


Abstract
Polyadenylation of RNA molecules in bacteria and chloroplasts has been implicated as part of the RNA degradation pathway. The polyadenylation reaction is performed in Escherichia coli mainly by the enzyme poly(A) polymerase I (PAP I). In order to understand the molecular mechanism of RNA polyadenylation in bacteria, we characterized the biochemical properties of this reaction in vitro using the purified enzyme. Unlike the PAP from yeast nucleus, which is specific for ATP, E.coli PAP I can use all four nucleotide triphosphates as substrates for addition of long ribohomopolymers to RNA. PAP I displays a high binding activity to poly(U), poly(C) and poly(A) ribohomopolymers, but not to poly(G). The 3′-ends of most of the mRNA molecules in bacteria are characterized by a stem–loop structure. We show here that in vitro PAP I activity is inhibited by a stem–loop structure. A tail of two to six nucleotides located 3′ to the stem–loop structure is sufficient to overcome this inhibition. These results suggest that the stem–loop structure located in most of the mRNA 3′-ends may function as an inhibitor of polyadenylation and degradation of the corresponding RNA molecule. However, RNA 3′-ends produced by endonucleolytic cleavage by RNase E in single-strand regions of mRNA molecules may serve as efficient substrates for polyadenylation that direct these molecules for rapid exonucleolytic degradation.
INTRODUCTION
Polyadenylation has been believed for a long time to be exclusively associated with eukaryotic mRNAs. Other RNAs, such as rRNAs and tRNAs, as well as RNAs in prokaryotes, were not considered to be polyadenylated. Indeed, most of these RNA molecules do not harbor a poly(A) tail at their 3′-end. Nevertheless, poly(A) tails have recently been detected in bacteria (1–8). The polyadenylated RNA accounts for only a very small fraction of the RNA population in the cell. This fraction increased several-fold in mutant bacterial cells that lack exoribonuclease(s) activity. On the other hand, in mutants in which RNA polyadenylation was inhibited due to the lack of poly(A) polymerase, the half-life of RNA molecules increased dramatically (reviewed in 1,2,9–11). These results suggested that in contrast to the nucleus and cytoplasm of eukaryotic cells, where the poly(A) tail is important for stability, maturation and translation of mRNA, the addition of poly(A) tails in bacterial mRNAs promotes their degradation. Taken together, polyadenylation of RNA molecules is part of the molecular mechanism of RNA degradation in bacteria (1,2,9–11). Similarly to bacterial cells, polyadenylation of RNA molecules during the degradation process has been described in chloroplasts and in mitochondria, cellular organelles that are believed to have arisen in an evolutionary manner from a prokaryotic ancestor (12–15).
The enzyme poly(A) polymerase I (PAP I) adds adenylate residues to the 3′-ends of RNAs using ATP as donor. It was purified from Escherichia coli cells in the early 1960s, well before the discovery of poly(A) in eukaryotic mRNAs (16). In E.coli, PAP I is a monomer of ~55 kDa, requiring Mg2+ ions for activity (17) and displaying some sequence homology to other poly(A) polymerases from yeast and mammals (18,19). In addition, it shows significant amino acid sequence homology to tRNA nucleotidyl transferase, a CCA-adding enzyme (18–22). Another poly(A) polymerase enzyme, PAP II, has been identified in E.coli, but its role in RNA metabolism remains to be elucidated (23). In contrast to the mechanism in eukaryotes, E.coli polyadenylation apparently does not depend on recognition sequences and does not require a multiprotein complex for activity (2). The polyadenylation of RNA molecules in E.coli and chloroplasts occurs at a variety of sites, including the mature 3′-end, but is most certainly an intermediate in processing and decay (2,12,24). PAP I does not purify with RNase E, PNPase, DEAE helicase and enolase in the degradosome, a high molecular weight protein complex that is believed to be important in RNA processing and degradation. Nevertheless, a recent publication suggested a transient interaction between the components of the degradosome, RNase E and DEAD RNA helicases, and PAP I (22).
In this work, we describe the biochemical characterization of the activity of E.coli PAP I. We found that the enzyme activity in vitro is not specific for ATP and that the protein binds poly(A), poly(U) and poly(C), but not poly(G), with high affinity. In addition, we show that a stem–loop structure inhibits the polyadenylation reaction and that the addition of 2–4 nt 3′ to the stem–loop overcomes this inhibitory effect.
MATERIALS AND METHODS
In vitro transcription of RNA
The plasmids used for in vitro transcription of parts of the mRNAs of the spinach chloroplast genes psbA (encoding the D1 protein of photosystem II) and petD (encoding subunit IV of the b6f complex) have been previously described (25). To generate a synthetic RNA corresponding to the 3′-end of threonine attenuator (thrA), the XbaI–EcoRI fragment of this E.coli gene was cloned into plasmid pBluescript KS+ (Stratagene) (26). To generate RNA molecules corresponding to the spinach chloroplast petD and the E.coli thrA mature 3′-ends (used in the experiment described in Fig. 4), the synthetic molecules described above were incubated with chloroplast soluble protein extract in an in vitro 3′-end processing reaction, as previously described, and the in vitro processed product was re-isolated by elution from a denaturing gel (25). In these RNA molecules, the 3′-end was localized to the end of the stem–loop structure (27). The stem–loop structure of the malE–malF intergenic region was PCR amplified using two oligonucleotides corresponding to nucleotides 1–17 and 106–94 of the intergenic region (GenBank accession no. M19202) (28). A single mutation A→C was introduced at the fifth nucleotide of the stem in order to strengthen the stem structure (28). The T7 promoter sequence was included in the first oligonucleotide in order to drive in vitro transcription. Two, four or six cytosine residues were added to the second oligonucleotide to generate RNA molecules with the addition of two, four or six residues 3′ to the stem–loop structure, as shown in Figure 5. RNAs were transcribed using T7 RNA polymerase and radioactively labeled with [α-32P]UTP to a specific activity of 8–10 × 103 and 8–10 × 104 c.p.m./fmol for polyadenylation and UV crosslinking experiments, respectively (29). The full-length transcription products were purified on 5% denaturing polyacrylamide gels.
Figure 4.
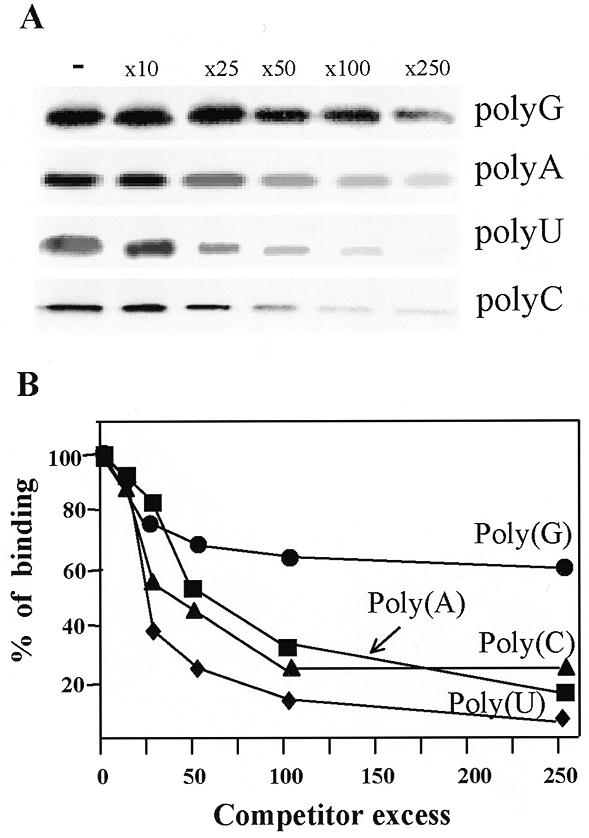
Binding of PAP I to different ribohomopolymers. (A) [32P]RNA was mixed with increasing amounts of the competitors poly(G), poly(A), poly(U) and poly(C) and 16.5 ng of PAP I. The competitor:[32P]RNA ratios were as follows: none (–), 10-fold (x10), 25-fold (x25), 50-fold (x50), 100-fold (x100) and 250-fold excess (x250). The mixture was immediately UV crosslinked and digested with RNase A and the proteins were then analyzed by SDS–PAGE and autoradiogaphy. (B) A graphical representation of the results of at least three independent competition experiments. The amount of [32P]RNA crosslinked to PAP I was quantified using a Fuji imaging analyzer and plotted as a function of the competitor excess.
Figure 5.
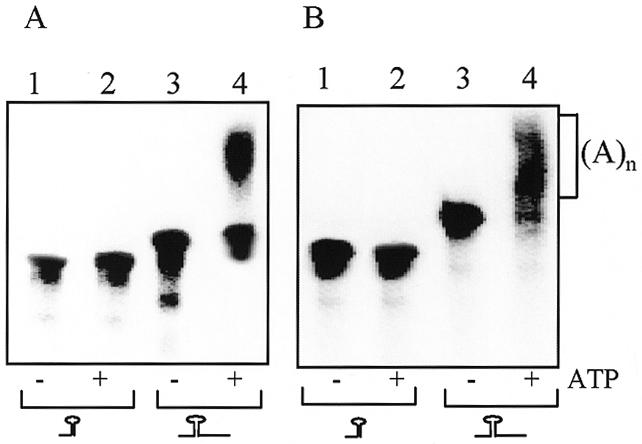
RNA terminating with a stem–loop structure is poorly polyadenylated. Synthetic transcribed [32P]RNA corresponding to the chloroplast gene petD (A) or the E.coli gene thrA (B) in either their 3′-end processed (lanes 1 and 2) or unprocessed precursor (lanes 3 and 4) forms were used in an in vitro polyadenylation assay. The RNAs were incubated for 35 min without (lanes 1 and 3) or with 1 mM ATP (lanes 2 and 4) and E.coli PAP I. Following incubation, the RNAs were isolated and analyzed by denaturing PAGE and autoradiography. A schematic representation of the RNA substrate is shown at the bottom.
In vitro RNA polyadenylation assay
In vitro RNA polyadenylation experiments were carried out as previously described (25). Briefly, in vitro synthesized RNA (2 fmol) was incubated with purified PAP I (Amersham Inc., catalog no. E2180Y) (0.8 µg/ml) or with yeast PAP (US Biochemicals, catalog no. E74225Y) (0.4 µg/ml) in buffer E (20 mM HEPES pH 7.9, 60 mM KCl, 12.5 mM MgCl2, 0.1 mM EDTA, 2 mM DTT and 17% glycerol) with the addition of 1 mM ATP for the times indicated in the figure legends. Following incubation, the RNA was isolated and analyzed by gel electrophoresis and autoradiography (25). For the experiment described in Figure 3, trace labeled RNA was incubated with PAP I in the presence of 0.5 mM ATP or 0.5 mM of all nucleotides and 1 × 106 c.p.m. [α-32P]ATP. The reaction was terminated by heat treatment (70°C for 5 min) before digestion with 25 µg RNase A and 200 U RNase T1 for 1 h at 37°C (25). The digestion products were resolved in 12% polyacrylamide gels containing 7 M urea.
Figure 3.

When incubated together PAP I incorporates all the nucleotides. Non-radioactive RNA of 260 nt was incubated with PAP and [α-32P]ATP for 40 min. The reaction was stopped by incubation at 70°C for 5 min and RNase A and RNase T1 (digesting at G, C and U but not A) were added for 1 h at 37°C following isolation of the RNA and analysis by denatured 12% PAGE and autoradiography (lane 2). In lane 3, non-radioactive CTP, GTP and UTP were added to the incubation with PAP I and the reaction proceeded as described for lane 2. The 260 nt radioactively labeled RNA is shown in lane 1 as a size marker. The polyadenylated RNA that was resistant to the ribonucleases and the digestion products produced when all nucleotides were used are indicated.
UV crosslinking assay
UV crosslinking of proteins to [α-32P]UTP-labeled RNA was carried out as previously described (30). Briefly, 3 fmol of RNA (240 000 c.p.m.) were incubated with 16.5 ng of PAP I in 15 µl of buffer E. In competition experiments, the ribohomopolymer was added to the protein before the [32P]RNA (30). Following 1.8 J UV irradiation in a UV crosslinking apparatus (Hoefer Inc.), the RNA was digested by 10 µg RNase A at 37°C for 1 h and the proteins were fractionated by SDS–PAGE. The label transferred from RNA to protein was detected by autoradiography and quantified using a Fuji imaging analyzer.
General methods
Purified PAP I from E.coli was obtained from Amersham Inc. This preparation apparently consisted of one polypeptide requiring no additional purification (Fig. 1). Purified PAP from yeast was obtained from US Biochemicals. SDS–PAGE fractionation and detection of PAP I by immunoblotting with specific antibodies (20) were performed as previously described (30). Protein concentration was determined using the Bio-Rad protein assay kit.
Figure 1.
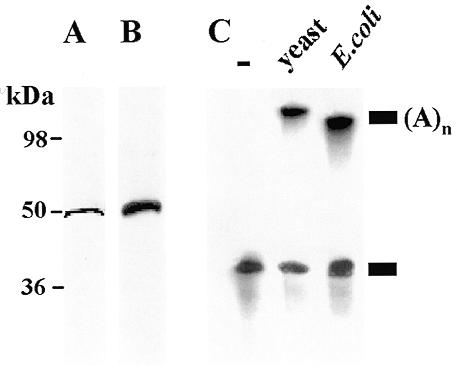
Characterization of E.coli PAP I. (A) A silver stained SDS–polyacrylamide gel of the PAP I fraction (50 ng). Molecular mass markers are shown on the left. (B) Western blot analysis. An aliquot of 30 ng of PAP I was separated by SDS–PAGE, transferred to a nitrocellulose membrane and allowed to react with antibodies raised against E.coli PAP I (20). (C) Detection of PAP I activity. In vitro transcribed [32P]RNA was incubated for 35 min without protein (lane –), with the addition of yeast PAP I (lane yeast) and with the addition of E.coli PAP I (lane E.coli). Following incubation, the RNA was isolated and analyzed by denaturing PAGE and autoradiography. A schematic representation of the non-polyadenylated and polyadenylated RNA molecules is shown on the right.
RESULTS
PAP I activity in vitro is not specific for ATP
In order to analyze whether or not PAP I activity is specific for ATP, we analyzed the in vitro activity of purified PAP I using different nucleotides as substrates. The purified PAP I consisted of one 55 kDa polypeptide that reacted with specific antibodies (Fig. 1A and B; 10,20). When the purified protein was incubated with in vitro transcribed RNA in the presence of ATP, the RNA was rapidly polyadenylated, similar to the reaction of PAP isolated from yeast (Fig. 1C). When purified PAP I was incubated with synthetic RNA as a substrate and each of the nucleotides was added, elongation activity was detected not only with ATP but also with GTP, UTP and CTP (Fig. 2A). Under the experimental conditions used, in the case of ATP and CTP elongation of RNA by ~500 nt was achieved within the first 5 min of incubation. This may suggest a processive activity of PAP I with these nucleotides, i.e. the enzyme, when bound to an RNA molecule, adds nucleotides without disconnecting from the elongated RNA between addition of each nucleotide (2). When using GTP or UTP, the length and quantity of the elongated RNA molecules increased during incubation, suggesting that either the PAP I activity using these nucleotides is not processive, or that the affinity for ATP and CTP is much lower (Fig. 2A). Indeed, when PAP I was incubated with increasing concentrations of ATP, saturation of the activity was observed using 0.5–1.0 mM ATP (Fig. 2B). However, when using GTP as substrate, no saturation was obtained even at a concentration of 2 mM and most of the RNA was not polymerized (Fig. 2B). The activity of yeast PAP under in vitro conditions has been previously characterized (31). This enzyme can add about 15 G residues and only a few U or C residues, but about 600 A nucleotides. Similar results were obtained in this work under the same experimental conditions for E.coli PAP I (Fig. 2C). Taken together, these results show that under the experimental conditions used here, whereby yeast PAP disclosed specific activity for ATP, the E.coli enzyme can use all four nucleotides. Next, we asked whether the enzyme, when incubated together with all four nucleotides, incorporates only adenosines or the other nucleotides as well. To this end, non-radioactive RNA was incubated with PAP I with the addition of either radioactive ATP or the four nucleotides including radioactive ATP. Following the polymerization reaction, the RNA molecules were digested with RNase A and RNase T1, which degrade the RNA molecule following G, U and C. Only elongated radioactive RNA was observed when ATP alone was used (Fig. 3, lane 2). However, short RNA molecules were observed when all four nucleotides were used, indicating that other nucleotides, in addition to adenosines, were incorporated into the polymerized molecules (Fig. 3, lane 3). Taking into account that mostly homologous poly(A) tails were obtained using RT–PCR of polyadenylated RNA molecules, why this enzyme activity is specific for ATP in bacterial cells remains to be elucidated (see Discussion).
Figure 2.
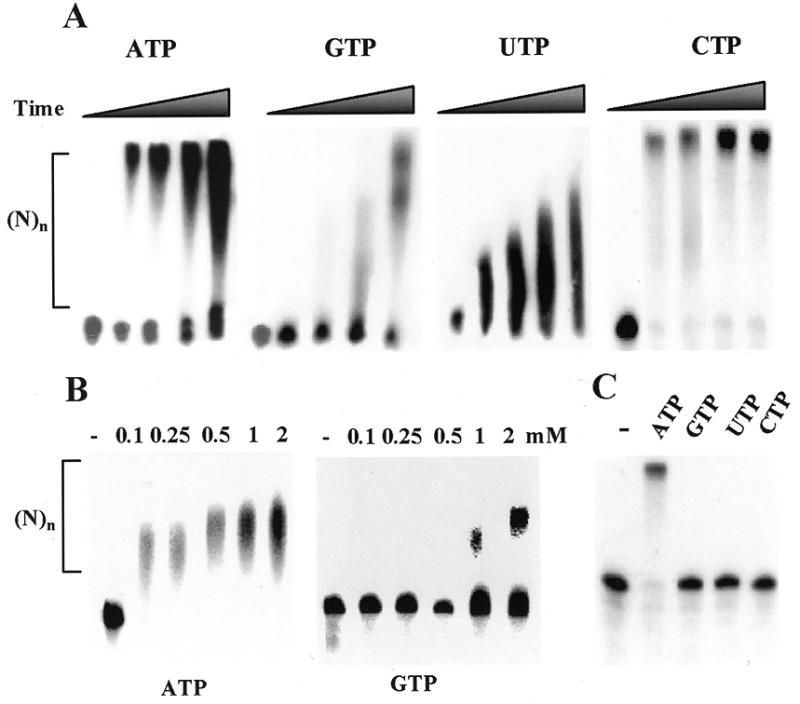
The E.coli PAP I activity is not specific for ATP. (A) Synthetic transcribed [32P]RNA of 380 nt was incubated with PAP I and 1 mM corresponding nucleotide. At 0, 5, 20, 40 and 60 min time points, the reaction was terminated and the RNA was isolated and analyzed by denaturing PAGE and autoradiography. (B) [32P]RNA as in (A) was incubated with PAP I for 40 min without (–) or with ATP or GTP at the concentrations shown in the figure. Following incubation, the reaction was terminated and the RNA was isolated and analyzed by denaturing PAGE and autoradiography. (C) PAP I isolated from yeast was incubated with synthetic transcribed [32P]RNA for 35 min without (–) or with the addition of 1 mM ATP, GTP, UTP or CTP. Following incubation, the RNA was isolated and analyzed as described in (A).
Binding affinities of PAP I to ribohomopolymers
One possibility for the differences in PAP I specificity in the cell compared to in vitro may be attributed to the binding affinity of the protein for different RNA molecules. In order to determine the binding affinity of PAP I for different ribohomopolymer RNA molecules, we carried out UV crosslinking competition experiments (30,32). Isolated PAP I was incubated with [32P]UTP-labeled RNA corresponding to the spinach chloroplast gene psbA. The RNA/protein mixture was UV crosslinked, followed by digestion of the unbound RNA with RNase. The label transferred from the RNA to protein was then analyzed by SDS–PAGE and autoradiography. Increased amounts of a competitor, a ribohomopolymer in this experiment, were used to define the I50 as the competitor concentration required to reduce the UV crosslinking signal to 50% (32). The results revealed a similar affinity of PAP I for poly(A), poly(U) and poly(C) and a reduced binding affinity for poly(G) (Fig. 4). With poly(U), poly(A) and poly(C), about 50 times competitor excess reduced binding by 50%, whereas with poly(G) the I50 was more than 250 times competitor excess (Fig. 4). On the one hand, these results are in agreement with those presented in Figure 2, showing that PAP I was able to polymerase the four nucleotides and not just adenosine. On the other hand, as previously described, only polyadenylated RNA molecules were found in bacteria by RT–PCR (1,2,10). Therefore, the specificity of the polyadenylation reaction in bacterial cells is not due to the intrinsic specificity of PAP I or a high binding affinity for poly(A).
RNAs terminating with a stem–loop structure are poorly polyadenylated by E.coli PAP I
In E.coli, polyadenylation of mRNA was detected both at the 3′-end and for the internal degradation products of endo- and exoribonucleases (2,24). In the chloroplast, where the RNA polyadenylation and degradation mechanisms are very similar to those of bacteria, a considerably greater number of polyadenylation sites were found inside the mRNA sequence and not at the mature 3′-end, which is characterized by a stem–loop structure (25). These polyadenylation sites were found to be the proximal products of endonucleolytic cleavage of mature full-length mRNA. Based on these results, it has been suggested that one of the functions of the stem–loop structure located at the 3′-end of most chloroplast genes is to prevent polyadenylation, which is followed by degradation (12). Most of the E.coli mRNAs are also characterized by a stem–loop structure at the 3′-end (9). In order to determine whether PAP I activity is modulated by the structure of the RNA, RNA molecules in which the stem–loop structure was located either at the 3′-end or in the middle of the molecule were tested in an in vitro polyadenylation assay. When these molecules, corresponding to the mature 3′-end of the spinach chloroplast gene petD or the E.coli gene thrA, were tested as substrates for PAP I, no polyadenylation was detected (Fig. 5). However, the same RNA molecules in which the stem–loop was not located at the 3′-end were efficiently polyadenylated (Fig. 5). These results suggest that, similarly to the situation in the chloroplast, E.coli PAP I is inhibited by a stem–loop structure located at the 3′-end.
We then wanted to determine what is the minimal number of nucleotides located 3′ to the stem–loop structure that is necessary to make the RNA a good substrate for PAP I. To do so, we constructed several RNA molecules differing in the number of nucleotides extending 3′ of the stem–loop that could serve as a ‘platform’ for PAP I. We used the long and stable malE–malF stem–loop structure, in which the nucleotide at position 5 from the base of the stem was modified from A to C in order to ensure stabilization of the stem structure (28). To this long and stable stem–loop structured RNA molecule, we added 2, 4 or 6 cytosine residues at the 3′-end. Cytosine was chosen over other nucleotides since visual inspection revealed that other nucleotides can potentially hybridize to the RNA molecule and thus not act as the ‘toe hold’ supposedly required for PAP I activity. Each of the radioactively labeled RNA molecules described above was incubated with E.coli PAP I and ATP following an analysis by denatured PAGE. The results showed that, similarly to other RNA molecules terminating with a stem–loop structure (Fig. 5), the malE–malF 3′-end was hardly adenylated by PAP I (Fig. 6, left). However, the addition of a ‘platform’ of two C residues to the stem increased the efficiency of the polyadenylation reaction several-fold and the addition of four and six cytosines resulted in an activity similar to that obtained with non-structured RNA (Fig. 6). These results demonstrated that a very short single-stranded ‘toe hold’, comprised of two or more nucleotides, is required for PAP I to efficiently polyadenylate an RNA molecule.
Figure 6.
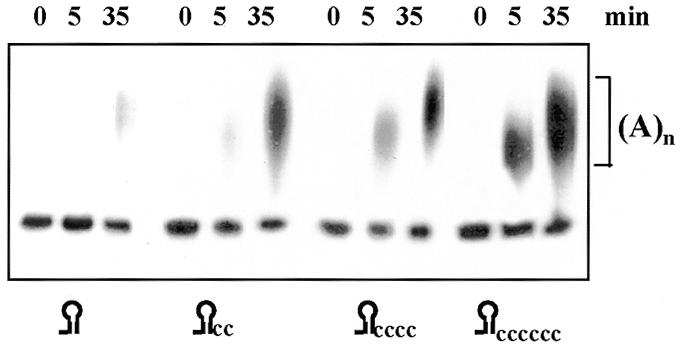
The addition of several nucleotides 3′ to the stem–loop is sufficient for efficient polyadenylation. [32P]RNA corresponding to the 3′-end of the E.coli malE mRNA with the addition of several nucleotides, as shown in the figure, was incubated with PAP I and ATP for the times indicated. Following incubation, the RNA was isolated and analyzed by denaturing PAGE and autoradiography.
DISCUSSION
Our work has demonstrated that E.coli PAP I, as a purified single protein, does not use adenosine preferentially as a substrate. In addition, it does not display a higher binding affinity for poly(A). These results were somewhat unexpected for two reasons. First, yeast PAP under the same experimental conditions displays specificity for adenosines (31; Fig. 2). Second, when polyadenylated RNAs from bacterial cells were cloned and sequenced using RT–PCR, poly(A) tails were mainly obtained (2 and reference therein), although poly(A) tails containing several nucleotides other than adenosine were obtained from cells during the stationary growth stage (33). Therefore, it seems that PAP I activity in bacterial cells mostly favors adenosines. On the other hand, polyadenylated RNAs that include, in addition to adenosines, ~25% guanosines as well as other nucleotides were isolated from the chloroplasts of higher plants (25). However, a purified fraction of PAP I from chloroplasts has not yet been analyzed for specificity for different nucleotides. What makes PAP I specific for adenosines inside the bacterial cell? Although the concentration of adenosine is higher than other nucleotides, this does not seem to account for the specificity of PAP I since RNA polymerase, for example, successfully uses all four nucleotides, and nucleotidyltransferase uses CTP in addition to ATP (18,19). It is possible that interaction with other proteins imposes the specificity of PAP I for adenosines. Recently, a possible weak and temporary interaction of PAP I with RNase E and the degradosome complex was reported (22). Eukaryotic PAP is involved in a multiprotein complex with other protein factors that, in several cases, are required for specific and proper activity (34). Clearly, more work is required to reveal the answer to the question why PAP I activity is specific for adenosine in the bacterial cell.
In addition to the PAP I used in this work, at least one more PAP, the product of the f310 gene, has been identified in E.coli cells, and named PAP II (23). These two enzymes seem to be dispensable, since even when one of the genes is inactivated the cells are still viable (23). Inactivation of both genes is lethal. The question as to what the biological functions of each enzyme are and whether or not these functions are complementary, different or the same, as well as the nature of the specificity of PAP II, are still under investigation.
Another bacterial enzyme exhibiting polyadenylation of RNA when assayed in vitro is polynucleotide phosphorylase (PNPase) (35). PNPase and RNase II are the two major exoribonucleases involved in the process of RNA degradation in bacterial cells. PNPase is included in the degradosome together with the endoribonuclease RNase E, a RNA helicase and enolase (11). When incubated in vitro, isolated PNPase displaces polymerase activity for all four nucleotides (35). However, it is believed that PNPase activity in the bacterial cell exclusively degrades RNA and polyadenylation is performed only (or almost exclusively) by the PAP enzymes and mostly by PAP I. This assumption is based on the fact that inactivation of the pnp gene, encoding PNPase, leads to an increase in RNA half-life and in accumulation of polyadenylated RNA in the bacterial cell (9). Inactivation of PAP I resulted in a 50% reduction in RNA polyadenylation and inactivation of both PAP I and PAP II was lethal, indicating that PNPase cannot replace the function of PAP in the cell (reviewed in 2). It is possible that the strict activity of PNPase as an exoribonuclease and not as a polymerase in the bacterial cell, in contrast to the situation under in vitro conditions, is due to the relatively high concentration of phosphate in the bacterial cell (35). However, other possibilities, such as modulation of the activity by interaction with other proteins, cannot be ruled out.
Another observation reported here is that a RNA molecule terminating with a stem–loop structure is a poor substrate for PAP I. Nevertheless, the addition of two or more nucleotides 3′ to the stem–loop structure allows efficient polyadenylation. Most of the mRNA molecules in bacterial cells are characterized by a stem–loop structure at their 3′-end and many also contain a tail of several nucleotides located 3′ of the stem–loop structure (9,10). For example, tRNAs that have a CCA ‘toe hold’ are known to be good substrates for PAP I (20–22). An RNA terminating with a stem–loop structure that is protected both from exonucleolytic degradation and polyadenylation can be produced as a result of exonucleolytic processing of a longer precursor by PNPase or RNase II. In this case, endonucleolytic cleavage by RNase E or another endoribonuclease will be necessary to continue degradation of the RNA. Polyadenylated tails were obtained at the 3′-end of several RNAs, such as RNA I, Sok antisense RNA, rpsO and lpp RNAs. All terminate with a stem–loop structure but have an additional 1–3 nt 3′ of the stem–loop. A detailed analysis of the polyadenylation sites located at both the 3′-end and internal RNase E cleavage sites was performed on the rpsO transcript (24). The 3′-end of this gene is characterized by a stem–loop structure and an additional 3 nt. In bacterial cells expressing RNase E, polyadenylation was observed more at internal cleavage sites than at the mature 3′-end (24). This is also the case with psbA RNA in the chloroplast, in which RNA polyadenylation and degradation seem to be similar to those in bacteria (25). These results suggest the possibility that one of the functions of the stem–loop structures located at the 3′-end of bacterial genes is stabilization of the corresponding RNA by serving as poor substrates for PAP I. Endonucleolytic cleavage of the transcript by RNase E generates a better substrate for PAP I, leading to the cascade of events of polyadenylation and exonucleolytic degradation.
One question that remains open regarding the mechanism of RNA degradation in bacteria is why PAP I is not a stable component of the degradosome. If degradation of an RNA molecule starts with endonucleolytic cleavage by RNase E, followed by polyadenylation, which attracts the exonuclease to the poly(A) tail, then why are the exo- and endoribonucleases associated with each other and not with PAP I? Several models have been suggested to answer this question (10). The RNA helicase is believed to be located in the degradosome in order to release the stem of stem–loop structures paving the way for PNPase. It is also possible that while unwinding the stem, a single-stranded RNA is formed, enabling PAP I to polyadenylate it, thus tagging it for exonucleolytic degradation. This hypothesis is strengthened by the recent observation of a possible interaction between PAP I and the degradosome (22).
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Dr A. J. Carpousis for the PAP I antibodies and Dr R. Rott for critical reading of the manuscript. This work was supported by the Israel Science Foundation administered by the Israel Academy of Science and Humanities, by a grant from the Israel–Japan Scientific Cooperation Research Foundation and by a grant from the Niedersachsen Foundation for Science and Art.
REFERENCES
- 1.Cohen S.N. (1995) Cell, 80, 829–832. [DOI] [PubMed] [Google Scholar]
- 2.Sarkar N. (1997) Annu. Rev. Biochem., 66, 173–197. [DOI] [PubMed] [Google Scholar]
- 3.Cao G. and Sarkar,N. (1992) Proc. Natl Acad. Sci. USA, 89, 7546–7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu F. and Cohen,S.N. (1995) Nature, 374, 180–183. [DOI] [PubMed] [Google Scholar]
- 5.O’Hara E.B., Chekanova,J.A., Ingle,C.A., Kushner,Z.R., Peters,E. and Kushner,R.S. (1995) Proc. Natl Acad. Sci. USA, 92, 1807–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hajnsdorf E., Braun,F., Haugel-Nielsen,J. and Regnier,P. (1995) Proc. Natl Acad. Sci. USA, 92, 3973–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Z., Pandit,S. and Deutscher,P. (1998) Proc. Natl Acad. Sci. USA, 95, 12158–12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dam Mikkelsen N. and Gerdes,K. (1997) Mol. Microbiol., 26, 311–320. [DOI] [PubMed] [Google Scholar]
- 9.Nierlich D.P. and Murakawa,G.J. (1996) Prog. Nucleic Acid Res. Mol. Biol., 52, 153–216. [DOI] [PubMed] [Google Scholar]
- 10.Coburn G.A. and Mackie,G.A. (1999) Prog. Nucleic Acid Res., 62, 55–108. [DOI] [PubMed] [Google Scholar]
- 11.Carpousis A.J., Vanzo,N.F. and Raynal,L.C. (1999) Trends Genet., 15, 24–28. [DOI] [PubMed] [Google Scholar]
- 12.Schuster G., Lisitsky,I. and Klaff,P. (1999) Plant Physiol., 120, 937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayes R., Kudla,J. and Gruissem,W. (1999) Trends Biochem. Sci., 24, 199–202. [DOI] [PubMed] [Google Scholar]
- 14.Lupold D.S., Caoile,A.G. and Stern,D.B. (1999) Plant Cell, 11, 1565–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gagliardi D. and Leaver,C.J. (1999) EMBO J., 18, 3757–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.August J.T., Ortiz,P.J. and Hurwitz,J. (1962) J. Biol. Chem., 237, 3786–3793. [PubMed] [Google Scholar]
- 17.Cao G.J. and Sarkar,N. (1992) Proc. Natl Acad. Sci. USA, 89, 10380–10384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin G. and Keller,W. (1996) EMBO J., 15, 2593–2603. [PMC free article] [PubMed] [Google Scholar]
- 19.Yue D., Maizels,N. and Weiner,A.M. (1996) RNA, 2, 895–908. [PMC free article] [PubMed] [Google Scholar]
- 20.Raynal L.C., Krisch,H.M. and Carpousis,A.J. (1996) Biochimie, 78, 390–398. [DOI] [PubMed] [Google Scholar]
- 21.Raynal L.C., Krisch,H.M. and Carpousis,A.J. (1998) J. Bacteriol., 180, 6276–6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raynal L.C. and Carpousis,A.J. (1999) Mol. Microbiol., 32, 765–775. [DOI] [PubMed] [Google Scholar]
- 23.Kalapos M.P., Cao,G.J., Kushner,S.R. and Sarkar,N. (1994) Biochem. Biophys. Res. Commun., 198, 459–465. [DOI] [PubMed] [Google Scholar]
- 24.Haugel-Nielsen J., Hajnsdorf,E. and Regnier,P. (1996) EMBO J., 15, 3144–3152. [PMC free article] [PubMed] [Google Scholar]
- 25.Lisitsky I., Klaff,P. and Schuster,G. (1996) Proc. Natl Acad. Sci. USA, 93, 13398–13403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rott R., Drager,R.G., Stern,D.B. and Schuster,G. (1996) Mol. Gen. Genet., 252, 676–683. [DOI] [PubMed] [Google Scholar]
- 27.Stern D.B. and Gruissem,W. (1987) Cell, 51, 1145–1157. [DOI] [PubMed] [Google Scholar]
- 28.Blum E., Carpousis,A.J. and Higgins,C.F. (1999) J. Biol. Chem., 274, 4009–4016. [DOI] [PubMed] [Google Scholar]
- 29.Lisitsky I. and Schuster,G. (1999) Eur. J. Biochem., 261, 468–474. [DOI] [PubMed] [Google Scholar]
- 30.Lisitsky I., Kotler,A. and Schuster,G. (1997) J. Biol. Chem., 272, 17648–17653. [DOI] [PubMed] [Google Scholar]
- 31.Martin G. and Keller,G. (1998) RNA, 4, 226–230. [PMC free article] [PubMed] [Google Scholar]
- 32.Lisitsky I., Liveanu,V. and Schuster,G. (1994) Nucleic Acids Res., 22, 4719–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao G. and Sarkar,N. (1997) Biochem. Biophys. Res. Commun., 239, 46–50. [DOI] [PubMed] [Google Scholar]
- 34.Wahle W. and Keller,W. (1996) Trends Biochem. Sci., 21, 247–250. [PubMed] [Google Scholar]
- 35.Littauer U.Z. and Grunberg-Manago,M. (1999) The Encyclopedia of Molecular Biology. John Wiley & Sons, New York, NY, pp. 1–7.