (CSD) at its N-terminus, similar to bacterial cold-shock proteins and to eukaryotic Y-box proteins (1). In bacteria, the CSD constitutes the sole component of the cold-shock family of proteins. Some members of this family, such as the cold-shock proteins CspA and CspB, have been demonstrated to bind RNA with low affinity and broad specificity (2,3). The eukaryotic Y-box proteins are composed of a conserved N-terminal CSD flanked by variable C-terminal domains. The latter include the basic and acidic islands present in several vertebrate Y-box proteins (4), the zinc finger motif in Lin-28 from Caenorhabditis elegans (5) and the RGG motif in the Drosophila YPS protein (6). Although the function of many Y-box proteins remains unclear, some have been demonstrated to be involved in the regulation of gene expression at the transcriptional as well as post-transcriptional level. For example, one of the best studied Y-box proteins is Xenopus laevis FRGY2, which functions in translational repression of mRNA in the oocyte. FRGY2 exhibits sequence-specific recognition of RNA mediated by its CSD (7). Another CSD-containing protein, the human Unr protein which is possibly involved in the initiation of translation of cellular mRNAs, also binds RNA through sequence-specific interactions (8).
In addition to its N-terminal CSD, the T.brucei Y-box protein RBP16 contains a C-terminal domain rich in glycine and arginine residues, resembling the RGG RNA-binding motif (1,8). In this respect, it appears to be a primitive member of the invertebrate class of Y-box proteins containing auxiliary RGG motifs (9). The RGG motif is defined as closely spaced Arg–Gly–Gly repeats separated by other, often aromatic, amino acids (10). It is usually present in combination with other RNA-binding motifs. RGG-mediated binding has been shown, in many cases, to be non-specific (11,12). This motif is thought to function by increasing the overall RNA affinity of proteins containing additional RNA-binding domains (10). However, in some cases, RGG motifs have been shown to confer sequence-specific binding (13).
Previous experiments from this laboratory showed that RBP16 binds U- and G-containing RNA homopolymers, but cannot bind efficiently to RNA polymers composed of C or A (1). Moreover, RBP16 binds both single-stranded RNA and DNA. Further experiments confirmed that RBP16 forms multiple, stable complexes with small RNA molecules known as guide RNAs (gRNAs) in vitro via the gRNA oligo(U) tail. In addition, immunoprecipitation experiments demonstrated an association between RBP16 and gRNAs within T.brucei mitochondria (1). gRNAs are involved in kinetoplastid RNA (kRNA) editing, a remarkable RNA processing mechanism characterized by the site-specific insertion and deletion of uridylate residues into pre-mRNAs. This process is required for the creation of functional mRNA molecules, as it often creates start codons, stop codons and entire open reading frames (14,15). gRNAs contain the genetic information that specifies uridylate insertion and deletion at multiple sites in the pre-mRNA (16,17). Demonstration of an RBP16–gRNA interaction in vitro and in vivo suggests a role for this protein in kRNA editing. In addition, RBP16 binds rRNAs in vivo, presumably via the oligo(U) tails present on these molecules. This suggests that RBP16 may also be involved in RNA translation (1). To gain further insight into the interaction between RBP16 and its RNA targets, we used gel retardation and UV cross-linking competition assays to (i) characterize the molecular interactions involved in the binding of RBP16 to gRNA, (ii) determine the minimal sequence length necessary for the binding of RBP16 and (iii) examine in greater detail the sequence specificity of RBP16–RNA interactions. Our results lead to a model involving both sequence specific and non-specific binding, and suggest roles for the CSD and RGG domains of RBP16 in RNA binding.
MATERIALS AND METHODS
Plasmids and nucleic acids
A construct encoding the gRNA gA6[14] with a 17 nt 3′ oligo(U) tail was previously described (18). A gA6[14] construct with a 6 nt 3′ oligo(U) tail (gA6[14]U6) was generated by PCR of gA6[14] cloned into pBluescribe (18). gA6[14] lacking an oligo(U) tail and 10 (gA6[14]NTdel10), 20 (gA6[14]NTdel20) or 30 (gA6[14]NTdel30) additional 3′ nt were generated by PCR of the gA6[14] plasmid (18), using the following antisense oligonucleotides: gA6[14]NTdel10, 5′-ATCACTGTCAAAATCTGATTCG-3′; gA6[14]NTdel20, 5′-AAATCTGATTCGTTATC-GGAG-3′; and gA6[14]NTdel30, 5′-CGTTATCGGAGTTATA-GTATAT-3′. For all three PCRs, T7 primer (5′-GTAATACG-ACTCACTATAGGGC-3′) was used as the sense oligonucleotide. The transcription template for the unedited version of the RPS12 RNA with or without a 20 nt 3′ poly(A) tail were obtained by PCR of a pBluescribe construct containing the gene coding for the unedited version of RPS12. CR6-5′ T7 (5′-TG-TAATACGACTCACTATAGGGCTAATACACTTTTGATA-ACAAACTAAAGTAAA-3′) was used as the sense oligonucleotide, and CR6-3′ U (5′-AAAAACATATCTTATTCT-3′) and CR3′-A20 (5′-TTTTTTTTTTTTTTTTTTTTAAAAACATATCTTAT-3′) were used as the antisense oligonucleotides for the amplification of the unedited version of RPS12 without and with a 3′ poly(A) tail, respectively. RNAs were synthesized in vitro using the Ambion Megascript kit and gel purified on 6% acrylamide/7 M urea gels. The ‘tetra U-scanmer’ oligonucleotides, with the exception of numbers 17A, 17B, 18 and 19 (Fig. 5), were generously provided by Dr Paul D. Gershon (Texas A & M University, Houston, TX) (19). The U7 homopolymer was generously supplied by Dr Margaret Hollingsworth (SUNY Buffalo, Buffalo, NY). All the other oligonucleotides were either obtained from Oligos Etc. Inc. (Wilsonville, OR) or Integrated DNA Technologies (Coralville, IA).
Figure 5.

Effect of the positioning of a tetra-uridylate patch within an oligonucleotide on the binding of RBP16. (A) Sequences of the oligonucleotides used to assess the importance of the position of a stretch of four uridines within the molecule (19). (B) Gel retardation assay of oligonucleotides 1–16, U34 and dC34, whose sequences were given in (A). (C) Gel retardation assay of oligonucleotides 17–19 and U10, whose sequences were given in (A). For both (B) and (C) the oligonucleotides were incubated with 2.5 µM RBP16. The complexes were separated on a native 8% polyacrylamide gel in 50 mM Tris–glycine (pH 8.8). The bands corresponding to the protein–oligonucleotide complex and the free oligonucleotide were quantified by densitometry. The affinity of RBP16 for each oligonucleotide is expressed as a percentage of the total oligonucleotide bound to RBP16 [(bound oligonucleotide/bound + free oligonucleotide) × 100]. Values shown represent the average of three to six separate experiments. (D) Binding of RBP16 to oligonucleotides 18 and 19. Purified RBP16 (600 ng) was cross-linked to [32P]-labeled gA6[14] (5 fmol) in the absence of competitor or in the presence of increasing amounts of unlabeled oligo 18 or 19. The competitors were used at molar excesses ranging from 5000- to 20 000-fold as compared to the labeled gA6[14]. The cross-linked proteins were then separated and analyzed as described in Figure 4.
The uridylate homopolymers, the ‘tetra U-scanmers’, as well as RPS12U and RPS12U+20A, were 5′-labeled with 12.5 µCi of [γ-32P]ATP (3000 Ci/mmol, 5 µCi/µl) (NEN) using 10 U of T4 polynucleotide kinase (Gibco BRL) for 10 min at 37°C. Nucleic acids were extracted with phenol/chloroform/isoamyl alcohol (25:24:1), precipitated with salt and ethanol, and resuspended in 10 µl of DEPC-treated water. Synthetic gA6[14] and its 3′ deletion derivatives were internally radiolabeled with [α-32P]UTP (800 Ci/mmol, 10 µCi/µl) (NEN) during the in vitro transcription of the appropriate DNA template with T7 RNA polymerase (Ambion). The transcripts were then gel purified on 6% acrylamide/7 M urea.
Bacterial expression and purification of MBP-RBP16
RBP16 was expressed as a maltose-binding fusion protein (MBP) in Escherichia coli and purified by amylose and poly(U)–Sepharose chromatography as previously described (1). This fusion protein was used in all the experiments reported in this paper and, for simplicity, will be referred to as RBP16.
Gel retardation assays
The 15 µl standard reaction mixture contained 6 mM HEPES (pH 7.5), 30 mM KCl, 2 mM MgCl2, 0.5 mM DTT, 1.5 mM ATP, 5 mM creatine phosphate, 0.1 mM EDTA, 10 µg/ml torula yeast RNA, 6% glycerol, and the indicated amount of radiolabeled RNA ligands and purified RBP16. Following incubation for 20 min at room temperature, RBP16–RNA complexes were separated by electrophoresis on a native 4% polyacrylamide (8% for the ‘tetra U-scanmers’) (2.7% bis-acrylamide) gel in 50 mM Tris–glycine (pH 8.8) at 4°C. The gels were fixed in 10% methanol/10% acetic acid for 30 min, dried and exposed to a film (Kodak X-OMAT Blue XB-1).
UV cross-linking experiments
Purified RBP16 (600 ng) was incubated with synthetic gA6[14] (5 fmol) internally labeled with [α-32P]UTP (800 Ci/mmol, 10 µCi/µl) (NEN) under the same conditions as described above for the gel retardation assays. Following incubation, the protein was UV cross-linked to the RNA as described (18). Competitor RNAs were added to the reactions prior to the addition of protein.
Chemical modification of RBP16 by phenylglyoxal
Thirty-four pmol of RBP16 in 0.1 M sodium bicarbonate (pH 8.5) were incubated with increasing amounts (0 to 500-fold molar excess) (reagent/protein) of phenylglyoxal (PGO) (Sigma) for 30 min at room temperature in a final volume of 10 µl. Samples were then diluted 20-fold with DEPC-treated water and the PGO removed by ultrafiltration using Centricon-10 concentrators following the manufacturer’s instructions. Samples were concentrated to 50 µl, and 10 µl was used in subsequent UV cross-linking experiments with internally radiolabeled gA6[14] as described above. In control experiments, PGO in 0.1 M sodium bicarbonate (pH 8.5) was incubated for 30 min at room temperature, diluted 20-fold with DEPC-treated water and the PGO subsequently removed as described above. The retentate was then incubated with 10 µl of RBP16 which had been previously incubated in 0.1 M sodium bicarbonate (pH 8.5) without PGO and concentrated as described above, followed by UV cross-linking to [32P]-labeled gA6[14] (5 fmol). These control experiments tested the possibility that residual PGO present during the cross-linking negatively affected RNA binding. Additionally, they controlled for any loss of RBP16 protein during the concentration procedure.
Oxidation of the 3′ OH of gA6[14]
The terminal 3′ hydroxyl of gA6[14] was oxidized by sodium periodate treatment as previously described (20). The modified RNA was then precipitated with salt and ethanol, followed by gel purification on 6% acrylamide/7 M urea. The efficiency of the 3′ end modification was >99%, as determined by [5′-32P]pCp labeling (20).
Quantification of the binding of RBP16 to RNA substrates
The UV cross-linking signals were visualized by autoradiography and quantified using a Bio-Rad model GS-700 imaging densitometer and the Molecular Analyst software v.1.5.
RESULTS
Nature of the molecular interactions involved in RBP16–RNA binding
To gain insight into the nature of the interactions involved in the binding of RBP16 to gRNA molecules, the effects of increasing amounts of KCl, MgCl2 and heparin on RBP16–gRNA binding were determined. It has been previously shown that, at a protein concentration of 2.5 µM, one RBP16–gA6[14] complex is formed while at protein concentrations of 5 µM and above, a second complex with a slower mobility is formed (1). We have termed the faster and slower migrating complexes A and B, respectively. The presence of these two complexes is specific to the RBP16–gA6[14] interaction as only one protein–RNA complex is observed with other substrates such as oligoribonucleotides composed solely of U residues [oligo(U)s] (data not shown). As shown in Figure 1B, the formation of both RBP16–gA6[14] complexes is quite resistant to high KCl concentrations when assessed by gel retardation. At low protein concentration (2.5 µM), the formation of the RBP16–gA6[14] complex A was inhibited by only 25 and 50% at KCl concentrations of 150 and 500 mM, respectively, compared to our standard binding condition of 30 mM KCl. At higher protein concentration (7.5 µM), the formation of complex A was still fairly insensitive to KCl (40% inhibition at 150 and 500 mM KCl) while complex B was inhibited by 55% at a KCl concentration of 150 and 250 mM. In the experiment shown in Figure 1B, complex B formation was inhibited by 10% at 500 mM KCl; however, we generally observed a 50–60% decrease in complex B formation at 500 mM KCl. Thus, both complex A and B were fairly resistant to KCl concentration and, even at 1 M, exhibited only 40 and 60% inhibition, respectively (data not shown). The polyanion heparin interfered with the formation of both complexes, especially complex A, but only at moderate to high concentrations (1000–13 000 µg/ml) (Fig. 1C). The ability of heparin, but not KCl, to completely inhibit RBP16–RNA binding may reflect the presence of multiple negative charges per heparin molecule. Taken together, these results demonstrate that electrostatic interactions do not play a prominent role in the binding of RBP16 to gRNAs, but do, nevertheless, contribute to the formation of these protein–gRNA complexes.
Figure 1.
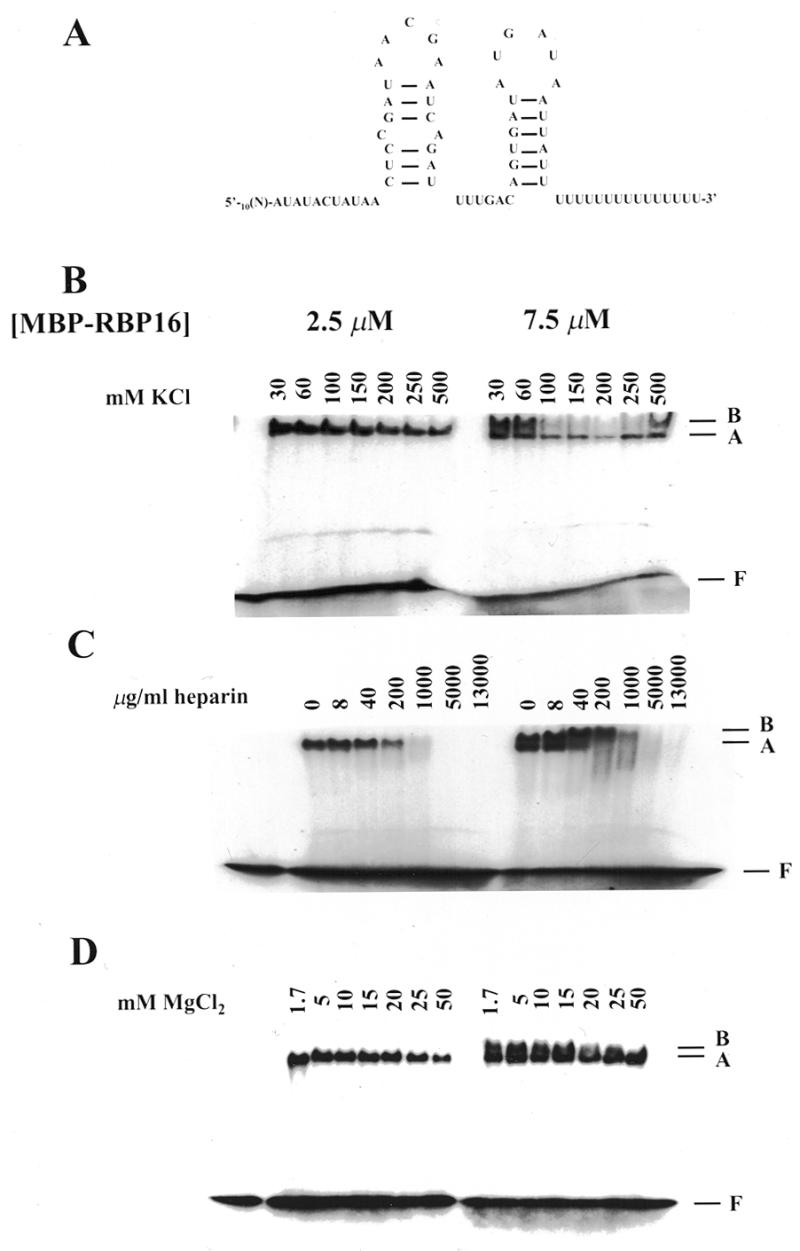
Binding properties of RBP16. (A) Nucleotide sequence (41) and experimentally determined secondary structure (24) of gA6[14]. Note that this structure was not verified in our experiments. [32P]-labeled gA6[14] (10 fmol) was incubated with 2.5 and 7.5 µM RBP16 in the presence of increasing amounts of (B) KCl, (C) heparin and (D) MgCl2. The resulting protein–gRNA complexes were separated on a native 4% polyacrylamide gel in 50 mM Tris–glycine (pH 8.8). The position of the faster and slower migrating complexes is indicated by A and B, respectively. F represents the free gRNA.
The effect of MgCl2 on the formation of the RBP16–gA6[14] complexes was also assessed. It has been suggested that Mg2+ stabilizes the protein–RNA conformation by alleviating the repulsive effects of the phosphate backbone and by stabilizing functional groups (8,21). Increasing the concentration of MgCl2, at a protein concentration of 2.5 µM, did not have a significant effect on the stability of the RBP16–gA6[14] A complex, although at the highest concentration tested (50 mM) the formation of this complex was inhibited by ~40% (Fig. 1D). At 7.5 µM RBP16, complex A was still quite stable up to 50 mM MgCl2. On the other hand, complex B was more sensitive to high concentrations of MgCl2. In fact, the increase of MgCl2 led to a gradual shift from complex B to complex A, probably reflecting a progressive conformational switch from the complex B structure to the complex A structure (Fig. 1D).
The predicted amino acid sequence of RBP16 revealed the presence of a CSD at its N-terminus and a region rich in glycine and arginine at its C-terminus. Both domains have arginine residues that may be crucial in the binding of RBP16 to RNA, based on comparisons to proteins with similar domains (10,22). This prompted us to assess the importance of arginine residues in the binding of RBP16 to gA6[14] by chemical modification with PGO, known to rapidly and specifically react with arginine residues in proteins, especially in the bicarbonate–carbonate buffer system used in our experiments (23). Treatment of RBP16 with a 25-fold molar excess of PGO totally prevented the binding of RBP16 to gA6[14] (Fig. 2). Since the MBP–RBP16 fusion protein contains a total of 29 arginines (13 from RBP16 and 16 from MBP), and the stoichiometry of the reaction involves the reaction of 2 mol of PGO with 1 mol of arginine (10), at a molar excess of 25:1 (PGO:protein), approximately half of the arginines/molecule are modified. This suggests that one or more particular arginine residues are preferentially modified and that these arginines are essential to the binding of RBP16 to gA6[14]. Inhibition of the RBP16–gA6[14] interaction by PGO treatment of the protein clearly indicates that one or more arginine residues are involved in this binding, most likely through hydrogen bonding (10).
Figure 2.
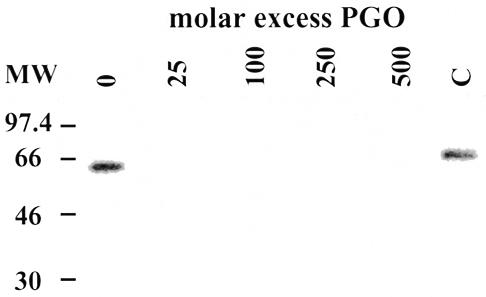
Effect of phenylglyoxal treatment of RBP16 on its binding to gA6[14]. RBP16 (34 pmol) was incubated in the presence of increasing amounts of PGO for 30 min at room temperature. PGO was removed by ultrafiltration and RBP16 molecules subjected to UV cross-linking to [32P]-labeled gA6[14] (5 fmol). The cross-linked proteins were then separated by SDS–PAGE on a 12.5% gel and visualized by autoradiography. C indicates a control performed to test the effect of any residual PGO in cross-linking reactions (see Materials and Methods).
Minimal binding site of RBP16
gA6[14] molecules with different lengths of sequence deleted from their 3′ ends were used to assess RNA length and sequence requirements for the formation of the A and B complexes (Fig. 3). All T.brucei gRNAs analyzed so far have been shown to adopt a similar secondary structure, despite having variable primary sequence (24). This secondary structure is characterized by two imperfect stem–loop elements separated by a single-stranded region. Both the 5′ and 3′ ends of the molecules are in a single-stranded conformation (Fig. 1A). Studies have revealed that the binding of gBP21 to gRNAs probably involves recognition of a stem–loop secondary structure element (24). We did not experimentally verify the secondary structures of the gA6[14] derivatives in these experiments. However, some of the deletions performed would necessarily destroy the ability to form one or both stem–loop structures (Fig. 3). We assessed the ability of gRNAs harboring 3′ deletions to form the A and B complexes. Removal of all but six residues in the 3′ U tail did not prevent the formation of complex A at low protein concentration (2.5 µM), nor did it block the formation of both complexes at 7.5 µM protein (compare Figs 3A and 1B). Both A and B complexes were also formed with gA6[14] from which an additional 10 nt had been removed from the 3′ end (Fig. 3B, gA6[14]NTdel10), suggesting that the 3′ stem–loop is not critical for binding. It is also noteworthy that a gradual shift from complex A to complex B is observed with the gA6[14] derivative gA6[14]NTdel10 as the salt concentration is increased. This most likely represents a gradual conformational change in the RNA associated with the increase in KCl concentration leading to altered mobility of the complex in a native gel. When purified RBP16 was incubated with radiolabeled gA6[14] lacking 37 nt at the 3′ end (gA6[14]NTdel20), no complex B was observed at 7.5 µM RBP16 (Fig. 3C). This suggests that either the binding sequence needed for the formation of complex B is located within the 3′ half of the gA6[14] molecule or that 42 nt is below the minimum length required for complex B formation. Finally, although neither complex was produced when RBP16 was incubated with gA6[14] lacking 47 nt at the 3′ end (gA6[14]NTdel30) at low protein concentration (2.5 µM), the A complex was formed at higher RBP16 concentration (7.5 µM) (Fig. 3D). From these experiments, we conclude that 32 nt, the length of the shortest substrate tested, can support RBP16 binding, and that gRNA structure is not a critical binding determinant. In addition, the gradual shift from A to B complex observed with gA6[14]NTdel10 suggests that complex A and complex B represent two complexes with different conformations, as opposed to representing one and two RBP16 molecules bound to gA6[14]. Additional experiments are, however, needed to clarify this point.
Figure 3.

Effect of gA6[14] 3′ deletions on the binding by RBP16. [32P]-labeled gA6[14] derivatives (10 fmol) were incubated with 2.5 and 7.5 µM RBP16 in the presence of increasing amounts of KCl. The resulting protein–RNA complexes were separated on a native 4% polyacrylamide gel in 50 mM Tris–glycine (pH 8.8). The sequence and potential secondary structure of each RNA is shown on the right. (A) gA6[14]U6, (B) gA6[14]NTdel10, (C) gA6[14]NTdel20 and (D) gA6[14]NTdel30. Lane 1, 30 mM KCl; lane 2, 60 mM KCl; lane 3, 100 mM KCl; lane 4, 150 mM KCl; lane 5, 200 mM KCl; lane 6, 250 mM KCl; lane 7, 500 mM KCl; lane 8, 1000 mM KCl. Note that in (C) and (D) the identity of the single complex as complex A was confirmed by comparison to the mobility of complexes formed with full length gA6[14].
To further investigate the minimum number of residues required for RBP16 RNA binding we used oligoribonucleotides composed solely of U residues [oligo(U)s] of different lengths. The use of homopolymers allowed us to eliminate the effects of RNA sequence and/or structure on RBP16 binding. Gel retardation experiments revealed that the migration of all the oligo(U)s tested was retarded by 1 µM RBP16 to varying degrees (data not shown). To quantify the relative affinities of RBP16 for oligo(U)s of different lengths, we employed UV cross-linking competition assays (1). Competition with full-length gA6[14] was included as a reference. The results of the UV cross-linking competition assays presented in Figure 4 show that, although RBP16 could bind to oligonucleotides as small as U4, its affinity increased with the length of the oligo(U) ribonucleotide. A striking increase in binding affinity was observed when the oligo(U) length was increased from seven to ten. The molar excess of U10 and U15 needed to compete the binding of RBP16 to gA6[14] by 50% was similar to that for gA6[14] (Fig. 1B). Increasing the length of the ribonucleotide to 20 and 40 Us led to a 4- to 6-fold increase in the ability to compete the binding of gA6[14]. On the other hand, the binding of gA6[14] could not be competed by >30% with U4 and U7 even at a molar excess of 40 000-fold, indicating that the binding of RBP16 to these two ribonucleotides is very weak compared to the binding to gA6[14] or longer oligo(U)s. Thus, the minimal number of uridylate residues required for efficient binding by RBP16 is ~10 nt when assayed by UV cross-linking competition. Since the gRNA oligo(U) tail is, on average, 10 to 15 nt long, and a U10 oligonucleotide competes gA6[14] binding nearly as efficiently as gA6[14] itself, this supports the model that the U tail is the primary binding determinant in RBP16–gRNA interaction. Another striking observation is that, although a significant decrease (30%) in the binding of RBP16 to gA6[14] is observed at 10 000-fold molar excess of U4 and U7, addition of increasing amounts of either one of these competitors does not result in a further reduction of the signal intensity. This suggests that there are two types of RBP16–gA6[14] interactions, one of which cannot be readily competed by smaller oligonucleotides such as U4 and U7.
Figure 4.

(A) Determination of the RBP16 minimal binding site. Purified RBP16 (600 ng) was cross-linked to [32P]-labeled gA6[14] (5 fmol) in the absence of competitor or in the presence of increasing amounts of unlabeled competitor. The competitors were used at molar excesses ranging from 2000- to 40 000-fold as compared to the labeled gA6[14]. The cross-linked proteins were then separated on a 12.5% SDS–PAGE gel and visualized by autoradiography. The UV cross-linking signals were quantified by densitometry. (B) Molar excess required to achieve 50% inhibition of the RBP16 binding to [32P]-labeled gA6[14] (MEI50).
Effect of the positioning of a tetra-uridylate patch within a 34 nt oligonucleotide
Gel retardation and UV cross-linking competition assays demonstrated that RBP16 can bind, to some extent, a stretch of uridylates as small as four Us (Fig. 4 and data not shown). We wanted to determine whether the position of this tetra-uridylate patch within the molecule was critical for the efficient binding by RBP16. We were especially interested in whether the U stretch has to be part of a 3′ U tail or whether it can be internal as found in many mitochondrial mRNAs. For this purpose, we used a set of 34mer oligonucleotides containing four adjacent uridylates placed at various positions within the oligonucleotide. The rest of the oligonucleotide was composed of oligo(dC), except for the extreme 3′ nt, which contained a ribose moiety (19). The position of the tetraU patch was moved from the 5′ to the 3′ end in dinucleotide increments (Fig. 5A). Since C is bound very inefficiently by RBP16 (1; see also Fig. 5B), embedded tetraU stretches in oligos otherwise completely composed of C residues allowed us to determine the importance of the positioning of the uridylate residues for the binding by RBP16. Binding to these oligonucleotides is expected to depend primarily on the position of the tetraU patch. The oligonucleotides were 5′ end labeled with [γ-32P]ATP and used to determine the binding ability of RBP16 by gel retardation assays. As shown in Figure 5B, only oligonucleotides in which the uridylate residues are located within the 3′ half of the molecule (oligo 8–14) were efficiently bound by RBP16. The weaker binding observed with oligo 15 and 16 indicates that, in addition to the distance from the 5′ end, the distance from the 3′ end of the molecule, although less critical, is also important for the efficient binding of RBP16. Interestingly, however, oligonucleotide 10 with the U patch located in the 3′ half of the molecule did not bind RBP16 with high affinity. This suggests that, in addition to a preference for the 3′ half of a 34mer oligonucleotide, more subtle effects of a tetraU patch positioning are important in the RBP16–gA6[14] interaction.
The preference of RBP16 for oligonucleotides with a U stretch located in the 3′ half could be explained by one of two models. First, the tetraU patch may need to be at least 14 nt from the 5′ end of the molecule. Second, the binding determinant may need to be >4 but <18 nt from the 3′ end to bind effectively to the RNA molecule. To test whether the distance from the 5′ or 3′ end is critical for the binding by RBP16, we performed a gel retardation assay using two additional oligonucleotides. In oligo 18, the tetraU patch is located 14 nt from the 5′ end and 22 nt from the 3′ end. In oligo 19, the uridylate residues are located 8 nt from the 5′ end and 16 nt from the 3′ end (Fig. 5A). If binding requires 14 or more residues 5′ of the tetraU patch, we would expect RBP16 to bind only oligo 18. This is because in oligo 18 the tetraU patch is sufficiently far from the 5′ end to support binding (see oligo 8), while the extended 3′ sequence should be irrelevant. On the other hand, if binding requires that the tetraU patch be 16 or less residues from the 3′ end, we would expect only oligo 19 to bind since the tetraU patch is positioned close enough to the 3′ end to support binding (see oligo 8), and the short sequence 5′ of the tetraU patch should be irrelevant. As shown in Figure 5C, the binding of oligo 18 by RBP16 (2.5 µM) was significantly better than oligo 19. The relative affinity of RBP16 for these two oligonucleotides was also assessed by their ability to compete the binding of RBP16 to gA6[14] in a gel retardation assay (Fig. 5D). The molar excess of oligo 18 required to compete the binding of RBP16 to gA6[14] by 50% was ~3000-fold, while a 9000-fold molar excess of oligo 19 was necessary to achieve the same competition. More importantly, the binding to gA6[14] could be entirely competed by a 5000-fold molar excess of oligo 18 while oligo 19 could not compete the binding of RBP16 to gA6[14] by more than 60% even at a 20 000-fold molar excess. These results indicate that RBP16 requires at least 14 nt of non-specific sequence 5′ of the tetraU stretch to achieve efficient binding to its RNA substrate, and that the distance from the 3′ end of the RNA is important, but not as critical, to the binding. The conclusion that the distance from the 3′ end of the RNA plays a role in the binding by RBP16 is strengthened by the fact that oligo 18 is a better substrate than oligo 8. In both oligonucleotides, the tetraU patch is located 14 nt from the 5′ end, but oligo 18 has a longer 3′ stretch of Cs (22 nt) than oligo 8 (16 nt). The longer 3′ end in oligo 18 possibly allows for additional interactions with the protein. Finally, as shown in Figure 5C, RBP16 could bind to oligonucleotide 8 better than to oligonucleotide 19. The fact that, in both oligonucleotides, the tetraU patch is located 16 nt from the 3′ end, but that oligo 8 has a 14 nt 5′ end (compared to an 8 nt 5′ end for oligo 19) reinforces our conclusion that, although RBP16 needs non-specific sequences 5′ and 3′ of the tetraU binding site, a minimal distance of 14 nt from the 5′ end is more critical than the distance from the 3′ end.
We next tested the effect of increasing the length of a U patch located in an unfavorable position. Increasing the U patch length from 4 to 8 nt did not increase binding (Fig. 5C, compare oligo 17A to oligo 3), indicating that the poor binding efficiency to oligos 1–7 is probably due to the positioning rather than the number of uridylate residues. However, increasing the length of the U patch to 10 nt resulted in a binding efficiency comparable to oligos 8–14 (Fig. 5C, oligo 17B). This suggests that, given the minimal number of uridylate residues required for efficient binding, RBP16 can overcome the negative effect of the close proximity to the 5′ end, possibly by making non-specific contacts with the rest of the molecule.
Surprisingly, U10 was weakly bound by RBP16 when assayed by gel retardation, although this oligonucleotide represents the minimal length required for efficient binding by RBP16, as determined by UV cross-linking competition. This could be explained by the fact that the interaction between RBP16 and U10 is stabilized by the UV cross-linking, thereby allowing RBP16 to remain covalently bound to its substrate throughout its migration in the gel. Conversely, during the gel retardation assay, the initial RNA–protein interaction may be disrupted as the complex migrates in the gel, leading to a gradual dissociation of the protein from the RNA–protein complex. In this regard, UV cross-linking would have a similar stabilizing effect on the binding of RBP16 to a substrate as do 5′ and 3′ non-specific sequences.
Binding of RBP16 to the unedited version of RPS12 RNA
The results presented above indicate that, although the 3′ U tail is the major binding determinant for gRNAs, it is not absolutely required for RBP16 binding. The results therefore suggest that mRNA could serve as a binding substrate. To examine this possibility directly, we examined the interaction of RBP16 with the ribosomal protein S12 (RPS12) RNA by gel retardation assay. The unedited RPS12 RNA (RPS12U) is 221 nt long and contains several U stretches of at least 4 nt. In addition, the G-richness of this mRNA could also contribute to RBP16 binding (25). In gel retardation experiments, an RBP16–RPS12U protein–RNA complex was observed at a protein concentration of 1 µM (data not shown). To quantify binding of RPS12U relative to a gRNA, a cross-linking competition assay was performed (Fig. 6). In addition, we tested the effect of adding a 20 nt poly(A) tail to RPS12U RNA to mimic a large fraction of the native RNA which possesses a poly(A) tail of approximately this length (26). Both RPS12U RNAs bound RBP16 slightly better than did gA6[14] gRNA (Fig. 6B). Addition of a poly(A) tail appeared to have no effect on binding. However, the use of lower concentrations of competitor RNAs might reveal subtle differences in binding efficiency between polyadenylated and non-polyadenylated RNA. These experiments confirm the results obtained with the 34mer oligonucleotides indicating that, for RBP16 binding, the U stretch does not have to be part of a U tail, but can be located within the RNA molecule.
Figure 6.

Binding of RBP16 to the unedited version of RPS12 RNA. (A) Purified RBP16 (600 ng) was cross-linked to [32P]-labeled gA6[14] (5 fmol) in the absence of competitor or in the presence of increasing amounts of unlabeled unedited RPS12 RNA without or with a 20 nt poly(A) tail. The competitors were used at molar excesses ranging from 5000- to 20 000-fold as compared to the labeled gA6[14]. The cross-linked proteins were then separated and analyzed as described in Figure 4. (B) Molar excess required to achieve 50% inhibition of the RBP16 binding to [32P]-labeled gA6[14] (MEI50).
Modification of the 3′ terminus of gA6[14]
It has been reported that gRNAs which have been modified either by the addition of a 3′ phosphate group or oxidized by treatment with periodate do not support in vitro U deletion editing (27). A recently proposed model suggested that the 3′ OH is necessary for interaction with a protein that stabilizes gRNA U tail binding to purine-rich sequences upstream of the editing site (27). The role of the RNA substrate 3′ OH in RBP16 binding was assessed by gel retardation and UV cross-linking competition assays. gA6[14] was either phosphorylated at its 3′ end using [5′-32P]pCp (28), or its terminal 3′ OH was oxidized by periodate treatment (20). In gel retardation assays, both periodate-treated and 3′ pCp-containing molecules bound RBP16 as efficiently as unmodified gA6[14] (data not shown). The affinity of RBP16 for the periodate-oxidized form of gA6[14] was further characterized by determining its ability to compete the binding of RBP16 to gA6[14] by UV cross-linking. As shown in Figure 7, the molar excess of oxidized-gA6[14] needed to achieve a 50% inhibition of the binding of RBP16 to intact gA6[14] is similar to the amount of gA6[14] needed to achieve the same level of inhibition. Taken together, these results clearly indicate that the terminal 3′ OH is not required for the binding of RBP16 to gA6[14].
Figure 7.

Binding of RBP16 to periodate-treated gA6[14]. (A) Purified RBP16 (600 ng) was cross-linked to [32P]-labeled gA6[14] (5 fmol) in the presence of increasing amounts of unlabeled untreated or periodate-treated gA6[14]. The competitor was used at molar excesses ranging from 5000- to 18 000-fold as compared to the labeled gA6[14]. The cross-linked proteins were then separated and analyzed as described in Figure 4. (B) Molar excess required to achieve 50% inhibition of the RBP16 binding to [32P]-labeled gA6[14] (MEI50).
DISCUSSION
In this report, the molecular interactions and sequence requirements for RBP16 RNA binding were analyzed. The RBP16–gA6[14] complex is relatively insensitive to high salt concentration indicating that ionic interactions do not play a prominent role in the binding of RBP16. This is in contrast to the demonstrated importance of ionic contacts in RNA binding by proteins such as the cold-shock protein CspA in E.coli (29), the U1A spliceosomal protein (30), as well as the gRNA-binding protein gBP21 in T.brucei (31,32). Our results suggest that interactions such as hydrogen bonding and hydrophobic interactions are likely to be involved in the interaction of RBP16 with gRNAs. The predicted amino acid sequence of RBP16 revealed the presence of a RNP1 RNA-binding motif (10), a component of all CSDs, at its N-terminus. In the case of the cold-shock protein CspB in Bacillus subtilis, aromatic residues located on the surface of the protein are believed to stabilize CspB–nucleic acid complex formation by allowing hydrophobic interactions with ssDNA or RNA (10,33). Moreover, the specific binding of the U1 snRNP protein, U1A, to its target RNA has been shown to involve the aromatic residues present in the RNP1 motif of the protein through direct and water-mediated hydrogen bonds (22,29). Two phenylalanines are found within the RBP16 RNP1 motif. Furthermore, many other phenylalanine and histidine residues are also present throughout the RBP16 molecule. Comparison of RBP16 to other RNP1-containing proteins suggests that these amino acids may make an important contribution to the binding of RBP16 to gRNAs.
The binding of RBP16 to gA6[14] was completely abolished by PGO treatment of the protein. PGO specifically modifies arginine residues by interacting with α-amino groups (23). Although PGO has been shown to react with both arginine and lysine residues under certain conditions, the reaction of PGO with arginine is remarkably accelerated and very specific in bicarbonate–carbonate buffer systems such as the one used in the present study. Thus, our results indicate the involvement of one or more arginine residues in the RBP16–gA6[14] interaction. The N-terminal CSD of RBP16 contains several arginines, as well as lysines, that may be involved in the binding to gRNAs. In CSD-containing proteins, arginine and lysine residues located on the β-sheet surface are known to interact extensively with the RNA (22). In CspB, for example, two lysine residues (K7, K13) located on the surface of such a β-sheet participate in single-stranded (ss) DNA binding. These residues create an attractive potential for nucleic acid interaction that overcomes the charge repulsion between the acidic CspB and the ssDNA (33). RBP16 also possesses a C-terminal half rich in glycine and arginine residues similar to an RGG RNA-binding motif (1,10). Within RGG domains, arginine residue side chains have been shown to directly interact with the RNA through hydrogen bonding and ring stacking (34,35). This suggests that arginines present in the C-terminal region of RBP16 may potentially be located on the surface of the protein and participate in RNA binding.
gRNA secondary structure does not appear to be a major binding determinant for RBP16, since gRNAs harboring deletions that would abolish stem–loop formation are still bound by the protein. In fact, our results indicate that as few as 10 uridylate residues can mediate efficient binding by RBP16, provided that the initial interaction is stabilized by UV cross-linking. Importantly, UV cross-linking experiments showed that competition of the binding of RBP16 to gA6[14] achieved by U10 and U15 homopolymers is within the range of the competition observed with gA6[14] itself. As the average length of the oligo(U) tail of gRNAs is 10–15 nt (36), this suggests that the oligo(U) tail accounts for most of the initial binding by RBP16. This is in agreement with the published results of Hayman and Read (1) who identified the oligo(U) tail as the primary determinant involved in RBP16–gRNA binding. Gel retardation experiments demonstrated that non-specific sequences are necessary to stabilize binding to a uridylate stretch of 10 residues in the absence of UV cross-linking. Nevertheless, these gel retardation experiments reinforce the conclusion that U10 is a critical length for RBP16 binding since the absolute requirement for 14 nt 5′ of a U stretch is abolished in the presence of a U stretch of 10 nt (Fig. 5, oligo 17B). As previously reported, RBP16 binds both gRNAs and rRNAs in vivo (1). Ten nucleotides is also within the range of the post-transcriptionally added 3′ poly(U) tail on mitochondrial rRNAs. The 9S rRNA has a tail of precisely 11 nt, while 12S has a heterogeneous tail of 2 to 17 uridines (37). Thus, the oligo(U) tail appears to act as a major binding determinant for RBP16 to gRNAs and has the potential to do so for at least some rRNAs. Binding to 12S rRNAs with U tails of <10 nt may be facilitated by non-specific contacts with upstream nucleotides (see below). In addition, while 3′ U tails play an important role in RBP16–RNA interaction, the 3′ OH group is not essential for binding.
RBP16 is also capable of binding some RNAs lacking U tails, as shown by gel retardation experiments performed with the unedited version of RPS12 RNA. The longest contiguous stretch of U residues in this mRNA is six (25). Based on UV cross-linking competition assays, six Us is not sufficient for high-affinity RBP16 binding. Results of the gel retardation assay with the ‘tetra U-scanmer’ oligonucleotides indicated that RBP16 requires at least 14 and 4 nt of non-specific sequence 5′ and 3′, respectively, of the binding site to achieve efficient binding, given a stretch of Us shorter than 10 nt. Therefore, RBP16 binding to mRNAs may be mediated by a combination of specific interactions with U stretches and non-specific contacts 5′ and 3′ of U-rich regions. This is reminiscent of the RNA binding properties of VP55, the vaccinia virus poly(A) polymerase catalytic subunit, which requires both U stretches as well as upstream base-independent contacts for optimal interaction (38).
Based on the results presented in this paper and the known properties of CSD and RGG domains, we propose the following working model for the RBP16–RNA interaction (Fig. 8). In the majority of CSD-containing RNA-binding proteins, the CSD provides the specificity and is involved in high-affinity binding. Since RBP16 exhibits a strong preference for poly(U) (1) and the gRNA oligo(U) tails appears to be a major RBP16 binding determinant (1 and this paper), we suggest that the CSD of RBP16 mediates sequence-specific binding to gRNA oligo(U) tails (Fig. 8A, solid arrows). However, since RBP16 forms a stable complex with U10 only when adjacent non-specific sequences are present on the RNA substrate (Fig. 5), this suggests that weak non-specific contacts also play a role in the RBP16–gRNA interaction (Fig. 8A, dotted lines). While the CSDs of eukaryotic Y-box proteins generally mediate sequence-specific RNA–protein interactions (10), binding by RGG boxes has often been shown to be non-specific, and this motif is thought to facilitate binding by adjacent RNA-binding domains within the same molecule (10). Thus, we predict that, in the case of the full length gA6[14], the RGG domain stabilizes the initial contact between RBP16 and the oligo(U) tail through non-specific interactions with the encoded region of the gRNA.
Figure 8.

Proposed model for the interaction of RBP16 with RNA. RBP16 possesses two putative RNA-binding domains: an N-terminal CSD and a C-terminal RGG domain (RGG). (A) Binding of RBP16, through its CSD and RGG domains, to full-length gA6[14]. Although the U tail acts as the major binding determinant, weak non-specific interactions through the RGG are required to stabilize the initial RNA–protein interaction. (B) Binding of RBP16, through both RNA-binding domains, in the presence of a sub-optimal binding site and non-specific upstream or downstream sequence. In this case, the binding to the uridine stretch is stabilized by non-specific contacts between the upstream or downstream nucleotide sequence and the RGG domain of RBP16. Although both non-specific upstream or downstream sequences are needed, the upstream sequence is more critical than the downstream sequence (indicated by four arrows versus two arrows). (C) Weak binding of RBP16, through its CSD or its RGG domain, to a sub-optimal binding site in the absence of a non-specific upstream sequence.
Our results demonstrate that RBP16 can bind to a stretch of uridylates as small as four Us (Fig. 4) provided that at least 14 nt of non-specific sequence are present upstream of the sub-optimal U stretch. RBP16 binding is also enhanced by the presence of 4 nt of non-specific sequence downstream of the sub-optimal binding site, although this requirement is not as critical as the upstream sequence. These 5′ and 3′ base-independent contacts are presumably required in order to allow non-specific interactions between RBP16 and the RNA substrate via the RGG domain (Fig. 8B). Binding to RNAs with sub-optimal U stretches has more stringent requirements for upstream non-specific contacts than does binding to RNAs with U stretches of 10 nt. For example, the requirement for 14 nt 5′ of a U4 stretch is apparently invariable, while this requirement is relaxed in the presence of 10 Us. The ability of RBP16 to bind to RNAs containing very short U stretches through both RNA-binding domains would explain the binding to some gA6[14] derivatives such as gA6[14]NTdel10 and gA6[14]NTdel20, as well as to the 12S rRNA with U tails <10 nt, and to many mRNAs. As depicted in Figure 8C, in the absence of a non-specific sequence upstream of a sub-optimal U stretch, RBP16 would still be capable of binding to the RNA molecule, but this binding would be very weak as it would occur solely through the RGG domain, or through weak interactions with the CSD that are not stabilized by contacts with the RGG domain. This model would explain the modest ability of U4 to compete gRNA binding in UV cross-linking experiments in which the very weak binding is stabilized by the cross-linking process. Thus, our results suggest that binding of RBP16 to native mitochondrial RNAs involves contacts with both the CSD and RGG domains. In addition to the role played by RGG domains in RNA binding, this domain has also been shown to mediate protein–protein interactions (39,40). Experiments are currently underway in our laboratory to directly determine the involvement of both the CSD and the arginine/glycine-rich domains of RBP16 in both RNA–protein and protein–protein interactions.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Mark Hayman, Kevin Militello and Dr Edward Niles for critical reading of the manuscript. We also thank Dr Margaret Hollingsworth for providing U7 and Dr Paul Gershon for his generous gift of the ‘tetraU-scanmers’. This work was supported by NIH grant GM53502 to L.K.R. who is the recipient of a Burroughs Wellcome New Investigator in Parasitology Award.
REFERENCES
- 1.Hayman M.L. and Read,L.K. (1999) J. Biol. Chem., 274, 12067–12074. [DOI] [PubMed] [Google Scholar]
- 2.Jiang W., Hou,Y. and Inouye,M. (1997) J. Biol. Chem., 272, 196–202. [DOI] [PubMed] [Google Scholar]
- 3.Schindelin H., Marahiel,M.A. and Heinemann,U. (1993) Nature, 364, 164–168. [DOI] [PubMed] [Google Scholar]
- 4.Wolffe A.P. (1994) Bioessays, 16, 245–251. [DOI] [PubMed] [Google Scholar]
- 5.Moss E.G., Lee,R.C. and Ambros,V. (1997) Cell, 88, 637–646. [DOI] [PubMed] [Google Scholar]
- 6.Thieringer H.A., Singh,K., Triverdi,H. and Inouye,M. (1997) Nucleic Acids Res., 25, 4764–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsumoto K., Meric,F. and Wolffe,A.P. (1996) J. Biol. Chem., 271, 22706–22712. [DOI] [PubMed] [Google Scholar]
- 8.Triqueneaux G., Velten,M., Franzon,P., Dautry,F. and Jacquemin-Sablon,H. (1999) Nucleic Acids Res., 27, 1926–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salvetti S., Batistoni,R., Deri,P., Rossi,L. and Sommerville,J. (1998) Dev. Biol., 201, 217–229. [DOI] [PubMed] [Google Scholar]
- 10.Burd C. and Dreyfuss,G. (1994) Science, 265, 615–621. [DOI] [PubMed] [Google Scholar]
- 11.Ghisolfi L., Kharrat,A., Joseph,G., Amalric,F. and Erard,M. (1992) Eur. J. Biochem., 209, 541–548. [DOI] [PubMed] [Google Scholar]
- 12.Nadler S.G., Merrill,B.M., Robert,W.J., Keating,K.M., Lisbin,M.J., Barnett,S.F., Wilson,S.H. and Williams,K.R. (1991) Biochemistry, 30, 2968–2976. [DOI] [PubMed] [Google Scholar]
- 13.Kiledjian M. and Dreyfuss,G. (1992) EMBO J., 11, 2655–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stuart K., Allen,T.E., Heidmann,S. and Seiwert,S.D. (1997) Microbiol. Mol. Biol. Rev., 61, 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alfonzo J.D., Thiemann,O. and Simpson,L. (1997) Nucleic Acids Res., 25, 3751–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blum B., Bakalara,N. and Simpson,L. (1990) Cell, 60, 189–197. [DOI] [PubMed] [Google Scholar]
- 17.Corell R.A., Feagin,J.E., Riley,G.R., Strickland,T., Guderian,J.A., Myler,P.J. and Stuart,K. (1993) Nucleic Acids Res., 21, 4313–4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Read L.K., Göringer,H.U. and Stuart,K. (1994) Mol. Cell. Biol., 14, 2629–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng L. and Gershon,D. (1997) EMBO J., 16, 1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seiwert S.D., Heidmann,S. and Stuart,K. (1996) Cell, 84, 831–841. [DOI] [PubMed] [Google Scholar]
- 21.Kalurachchi K. and Nikonowicz,E.P. (1998) J. Mol. Biol., 280, 639–654. [DOI] [PubMed] [Google Scholar]
- 22.Graumann P. and Marahiel,M.A. (1996) Bioessays, 18, 309–315. [DOI] [PubMed] [Google Scholar]
- 23.Lundblad R.L. and Noyes,C.M. (1984) Chemical Reagents for Protein Modification. CRC Press, Boca Raton, FL, Vol. II, pp. 1–46.
- 24.Schmid B., Riley,G.R., Stuart,K. and Göringer,H.U. (1995) Nucleic Acids Res., 23, 3093–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Read L.K., Myler,P.J. and Stuart,K. (1992) J. Biol. Chem., 267, 1123–1128. [PubMed] [Google Scholar]
- 26.Militello K.T. and Read,L.K. (1999) Nucleic Acids Res., 27, 1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burgess M.L.K., Heidmann,S. and Stuart,K. (1999) RNA, 5, 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seiwert S.D. and Stuart,K. (1994) Science, 266, 114–117. [DOI] [PubMed] [Google Scholar]
- 29.Schindelin H., Jiang,W., Inouye,M. and Heinemann,U. (1994) Proc. Natl Acad. Sci. USA, 91, 5119–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oubridge C., Ito,N., Evans,P.R., Teo,C.H. and Nagai,K. (1994) Nature, 372, 432–438. [DOI] [PubMed] [Google Scholar]
- 31.Köller J., Müller,U.F., Schmid,B., Missel,A., Kruft,V., Stuart,K. and Göringer,H.U. (1997) J. Biol. Chem., 272, 3749–3757. [DOI] [PubMed] [Google Scholar]
- 32.Herman T., Schmid,B., Heumann,H. and Göringer,H.U. (1997) Nucleic Acids Res., 25, 2311–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schröder K., Graumann,P., Schnuchel,A., Holak,T.A. and Marahiel,M.A. (1995) Mol. Microbiol., 16, 699–708. [DOI] [PubMed] [Google Scholar]
- 34.Draper D.E. (1995) Annu. Rev. Biochem., 64, 593–620. [DOI] [PubMed] [Google Scholar]
- 35.Nagai K., Oubridge,C., Jessen,T.H., Li,J. and Evans,P.R. (1990) Nature, 348, 515–520. [DOI] [PubMed] [Google Scholar]
- 36.Blum B. and Simpson,L. (1990) Cell, 62, 391–397. [DOI] [PubMed] [Google Scholar]
- 37.Adler B.K., Harris,M.E., Bertrand,K.I. and Hajduk,S.L. (1991) Mol. Cell. Biol., 11, 5878–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng L., Beigelman,L., Matulic-Adamic,J., Karpeisky,A. and Gershon,P.D. (1997) J. Biol. Chem., 272, 31542–31552. [DOI] [PubMed] [Google Scholar]
- 39.Bouvet P., Diaz,J.-J., Kindbeiter,K., Madjar,J.-J. and Amalric,F. (1998) J. Biol. Chem., 273, 19025–19029. [DOI] [PubMed] [Google Scholar]
- 40.Cartegni L., Maconi,M., Morandi,E., Cobianchi,F., Riva,S. and Biamonti,G. (1996) J. Mol. Biol., 259, 337–348. [DOI] [PubMed] [Google Scholar]
- 41.Koslowsky D.J., Riley,G.R., Feagin,J.E. and Stuart,K. (1992) Mol. Cell. Biol., 12, 2043–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]