


Abstract
People afflicted with certain rheumatological autoimmune diseases produce autoantibodies directed against a select group of proteins such as the La autoantigen. Biochemical studies have revealed La to be a promiscuous RNA-binding protein that appears to play a role in a variety of intracellular activities such as processing and/or transport of RNA polymerase III precursor transcripts and translational regulation from internal ribosome entry sites (IRES). We have previously identified an RNA-binding protein that is a Drosophila melanogaster homolog of La (D-La) and shown that early transcript accumulation throughout the embryo is later refined to be most prevalent in the visceral mesoderm, gut, gonads and salivary glands. Here we report the first in vivo genetic characterization of a La homolog in a multicellular eukaryote. Lethality was observed in homozygous larvae harboring a small chromosomal deletion that removed the D-La gene, which was rescued by an inducible D-La cDNA transgene. This implies that D-La confers essential functions for larval development. In addition, loss of D-La function gives rise to defects in embryonic midgut morphogenesis; one of the midgut defects correlates with loss of Ultrabithorax (Ubx) expression along the second midgut constriction. Finally, genetic interactions between chromosomal deficiencies that remove D-La and certain Ubx alleles were demonstrated in adults. Our results support the hypothesis that D-La provides essential functions for proper Drosophila development and imply that the conserved La family of proteins may perform critical developmental functions in higher eukaryotes.
INTRODUCTION
The RNA-binding protein La/SSB is one of the major antigens against which autoantibodies are targeted in human rheumatological autoimmune diseases such as systemic lupus erythematosus (SLE) and Sjogren’s syndrome. La normally localizes to the nuclear or cytoplasmic compartments of the cell, where it exists as part of a ribonuclear protein (RNP) complex of unknown function (1,2). Biochemical and molecular studies have indicated that La can bind to the 3′-end of small nuclear RNA (snRNA) precursors transcribed by RNA polymerase III. Examples of this association include precursors of tRNA, 4.5S RNA, 5S RNA, 7S RNA and U6 RNA (3,4), where La binds to a short tract of uridylate residues (5–7). The significance of this binding is unknown but, as a consequence, La may function in the termination, processing and/or nuclear export of RNA polymerase III precursor transcripts. La has also been implicated in transcript release and transcription reinitiation by RNA polymerase III (8,9).
Other studies have connected La to the regulation of translation. For example, La can bind to and alleviate the translational repression normally mediated by the secondary structure of the TAR sequence located within the first 111 nt of HIV-1 mRNA (10,11). This binding is direct and specific, since La–TAR RNP complexes can be identified in HIV-1-infected lymphocytes. In addition, the specific binding of La to transcripts containing TAR sequences fused to a reporter gene can relieve TAR-mediated translational inhibition in reticulocyte lysates (10,11).
La has also been implicated in regulating translational initiation from internal ribosome entry sites (IRES). Translation of mRNA is usually dependent on recognition of the 5′-cap structure and subsequent scanning by the ribosome (12). However, viruses with segmented RNA genomes that lack caps use IRESs to properly recruit the ribosome and initiate translation. La-enhanced internal initiation of translation at IRESs has been reported for certain uncapped picornaviruses such as poliovirus (13). In addition, certain cellular mRNAs possess relatively long 5′-untranslated regions (UTRs) or extensive secondary structures that prevent cap-dependent translational read-through by the ribosome. In such cases, translational initiation can occur through an IRES (14,15). UV crosslinking experiments and competition assays have revealed that La can bind to IRESs and that this binding can enhance IRES-mediated translation (16). Functional IRESs have been identified by assaying the expression of dicistronic constructs in transfected cells. Examples of mRNAs with a functional IRES include those encoding human fibroblast growth factor (fgf2) (17), human insulin growth factor (18), human eIF-4G (19), human immunoglobulin heavy chain binding protein (Bip) (20,21) and the Drosophila homeotic gene Antennapedia (22). Moreover, mRNA encoded by the Drosophila homeotic gene Ultrabithorax (Ubx) has been shown to contain an IRES that can function in transfected cells and in transgenic flies (23).
In summary, biochemical and molecular data have shown La to be an RNA-binding protein implicated in many aspects of RNA metabolism. It has been proposed that La can do this by acting as a molecular chaperone that stabilizes the tertiary structure of RNA (24). This stabilization is thought to make RNA more accessible to proteins that mediate diverse processes such as RNA transport, processing and translation. A genetic analysis of La protein function would provide an entry point to establish the in vivo role of La in these RNA metabolic and regulatory pathways. To date, genetic analysis has only been performed with a yeast La homolog in Saccharomyces cerevisiae. Unfortunately, yeast cells containing a null allele of the gene encoding the La protein are viable, suggesting that either La is not essential in yeast or that another protein(s) plays a functionally redundant role (25).
To address the consequences of genetically altering the activity of the La gene in a higher multicellular eukaryote, we have focused on the fruit fly Drosophila melanogaster. Previously, we reported the identification of cDNA clones from D.melanogaster that encode a protein homologous throughout its entire length to vertebrate La (26). Like its vertebrate homologs, D-La can bind RNA in vitro, presumably through its putative RNA recognition motif (RRM) which harbors RNP1 and RNP2 consensus sequences. Whole mount in situ hybridization experiments revealed that D-La transcripts are not present in all ovarian tissues and early expression throughout the embryo is later refined to be most prevalent in the visceral mesoderm, gonads, gut and salivary glands. In the present study, we provide genetic evidence suggesting that D-La is encoded by an essential gene that functions at multiple stages of Drosophila development. Our results imply that the activity of the conserved La protein family is essential for proper development of higher eukaryotes.
MATERIALS AND METHODS
Drosophila strains
Fly strain l(2)08327 was obtained from the collection of Allan Spradling (Carnegie Institution). Df(2L)11A was obtained from Lynn Manseau (University of Arizona). In situ hybridization of l(2)08327 larval salivary gland polytene chromosomes revealed the location of P-element insertions at 27A1,2, 38C1,2 and 47A11,14. All other stocks were either generated in this study or obtained from the Bloomington Drosophila Stock Center.
Recombination
Since D-La maps to the 38C region (26), we used genetic recombination to remove two undesirable P-element insertions in l(2)08327. First, we recombined the 38C1,2 and 47A11,14 insertions onto a chromosome marked with the dominant marker Sp (at 28A1,B4) to remove the P-element insertion at 27A1,2. We then used recombination to remove the 47A11,14 insertion by following loss of the recessive marker cn (at 43E6,8). The location of the P-element insertion in resulting stocks was confirmed by genomic Southern blot analysis and in situ hybridization to polytene chromosomes (data not shown). To simplify phenotypic analysis following local P-element transposition, we also removed the Sp marker by recombination with a wild-type chromosome. One of derived lines, fs(2)P8, was used in the local transposition screen.
Screening for P-element-derived D-La alleles
A screen for local transposition was performed (27) as follows. Female fs(2)P8/CyO; ry506 flies were crossed to male Sp cn/CyO; Sb Δ 2-3/TM3 flies. Virgin females of genotype fs(2)P8/CyO; Sb Δ 2-3/ry506 were selected and mated with bTft/CyO; ry506 males. Offspring that lacked the Sb marker but carry ry+ were candidates for further analysis. Approximately 1200 individuals were then each mated to several Df(2L)pr21/CyO flies that carried a tester chromosome containing a large deletion in the 38C region around D-La. A total of 27 crosses failed to give straight winged P, ry+/Df(2L)pr21 progeny, representing new P-element-derived alleles that are lethal over the Df(2L)pr21 deficiency. Male flies of the genotype P, ry+/CyO were then crossed to another tester chromosome Df(2L)11A, which represents a small overlapping deficiency in 38C (L.Manseau, personal communication; see also Fig. 1). Only five of the crosses failed to produce P, ry+/Df(2L)11A (straight winged) flies. Oligonucleotide primers directed against the ends of the P-element included P-3 (5′-GCAAGCATACGTTAAGTGGATG) and pBRI (5′-GA-ATTCATACTTCGGTAAGCTTCGGCTATCGACG).
Figure 1.

Genomic map of the region containing RhoGEF, D-La and spire. Sequence data for this region is available from P1 clone AC002503. The breakpoints of deficiencies Df(2L)11A and Df(2L)pr21 are shown below and represented by dark rectangles. The structures of Df(2L)PJ17, Df(2L)PJ19 and f(2L)PJ2 are also shown. Arrows indicate that the exact deletion end point was not precisely determined.
Phenotypic analysis
Two balanced D-La deficiency stocks, Df(2L)PJ17 and Df(2L)PJ19, were selected for genetic analysis. Lethality tests were performed with the Df(2L)PJ17 and Df(2L)PJ19 alleles by crossing females from each of these stocks with corresponding mutant/wild-type males. We also crossed +/CyO females to wild-type males as a control. For analysis of mutant embryonic phenotypes, Df(2L)PJ17 and Df(2L)PJ19 were placed over a marked balancer chromosome [CyO, P(ftz-lacZ)]. Homozygous mutant embryos were distinguished by the lack of β-galactosidase expression when stained with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal).
β-Galactosidase staining
Appropriately aged embryos were dechorionated with 100% Clorox for 3 min and fixed with 2.5% formaldehyde and heptane for 10 min. Fixed embryos were then devitellinized by shaking vigorously in a heptane/methanol mixture for 1 min. After heptane was removed, embryos were rehydrated in 0.5% Triton X-100 in phosphate-buffered saline (PBT). The PBT was then replaced with staining buffer containing 10 mM sodium phosphate, pH 7.2, 150 mM NaCl, 1 mM MgCl2, 3 mM K4[Fe(II)CN6], 3 mM K3[Fe(III)CN6], 0.3% Triton X-100, 0.2% X-Gal. Embryos were stained overnight. Stained samples were then rinsed with PBT and mounted in Polyaquamount (Polysciences).
Whole mount antibody staining
Immunolocalization of Ubx protein in embryos was performed as previously described (28,29). The monoclonal mouse anti-Ubx antibody FP3.38 (30) was used at a 1:20 dilution and the horse/goat anti-mouse secondary antibody (Vector Laboratories) was diluted 1:500. Histochemical reactions were performed using the DAB substrate kit (Vector Laboratories), as recommended by the manufacturer. Stained embryos were mounted in Polyaquamount (Polysciences).
Transgene construction and germline transformation
A P-element transposon containing D-La was constructed and introduced into the Drosophila germline (31). The transposon was constructed as follows. A 1.4 kb D-La cDNA fragment was released from pBluescript (26) with restriction enzymes ClaI and SacI. The DNA fragment was then purified and inserted into the ClaI and SacI sites of plasmid pSP73 (Promega). The 318 bp hsp70 promoter (–256 to +62) was then excised from HIC-L (32) by BamHI + ClaI digestion and ligated into the BglII and ClaI sites of the D-La-containing pSP73 construct described above. The hsp70-D-La transgene cassette was then excised from pSP73 with XbaI and inserted into the XbaI site of P-element vector pRosy Rhino (33). Germline transformation was performed by injecting this construct into cn; ry506 embryos together with plasmid pπ25.7wc, which provides transposase (34). A transformed line containing a single transposon insertion on the third chromosome was selected for rescue experiments.
RESULTS
Generation of D-La loss of function mutants
Based on its putative involvement in several distinct cellular processes, we assumed that D-La is encoded by an essential gene. Though mutations in essential genes located in the vicinity of D-La at 38C1,2 have been reported, lethal saturation screens uncovering all possible complementation groups within this region have not been conducted. We thus decided to generate mutations for phenotypic analysis by conducting a P-element transposition screen (35) over a series of overlapping deficiencies that completely remove the D-La gene. Figure 1 schematically depicts the boundaries of two available deficiencies, Df(2L)11A and Df(2L)pr21, that completely removed the 38C1,2 region. Quantitative genomic Southern blot analysis confirmed that D-La was completely removed from Df(2L)pr21 (data not shown). An available P-element insertion l(2)08327, in the 38C1,2 region, was obtained from the collection of Allan Spradling (Carnegie Institution) and subsequent molecular analysis indicated that the D-La transcription unit was not disrupted (data not shown). In addition to the P-element (marked with ry+) at 38C1,2, this chromosome also harbored two additional insertions at 27A1,2 and 47A11,14. Hence, before using this stock for local P-element transposition, we used genetic recombination to remove the two undesirable P-elements and generated a new stock containing only a single P-element insertion at 38C1,2. The resulting stock, which we refer to as fs(2)P8, was no longer homozygous lethal, but was female sterile due to a single P-element insertion that could not be complemented by mutant alleles of the female sterile gene spire (36,37).
DNA sequences flanking the transposon insertion in fs(2)P8 were isolated, sequenced and found to be located in the first exon of spire, as deduced from P1 clone AC002503, placing the P-element less than 40 kb away from D-La (Fig. 1). Since mobilized P-elements tend to reinsert locally in the vicinity of the original insertion (35), we attempted to mobilize the P-element in fs(2)P8 in an effort to generate insertional mutations in D-La. Five of the 1200 jumps were lethal over the overlapping deficiencies Df(2L)11A and Df(2L)pr21 that completely removed the D-La gene. To confirm that these five lethal lines contain P-elements, genomic DNA was digested with EcoRI and SalI and probed with the left end of the ry gene (ryL) (Fig. 2A). This probe hybridizes to a 3 kb EcoRI–SalI fragment of the endogenous ry gene as well as to fragments containing a ry transgene in the P-element (38). Since this SalI site is included in the ry transgene, the ryL probe detects fragments from EcoRI on the P-element to the next flanking genomic SalI or EcoRI site. This analysis indicated that all five lines contain P-element insertions (Fig. 2B). Moreover, the P-elements in PJ2, PJ17 and PJ19 appear to be located in the same position as in the original P-element in fs(2)P8. PJ20 and PJ25 contain new P-element insertions in the 38C region, but subsequent molecular analysis indicated that neither disrupted the D-La transcription unit (data not shown). This analysis also revealed that fs(2)P8, PJ2 and PJ17 display a band migrating below the 3 kb endogenous ry fragment (Fig. 2B) which may represent a P-element inserted within a P-element or a partially deleted ry gene inserted somewhere between Sp and cn on chromosome II. We favor the first possibility since there is no evidence for a second P-element in chromosomal in situ hybridization experiments. Nevertheless, since this second insertion is also in the parental fs(2)P8 strain, it clearly does not contribute to any detectable phenotype.
Figure 2.
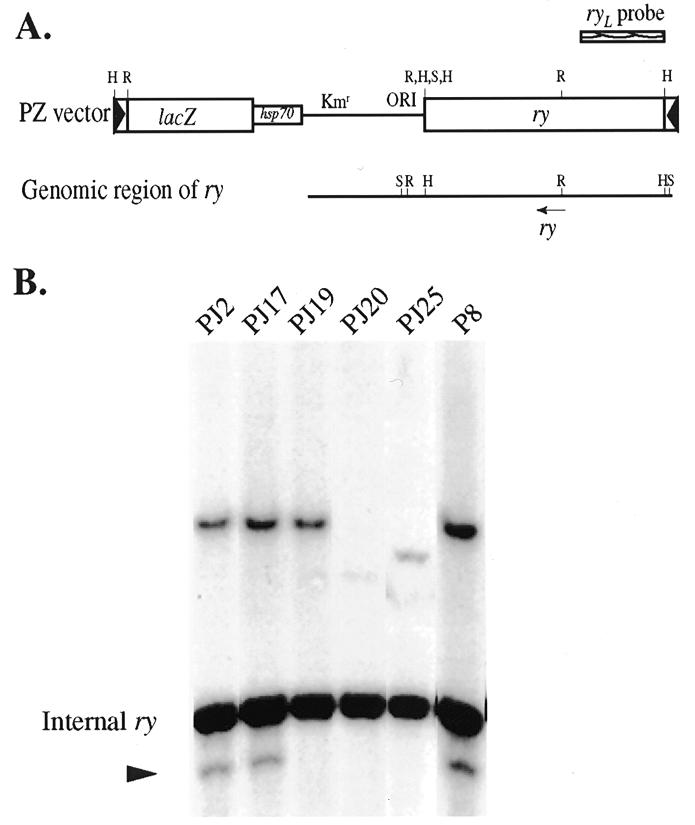
Southern blot analysis of genomic DNA from lines generated by local P-element transposition. (A) Restriction maps of the PZ vector and the genomic region of ry as well as the ryL probe. R, EcoRI; H, HindIII; S, SalI. (B) Southern blot analysis. Genomic DNA was digested with EcoRI and SalI and probed with ryL. Each lane contained ~4 µg of genomic DNA. A small band representing a second P-element is indicated by an arrowhead.
The D-La gene is located between positions 44077 and 42468 on the P1 clone AC002503 (Fig. 1). Since PJ2, PJ17 and PJ19 represent lethal alleles that were indistinguishable (in Southern blots using the ryL probe) from the female sterile fs(2)P8 insertion, we concluded that the lethality must result from deletions adjacent to the original P-element insertion. To determine if any of these putative deletions affected D-La, a series of primers along the AC002503 clone were designed and used together with P-element primers in PCR amplification experiments. The most interesting result was obtained with PJ17 genomic DNA, when primer pair pBRI (at the 5′-end of the P-element, facing proximally) and RhoGEF45180 (between positions 45180 and 45161 on AC002503) were used, which generated a fragment of ~1.2 kb. Sequencing of the generated PCR product revealed that the deletion broke at the fourth amino acid of D-La. At the opposite end of the P-element, when primer pair P-3 (at the 3′-end, facing distally) and 4877 (between positions 4877 and 4897 on AC002503) were used, a fragment of ~750 bp was generated. Sequencing of this PCR product confirmed that the P-element in PJ17 was still inserted at position 5611, exactly as in fs(2)P8, from which it was derived. Therefore, in Df(2L)PJ17 the genomic region from 5612 to 44006 is deleted, removing all of the coding region of spire and D-La. The deletions in Df(2L)PJ2 and Df(2L)PJ19 contain the same distal break point but extend past D-La and RhoGEF (Fig. 1).
D-La deficiencies are lethal
Molecular analysis indicated that the adjacent genes spire and D-La (which overlap by 6 bp at their 3′-ends) are disrupted in deficiencies Df(2L)PJ2, Df(2L)PJ17 and Df(2L)PJ19. Since spire is not an essential gene (36,37) and is located next to D-La, then the lethality associated with Df(2L)PJ17 apparently results from either a deletion of D-La, an unknown overlapping transcription unit or from a spontaneously arising secondary lethal mutation located outside the 38C1,2 region. To examine the latter possibility, we performed complementation tests among fs(2)P8, Df(2L)PJ17 and Df(2L)PJ19. The finding that Df(2L)PJ17 and Df(2L)PJ19 could not complement the female sterility of fs(2)P8 is consistent with the molecular data that spire is disrupted in both deficiency chromosomes. However, fs(2)P8 can complement the lethality of both Df(2L)PJ17 and Df(2L)PJ19, which are derived independently and contain deletions in spire and D-La, whereas Df(2L)PJ17 and Df(2L)PJ19 cannot complement each other (Table 1). These results support the hypothesis that the lethality associated with Df(2L)PJ17 is due to removal of DNA between D-La and spire and not the result of secondary mutations arising outside the chromosomal deletion at 38C1,2.
Table 1. Complementation tests.
| Genotypes | Straight wing progeny | Curly wing progeny |
|---|---|---|
| P8/CyO × PJ17/CyO | 5 | 38 |
| P8/CyO × PJ19/CyO | 14 | 54 |
| PJ17/CyO × PJ19/CyO | 0 | 100 |
The number of progeny obtained are indicated under the observed phenotype.
To determine the stage at which D-La may be required for viability, a lethality test was conducted (as described in Materials and Methods). Since only 25% of the offspring derived from the test cross are homozygous mutant for D-La, lethality is defined as the stage whose survival rate is proportionally decreased. As shown in Table 2, the lethality associated with both Df(2L)PJ17 and Df(2L)PJ19 occurs at the larval stage. Based on the zygotic visceral mesoderm expression of D-La, mutant embryos would be expected to hatch but may die later as feeding larvae because of possible associated gut abnormalities. Examination of homozygous mutant D-La embryos (as revealed by lack of β-galactosidase staining of the lacZ marked balancer chromosome) indicated that most of the embryos displayed a gut phenotype resembling a failure of the three midgut constrictions (Fig. 3).
Table 2. Lethality test.
| Stage | Oregon R | PJ17 | PJ19 |
|---|---|---|---|
| Embryo | 340 | 365 | 391 |
| Larvae | 328 (96%) | 351 (96%) | 367 (95%) |
| Pupae | 281 (86%) | 241 (68%) | 204 (56%) |
| Adults | 273 (97%) | 233 (97%) | 194 (95%) |
For each column, the first row represents the actual number of embryos sampled and the following rows refer to the number that survived to the next developmental stage (with percent survival indicated).
Figure 3.
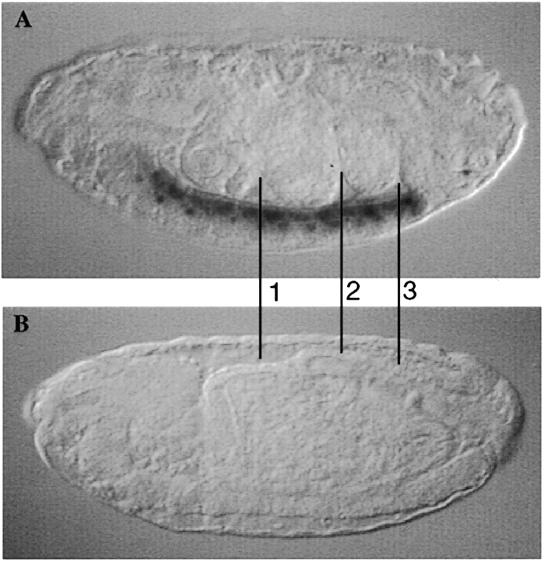
Loss of D-La function gives rise to embryonic midgut morphogenesis defects. This allele is balanced over CyO ftz-lacZ which enables the identification of Df(2L)PJ19 homozygotes by the absence of X-gal staining in the central nervous system. The three midgut constrictions that normally occur in wild-type embryos (lines in A) are absent in Df(2L)PJ19 homozygotes (B). Similar results were observed with Df(2L)PJ17 embryos. Embryos were viewed laterally with anterior on the left using a 20× objective and 10× ocular lens.
Genetic rescue of Df(2L)PJ17 lethality
Assuming that loss of D-La is responsible for the associated lethality, we attempted to rescue Df(2L)PJ17 and Df(2L)PJ19 by introducing chromosomes that carried a D-La cDNA under control of the hsp70 promoter. After examining over 5000 individual flies for each strain maintained at 25°C, we did not observe any Df(2L)PJ17 or Df(2L)PJ19 escapers. To induce expression of the hsp70-D-La cDNA, we employed a protocol which administered two heat shock treatments per day over a 2 week period. As a negative control, we performed identical heat shock induction experiments and monitored the emergence to adulthood of 1691 Df(2L)PJ17 and 1123 Df(2L)PJ19 mutants that lacked D-La cDNA transgenes. Homozygous escapers were not observed in these negative control groups. Thus, any homozygous (straight wing) flies carrying an inducible D-La cDNA transgene that emerge from our heat shock induction experiments would represent rescued adults. We did not observe any rescue of Df(2L)PJ19 (out of a total of 881 flies examined). However, a total of three straight wing adults out of 682 flies examined were rescued in Df(2L)PJ17 by the D-La cDNA transgene. Among these rescued flies, one was female and two were male. All three flies died within a week after eclosion in the absence of additional heat shock. As expected, no offspring were generated from the rescued female (which was still homozygous for the female sterile spire deletion) when crossed to wild-type males. These results support the hypothesis that the larval lethality associated with Df(2L)PJ17 is due to a loss of D-La function and suggest that expression of D-La is also required in adults.
Deficiencies lacking D-La fail to express Ubx in the second midgut constriction
We have shown that deficiencies in D-La function give rise to a midgut phenotype in the embryo. Since vertebrate La has been implicated in regulating translational initiation from IRESs, we determined whether any of the genes known to function in Drosophila midgut morphogenesis contained such an element. A good candidate was Ubx, which is expressed along and required for formation of the second midgut constriction (39) and whose translation is mediated by internal ribosome entry mechanisms (23). To test whether D-La may regulate expression of Ubx in this region, we examined the distribution of Ubx protein in wild-type and mutant embryos. In wild-type embryos, both Ubx mRNA and protein can be detected in the second midgut constriction (Fig. 4A and B). However, in homozygous mutant embryos (as revealed by lack of β-galactosidase staining of the lacZ marked balancer chromosome) expression of Ubx was lost (Fig. 4E and F), resulting in gut constriction defects during embryogenesis. The penetrance of this midgut phenotype is 73% (72 of 98 homozygous stage 15–16 D-La mutant embryos). The constrictions and Ubx expression in the remaining mutant embryos (27%) were unaffected.
Figure 4.
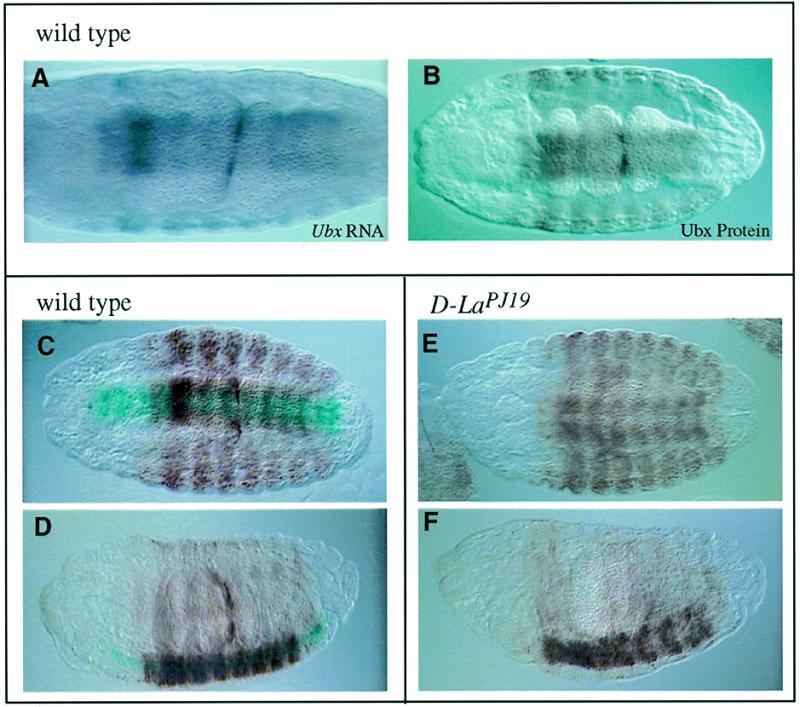
Ubx expression is altered in D-La mutant embryos. (A) Ubx mRNA (purple staining) can be detected in the second midgut constriction of wild-type embryos. (B) Ubx protein (brown staining) is evident in the second midgut constriction in wild-type embryos. (C and D) In wild-type embryos (as distinguished by X-Gal staining in blue), Ubx protein can be detected in the second midgut constriction. (E and F) In homozygous mutant D-LaPJ19 embryos, Ubx protein cannot be detected in the region that normally forms the second midgut constriction and all three constrictions fail to form. All embryos are oriented with the anterior on the left and viewed with a 20× objective and 10× ocular lens, except in (A) where a 40× objective was used. The embryos in (A), (B), (C) and (E) are viewed from the dorsal side, embryos in (D) and (F) are viewed laterally.
The results suggesting that D-La is required for expression of Ubx in the second midgut constriction imply that D-La may regulate translational initiation of Ubx directly from the IRES. If D-La is required for the IRES-dependent translation of Ubx, we would expect to see an effect on translation of Ubx protein in D-La deletion embryos, but no effect on Ubx transcription in the second midgut constriction region. When we examined D-La mutant embryos for Ubx mRNA and protein simultaneously, neither was detected and we did not observe any embryos that expressed Ubx mRNA in the absence of Ubx protein (data not shown). Thus, instead of supporting a direct involvement of D-La in regulating translational initiation from the Ubx IRES, these results are more consistent with a role in stabilizing Ubx transcripts.
Genetic interactions in adults between D-La deficiencies and Ubx alleles
Certain Ubx alleles, when maintained as heterozygotes, display ectopic bristles on adult halteres. One such allele, Ubx9.22, which contains a deletion that originates in the third intron and extends into the 3′ exon, does not produce any of the Ubx protein isoforms (40). Flies that are Ubx9.22/+ contain an average of 0.9 bristles/haltere (41). To determine whether a D-La allele can enhance or suppress this phenotype, we crossed Df(2L)PJ17/CyO females to Ubx9.22/MKRS males and examined the halteres of adult progeny with genotype Df(2L)PJ17/+; Ubx9.22/+. An examination of 203 adults revealed that flies heterozygous for both Df(2L)PJ17 and Ubx9.22 display an average of 3.5 bristles/haltere, thus enhancing the Ubx9.22 adult phenotype. This is consistent with the deficiency screen of Burnette et al. (41), who reported an average of 2.9 bristles/haltere for flies heterozygous for both Ubx9.22 and Df(2L)TW84, a large deficiency that covers the D-La region. These results imply that dominant genetic interactions between D-La and Ubx can occur in adult structures. In addition, RT–PCR analysis of Ubx9.22 and Df(2L)PJ17 double heterozygotes did not indicate any alterations in the relative accumulation of Ubx isoforms (A.R.Hatton and A.J.Lopez, personal communication), suggesting that D-La does not affect the splicing of Ubx transcripts.
DISCUSSION
The goal of this study was to address the consequences of genetically altering the activity of La in a higher multicellular eukaryote. To accomplish this aim, we focused on genetic assessment of D-La, a La homolog of D.melanogaster. In generating D-La null alleles, PCR analysis and DNA sequencing confirmed that Df(2L)PJ17 contained a deletion which removed both spire and the adjacent D-La gene. Since spire is not a lethal gene (36,37), this implied that the lethality of Df(2L)PJ17 was caused by loss of D-La function. This was confirmed in genetic rescue experiments by crossing into Df(2L)PJ17, a transgenic line which contained an inducible hsp70-D-La cDNA construct on the third chromosome. The fact that adults rescued with hsp70-D-La cDNA died after cessation of heat shock induction indicates that ongoing D-La expression is required even during adulthood. This low frequency of rescue is not surprising considering that the cDNA transgene was driven by a hsp70 promoter and thus required a continuous induction scheme over the entire Drosophila life cycle. Moreover, since D-La is a promiscuous RNA-binding protein, its induced overexpression (which is a consequence of the rescue assay) may be detrimental to several different RNA metabolic pathways and this in turn can be responsible for the low rescue frequency. In support of this, we have noted loss of viability in heat shocked lines that contain wild-type copies of the endogenous D-La gene and a hsp70-D-La-cDNA transgene (data not shown). Df(2L)PJ19, which contains an extended proximal deletion beyond D-La (Fig. 1), was not rescued by crossing in transgenic lines that contain a hsp70-D-La cDNA, suggesting the presence of additional essential genes in the deleted region.
In addition to larval lethality, we have shown that deficiency homozygotes that lack D-La function give rise to embryonic midgut phenotypes. Since Ubx mRNAs contain a functional IRES (23) and are expressed and required for the second midgut constriction (39), it seemed likely that D-La may be involved in controlling the translation of Ubx. In support of this hypothesis, vertebrate La has been shown to directly bind and regulate translation through IRES elements (10,11,16). Consistent with this interpretation, our analysis revealed that 73% of homozygous stage 15–16 D-La mutant embryos did not express Ubx protein. However, when D-La mutant embryos were examined for mRNA and protein simultaneously, we did not find any embryos that expressed Ubx mRNA in the absence of Ubx protein.
Rather than directly regulating Ubx translation through the IRES, our results are more supportive of the possibility that D-La may indirectly control translation by preventing the turnover of Ubx transcripts. Under this scenario, D-La may bind and stabilize Ubx mRNA by acting as a molecular chaperone. In contrast to what is observed in D-La mutants, Ubx mRNA is stable and can be clearly detected along the second midgut constriction of wild-type embryos. If D-La is functioning as a universal RNA chaperone, it is not surprising that a hsp70-D-La cDNA construct was capable of rescuing defects associated with D-La loss of function without introducing many adverse gain of function phenotypes. The RNA chaperone hypothesis is also consistent with the RNA-binding promiscuity associated with La family members and their putative functional role in several unrelated post-transcriptional processes (such as TAR- and IRES-mediated translational control as well as processing of RNA polymerase III precursor transcripts). This model also attractively explains how La can function in various aspects of RNA metabolism. However, the relatively discrete embryonic midgut phenotype associated with D-La loss of function is somewhat at odds with the RNA chaperone hypothesis. One possibility is that although D-La may stabilize a variety of RNAs and increase the efficiency of many RNA metabolic processes, it is not essential for most aspects of RNA metabolism. This is consistent with the lack of lethality in yeast knockouts. Specific phenotypes in higher organisms may be explained by proposing that, in addition to acting as an RNA chaperone, La might specifically interact with or recruit other essential proteins in particular processes. For example, D-La may be controlling the stability of the Ubx midgut-specific isoform (40) by recruiting an essential unknown factor. The absence of D-La protein may allow otherwise sequestered nucleases to become available to degrade Ubx mRNA. Rather than a direct interaction between D-La and Ubx mRNA, our expression, rescue and haploinsufficiency data could be alternatively interpreted as indirect effects of D-La in the regulation of nuclease activity or localization.
Based on the results presented here, and the overwhelming evidence that La proteins directly bind to many RNAs, we favor the hypothesis that D-La may stabilize Ubx mRNA and facilitate its efficient translation. Whether this putative direct regulation of stability and indirect regulation of translational efficiency operates through the IRES remains to be demonstrated. Genetic analysis of a yeast La homolog supports this speculation. One of two yeast homologs is encoded by the LHP1 gene (25), which, like mammalian La, binds nascent polymerase III transcripts and is involved in their processing. In wild-type yeast cells, Lhp1p is involved in the endonucleolytic removal of the 3′-trailer sequence of pre-tRNASer. In the absence of Lhp1p, the 3′-trailer sequence of pre-tRNASer is removed by an exonuclease(s). Though LHP1 is normally not required for viability, when a second mutation that disrupts the anticodon stem structure of pre-tRNAs occurs, Lhp1p becomes necessary for survival. An additional mutation that restores base pairing of the anticodon stem structure eliminates the requirement for Lhp1p (42). These data suggest that the yeast La homolog may function to stabilize the conformation of yeast pre-tRNA so as to allow pre-tRNA processing to occur in a specific pathway.
Interestingly, the midgut phenotype associated with D-La mutant embryos is not fully penetrant (i.e. 73%) whereas larval lethality is observed in all homozygotes. One explanation to account for this difference is that maternal D-La may compensate for loss of the zygotic function in the embryo. Maternal D-La transcripts may persist and contribute functionally after gastrulation and therefore partially complement the requirement for zygotic D-La. Since the zygotic expression of D-La and Ubx overlap only in the central midgut, it is possible that a maternally derived source of D-La may regulate Ubx elsewhere in the embryo, but this will have to await the analysis of D-La germline clones. Thus, the incomplete penetrance (73%) of the embryonic midgut constriction phenotype associated with Df(2L)PJ17 homozygotes may be due to stochastic compensation by maternally derived D-La transcripts in the embryonic midgut. This maternal activity may be insufficient or may fail to persist later in development to compensate for lack of the zygotic function during larval development. Other possibilities are that translation of Ubx is controlled differentially in different tissues (supported by the observation that its translation in the ventral cord is not affected in the absence of D-La) or that other molecules can partially compensate for loss of D-La function. It is also possible that other RNA metabolic processes, such as expression and maturation of RNA polymerase III transcripts and translational regulation of other loci besides Ubx, may be affected by the D-La insufficiency.
To further examine its putative role as an RNA chaperone functioning in a variety of post-transcriptional processes, future experiments with D-La will focus on performing genetic screens to identify interacting genes and downstream targets.
Acknowledgments
ACKNOWLEDGEMENTS
We thank L. Manseau, A. R. Hatton and A. J. Lopez for communicating unpublished data and for helpful discussions. We also thank C. R. Chen for technical assistance. This work was supported by grants to P.P.T. from the Markey Charitable Trust-Sponsored PHRI New Initiative Program and the National Institutes of Health (HG01783).
REFERENCES
- 1.Bachmann M., Falke,D., Schroder,H.C. and Muller,W.E.G. (1989) J. Gen. Virol., 70, 881–891. [DOI] [PubMed] [Google Scholar]
- 2.Bachmann M., Pfeifer,K., Schroder,H.C. and Muller,W.E.G. (1989) Mol. Cell. Biochem., 85, 103–114. [DOI] [PubMed] [Google Scholar]
- 3.Rinke J. and Steitz,J.A. (1982) Cell, 29, 149–159. [DOI] [PubMed] [Google Scholar]
- 4.Rinke J. and Steitz,J.A. (1985) Nucleic Acids Res., 13, 2617–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy R., Henning,D., Tan,E. and Busch,H. (1983) J. Biol. Chem., 258, 8352–8356. [PubMed] [Google Scholar]
- 6.Mathews M.B. and Francoeur,A.M. (1984) Mol. Cell. Biol., 4, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stefano J.E. (1984) Cell, 36, 145–154. [DOI] [PubMed] [Google Scholar]
- 8.Maraia R.J., Kenan,D.J. and Keene,J.D. (1994) Mol. Cell. Biol., 14, 2147–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maraia R.J. (1996) Proc. Natl Acad. Sci. USA, 93, 3383–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang Y.N., Kenan,D.J., Keene,J.D., Gatignol,A. and Jeang,K.T. (1994) J. Virol., 68, 7008–7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Svitkin Y.V., Pause,A. and Sonenberg,N. (1994) J. Virol., 68, 7001–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kozak M. (1989) J. Cell Biol., 108, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meerovitch K., Svitkin,Y.V., Lee,H.S., Lejbkowicz,F., Kenan,K.J., Chan,E.K.L., Agol,V.I., Keene,J.D. and Sonenberg,N. (1993) J. Virol., 67, 3798–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pelletier J. and Sonenberg,N. (1988) Nature, 334, 320–325. [DOI] [PubMed] [Google Scholar]
- 15.Jang S.K., Davies,M.V., Kaufman,R.J. and Wimmer,E. (1989) J. Virol., 63, 1651–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ali N. and Siddiqui,A. (1997) Proc. Natl Acad. Sci. USA, 94, 2249–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vagner S., Gensac,M.C., Maret,A., Bayard,F., Amalric,F., Prats,H. and Prats,A.C. (1995) Mol. Cell. Biol., 15, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teerink H., Voorma,H.O. and Thomas,A.A. (1995) Biochim. Biophys. Acta, 1264, 403–408. [DOI] [PubMed] [Google Scholar]
- 19.Gan W. and Rhoads,R.E. (1996) J. Biol. Chem., 271, 623–626. [DOI] [PubMed] [Google Scholar]
- 20.Macejak D.G. and Sarnow,P. (1991) Nature, 353, 90–94. [DOI] [PubMed] [Google Scholar]
- 21.Sarnow P. (1989) Proc. Natl Acad. Sci. USA, 86, 5795–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oh S.-K., Scott,M.P. and Sarnow,P. (1992) Genes Dev., 6, 1643–1653. [DOI] [PubMed] [Google Scholar]
- 23.Ye X., Fong,P., Iizuka,N., Choate,D. and Cavener,D. (1997) Mol. Cell. Biol., 17, 1714–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pannone B.K., Xue,D. and Wolin,S.L. (1998) EMBO J., 17, 7442–7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoo C.J. and Wolin,S.L. (1994) Mol. Cell. Biol., 14, 5412–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bai C., Li,Z. and Tolias,P.P. (1994) Mol. Cell. Biol., 14, 5123–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robertson H.M., Preston,C.R., Phillis,R.W., Johnson-Schlitz,D.M., Benz,W.K. and Engels,W.R. (1988) Genetics, 118, 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xue F. and Cooley,L. (1993) Cell, 72, 681–693. [DOI] [PubMed] [Google Scholar]
- 29.Stroumbakis N.D., Li,Z. and Tolias,P.P. (1996) Mol. Cell. Biol., 16, 192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White R.A.H. and Wilcox,M. (1985) EMBO J., 4, 2035–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubin G.M. and Spradling,A.C. (1982) Science, 218, 348–353. [DOI] [PubMed] [Google Scholar]
- 32.Kraus K.W., Lee,Y.H., Lis,J.T. and Wolfner,M.F. (1988) Mol. Cell. Biol., 8, 4756–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown N.H. (1994) Development, 120, 1221–1231. [DOI] [PubMed] [Google Scholar]
- 34.Karess R.E. and Rubin,G.M. (1984) Cell, 38, 135–146. [DOI] [PubMed] [Google Scholar]
- 35.Zhang P. and Spradling,A.C. (1993) Genetics, 133, 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manseau L.J. and Schüpbach,T. (1989) Genes Dev., 3, 1437–1452. [DOI] [PubMed] [Google Scholar]
- 37.Wellington A., Emmons,S., James,B., Calley,J., Grover,M., Tolias,P. and Manseau,L. (1999) Development, 126, 5267–5274. [DOI] [PubMed] [Google Scholar]
- 38.Delidakis C. and Kafatos,F.C. (1987) J. Mol. Biol., 197, 11–26. [DOI] [PubMed] [Google Scholar]
- 39.Tremml G. and Bienz,M. (1989) EMBO J., 8, 2677–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weinzierl R.O.J., Axton,J.M., Ghysen,A. and Akam,M.E. (1987) Genes Dev., 1, 386–397. [Google Scholar]
- 41.Burnette J.M., Hatton,A.R. and Lopez,A.J. (1999) Genetics, 151, 1517–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoo C.J. and Wolin,S.L. (1997) Cell, 89, 393–402. [DOI] [PubMed] [Google Scholar]