


Abstract
RAD26, the yeast homologue of human CSB, has an essential role in transcription-coupled repair (TCR). We have mapped the requisite of Rad26 for nucleotide excision repair (NER) within the different regions of the yeast Saccharomyces cerevisiae MFA2 gene at nucleotide resolution. Our results show that Rad26 is dispensable for enhanced NER in both the MFA2 upstream promoter, except in the TATA box region, and for enhanced NER in both strands of the active gene at a site close to the transcription termination region. As expected, it is not needed for repair of regions downstream of where transcription terminates. However, it is required for TCR in the transcription initiation and elongation regions. Our data support the hypothesis that Rad26 is required for the interchange between holo-TFIIH and a putative repairosome containing core TFIIH and other NER proteins. Close to the end of transcription, hotspots for the repair of CPDs in both the transcribed strand and the non-transcribed strand occur. This enhanced repair is independent of Rad26. Hence, TFIIH may take a form favourable for forming a repairosome without Rad26 assistance; here the organisation of the DNA during the termination of transcription may facilitate access of a repair complex to enable enhanced repair of both strands.
INTRODUCTION
Nucleotide excision repair (NER) is a complex process used by cells to remove a broad spectrum of structurally unrelated lesions. This includes the major class of lesion induced by UV light, namely cyclobutane pyrimidine dimers (CPDs) (1–3). DNA damage is removed by NER through two incisions of the damaged strand on both sides of the lesion, followed by gap repair synthesis and ligation. In eukaryotes, this process requires more than 20 different polypeptides and the damage is excised in a 24–32 nt long fragment. NER has been highly conserved and homologous genes are found in organisms ranging from yeast to humans. The human proteins required for the dual incisions include seven xeroderma pigmentosum (XP) proteins, trimeric replication protein A (RPA) and the multisubunit general transcription factor TFIIH.
The rate of NER is heterogeneous within the genome. For genes actively being transcribed by RNA polymerase II, CPDs in the transcribed strand (TS) are repaired faster than CPDs in the non-transcribed strand (NTS) (4–10). This preferential repair of the TS is referred to as transcription-coupled repair (TCR). The slower repair of the NTS, or that of both the TS and NTS of inactive genes, is termed global repair. In the yeast Saccharomyces cerevisiae global repair requires both the RAD7 and RAD16 genes (11).
In humans several gene products implicated in TCR are defective in three rare hereditary disorders: XP, trichothiodystrophy (TTD) and Cockayne syndrome (CS) (1,12). Two of the complementation groups of CS (CS-A and CS-B) exhibit no defect in global repair, but they are severely impaired in TCR (13,14). Mutation analysis has shown that CSB is selectively involved in TCR (15). In S.cerevisiae, a gene homologous to CSB was identified and designated RAD26 (16). The rad26 disruption mutant is not sensitive to UV, cis-platinum or X-rays and grows normally, indicating that the gene does not have an essential function. CPD repair in the rad26 mutant at the gene level revealed that preferential repair of UV-induced CPDs in the TS of the active RPB2 gene is severely impaired (16). However, using high resolution repair analysis, Tijsterman et al. (17) showed that Rad26 is not required for efficient repair of the transcription initiation region, but only for the region concerned with the elongation stage of RNA polymerase II transcription.
We have established a model system in S.cerevisiae that permits the dissection of DNA repair at nucleotide resolution in the MFA2 gene. It entails a simple, efficient end-labelling system and has been used by us to investigate the removal of UV-induced CPDs in the MFA2 control or coding sequences (9,10). We previously mapped NER of CPDs in a rad16 strain and confirmed at nucleotide resolution that Rad16 is necessary for NER in the NTS of the MFA2 gene. However, in the non-transcribed upstream control region of the MFA2 TS, we detected NER in a rad16 mutant. Therefore, Rad16 is not absolutely required for the repair of CPDs in this upstream non-transcribed region (9). Here we report experiments that examine the role of Rad26 in repair of the MFA2 gene in MATa cells, where MFA2 is transcriptionally active.
MATERIALS AND METHODS
UV irradiation and DNA extraction
The rad26 (MATa rad26::HIS3 ade2 his3-532 trp1-289 ura3-52) mutant strain was grown in yeast complete medium at 28°C overnight to a density of ~4 × 107 cells/ml. UV irradiation of cells, DNA isolation and its incubation with T4 endonuclease V were all undertaken as described by Reed et al. (18). The RAD26 strain (MATa ade2-1 trp1-1 can1-100 leu2-3 112 his 3-11, 15 ura3) was similarly processed.
Probes/primers used for the analyses
For fragments obtained by restriction with RsaI: probe/primer 1, 5′-biotin-gatagcttttttACACCATCTACTACATAATTAATTGATAGTTTCCT-3′ (the sequence in lower case is an overhang modification); probe/primer 2, 5′-biotin-gatagctttttt-ACGGACTTGATGCACGTGAAAAACCATTATTTAAA-3′; primer 3, 5′-gatagcACGGACTTGATGCACGTGAAAAACCATTAT-3′; primer 4, 5′-gatagcACACCATCTACTACATAATTAATTGATAGT-3′. For fragments obtained by restri-ction with MnlI: probe/primer 1, 5′-biotin-gatagcttttttAAAGC-GAGAGGAAAAAGCTGTTGCATTACC-3′; probe/primer 2, 5′-biotin-gatagcttttttCTTTTCAGAGGATTTATCCTTCTGAG-TGGC-3′; primer 3, 5′-gatagcCTTTTCAGAGGATTTATCCTTCTGAGTGGC-3′; primer 4, 5′-gatagcAAAGCGAGAGGAAAAAGCTGTTGCATTACC-3′. For fragments obtained by restriction with AluI: probe/primer 1, 5′-biotin-gatagctttttt-CTATCATCTTCATACAACAATAACTACCA-3′; probe/primer 2, 5′-biotin-gatagcttttttCTAATGATGAGAGAATTGGAATAAATTAGT-3′; primer 3, 5′-gatagcCTAATGATGAGAGAATTGGAATAAATTAGT-3′; primer 4, 5′-gatagc-CTATCATCTTCATACAACAATAACTACCA-3′.
Purification and labelling of MFA2 fragments
An aliquot of 30 µg of yeast DNA was isolated for each sample from cells that were unirradiated or irradiated with 125 J/m2 UV at 254 nm and sampled either immediately or after 1, 2, 3 or 4 h repair times. DNA was restricted with 60 U of restriction enzyme (RsaI, MnlI or AluI; Promega) at 37°C for 1 h in a total reaction volume of 200 µl. The enzyme was removed by phenol/chloroform extraction and the DNA was then precipitated using 20 µl of 3 M NaAc and 220 µl of isopropanol. After centrifugation, the DNA pellets were resuspended in 100 µl of TE buffer. T4 endonuclease V was used to cut the CPDs at 37°C for 1 h. T4 endonuclease was removed by phenol/chloroform extraction followed by chloroform extraction. To the samples was added 1 pmol of biotinylated probe 1 to detect damage in the TS. The samples were denatured at 95°C for 5 min and then incubated at 60°C for 15 min to allow the probe to anneal to the MFA2 fragments (the length of the MFA2 fragment annealed to the probe will depend on the position of CPDs within the fragment). To each sample were added 100 µg of washed Dynabeads, which were mixed well. The mixtures were kept at room temperature for 15 min to allow the streptavidin-coated Dynabeads to bind to the biotin molecule at the 5′-end of the probe. The tubes were placed in a Dynal magnetic particle concentrator (MPC) and the beads with the attached MFA2 fragments were attracted to the wall of the tube. The MPC was used at subsequent steps to attract the fragments to the wall of the tube following the various washing and labelling steps. The supernatant containing the MFA2 NTS fragments and other genomic DNA fragments was removed to a fresh tube for purification of the MFA2 NTS following the same procedure as for the TS except using probe 2. The beads bound with MFA2 fragments were washed with 60 µl of washing buffer (5 mM Tris–HCl, pH 7.5, 0.5 mM EDTA, 1.0 M NaCl) at 60°C for 5 min and the buffer removed. The second and third washes were performed at room temperature with 60 µl of water. The MFA2 fragments were end-labelled with six [α-32P]dATP residues by T7 DNA polymerase (Amersham) using the six-T overhang in the probe as template. The labelling reaction was complete after 10 min at 37°C. The labelling medium was removed while the beads with the bound MFA2 fragments were kept on the wall of the Eppendorf tube. The order of retrieval of TS and NTS has no effect on the labelling result, e.g. probe 2 can be used to retrieve the NTS before TS purification.
Following two washes with 30 µl of TE, the labelled MFA2 fragments were eluted from the Dynabeads at room temperature by the addition of 3 µl of formamide loading buffer (95% formamide, 20 mM EDTA, 0.05% bromophenol blue). The eluted fragments were resolved by electrophoresis on a 6% denaturing polyacrylamide gel. The running buffer was TBE (89 mM Tris base, 89 mM boric acid, 2.5 mM Na2EDTA·2H2O) and electrophoresis was carried out in a field of 40 V/cm.
Sequencing of the MFA2-containing fragments and quantification of DNA damage and repair
RESULTS
Examining CPDs at the level of the nucleotide
To examine the repair of UV-induced CPDs in vivo at nucleotide resolution, cells were irradiated with UV and DNA was extracted as described in Materials and Methods. It should be noted that S.cerevisiae is quite UV resistant and repair is clearly measurable after the 125 J/m2 dose applied here and in our previous experiments. RsaI digestion yields an 869 bp MFA2 fragment covering the whole MFA2 transcription region, its upstream promoter region and its downstream transcription termination region. After restriction enzyme digestion and cutting at CPDs, MFA2 fragments were purified and labelled. Labelled fragments were eluted from the Dynabeads and loaded onto a sequencing gel. Typical autoradiographs from experiments examining CPD repair in the TS and NTS of RsaI restriction fragments of a RAD26 wild-type strain are shown in Figure 1. It is important to note that many repair trends cannot be easily distinguished by visual examination of these gels. Up to 50% variation can occur in loadings per lane. Thus repair is only quantifiable by analysis of total radioactivity versus that in given bands of a lane as estimated from the phosphorimage. Our images are shown to illustrate the quality of our gels and to emphasise that no spurious bands occur at non-CPD sites, which would be evidence of incomplete polymerisation during the end-labelling. The U lanes have a strong band at the top of the autoradiograph representing the intact MFA2 fragments. In other lanes (0–3), bands representing smaller fragments due to cutting at CPD sites emerged at the di- or poly-pyrimidine positions referenced from the sequencing ladder. The signal at a specific band compared to the total activity in a lane is indicative of the frequency of damage at that site and its decrease after a certain repair time is indicative of CPD repair. The putative TATA sequences and Mcm1 binding site are marked in dark grey, located in the control region at –83 and –209, respectively. Due to analysis being undertaken by a phosphorimager, the efficiency and accuracy of the gel quantification are greatly improved compared to our previous analysis of rad16 (9). Now we are able to focus accurately on repair trends for closely spaced CPDs within the control and coding regions. The quantified results for the RAD26 strain are shown in Figure 3. The repair of CPDs in the upstream control region of RAD26 is enhanced towards the transcription start point, except for the TATA box region, where CPD repair is slow compared with other CPDs nearby. In the upstream region close to the transcription start point, the rate for repair of a group of CPDs at –4 to –8 is enhanced to the TCR level observed in the MFA2 transcribed region. In the region after the termination of transcription, the repair rate drops.
Figure 1.
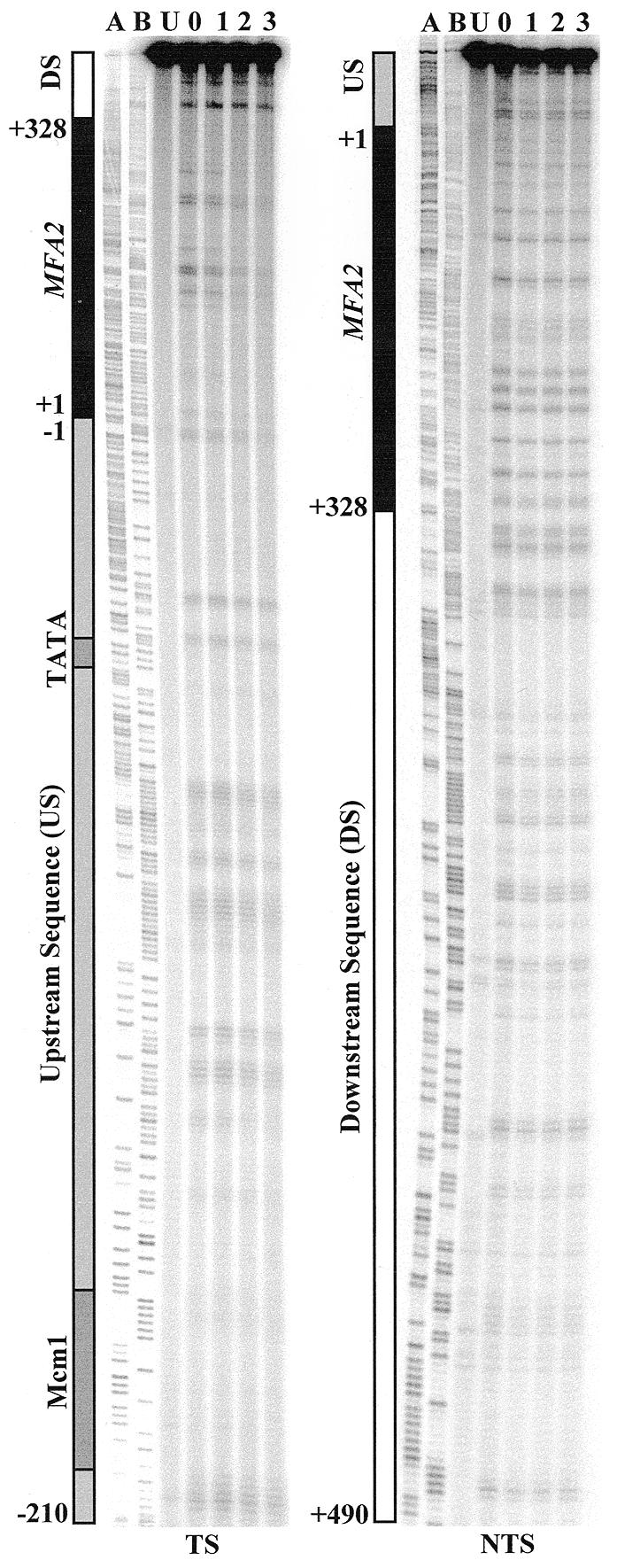
Typical autoradiographs depicting UV-induced CPDs in the TS and NTS of a RAD26 wild-type strain. CPD repair was examined in the RsaI restriction fragment, which contains the MFA2 transcribed region (black bar), its upstream sequence (US, grey bar) and downstream sequence (DS, white bar). The Mcm1 binding site and the TATA box are indicated by dark grey shading in the control region. Numbers on the left of the bars denote the nucleotide position in relation to the MFA2 transcription start point. Ladder A, G+A; ladder B, C+T. Lane U, DNA from unirradiated cells; lane 0, DNA from cells receiving 125 J/m2 UV and extracted immediately; lanes 1–3, DNA from cells that received UV but which were incubated afterwards in medium for these times (h) before extraction.
Figure 3.

CPD repair in RAD26 and rad26 cells. The autoradiographs were quantified with ImageQuant software (Molecular Dynamics). The time for repair of 50% of CPDs (t50%) at a given site was calculated or extrapolated. The t50% for slowly or unrepaired CPDs (t50% ≥ 10 h) is shown at the same level on the graph. Open diamonds, CPDs in the TS; closed diamonds, CPDs in the NTS. The horizontal bars at the bottom show the different regions of the RsaI fragment containing the MFA2 gene, and are as in Figure 1. Numbers beneath the bars denote the nucleotide position in relation to MFA2.
CPD repair in the MFA2 TS of the rad26 strain
Figure 2a depicts typical images detecting CPDs in the TS of the RsaI restriction fragment in DNA isolated from the rad26 mutant strain. Adjacent CPD bands in the whole control region were well resolved as distinct bands on the sequencing gel. In contrast, due to the length of the fragment being analysed, CPDs in the transcribed region and transcription termination region at the top of the gel are not always resolved into discrete bands. Using different enzymes and probes, these CPDs can be resolved as distinct bands. MnlI digestion yields a 225 bp MFA2 fragment, which permits a detailed analysis of CPD repair around the transcription start site (Fig. 2b). AluI restriction produces a 247 bp fragment containing the MFA2 transcribed sequence, and CPDs on this fragment are well separated on a sequencing gel (autoradiographs not shown).
Figure 2.

Typical autoradiograph depicting UV-induced CPDs in a rad26 strain. (a) CPD repair in the TS of the RsaI restriction fragment, which contains the whole MFA2 transcribed region and its upstream promoter and downstream transcription termination regions. (b) CPD repair in the TS of the MnlI fragment, which covers the sequences around the transcription initiation point. Lane U, DNA from unirradiated cells; lane 0, DNA from cells receiving 125 J/m2 UV and extracted immediately; lanes 1–4, DNA from cells that received UV but which were incubated afterwards in medium for these times (h) before extraction.
Analysis of the different restriction fragments on the above sequencing gels gave a comprehensive picture of CPD repair for the whole MFA2 TS (upstream control, transcription and downstream transcription termination regions). The data are presented in Figure 3 and are the averages of at least two experiments with each restriction enzyme (RsaI gives the whole region, whereas MnlI and AluI duplicate smaller regions within the RsaI fragment). Most of the CPDs on the TS were repaired at a similar rate, with a t50% of 4 h. However, heterogeneous repair is observed in some regions. Here, we report differences in repair for specific single CPDs or for more than one CPD in a poly-pyrimidine tract which are consistently observed in all experiments.
Progressing from the Mcm1 binding site to the transcription start point, repair of CPDs shows an increasing trend, from a t50% of 4 h at –216TTTTTTC–210 to a t50% of 2 h at –8TTTT–4, except for the TATA box area, where two group of CPDs were found with a t50% of 4 h. As shown in the upper histograms of Figure 3, for the region close to the transcription start point, CPDs at position +5CTC+7 are repaired with a t50% of 4 h, which matches the average repair rate for the whole of the transcription region. Fast repair of CPDs occurs in the region close to where transcription terminates, and a t50% of 2 h is observed at +313TT+314 and +316TCT+318. After the termination of transcription (downstream of +329), repair of CPDs was detected at the same rate as the average for the transcription region and for the NTS (t50% 4 h).
CPD repair in the MFA2 NTS of the rad26 strain
Analysis of the autoradiographs detecting CPDs in the MFA2 NTS of the rad26 strain (autoradiographs not shown) shows that (Fig. 3, lower histograms) although the majority of CPDs were repaired with a t50% of 3–4 h, heterogeneity exists for CPD repair. Three CPDs at +309CTTT+312 were repaired with a t50% of 1.8 h: this is the region where transcription of the complementary strand terminates. Thus, enhanced repair in this termination region has been observed in both the RAD26 wild-type and in the rad26 mutant when MFA2 is transcriptionally active. It should be noted that, as previously shown, CPDs further downstream of the MFA2 NTS region but which are still within the RsaI restriction fragment are in the end of an ORF running in the opposite direction. Here, enhanced repair of CPDs occurs due to TCR. This region will not be discussed further as it is not part of the MFA2 gene.
DISCUSSION
The NER pathway consists of two sub-pathways: TCR and global genome repair. The former sub-pathway operates on NER substrates in the TS of active genes so that they are removed at a faster rate compared to substrates removed via global repair of non-transcribed DNA. Although the NER model is widely accepted and nearly 20 NER essential repair proteins have been characterised, the molecular mechanisms of both NER sub-pathways in eukaryotes are still poorly understood. van Gool et al. (16) cloned and characterised RAD26, the yeast counterpart of human CSB. They showed that disruption of RAD26 gave viable cells that grow normally. However, preferential repair of CPDs in the TS of the active RPB2 gene is severely impaired in a rad26 mutant. This finding indicates that the yeast protein Rad26 is essential for TCR. Thus, the CSA, CSB and Rad26 proteins have been considered as TCR factors. However, the mechanism of their participation in TCR still remains to be elucidated.
In previous studies (9,10), we showed that the NER of CPDs in the MFA2 TS was enhanced in the upstream Mcm1 binding region. This is ~200 nt away from the MFA2 transcription start point, and in RAD26 cells, the closer to the transcription start point, the quicker the repair observed, except for CPDs in the TATA box region, where repair slows down. This enhanced repair rate was greatest at –8 to –4 and continued into the transcribed region, slowing down where transcription terminates. Our results in this study show that in the MFA2 upstream promoter region, similar enhanced repair generally occurred in the rad26 mutant as in the wild-type (9,10). However, slower repair in rad26 does occur for CPDs in the TATA box region. Here, t50% in RAD26 cells is 3–4 h and in rad26 cells is 4–5 h. The enhanced repair rate for the transcribed region in the wild-type (t50% 2 h) dropped in the rad26 mutant to the average repair level observed in the NTS region (t50% 4 h). In the region close to where transcription ends, CPDs at +313TT+314 and +316TCT+318 were repaired in rad26 cells as in the wild-type, i.e. at a t50% of 2 h. These data indicate that Rad26 does not influence NER of CPDs in the MFA2 upstream control region with the exception of those in the TATA box region. This result may be due to rad26 having a somewhat slower rate of transcription compared to the wild-type: a fact that may also influence the TCR result. However, no detectable significant difference in transcription of the RPB2 gene occurs in rad26 (J.Brouwer, personal communication). Thus, the TCR defect cannot be attributed solely to reduced transcription. For MFA2, Rad26 is required for TCR in the initiation region of the transcription unit and its downstream transcription sequences, except for some sites close to the end of transcription.
RNA polymerase II promoters are composed of core and regulatory regions. The core regions comprise a TATA box and the transcription start site. TATA box binding protein (TBP) and other TBP-associated factors (TAF) bind the TATA box in the first step of forming a transcription initiation complex that comprises RNA polymerase II, TBP, TAFs, TFIIB, TFIIE, TFIIF, TFIIH and other transcription factors. This TATA box binding of the complex may impair the accessibility of the NER proteins to CPDs close to this binding region, thus accounting for the reduced NER in this region. Among the transcription factors mentioned above, TFIIH plays a dual role in both transcription and NER and this may explain the TCR pathway. Yeast TFIIH exists in two different forms. Core TFIIH is active in NER and consists of five subunits, including the repair protein Rad3, TFB1 and Ssl1; holo-TFIIH is active in transcription and consists of core TFIIH and the CTD kinase complex, TFIIK, which is essential for transcription and dispensable for NER (19–21).
The results at the level of the nucleotide in our study indicate a significant role for Rad26 in TCR operating on the whole of the MFA2 transcribed region. In vitro experiments showed that active NER significantly limits the extent of RNA polymerase II transcription. This inhibition of transcription is relieved by supplementing holo-TFIIH, but not core TFIIH. Furthermore, RAD26 is required for this NER-dependent transcription (22). When RNA polymerase II stalls at a damage site, it is possible that holo-TFIIH is disassembled to the core TFIIH and TFIIK and then core TFIIH is recruited to form a repairosome with other repair proteins. Whether holo-TFIIH dissociates from the RNA polymerase II complex before forming a repairosome is not clear. Tijsterman et al. (17) suggested a putative role of Rad26 in the recruitment of TFIIH in the repairosome. They found that Rad26 is not needed for efficient TCR of CPDs positioned within approximately 50 bases from the transcription initiation site in the transcribed strand of the RPB2 and URA3 genes. Thus they proposed that this protein is only needed for TCR in the elongation stage of RNA polymerase II transcription, but that it is not needed for TCR in the transcription initiation region. In contrast, a requisite for Rad26 in efficient TCR of the transcription initiation region and the elongation region of the MFA2 gene was observed in our study.
It should be borne in mind that the differences between the regions where TCR operates for MFA2 versus RPB2 and URA3 could reflect the extent of transcription from these loci. It is possible that TCR profiles for lowly transcribed genes differ from those for genes where transcription is greater. Experiments examining TCR of the Escherichia coli tRNA Tyr gene have demonstrated differences in the extent of TCR in relation to the amount of transcription (23).
In the MFA2 upstream control region, NER proceeds with characteristics different from both global repair and TCR, since (i) we detected that the global repair protein Rad16 is not absolutely required for this region and (ii) the quick repair of CPDs close to the transcription initiation point is not due to TCR because the sequence is not transcribed (9; this paper). Therefore, we speculate that an interim repair pathway exists for repairing such a region where a global repair to TCR transition occurs. For the MFA2 gene, CPD repair in this region is mainly Rad26-independent, the exception being CPDs in the TATA box region. Here, it is possible that there are roles in repair for nucleosome positioning and/or histone acetylation and binding of regulatory or transcriptional proteins. Studies on repair performed in conjunction with footprinting will assist in resolving this aspect. Once transcription starts, Rad26 is required for TCR, presumably via the interchange between holo-TFIIH and the core TFIIH.
Surprisingly, faster repair is observed for CPDs in the region (+313TT+314 and +316TCT+318) close to the end of transcription. Here, TFIIH may take a form favourable for forming a repairosome without Rad26 assistance. However, we cannot rule out the possibility that the organisation of DNA into nucleosomes/chromatin or its conformation with respect to the termination of transcription can create faster repair spots. CPDs in the same region of the MFA2 NTS (+309CTTT+312) are repaired faster than CPDs nearby in the same strand. Hence, any mechanism must account for enhanced NER irrespective of the strand involved. Mapping DNA repair at the level of the nucleotide with respect to the position of nucleosomes and with respect to the ability to terminate transcription of MFA2 at this site will address this issue.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Dr S. Reed for supplying the yeast strains and Dr A. Scott for reading of the manuscript. This work was supported by the MRC (G99 00118).
REFERENCES
- 1.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. American Society of Microbiology Press, Washington, DC.
- 2.Sancar A. (1996) Annu. Rev. Biochem., 65, 43–81. [DOI] [PubMed] [Google Scholar]
- 3.Wood R.D. (1996) Annu. Rev. Biochem., 65, 135–167. [DOI] [PubMed] [Google Scholar]
- 4.Mellon I. and Hanawalt,P.C. (1989) Nature, 342, 95–98. [DOI] [PubMed] [Google Scholar]
- 5.Mellon I., Spivak,G. and Hanawalt,P.C. (1987) Cell, 51, 241–249. [DOI] [PubMed] [Google Scholar]
- 6.Smerdon M.J. and Thoma,F. (1990) Cell, 61, 675–684. [DOI] [PubMed] [Google Scholar]
- 7.Leadon S.A. and Lawrence,D.A. (1992) J. Biol. Chem., 267, 23175–23182. [PubMed] [Google Scholar]
- 8.Verhage R., Zeeman,A.M., Degroot,N., Gleig,F., Bang,D.D., Vandeputte,P. and Brouwer,J. (1994) Mol. Cell. Biol., 14, 6135–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teng Y., Li,S., Waters,R. and Reed,S.H. (1997) J. Mol. Biol., 267, 324–337. [DOI] [PubMed] [Google Scholar]
- 10.Teng Y.M., Longhese,M., McDonough,G. and Waters,R. (1998) J. Mol. Biol., 280, 355–363. [DOI] [PubMed] [Google Scholar]
- 11.Terleth C., Schenk,P.W., Vansluis,C.A. and Vandeputte,P. (1990) Mutagenesis, 5, 78. [Google Scholar]
- 12.Boyer J.C., Kaufmann,W.K., Brylawski,B.P. and Cordeirostone,M. (1990) Cancer Res., 50, 2593–2598. [PubMed] [Google Scholar]
- 13.Venema J., Mullenders,L.H.F., Natarajan,A.T., Vanzeeland,A.A. and Mayne,L.V. (1990) Proc. Natl Acad. Sci. USA, 87, 4707–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Hoffen A., Natarajan,A.T., Mayne,L.V., Vanzeeland,A.A., Mullenders,L.H.F. and Venema,J. (1993) Nucleic Acids Res., 21, 5890–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Troelstra C., Vangool,A., Dewit,J., Vermeulen,W., Bootsma,D. and Hoeijmakers,J.H.J. (1992) Cell, 71, 939–953. [DOI] [PubMed] [Google Scholar]
- 16.van Gool A.J., Verhage,R., Swagemakers,S.M.A., Vandeputte,P., Brouwer,J., Troelstra,C., Bootsma,D. and Hoeijmakers,J.H.J. (1994) EMBO J., 13, 5361–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tijsterman M., Verhage,R.A., van de Putte,P., Tasseron-de Jong,J.G. and Brouwer,J. (1997) Proc. Natl Acad. Sci. USA, 94, 8027–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reed S.H., Boiteux,S. and Waters,R. (1996) Mol. Gen. Genet., 250, 505–514. [DOI] [PubMed] [Google Scholar]
- 19.Feaver W.J., Svejstrup,J.Q., Bardwell,L., Bardwell,A.J., Buratowski,S., Gulyas,K.D., Donahue,T.F., Friedberg,E.C. and Kornberg,R.D. (1993) Cell, 75, 1379–1387. [DOI] [PubMed] [Google Scholar]
- 20.Svejstrup J.Q., Wang,Z.G., Feaver,W.J., Wu,X.H., Bushnell,D.A., Donahue,T.F., Friedberg,E.C. and Kornberg,R.D. (1995) Cell, 80, 21–28. [DOI] [PubMed] [Google Scholar]
- 21.Svejstrup J.Q., Vichi,P. and Egly,J.M. (1996) Trends Biochem. Sci., 21, 346–350. [PubMed] [Google Scholar]
- 22.You Z.Y., Feaver,W.J. and Friedberg,E.C. (1998) Mol. Cell. Biol., 18, 2668–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li S.S. and Waters,R. (1997) J. Mol. Biol., 271, 31–46. [DOI] [PubMed] [Google Scholar]