

Abstract
75Cancer research has found in the recent years a formidable ally in liquid biopsy, a noninvasive technique that allows the study of circulating tumor cells (CTCs) and biomolecules involved in the dynamics of cancer spread like cell-free nucleid acids or tumor-derived extracellular vesicles. However, single-cell isolation of CTCs with high viability for further genetic, phenotypic, and morphological characterization remains a challenge. We present a new approach for single CTC isolation in enriched blood samples using a liquid laser transfer (LLT) process, adapted from standard laser direct write techniques. In order to completely preserve the cells from direct laser irradiation, we used an ultraviolet laser to produce a blister-actuated laser-induced forward transfer process (BA-LIFT). Using a plasma-treated polyimide layer for blister generation, we completely shield the sample from the incident laser beam. The optical transparency of the polyimide allows direct cell targeting using a simplified optical setup, in which the laser irradiation module, standard imaging, and fluorescence imaging share a common optical path. Peripheral blood mononuclear cells (PBMCs) were identified by fluorescent markers, while target cancer cells remained unstained. As a proof of concept, we were able to isolate single MDA-MB-231 cancer cells using this negative selection process. Unstained target cells were isolated and culture while their DNA was sent for single-cell sequencing (SCS). Our approach appears to be an effective approach to isolate single CTCs, preserving cell characteristics in terms of cell viability and potential for further SCS.
Keywords: Liquid biopsy, Single-cell isolation, Laser-induced forward transfer, Laser direct write
1. Introduction
Cancer cells released from a solid tumor and traveling in the peripheral blood are known as circulating tumor cells (CTCs). Although their existence was reported in 1869[1], they have only become a central subject in oncological research in the last few years, mainly 76in coincidence with the deployment of several different techniques for CTCs analysis[2]. Directly linked with cancer metastasis, CTCs detection and characterization alone or with other cancer biomarkers is considered a valuable tool in disease prognosis and progression monitoring[3-5].
It has been extensively reported that metastatic dynamics of solid tumors is linked to the circulation of tumor cells, leaving the primary tumor and traveling through the peripheral blood to colonize other tissues[6-8]. In addition, these cells appear to be responsible for more complex tumor dissemination processes like those based on tumor self-seeding[9]. Therefore, an accurate monitoring of CTCs and specially a full liquid biopsy in cancer patients is of great importance for clinical oncology[10].
Isolation of CTCs from body fluids is a liquid biopsy method that can be used both for early diagnosis, which may lead to a selection of more suitable treatments, as well as for real-time monitoring of the disease progression[11]. Molecular analysis at the single-cell level by single-cell sequencing (SCS) allows the complete characterization of the amplified genetic material, offering a huge advantage for the characterization of both cell populations with limited number or singular cell populations[12-14]. Moreover, a comprehensive phenotypic profiling of heterogeneous CTCs at single-cell resolution would have an important impact on cancer management[15-17]. Therefore, the appropriate combination of liquid biopsy with SCS would be useful to monitor the patient’s response to a particular treatment or the disease evolution. Once the prognostic or predictive role of these biomarkers is confirmed, they could be used in developing targeted therapies[18-20]. However, the effective capture of CTCs with different phenotypic characteristics and with the potential of subpopulation sorting is still a goal that is not completely fulfilled.
Although several technologies have been developed or adapted for CTCs isolation in the last few years, cell isolation remains a challenge, in particular when good cell viability is required. The main problem is related to the very low concentration of CTCs in cancer early stages (1 CTC per 106–109 blood cells), making it challenging in terms of sensitivity and specificity of the detection technologies[5,21]. Due to the low concentration of CTCs, enrichment-based methods for isolation are currently preferred for CTCs detection and analysis.
Different enrichment methods based on physical and/or biological properties have been proposed, and in the last years, a number of reviews[3,18,19,22-25] have summarized the pros and cons of the available technologies and the challenges of implementation in clinical practice[26,27]. Immunomagnetic techniques for positive and negative selection of CTCs are probably the most common approaches for enrichment[28], with CELLSEARCH® being the only FDA-approved test for enumeration of CTCs of epithelial origin[29]. However, immunomagnetic a-EpCAM positive selection approaches need to seed the sample with ferromagnetic particles, thus implying the direct tagging of CTCs, which is not compatible with the idea of maintaining the CTCs as unaltered as possible. In addition, some works defend the clinical utility of non-EpCAM assays due to the limitations of the immunomagnetic techniques to detect nonepithelial CTCs of clinical relevance in many tumors[30].
On the other hand, biophysical methods for enrichment have the advantage of avoiding labeling, addressing the separation of CTCs through their distinctive physical properties (morphological, electrical, and mechanical). Among them, size-based isolation methodologies (due to the larger size of CTCs, 12–25 μm, compared with surrounding cells) have a huge potential in maintaining the CTCs unmodified[2,31]. Microfluidic devices, microfiltration, dielectrophoresis, and other related techniques including approaches based on advanced lab-on-a-chip concepts or even advanced programmable materials[32–37] appear as well suited as enrichment methods. However, in many of these approaches, the subsequent CTCs isolation from the enriched liquid remains a challenge. There is an unmet need to increase the purity and concentration of CTCs fraction, as well as to maintain cellular viability in order to facilitate both the molecular characterization and the possibility of performing in vitro studies.
For the detection and selection of the appropriate cells for isolation, the technologies are intimately linked to the enrichment process. Moreover, standard optical technologies (especially those related to fluorescence detection for negative or positive selection) are relevant for accurate selection of appropriate targets for final isolation and sequencing[38].
2. Lasers in CTCs research
Laser technology is currently a widely used tool in CTCs studies. Used as a coherent light source for illumination in microscopy and fluorescence set-ups, especially for detection, laser technology is employed as well for subpopulation separation in approaches for CTCs isolation and sorting using flow cytometry[21,39]. However, lasers could play a fundamental role in the most challenging step in CTCs analysis: isolation of viable cells. In fact, laser capture microdissection (LCM), a widely used technique for cell isolation of complex tissue[40,41] has been proposed for CTCs isolation, both in combination with microfluidic devices for previous enrichment and as a part of sophisticated techniques that requires previous CTCs encapsulation in hydrogels[42,43]. However, these 77approaches, besides comprising an important number of complex processes in sample preparation, in some cases imply a final laser pressure catapulting process (LCP) of the sample in which direct irradiation, normally in the ultraviolet (UV) range, is unavoidable[44]. More recently sophisticated processes using laser induced isolation of microstructures with CTCs, and using IR laser light to transfer the functional structure have been proposed as well[45].
In this work, we propose a completely new approach for CTC isolation in the classical flow sequence for CTCs sample treatment of enrichment-identification-isolation but using a liquid laser transfer (LLT) process for the last step. This transfer approach is based on an adaptation of the blister-actuated laser-induced forward transfer (BA-LIFT) direct write technique that completely avoids the interaction between the laser photonic field and the cells to be isolated (Figure 1). This work demonstrates the feasibility of this approach with a first proof of concept, isolating cells of a breast cancer line (MDA-MB-231) in the negative selection and later SCS of the isolated cells.
Figure 1.
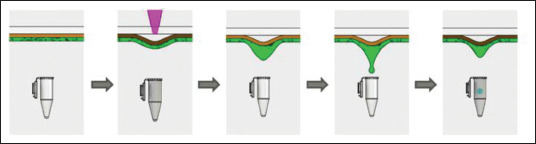
Liquid laser transfer for CTC isolation.
In our approach, we adapted the BA-LIFT technique, using a thick adhesive polyimide layer conveniently treated to improve hydrophilicity, to produce a droplet containing the previously identified and selected CTCs using a single laser pulse. The CTCs were recovered in a tube or culture well for further SCS and/or viability studies. The process is conceptually presented in Figure 1 and discussed in next sections.
2.1. Cell isolation using laser direct write
Laser direct write techniques have been considered for cell isolation in different fields[46], with those based in laser-induced forward transfer (LIFT) approaches being the most currently envisaged[47]. LIFT refers to a group of laser-based direct write techniques developed originally for art graphics and semiconductor industry and now applied in different technological and scientific fields. Due to the interest in many different technological and industrial sectors, an increasing number of reviews about the technology are available[48–53].
Most LIFT approaches use a pulsed laser beam to transfer material from a donor substrate to an acceptor surface or any other receptacle. In its simplest configuration, LIFT is performed irradiating the interface between a transparent substrate and the material to be transferred, previously deposited forming a thin layer onto the substrate surface. Conversion of laser energy into kinetic energy of part of the material allows the transfer of small volumes with high spatial accuracy, providing a printing method with spatial resolution in the order of 1 μm. The process dynamics is strongly dependent on the physical properties and thickness of the material to be transferred, the gap between acceptor and donor substrate, the configuration of the donor substrate, and the parameters used in the laser irradiation process[52]. The main advantage of the technique, if compared with other mature printing technologies like inkjet or extrusion, is the capacity to transfer material in an enormous range of viscosities and rheological behavior, from solid-state to low-density inks. In addition, the capability of the technique to transfer small volumes of material (as low as picoliters) and the high lateral spatial resolution (down to a few micrometers) to produce printed patterns are remarkable.
LIFT has attracted huge attention as bioprinting technique as well, and in the last 20 years, several works have demonstrated applications of 2D and 3D printing of biomolecules and living cells both for in vitro and in vivo approaches[52-61], including single cell isolation[46,47] and using different laser pulse duration regimes[62]. In addition, it has been extensively shown that LIFT is one of the bioprinting techniques with the highest cell viability, even better than the widely applied extrusion or inkjet printing methods. It guarantees in general the DNA integrity after the transfer process, despite the fact that the cells could suffer some mechanical stresses due to the dynamics of the liquid transfer process, has it has been published in a very recent work even for cancer cell lines[63]. However, the fact that direct laser irradiation and/or heating of the media containing the cells could affect cell functionality in any extent beyond DNA integrity has been a constant concern in this field, and a number of approaches envisaged the 78problem of avoiding direct laser irradiation of the cells. Therefore, set-ups comprising a transparent donor substrate coated with an energy absorption layer, acting as a sacrificial layer, have been proposed to avoid direct interaction between the laser light and the liquid media containing living cells. A common approach is to use a thin sacrificial absorption layer made of a biocompatible metallic material (titanium, silver, gold, etc.)[52,53,64-67]. This thin film acts as a dynamic release layer, whose thermal expansion after laser irradiation propels the liquid generating a jet that finally collapses, producing the desired droplet transfer. Although good cell viability after printing has been demonstrated with sacrificial metallic layers, some heating and residual contamination of the liquid is unavoidable. For that reason, when using very sensitive biological samples[68] or when a fundamental goal is to maintain the cells as intact as possible, other approaches can be envisaged. In our work, we consider it is a priority to protect the cells not only from direct laser irradiation but also from a possible residual heating by designing a specific approach based on the standard BA-LIFT process.
2.2. Adaption of the BA-LIFT technique for single-cell isolation
Experimental set-up BA-LIFT uses an intermediate layer between the donor substrate and the liquid but made of a polymer that, under laser irradiation, partially vaporizes creating a vapor bubble that induces an elastic and plastic deformation in the remaining material (Figure 2). Consequently, a blister in the viscoelastic material is produced whose dynamics induces mechanical propulsion of the liquid. The process has been widely studied[53,69-72] mainly for polyimide sacrificial layers with thickness up to 10 μm deposited by spin-coating. Some of the authors of this work have previously discussed the potential of this approach for cell identification and sorting with very good survival results for different types of cells, including mouse hematopoietic progenitor stem cells[73].
Figure 2.
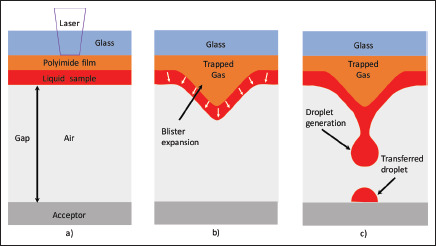
The physical basis of BA-LIFT transfer. The pulsed UV laser ablates a small portion of the polyimide film (a), generating a hot gas that produces a plastic deformation in the remaining polymer layer (b). The blister generated in this way pushes the liquid layer, leading to a jet formation that, after collapsing by surface tension, produces the droplet that is transferred to an acceptor substrate or any other receptacle (c). In this case, the droplet contains the selected cell.
In our approach, described in detail in the Methods section, we used a donor structure made with a commercial polyimide film with a thickness of 30 μm. The main advantage of using thick polyimide films is its high absorption in the UV range, efficiently absorbing the laser beam and avoiding direct cell irradiation or even heating of the liquid sample[62,69,73]. On the other hand, the use of polyimide presents other advantages: the material is partially transparent to the visible light so fluorescence-vision and techniques for cell recognition and selection can be used, even in a compact optical arrangement in which fluorescence and vision arrangements share the same optical path that the laser beam used to produce the liquid transfer[73].
3. Methods
3.1. Cell isolation using laser direct write
MDA-MB-231 cell line (derived from breast cancer), obtained from ATCC (American Type Culture Collection (ATCC), Manassas, VA) was cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cells were maintained in a humidified atmosphere of 5% CO2 in air. Following culture, cells were harvested using 790.05% trypsin, and used only when its viability exceeded 90% as assessed by Trypan Blue exclusion.
3.2. Analytical blood samples
Analytical samples were prepared to simulate cancer samples in experiments that rely on knowing the starting concentration of target cells. Peripheral blood was obtained from healthy donors. To collect their samples as part of the C.0005266 collection (“Research in Cancer”) for biomedical research purposes, the informed consent was previously approved by the corresponding Ethics and Scientific Committees, obtained from the donor prior to specimen collection.
Blood was drawn into EDTA tubes (Vacutainer BD, Franklin Lkes, NJ) and subsequently spiked with on cancer cell lines. MDA-MB-231 was used both for analytical instrument performance validation experiments and for TP53 and BRAF assay validations because this line harbors a known homozygous TP53 c.839G>A p.R280K mutation and heterozygous BRAF c.1391G>T p.G464V mutation. Cells suspended in a stock solution were first counted on Neubahuer chamber and then spiked into 7.5 mL whole blood.
3.3. Sample preparation
Whole blood samples were added with different concentrations of MDA-MB-231 cell line (1000, 10,000, 100,000, and 1,000,000 cells).
These blood tubes were processed to recover the fraction of peripheral blood mononuclear cells from where the MDA-MB-231 cell line was also identified by density. LeucoSep tubes (Greiner Bio-One, Monroe, NC) were prepared by adding 15 mL of Ficoll-Paque Plus (d = 1.077gr / mol; GE Healthcare, Pittsburgh, PA). The peripheral blood mononuclear cell fraction, together with the MDA-MB-231, was recovered and resuspended in 500 μL of MACS buffer. Cell count was performed and the cell pellet was resuspended in 80 μL of buffer MACs and 20 μL of CD45 MicroBeads per 107 total cells. The mixture was incubated for 15 min at 4°C. Then, the sample was added to MS column (MACS, Miltenyi Biotec). In this study, the relationship between MDA-MB-231 and PBMC does not reflect the concentration of CTC in the blood (1 CTC / 106–109 PBMCs), because as proof of concept, we only wanted to confirm that the technology is able to discriminate between different subpopulations.
3.4. Staining
Cells were stained with a phycoerythrin-conjugated monoclonal antibody against CD45 (a-CD45/PE, Biolegend, 368510). Cells were washed two times using 200 μL 1× phosphate-buffered saline (PBS), and then stained with 0.05 μg (2 μL) of a-CD45/PE for 30 min at 4°C in the dark. After that, cells were washed two times with 200 μL 1× PBS, resuspended in a suitable volume of 1× PBS, and stored shortly at 4°C before use.
3.5. Donor substrate preparation
Donor substrate was made from a soda lime glass microscope slide (Thermo Scientific AAAA000001##02E) and laser-cut to a final dimension of 12.5 × 12.5 mm each. Two layers of commercial polyimide tape were adhered to the slide covering all of its surfaces: the first one (Caplinq PIT0.5S-UT/19) with a thickness of 30 μm and the second one (Caplinq PIT1S/19) with a thickness of 70 μm on the top. Once both tapes were added to the square slide, a 5-mm diameter hole was laser-cut at the center of the topmost tape and carefully removed to leave a hole with a depth equal to the thickness of the outermost tape (70 μm). This hole worked as a microwell to contain the liquid with the cells. All slides were cleaned thoroughly with 70% v/v ethanol and left under UV light before used. Slides were stored inside a class II biological safety cabinet kept in a sterile environment.
To assure a homogenous and thin layer of liquid in the circular well, and due to the wettability properties of polyimide, a surface treatment to improve hydrophilicity was needed before the liquid deposition that happened prior to the laser transfer process. Therefore, the sample was placed inside a standard vacuum desiccator (Duran 247824604) with both the lid and stopcock sealed with high vacuum grease (Dow Corning). Immediately after that, air was evacuated using a rotary vane pump (Busch 016-112) in 12 s to a pressure of approximately 1000 mTorr. The desiccator was placed inside a microwave source of 2450 MHz and a power of 700 W (LG MS-1924W/02), letting the air plasma formed act for 10 s on the sample. Plasma-activated donor substrate was left to cool down for 30 s, and 1.5 μL of spiked sample (MDA-MB-231+PBMCs) was placed inside the circular well and spread evenly along the whole area. Donor substrate was then turned upside down and placed on top of a 0.2-mL PCR tube (Fisherbrand™). The tube was fixed on a 96-well PCR plate and the sample was kept on its place using a 3D-printed fixture that adapt to the 96-well plate. Finally, the sample was taken to the laser system for processing.
3.6. Laser system
Laser system comprises a UV laser (Crylas FTSS355-Q4) emitting 1.3 ns single pulses at demand with a wavelength of 355 nm. Laser pulse energy was controlled in the range of 1 to 23 ± 2 μJ by means of a half-wave plate and a beam polarizer. Laser beam was focused onto the sample down to a beam waist at focus of 10 μm using a 10× microscope objective.
80Focusing optics were fixed and the beam was scanned along the sample by moving the sample using lineal servo XY axes (Parker) with an accuracy of 1 μm. The distance between the sample and the laser beam focal point was controlled using a manual micrometric Z stage with and accuracy of 10 μm. The laser was fired directly to the location of the desired cell, which was centered in the vision system; in that way, the possible mechanical damage induced by the shear stress in the edges of the affected volume of liquid during jet formation can be minimized.
Laser pulse energy of 5 μJ is typically used for transferring cells with single laser pulses, resulting in a mean laser fluence of 3.5 J/cm2. With these parameters, the volume of the transferred droplet containing the cell is around 50 pL.
3.7. Fluorescence system
In our experiments, the optical set-up used combines laser irradiation for LIFT process, in situ video recording and analysis of the sample fluorescence (Figure 3). The focusing optics are common to the different optical systems and consist of a 10× microscope objective, while the laser beam path, bright field microscopy and fluorescence microscopy are combined by means of appropriate dichroic mirrors. As it has been previously detailed, the laser source used for BA-LIFT is a UV, ns-pulsed laser, delivering 3.5 J/cm2 laser pulses, in terms of fluence, at the sample.
Figure 3.
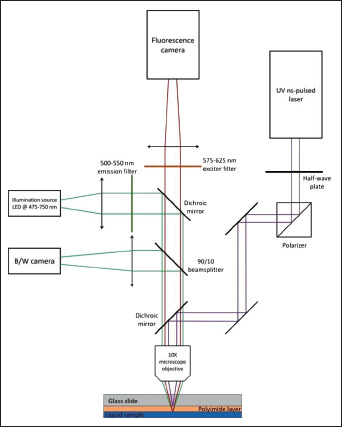
Optical set-up for laser transfer and cell identification and selection. Our arrangement combines laser irradiation for BA-LIFT process, in situ video recording and analysis of the sample fluorescence. The focusing optics are common to the different optical systems.
The microscope objective used for focusing the laser beam onto the samples was also used for obtaining in situ, real-time microscope images of the processed sample with two independent CMOS cameras, with one recording bright field B/W images (Thorlabs DCC1545M) and the other recording fluorescence images (Andor Zyla 5.5). UV laser and visible microscopy optical paths are combined by means of a UV dichroic mirror just before the focusing optics.
A LED source (Thorlabs M565L3) emitting at a wavelength range from 475 to 750 nm with peak intensity at 550 nm was used for illuminating the sample. LED light was initially filtered using a single band exciter filter (Semrock FF01-531/40) with high transmission from 500 to 550 nm and a fluorescence dichroic mirror (Semrock FF562-Di03) cutting at 562 nm. The reflected light returning from the sample was divided in two 81using a 90/10 beam splitter: 10% of the light intensity was deflected to B/W camera for recording a bright field image of the sample, and the other 90% light intensity passed through beam splitter, fluoresce dichroic mirror, and fluorescence emitter filter (Semrock FF01-593/40), which lets pass only wavelengths from the range of 575–625 nm, corresponding to the emission wavelength of the fluorophore used for staining the cells. Filtered fluorescence light was recorded using the fluorescence camera. Micro-Manager 2.0 beta software was used for the analysis of both bright field and fluorescence live images. Bright field images were taken as recorded, while an orange LUT (look-up table) was applied to the fluorescence image. Both images were compared, differentiating the nonstained cells and the stained fluorescent (Figure 4). Once the desired cells were localized, the sample was moved with respect to the fixed focusing optics by means of a motorized XY stage and manual Z stage in order to laser-point the target cell and transfer them using a single laser pulse (Figure 5).
Figure 4.
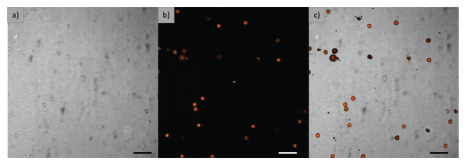
Cell detection. (a) Optical microscopy view, (b) fluorescence view, and (c) digital superposition image of both optical and fluorescence images. Dimension bar represents 100 μm.
Figure 5.

Cell isolation. (a) Detected nonstained cell. (b) Image after laser irradiation: the shadow on the left is due to blister formation after laser transfer process at the cell location. Dimension bar represents 100 μm.
3.8. Extraction and amplification of DNA
DNA of laser-isolated cells was extracted and amplified with the REPLI-g Single Cell kit (QIAGEN), which carries out isothermal genome amplification utilizing a uniquely processive DNA polymerase capable of replicating up to 100 kb without dissociating from the genomic DNA template. Phi 29 polymerase has a 3′–5′ exonuclease proofreading activity to maintain 1000-fold fidelity higher than that of Taq Polymerase during replication. Multiple displacement amplification technology was used in the presence of exonuclease-resistant primers to achieve high yields of DNA product.
Laser-isolated MDA-MB-231 DNA extraction was performed on 4 μL of PBSsc (REPLI-g Single Cell kit) in a PCR tube, accordingly to manufacturer’s instructions.
3.9. Polymerase chain reaction
DNA from laser/isolated MDA-MB-231 was quantified and amplified by PCR using primers for the exon 9 of TP53 82gene and for the exon 11 of BRAF gene, all of which include mutational hot spots.
The primers used were as follows:
-
• TP53 forward primer:
5'-GGCTTTGGGACCTCTTAACC-3'
-
• TP53 reverse primer:
5'-TAACTGCACCCTTGGTCTCC-3'
-
• BRAF forward primer:
5'-CACTTGGTAGACGGGACTCG-3'
-
• BRAF reverse primer:
5'-GTTTATTGATGCGAACAGTGA-3'
PCR products were analyzed by agarose gel electrophoresis coupled with fluorescence detection.
3.1. Sanger sequencing
DNA sequencing is considered the gold standard for detection of mutations. This method allows determination of the order of nucleotides in a target DNA sequence. The Sanger chain-termination method is commonly used in molecular diagnostic laboratories. In this method, the incorporation of a chemically modified nucleotide (dideoxynucleotide) terminates extension of the DNA strand at the point of incorporation. This results in a mixture of DNA fragments of various lengths. Each dideoxynucleotide (A, T, C, or G) was labeled with different fluorescent dye, allowing their individual detection. The newly synthesized and labeled DNA fragments were separated by size through capillary gel electrophoresis. The fluorescence was detected by an automated sequence analyzer (Applied Biosystems 3130xl y 3730xl genetic analyzer) and the order of nucleotides in the target DNA was depicted as a sequence electropherogram.
3.11. Immunofluorescence staining
After blocking by 10% FBS for 10 min, cells were incubated with a-CD45/PE clone (2D1) (Biolegend), then permeabilized with 0.2% Triton for 10 min, stained with anticytokeratin-FITC human clone (CK3-6H5) (Miltenyi Biotec) for 60 min, and finally mounted with Hoechst 33342.
Trypan Blue (Sigma) staining was used to determine viability of the transferred cells. The cells that are not stained by Trypan Blue are considered viable, whereas those stained by Trypan Blue were considered nonviable. In this study, we did not perform any other experiments to evaluate cell viability.
3.12. Cell viability and proliferation assay
Trypan Blue (Sigma) staining was used to determine viability right after the transference process. Cells without Trypan Blue staining were considered viable, whereas those stained with Trypan Blue were considered nonviable. CTC-stained cells were selected and isolated in 96-well plates containing RPMI medium (GibcoTM), 10% of FBS (Sigma) and 10% of acclimated medium (from established cultures from which cells were removed), under normal cell incubation environment (37°C, 5% CO2). Proliferation was observed after 9 days and maintained at day 14.
4. Results and discussion
4.1. Single CTC isolation using BA-LIFT and SCS
Our assessment experiments used a classic approach in which a CTC was defined as a CK-positive, CD45-negative, nucleated intact cell. In our case, MDA-MB-231 (cancer cell line from human breast carcinoma) simulates the characteristics of a true CTC. As indicated previously, the first step before CTC isolation comprises the enrichment of the sample. This step consists of cell blood subpopulation separation by Ficoll density gradient centrifugation; CTCs will be present in the peripheral blood mononuclear cell (PBMCs) fraction[74]. Depending on the approach followed for cell identification, the pellet should be fixated or not and the staining could be used for positive selection (EpCAM, Vimentin or another specific CTCs markers) or negative selection (CD45 leukocyte staining) (Figure 6).
Figure 6.
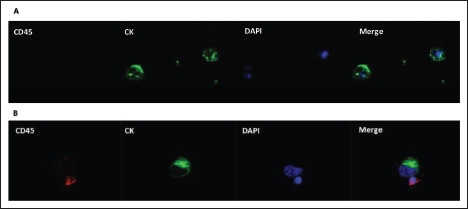
Double-immunofluorescence (CK/CD45) in MDA-MB-231 isolated using liquid laser transfer. (A) Cells were double-stained with anti-CK (clone CK3-6H5) human antibody conjugated with FITC and anti-CD45 (clone 2D1) human antibody conjugated with PE. Original magnification, 100×. (B) PBMCs were used as CD45 positive control. Abbreviation: CK, cytokeratin.
In this study, we performed a negative selection using a phycoerythrin-conjugated monoclonal antibody against CD45 (a-CD45/PE, clone 2D1, Biolegend). Nonfixated cells were incubated for 30 min at 4°C. Once the liquid sample has been prepared and deposited in the donor substrate, cells to be transferred were identified with the vision and fluorescence systems previously described.
MDA-MB-231 cells (negative for CD45) were transferred by shooting a single laser pulse at the cell location with a focused laser beam diameter of approximately 20 μm and an effective laser pulse energy of 5.5 μJ, resulting in a laser fluence of 3.5 J/cm2. This laser transfer system allows us to isolate the number of cells that we determine and collect them in the most suitable platform for the experiments.
For our proof of concept, a known number of MDA-MB-231 cells suspended in PBS were spiked into 7.5 mL of whole blood. We performed two types of transfer experiments. First, we addressed the problem of isolating a single cell in a tube. Second, we isolated 25 cells, which were transferred one by one into a tube. In both types of experiments, the collector tubes contain 4 μL of PBS. Once the cells were transferred, DNA was extracted and amplified with REPLI-g Single Cell kit (QIAGEN)[75], which contains an optimized Phi29 polymerase formulation as well as buffers and reagents for whole genome amplification 83(WGA) from single cells and very small samples, using multiple displacement amplification technology, which carries out isothermal genome amplification utilizing a uniquely processive DNA polymerase capable of replicating up to 100 kb without dissociating from the genomic DNA template. In the first step of the procedure, the cell sample was lysed, and the DNA was denatured. Once denaturation was stopped by the addition of neutralization buffer, a master mix containing buffer and DNA polymerase was added. The isothermal amplification reaction proceeded for 8 h at 30°C.
To confirm the origin of the isolated DNA, exon 9 of TP53 gene and exon 11 of BRAF gene (mutations described in this cell line) were amplified by PCR. Then, we sequenced the amplicons through Sanger sequencing[76]. Since DNA sequencing is considered the gold standard for detecting mutations, in this case, we did not use next-generation sequencing, because we intended to look for specific point mutation characteristic of the cell line used in the assay. As shown in Figure 7, the expected bands were confirmed for TP53 exon 9 and BRAF exon 11. Single PCR bands for each exon verified the specificity of the amplification before sequencing. A known homozygous TP53 exon 9 mutation, R280K, and a heterozygous BRAF exon 11 mutation, G464V, were identified in replicate samples of MDA-MB-231 cells (positive control)[77] but were not detected in PBMCs (wild type/negative control).
Figure 7.
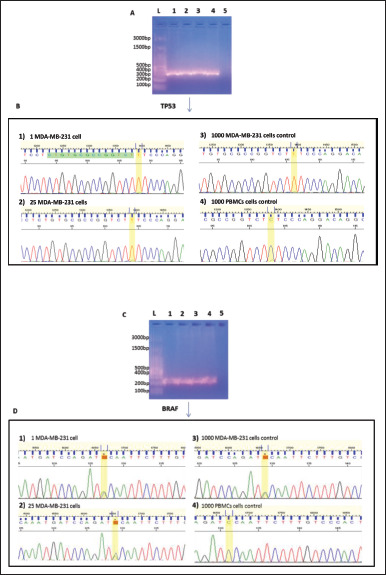
TP53 and BRAF mutations detected in MDA-MB-231 cells isolated by BA-LIFT technology. (A) Agarose gel of PCR reaction products from the amplification of exon 9 of the TP53 in a MDA-MB-231 cell (lane 1), 25 MDA-MB-231 cells (lane 2), 1000 MDA-MB-231 control (lane 3), 1000 PBMCs control (lane 4), and negative control (lane 5) compared to a DNA ladder (lane L). (B) Electropherogram showing the partial sequence of exon 9 of TP53 in a MDA-MB-231 cell (B.1), 25 MDA-MB-231 cells (B.2), 1000 MDA-MB-231 cells control (B.3), and 1000 PBMCs control (B.4). The presence of thymine and absence of guanine at position 839 of exon 9 of TP53 gene (homozygous) can be observed in cases B.1, B.2, and B.3. The mutation is not observed in B.4. (C) Agarose gel of PCR reaction products from the amplification of exon 11 of BRAF gene in a MDA-MB-231 cell (lane 1), 25 MDA-MB-231 cells (lane 2), 1000 MDA-MB-231 control (lane 3), 1000 PBMCs control (lane 4), and negative control (lane 5) compared to a DNA ladder (lane L). (D) Electropherogram showing the partial sequence of exon 11 of BRAF gene in a MDA-MB-231 cell (D.1), 25 MDA-MB-231 cells (D.2), 1000 MDA-MB-231 cells control (B.3), and 1000 PBMCs control (B.4). The presence of guanine and thymine at position 1391 of exon 11 of BRAF gene (heterozygous) can be observed in cases B.1, B.2 and B.3. The mutation is not observed in B.4.
We also transferred a sample of previously fixated MDA-MB-231 cells. After the transfer process, cells were stained with CK (a-CK/FITC, clone CK3-6H5, Miltenyi Biotec), CD45 (a-CD45/PE, clone 2D1, Biolegend) and DAPI for visualization and confirming that the subpopulations isolated correspond to MDA-MB-231.
4.2. Proliferation assays
Finally, in order to assess that after the transfer process, the cells were viable for in vitro experiments, transferred pellets of single cell and also transferred groups of 25 cells were stained with Trypan Blue, a dye to selectively stain dead tissues or cells blue. In all cases, the observed cells were alive. Also, to ensure CTCs viability preservation after the process, a proliferation assay was performed. CTCs were isolated in 96-well plates (see Methods section), and images were taken daily. After 9 days, the proliferation rate was lower than that of the control cells. After 14 days, however, confluence was reached (Figure 8).
Figure 8.
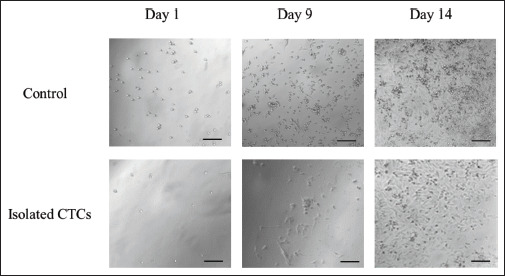
Isolated CTC proliferation assay. MDA-MB-231 were cultured in a 96-well plate with RPMI culture media enriched with FBS (10%) and acclimated media (10%)[78]. One microliter of control cells was taken from the sample prior to the laser isolation and cultured in the same conditions. Dimension bar represents 100 μm.
5. Conclusion
This work demonstrates that it is possible to preserve maximum CTCs integrity with negative selection. We believe that this approach opens up a new path not only for single-cell isolation but also for CTCs sorting, if the appropriate staining and imaging strategies are adapted and as this has been discussed for other cell lines in a previous work[73]. Although relevant aspects like throughput, process automation, etc. are not investigated in this paper, we must highlight the fact that the transfer process is based on a particular adaptation of a laser direct write technique, in many aspects which is a standard laser material processing approach. This undoubtedly offers the possibility of future use of the technique with all the potential of laser technology, in terms of precision, repeatability, throughput, and ease of integration in already existing platforms, as has been proven in medical and industrial fields. Regarding clinical applications, this new system would bring us closer to “real-time biopsy” based on CTC molecular characterization. Our technique is promising and this approach remains a key area of study for further intensive research. Since this novel technology 84would help to improve the molecular characterization of the tumor, it could be used to identify new treatment targets and select the most appropriate therapies for each stage of the disease. Evaluation of biomarker panels from a liquid biopsy may enlarge the molecular landscape of primary and metastatic lesions while offering the opportunity to evaluate molecular evolution of a tumor as a real-time film.
The approach followed in this work would preserve cells for any direct irradiation of the laser light, and the negative selection approach followed would maintain the CTCs free of any direct labeling, preserving the cells in perfect condition for further culture, characterization, or analysis (Figure 9). Therefore, this approach for CTCs isolation presents a relevant tool to deepen the study of single CTCs at molecular level.
Figure 9.
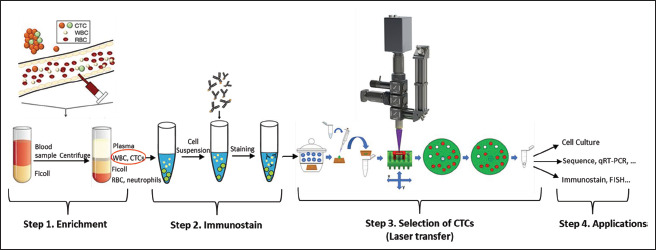
Complete enrichment-identification-isolation workflow. The proposed approach follows a standard enrichment-identification-isolation process but due to the negative selection and laser transfer using a BA-LIFT approach, the CTCs remain completely unaltered.
Acknowledgments
85None.
Funding
This work was partially supported by Comunidad de Madrid Government/Fondo Social Europeo , S2018/BAA-4480, Biopieltec-CM, and Spanish Ministry for Education FPU Grant 2015/05605.
Conflict of interest
Andres Muñoz has a consulting or advisory role at Sanofi, Celgene, Pfizer, BMS, Leo Pharma, Astra Zeneca, MSD, Incyte, Servier, Lilly, Roche, and Daiichi Sankyo; is in the Speakers’ Bureau of Rovi, Stada, Menarini, Bayer, Merck Serono, Amgen, Lilly, Astra Zeneca, Sanofi, BMS, Pfizer, and Daiichi Sankyo; and has been sponsored by Roche, Amgen, Merck Serono, Celgene, and Astra Zeneca to cover expenses in travel and accommodations. Sara Lopez-Tarruella has a consulting or advisory role at AstraZeneca, Novartis, Roche, Pfizer, Pierre Fabre, Lilly, Seagen, Daiichi Sankyo, Gilead Sciences, MSD, GlaxoSmithKline and Veracity; and is in the Speakers’ Bureau of Lilly. Miguel Martin has a consulting or advisory role at Roche/Genentech, Novartis, Pfizer, Lilly, AstraZeneca, Taiho Pharmaceutical, and PharmaMar; is in 86the Speakers’ Bureau of Lilly/ImClone, Roche/Genentech, and Pierre Fabre; has received honoraria from Roche/Genentech, Lilly, Pfizer, Novartis, and Pierre-Fabre; and has received research funding from Novartis, Roche, and PUMA.
Author contributions
Conceptualization: Carlos Molpeceres, Rocio Ramos-Medina, Miguel Martin
Formal analysis: Andres Marquez, Miguel Gomez-Fontela
Investigation: Andres Marquez, Miguel Gomez-Fontela, Rocío Candorcio-Simon, David Muñoz-Martin, Paula Romero
Methodology: Carlos Molpeceres, Rocio Ramos-Medina, Miguel Martin
Project administration: Maria del Monte-Millan
Resources: Maria del Monte-Millan
Supervision: Carlos Molpeceres, Rocio Ramos-Medina, Miguel Martin
Validation: Carlos Molpeceres, Rocio Ramos-Medina, Andres Marquez, Miguel Gomez-Fontela, Sara Lauzurica, Miguel Morales, David Muñoz-Martin, Miguel Martin
Visualization: Rocio Ramos-Medina, Andres Marquez
Writing – original draft: Carlos Molpeceres, Rocio Ramos-Medina
Writing – review & editing: Andres Muñoz, Sara Lopez-Tarruell, Tatiana Massarrah, Miguel Martin
Ethics approval and consent to participate
The human study protocol was approved by Comité de Ética de la Investigación con Medicamentos (CEIm), Hospital General Universitario Gregorio Marañón.
Consent for publication
Not applicable.
Availability of data
Not applicable.
References
- 1.Ashworth TR. A case of cancer in which cells similar to those in the tumours were seen in the blood after death. Aust Med J. 1869;14:146–149. [Google Scholar]
- 2.Ferreira MM, Ramani VC, Jeffrey SS. Circulating tumor cell technologies. Mol Oncol. 2016;10:374–394. doi: 10.1016/j.molonc.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poudineh M, Sargent EH, Pantel K, et al. Profiling circulating tumour cells and other biomarkers of invasive cancers. Nat Biomed Eng. 2018;2:72–84. doi: 10.1038/s41551-018-0190-5. [DOI] [PubMed] [Google Scholar]
- 4.Wan JCM, Massie C, GarciaCorbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–238. doi: 10.1038/nrc.2017.7. [DOI] [PubMed] [Google Scholar]
- 5.AlixPanabières C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer. 2014;14:623–631. doi: 10.1038/nrc3820. [DOI] [PubMed] [Google Scholar]
- 6.Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306. doi: 10.1038/nature17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pantel K, Brakenhoff RH, Brandt B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat Rev Cancer. 2008;8:329–340. doi: 10.1038/nrc2375. [DOI] [PubMed] [Google Scholar]
- 8.Pantel K, AlixPanabières C, Riethdorf S. Cancer micrometastases. Nat Rev Clin Oncol. 2009;6:339–351. doi: 10.1038/nrclinonc.2009.44. [DOI] [PubMed] [Google Scholar]
- 9.Kim MY, Oskarsson T, Acharyya S, et al. Tumor self-seeding by circulating cancer cells. Cell. 2009;139:1315–1326. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin D, Lesang S, Meng L, et al. Circulating tumor cells: biology and clinical significance. Signal Trans Targ Ther. 2021;6(1):404. doi: 10.1038/s41392-021-00817-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.AlixPanabières C, Pantel K. Circulating tumor cells: Liquid biopsy of cancer. Clin Chem. 2013;59:110–118. doi: 10.1373/clinchem.2012.194258. [DOI] [PubMed] [Google Scholar]
- 12.Wills QF, Mead AJ. Application of single-cell genomics in cancer: Promise and challenges. Hum Mol Genet. 2015;24:R74–R84. doi: 10.1093/hmg/ddv235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet. 2013;14:618–630. doi: 10.1038/nrg3542. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Navin NE. Advances and applications of single-cell sequencing technologies. Mol Cell. 2015;58:598–609. doi: 10.1016/j.molcel.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su H, Xingxing L, Hunag L, et al. 2021. Plasmonic alloys reveal a distinct metabolic phenotype or early gastric cancer Adv Mater 332007978 [DOI] [PubMed] [Google Scholar]
- 16.Li X, Kulkarni AS, Liu X, et al. Metal-organic framework hybrids aid metabolic profiling for colorectal cancer. Small Methods. 2021;5:2001001. doi: 10.1002/smtd.202001001. [DOI] [PubMed] [Google Scholar]
- 17.Wu L, Zhang Z, Tang M, et al. Spectrally combined encoding for profiling heterogeneous circulating tumor cells using a multifunctional nanosphere-mediated microfluidic platform. Angew Chem Int Ed. 2020;59:11240–11244. doi: 10.1002/anie.201914468. [DOI] [PubMed] [Google Scholar]
- 18.Chen XX, Bai F. Single-cell analyses of circulating tumor cells. Cancer Biol Med. 2015;12:184–192. doi: 10.7497/j.issn.2095-3941.2015.0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ortiz V, Yu M. Analyzing circulating tumor cells one at a time. Trends Cell Biol. 2018;28:764–775. doi: 10.1016/j.tcb.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawson DA, Kessenbrock K, Davis RT, et al. Tumour heterogeneity and metastasis at single-cell resolution. Nat Cell Biol. 2018;20:1349–1360. doi: 10.1038/s41556-018-0236-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kowalik A, Kowalewska M, Gózdz S. Current approaches for avoiding the limitations of circulating tumor cells detection methods—Implications for diagnosis and treatment of patients with solid tumors. Transl Res. 2017;18558-84:e15. doi: 10.1016/j.trsl.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 22.87Yu M, Stott S, Toner M, et al. Circulating tumor cells: Approaches to isolation and characterization. J Cell Biol. 2011;192:373–382. doi: 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esmaeilsabzali H, Beischlag TV, Cox ME, et al. Detection and isolation of circulating tumor cells: Principles and methods. Biotechnol Adv. 2013;31:1063–1084. doi: 10.1016/j.biotechadv.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 24.Hong B, Zu Y. Detecting circulating tumor cells: Current challenges and new trends. Theranostics. 2013;3:377–394. doi: 10.7150/thno.5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.RamosMedina R, Moreno F, LopezTarruella S, et al. Review: Circulating tumor cells in the practice of breast cancer oncology. Clin Transl Oncol. 2016;18:749–759. doi: 10.1007/s12094-015-1460-2. [DOI] [PubMed] [Google Scholar]
- 26.RamosMedina R, LopezTarruella S, Del MonteMillan M, et al. Technical challenges for CTC implementation in breast cancer. Cancers. 2021;13(18):46192021. doi: 10.3390/cancers13184619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.YuPing Y, Giret TM, Cote R, et al. Circulating tumor cells from enumeration to analysis: Current challenges and future opportunities. Cancers. 2021;13:2723. doi: 10.3390/cancers13112723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoeppener AELM, Swennenhuis JF, Terstappen LWMM. Immunomagnetic separation technologies, in. Minimal Residual Disease and Circulating Tumor Cells in Breast Cancer. 2012;195:43–58. doi: 10.1007/978-3-642-28160-0_4. (Eds. Ignatiadis M, Sotiriou C, Pantel K), Springer, Berlin, Heidelberg, [DOI] [PubMed] [Google Scholar]
- 29.Andree KC, van Dalum G, Terstappen LWMM. Challenges in circulating tumor cell detection by the CellSearch system. Mol Oncol. 2016;10:395–407. doi: 10.1016/j.molonc.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Austin RG, Huang TJ, Wu M, et al. Clinical utility of non-EpCAM based circulating tumor cell assays. Adv Drug Deliv Rev. 2018;125:132–142. doi: 10.1016/j.addr.2018.01.013. [DOI] [PubMed] [Google Scholar]
- 31.Hao SJ, Wan Y, Xia YQ, et al. Size-based separation methods of circulating tumor cells. Adv Drug Deliv Rev. 2018;125:3–20. doi: 10.1016/j.addr.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Hajba L, Guttman A. Circulating tumor-cell detection and capture using microfluidic devices. Trends Anal Chem. 2014;59:9–16. [Google Scholar]
- 33.Parker SG, Yang Y, Ciampi S, et al. A photoelectrochemical platform for the capture and release of rare single cells. Nat Commun. 2018;9:2288. doi: 10.1038/s41467-018-04701-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou J, Tu C, Liang Y, et al. Isolation of cells from whole blood using shear-induced diffusion. Sci Rep. 2018;8:9411. doi: 10.1038/s41598-018-27779-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valihrach L, Androvic P, Kubista M. Platforms for single-cell collection and analysis. Int J Mol Sci. 2018;19:807. doi: 10.3390/ijms19030807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miccio L, Cimmino F, Kurelac I, et al. Perspectives on liquid biopsy for label-free detection of “circulating tumor cells” through intelligent lab-on-chips. VIEW. 2020;1:20200034. [Google Scholar]
- 37.Hou J, Liu X, Zhou S. Programmable materials for efficient CTCs isolation: From micro/nanotechnology to biomimicry. VIEW. 2021;2:20200023. [Google Scholar]
- 38.Millner LM, Linder MW, Valdes R. Circulating tumor cells: A review of present methods and the need to identify heterogeneous phenotypes. Ann Clin Lab Sci. 2013;43:295–304. [PMC free article] [PubMed] [Google Scholar]
- 39.Bhagwat N, Dulmage K, Pletcher CH, et al. An integrated flow cytometry-based platform for isolation and molecular characterization of circulating tumor single cells and clusters. Sci Rep. 2018;8:5035. doi: 10.1038/s41598-018-23217-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.EmmertBuck MR, Bonner RF, Smith PD, et al. Laser capture microdissection. Science. 1996;274:998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- 41.Espina V, Wulfkhule JD, Calvert VS, et al. Laser-capture microdissection. Nat Protoc. 2006;1:586–603. doi: 10.1038/nprot.2006.85. [DOI] [PubMed] [Google Scholar]
- 42.Bevilacqua C, Ducos B. Laser microdissection: A powerful tool for genomics at cell level. Mol Aspects Med. 2018;59:5–27. doi: 10.1016/j.mam.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 43.Park ES, Yan JP, Ang RA, et al. Isolation and genome sequencing of individual circulating tumor cells using hydrogel encapsulation and laser capture microdissection. Lab Chip. 2018;18:1736–1749. doi: 10.1039/c8lc00184g. [DOI] [PubMed] [Google Scholar]
- 44.Vogel A, Horneffer V, Lorenz K, et al. Principles of laser microdissection and catapulting of histologic specimens and live cells. Methods Cell Biol. 2007;82:153–205. doi: 10.1016/S0091-679X(06)82005-4. [DOI] [PubMed] [Google Scholar]
- 45.Kim O, Lee D, Lee AC, et al. 2019. Single cell genomics: Whole genome sequencing of single circulating tumor cells isolated by applying a pulsed laser to cell capturing microstructures Small 151902607 [DOI] [PubMed] [Google Scholar]
- 46.Schiele NR, Corr DT, Huang Y, et al. Laser-based direct-write techniques for cell printing. Biofabrication. 2010;2:032001. doi: 10.1088/1758-5082/2/3/032001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deng Y, Renaud P, Guo Z, et al. Single cell isolation process with laser induced forward transfer. J Biol Eng. 2017;11:2. doi: 10.1186/s13036-016-0045-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arnold CB, Serra P, Piqué A. Laser direct-write techniques for printing of complex materials. MRS Bull. 2007;32:23–32. [Google Scholar]
- 49.Hon KKB, Li L, Hutchings IM. Direct writing technology— Advances and developments. CIRP Ann. 2008;57:601–620. [Google Scholar]
- 50.Serra P, Duocastella M, FernándezPradas JM, et al. Liquids microprinting through laser-induced forward transfer. Appl Surf Sci. 2009;255:5342–5345. [Google Scholar]
- 51.FernándezPradas JM, Florian C, CaballeroLucas F, et al. Laser-induced forward transfer: Propelling liquids with light. App Surf Sci. 2017;418:559–564. [Google Scholar]
- 52.Morales M, MunozMartin D, Marquez A, et al. Advances in Laser Materials Processing: Technology, Research and Applications. 2018 Ed. Jonhatan Lawrence Woodhead Publishing, an imprint of Elsevier. [Google Scholar]
- 53.Piqué A, Serra P. Laser Printing of Functional Materials: 3D Microfabrication, Electronics and Biomedicine. Wiley-VCH Verlag GmbH & Co KGaA; 2018. [Google Scholar]
- 54.Barron JA, Wu P, Ladouceur HD, et al. Biological laser printing: A novel technique for creating heterogeneous 3-dimensional cell patterns. Biomed Microdevices. 2004;6:139–147. doi: 10.1023/b:bmmd.0000031751.67267.9f. [DOI] [PubMed] [Google Scholar]
- 55.88Hopp B, Smausz T, Kresz N, et al. Survival and proliferative ability of various living cell types after laser-induced forward transfer. Tissue Eng. 2005;11:1817–1823. doi: 10.1089/ten.2005.11.1817. [DOI] [PubMed] [Google Scholar]
- 56.Colina M, Serra P, FernándezPradas JM, et al. DNA deposition through laser induced forward transfer. Biosens Bioelectron. 2005;20:1638–1642. doi: 10.1016/j.bios.2004.08.047. [DOI] [PubMed] [Google Scholar]
- 57.Duocastella M, Colina M, FernándezPradas JM, et al. Study of the laser-induced forward transfer of liquids for laser bioprinting. Appl Surf Sci. 253:7855–7859. [Google Scholar]
- 58.Ovsianikov A, Gruene M, Pflaum M, et al. Laser printing of cells into 3D scaffolds. Biofabrication. 2010;2:014104. doi: 10.1088/1758-5082/2/1/014104. [DOI] [PubMed] [Google Scholar]
- 59.Guillemot F, Souquet A, Catros S, et al. Laser-assisted cell printing: Principle, physical parameters versus cell fate and perspectives in tissue engineering. Nanomedicine (Lond) 2010;5:507–515. doi: 10.2217/nnm.10.14. [DOI] [PubMed] [Google Scholar]
- 60.Ali M, Pages E, Ducom A, et al. Controlling laser-induced jet formation for bioprinting mesenchymal stem cells with high viability and high resolution. Biofabrication. 2014;6:045001. doi: 10.1088/1758-5082/6/4/045001. [DOI] [PubMed] [Google Scholar]
- 61.Gruene M, Pflaum M, Deiwick A, et al. Adipogenic differentiation of laser-printed 3D tissue grafts consisting of human adipose-derived stem cells. Biofabrication. 2011;3:015005. doi: 10.1088/1758-5082/3/1/015005. [DOI] [PubMed] [Google Scholar]
- 62.Zhang J, Geiger Y, Sotier F, et al. Extending single cell bioprinting from femtosecond to picosecond laser pulse durations. Micromachines. 2021;12:1172. doi: 10.3390/mi12101172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karakaidos P, Kryou C, Simigdala N, et al. Laser bioprinting of cells using UV and visible wavelengths: A comparative DNA damage study. Bioengineering. 2022;9:378. doi: 10.3390/bioengineering9080378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Catros S, Fricain JC, Guillotin B, et al. Laser-assisted bioprinting for creating on-demand patterns of human osteoprogenitor cells and nano-hydroxyapatite. Biofabrication. 2011;3:025001. doi: 10.1088/1758-5082/3/2/025001. [DOI] [PubMed] [Google Scholar]
- 65.Bourget JM, Kerouredan O, Medina M, et al. Patterning of endothelial cells and mesenchymal stem cells by laser-assisted bioprinting to study cell migration. Biomed Res Int. 2016:3569843. doi: 10.1155/2016/3569843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vinson BT, Phamduy TB, Shipman J, et al. Laser direct-write based fabrication of a spatially-defined, biomimetic construct as a potential model for breast cancer cell invasion into adipose tissue. Biofabrication. 2017;9:025013. doi: 10.1088/1758-5090/aa6bad. [DOI] [PubMed] [Google Scholar]
- 67.Koch L, Deiwick A, Franke A, et al. Laser bioprinting of human induced pluripotent stem cells—The effect of printing and biomaterials on cell survival, pluripotency, and differentiation. Biofabrication. 2018;10(3):035005. doi: 10.1088/1758-5090/aab981. [DOI] [PubMed] [Google Scholar]
- 68.Kattamis NT, Purnick PE, Weiss R, et al. Thick film laser induced forward transfer for deposition of thermally and mechanically sensitive materials. Appl Phys Lett. 2007;91:171120. [Google Scholar]
- 69.Brown MS, Kattamis NT, Arnold CB. Time-resolved study of polyimide absorption layers for blister-actuated laser-induced forward transfer. J Appl Phys. 2010;107:083103. [Google Scholar]
- 70.Brown MS, Kattamis NT, Arnold CB. Time-resolved dynamics of laser-induced micro-jets from thin liquid films. Microfluid Nanofluid. 2011;11:199–207. [Google Scholar]
- 71.Brown MS, Brasz CF, Ventikos Y, et al. Impulsively actuated jets from thin liquid films for high-resolution printing applications. J Fluid Mech. 2012;709:341–370. [Google Scholar]
- 72.Turkoz E, Perazzo A, Kim H, et al. Impulsively induced jets from viscoelastic films for high-resolution printing. Phys Rev Lett. 2018;120:074501. doi: 10.1103/PhysRevLett.120.074501. [DOI] [PubMed] [Google Scholar]
- 73.Márquez A, GomezFontela M, Lauzurica S, et al. Fluorescence enhanced BA-LIFT for single cell detection and isolation. Biofabrication. 2020;12(2):025019. doi: 10.1088/1758-5090/ab6138. [DOI] [PubMed] [Google Scholar]
- 74.Warawdekar UM, Parmar V, Prabhu A, et al. A versatile method for enumeration and characterization of circulating tumour cells from patients with breast cancer. J Cancer Metastasis Treat. 2017;3:23. [Google Scholar]
- 75.Zhu Z, Qiu S, Shao K, et al. Progress and challenges of sequencing and analyzing circulating tumor cells. Cell Biol Toxicol. 2018;34:405–415. doi: 10.1007/s10565-017-9418-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kou R, Zhao J, Gogoi P, et al. Enrichment and mutation detection of circulating tumor cells from blood samples. Oncol Rep. 2018;39:2537–2544. doi: 10.3892/or.2018.6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ikediobi ON, Davies H, Bignell G, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5:2606–2612. doi: 10.1158/1535-7163.MCT-06-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muir W, Hildebrandt A, Riker A. Am J Botany. 1958;45(8):589–597. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.