


Abstract
Triplex-forming oligonucleotides (TFOs) modified with N,N-diethylethylenediamine can inhibit the expression of a reporter plasmid in Xenopus oocytes if the triplex is preformed prior to injection while unmodified oligonucleotides cannot. Here we show that merely forming a triplex in a reporter plasmid does not disrupt transcription, but when TFOs are targeted to sites within the transcribed region of a reporter gene then gene activity is inhibited. TFO-based inhibition did not lead to large scale degradation or mutation of the reporter plasmid, but dramatically lowered mRNA levels. Finally, we investigated the accessibility of a triplex target site on a reporter plasmid after injection into nuclei. We found that the site used for our previous studies was inaccessible to restriction endonuclease after injection into nuclei. This observation may explain why inhibition was dependent on forming the triplex before injection into oocytes. Based on the assumption that oligonucleotide association, like restriction enzyme access, was excluded by nucleosome formation, additional target sites were inserted so that all sites could not simultaneously be associated with the octamer core of a nucleosome. With multiple target sites prior association of the plasmid with nuclear proteins does not prevent oligonucleotide-mediated inhibition of gene activity.
INTRODUCTION
Oligonucleotide-based methods to inhibit gene expression are dependent on base-specific hydrogen bonding patterns for recognition of target sequences. When oligonucleotides form nucleic acid duplexes, Watson–Crick base pairing rules are followed. When oligonucleotides bind to duplex DNA to form triple helices, Hoogsteen or reverse Hoogsteen base pairing rules allow sequence-specific interaction of the oligonucleotide in the major groove of the duplex.
Target sites in a DNA duplex that can support triple helix formation are purine rich with hydrogen bonds formed between the polypurine strand and the oligonucleotide stabilizing the triplex. Triplex-forming oligonucleotides (TFOs) can compete for transcription factor binding sites, inhibit initiation of transcription and cause premature termination of elongation (1). There are a large number of examples of the efficacy of TFOs in vitro (2–4), but many fewer examples in vivo (1). In part, in vivo effectiveness is compromised by the very specific reaction conditions that exist inside a cell. These conditions include the presence of single-strand nucleases, a near neutral pH, high potassium (130 mM) and low Mg concentrations and competition for DNA binding provided by both relatively non-specific nucleic acid-binding proteins (like histones) and sequence-specific binding proteins (like transcription factors).
Chemical modification of deoxyoligonucleotides can help overcome many of these constraints. For example, oligonucleotide stability can be increased by a variety of backbone modifications (5–8) and pH-dependent hydrogen bonding can be overcome using modified pyrimidines (1,9). We have shown that modification of the phosphodiester linkages between nucleosides by oxidative amidation using N,N-diethylethylenediamine (DEED) produces cationic oligonucleotides that resist self-aggregation at physiological potassium concentrations and bind well to duplex targets in vitro (10). We have further studied the in vivo activity of DEED-modified oligonucleotides by monitoring the ability of these oligonucleotides to inhibit the expression of a chloramphenicol acetyltransferase (CAT) reporter plasmid injected into Xenopus oocytes. We found that the ability of DEED-modified oligonucleotides to inhibit gene expression was sequence dependent and target sequence orientation independent. Whereas we were able to show nearly complete repression of CAT activity using DEED-modified oligonucleotides, we saw no reduction in CAT activity using unmodified oligonucleotides (11). However, these studies pointed to an additional complication encountered in vivo, competition between the oligonucleotide and proteins that assemble DNA into chromatin. We found that if the reporter plasmid was allowed to assemble into chromatin prior to exposure to the oligonucleotide there was little reduction in gene activity (11).
In order to better understand both the possibilities and limitations of oligonucleotide-mediated triplex control of gene expression we have pursued the interactions of DEED-modified oligonucleotides with a plasmid in Xenopus oocytes. We have examined the consequence of altering target placement, moving the triplex binding site between the enhancer and promoter of the reporter plasmid and using a target site within the transcribed region of the test gene more than 500 nt after the transcription start site. We have also examined whether the formation of a triplex within the reporter plasmid leads to plasmid degradation or mutation, testing whether pathways involved in monitoring DNA damage might be involved in the reduction in gene activity we had observed. We proposed previously that the formation of a triplex within the transcribed region of a gene would cause arrest of transcription, and we have directly examined the levels of RNA made by the reported plasmid in the presence of an oligonucleotide-mediated triplex. In addition, we have re-examined the effect of chromatin formation on the efficacy of triplex-mediated inhibition to explore the issue of target sequence accessibility.
MATERIALS AND METHODS
Oligonucleotide synthesis
Oligonucleotides were synthesized on an Applied Biosystems PCR-Mate DNA synthesizer (Perkin Elmer Corp., Foster City, CA) using hydrogen phosphonate chemistry (12). All reagents used for automated synthesis were purchased from Glen Research (Sterling, VA). Oxidative amidation of hydrogen phosphonate diesters was performed manually with 3.3 ml of 10% DEED (Aldrich, Milwaukee, WI) in anhydrous carbon tetrachloride for 30–60 min (12). Details of synthesis and purification can be found in Dagle and Weeks (10). Purification using reverse phase HPLC before removal of the trityl blocking group and again post-trityl group removal in addition to removal of small molecule contamination using sequential NAP-5 column elutions (Pharmacia) was carried out prior to use in oocytes. A schematic of the modified phosphate linkage is shown in Figure 1.
Figure 1.

Schematic of phosphate modification. The structure of the phosphate modification used in this study. DEED phosphoramidate modification results in a positively charged TFO at neutral pH.
Preparation of plasmid
pCAT-control was purchased from Promega Biotech (Madison, WI). pCAT-control and all other plasmids used were amplified and purified after transformation into Escherichia coli strain DH5-α, using a Qiagen (Chatsworth, CA) midi preparation kit. For the construction of pCAT-target, pCAT-6target and pCAT-Eco72I, pCAT-control was linearized with StuI (Promega). For the construction of pCAT-AatII, pCAT-control was linearized with AatII (Promega) followed by treatment with calf intestinal alkaline phosphatase (Promega). Linearized pCAT-control was separated from undigested plasmid by gel electrophoresis on a 1% (w/v) agarose gel and purified using glass beads from Geneclean (Bio101, Vista, CA).
Preparation of target insert
Unmodified oligonucleotides containing the triplex target sequence were purchased from Gibco BRL (Gaithersburg, MD). Target duplexes were formed from a 1:1 mixture of unmodified complementary oligonucleotides in sterile water by denaturing at 80°C for 5 min and slow annealing at room temperature. The duplexes were phosphorylated with T4 polynucleotide kinase (Promega). Inserted sequences were as follows: pCAT-target and pCAT-AatII, AGTTTTGTGTCCC-CCTCTCAGGTGTCACAG; pCAT-Eco72I, AGTTTTGTG-TCCCCCTCTCACGTGTCACAG, where the single base alteration in the sequence (note the bold C) generates an Eco72I site. The pCAT-6target insert (216 bp long) was generated using two oligonucleotides purchased from Genosys (The Woodlands, TX). The first oligonucleotide (108 bp) contained three consecutive target sequences: GCAAGCTTAGTTTTG-TGTCCCCCTCTCAGGTGTCACAGAGTTTTGTGTCCCCC-TCTCAGGTGTCACAGAGTTTTGTGTCCCCCTCTCAGG-TGTCACAGGGATCCGGCG (target sequence AGTTTTGTGTCCCCCTCTCAGGTGTCACAG) and 18 bp of sequence to allow annealing to a second oligonucleotide. The second oligonucleotide (108 bp) also contained three target sequences and 18 bp of sequence complementary to the first oligonucleotide: GCAAGCTTAGTTTTGTGTCCCCCTCTCAGGTGTCACAGAGTTTTGTGTCCCCCTCTCAGGTGTCACAGAGTTTTGTGTCCCCCTCTCAGGTGTCACAGCGCCGGATC. These two oligonucleotides were mixed in a 1:1 ratio and briefly denatured by heating to 100°C for 5 min and incubated at room temperature for 20 min to allow the complementary 18 bp sequence to anneal. Annealed oligonucleotide was submitted to a DNA polymerase extension procedure using Taq polymerase (Promega). Briefly, 1 pmol of annealed oligonucleotide was incubated at 55°C for 30 min in 50 µl of 50 mM KCl, 10 mM Tris–HCl, pH 9.0, 0.1% Triton X-100, 0.02 mM dATP, dCTP, dGTP and dTTP, 6 mM MgCl2 and 1.25 U Taq DNA polymerase (Promega). After polymerization this mixture was sequentially extracted with an equal volume of phenol:chloroform (1:1) and with an equal volume of chloroform:isoamyl alcohol (24:1) and precipitated with ethanol. Schematics of the plasmids used and target sequences are presented in Figure 2.
Figure 2.

Schematic of plasmids and insertion site. Schematic of pCAT-control, pCAT-AatII, pCAT-Eco72I, pCAT-target and pCAT-6target. Linear representations of plasmids are not drawn to scale.
Characterization of pCAT-target clones
Successful ligation of the triplex target site into reporter plasmids was identified by colony hybridization. Colonies were transferred to Nytran filters (Schleicher & Schuell, Keene, NH) and the DNA crosslinked to the membrane by 120 mJ UV light for 10 s in a Stratalinker (Stratagene, La Jolla, CA). The membranes were incubated in hybridization buffer of 10% dextran sulfate, 7% SDS and 1.5× SSPE (0.27 M NaCl, 15 mM NaPO4, pH 7.7, 1.5 mM EDTA) at 65°C for 2 h. Target sequences were end-labeled as described above and added to the hybridization buffer for an 18 h incubation at 65°C. The membranes were then washed in 0.5× SSC (0.15 M NaCl, 15 mM sodium citrate, pH 7.0) and 0.1% SDS twice for 30 min each at 65°C and exposed to film for 5 h. Colonies containing the inserts were identified, the clones were isolated and sequenced (DNA Facility, University of Iowa) and a large scale plasmid preparation was then performed (Qiagen).
Collection and microinjection of oocytes
CAT activity from oocytes injected with a CAT reporter plasmid can vary by as much as 30-fold from one frog to another. Only frogs that produced enough CAT activity to be assayed in the time period appropriate for retaining a linear CAT response were selected. Once a frog was selected, multiple experiments (up to eight) were performed on its oocytes. Stage VI oocytes (13) were obtained from mature female frogs as described by Colman (14). Briefly, frogs were anesthetized by immersion in charcoal-filtered tap water and ice containing 0.1% 3-aminobenzoic acid ethyl ester (tricaine; Sigma Chemical Co., St Louis, MO). Following removal of the desired amount of ovary, the incision was sutured with 0-chromic gut (Ethicon, Somerville, NJ). The ovarian tissue was washed in OR2 (82.50 mM NaCl, 2.5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 1 mM Na2HPO4, 5 mM HEPES, pH 7.8) and the ovary was teased apart to expose the oocytes. The oocytes were incubated in 0.2% collagenase (Sigma Chemical Co.) to help weaken the surrounding connective tissue and follicle cells and then placed in OR2 at 18°C until injection (<24 h). Using watchmaker’s forceps, the translucent single layer of follicular cells was manually stripped away from the individual oocytes. The nuclei of defolliculated oocytes were injected with an Inject + Matic (Geneva, Switzerland) injector by inserting the injection needle held in a Sinker MK-1 micromanipulator (Singer Instruments, Somerset, UK). Injection volumes were typically 5 or 10 nl and generally 50 oocytes were injected to obtain enough oocytes for one sample. CAT plasmid (3.2 × 10–7 M) and TFO (1.8 × 10–4 M) were preincubated in 130 mM KCl, 1 mM MgCl2 and 20 mM HEPES, pH 7.5. Following injection, penicillin (0.5 U) and streptomycin (0.5 µg) were added to the OR2 and the oocytes were incubated for 24 h at 18°C. Equal numbers of oocytes were harvested and stored at –80°C for further analysis. For order of addition experiments oocytes were sequentially injected either first with plasmid or TFO and then secondly with either TFO or plasmid. The time between the two injections was ~30 min. After these injections, oocytes were incubated for 24 h prior to processing.
CAT assay
CAT assays were based on a combination of protocols provided by Promega and reported by Jones et al. (15) and detailed in Bailey et al. (11). Reagents were purchased from Promega except where noted. Oocytes (10–15) were homogenized by sonication in 200 µl 1× Reporter Lysis Buffer (Promega) with 1 mM PMSF. The extracts were cleared by centrifugation for 3 min at 4°C in a microcentrifuge (12 000 r.p.m.). Part of the extract (70 µl) was set aside for the β-galactosidase assay. CAT activity was assayed using n-butyryl CoA and [14C]chloramphenicol (0.05 mCi/ml; Amersham, Arlington Heights, IL) for 2–3 h at 37°C. Samples are extracted with ethyl acetate prior to chromatographic separation on a silica gel TLC plate (Whatman, Clifton, NJ). The silica plate chromatogram was developed using chloroform:methanol (97:3) as the mobile phase, for ~1 h in a closed chamber, removed and allowed to dry. Detection and quantitation of [14C]chloramphenicol was carried out using an Instant Imager (Packard Instrument Co., Meriden, CT) with % conversion = converted counts/total counts. Percent conversion included both the mono- and diacetylated forms. CAT activity in pCAT-control-injected oocytes was determined to be linear over a range of 10–75% conversion of chloramphenicol to acetylated chloramphenicol. All CAT activities were normalized using β-galactosidase values. To allow comparison of assays from different experiments, CAT activity from pCAT-target was set at 100%.
β-Galactosidase and luciferase assays
An independent measure of gene expression was provided by the inclusion of pSVβ-galactosidase control or pGL-2 (Promega) in all injections. β-Galactosidase assays were performed by a modified Promega protocol detailed in Bailey et al. (11). As indicated above, cell extracts made from injected oocytes were split into two aliquots to allow simultaneous evaluation of CAT activity and β-galactosidase or luciferase activity. Prior to assaying for β-galactosidase activity, the cell extract was extracted with an equal volume of chloroform:isoamyl alcohol (24:1) to remove lipid and other particulate matter that interfered with the absorbance readings. β-Galactosidase activity was quantitated by measuring the absorbance of the samples at 414 nm using a Microskan TCC/340 plate reader (Titertek, Huntsville, AL). Values obtained for β-galactosidase activity routinely varied by less than 10% and were used to normalize CAT activity. Luciferase assays were performed according to a Promega procedure. Light produced for 10 s was measured using a luminometer (Analytical Luminescence Laboratory, Sparks, MD). Each luciferase assay was performed three times, each with 1 µl of the same extract, and the average of the three trials obtained.
Northern blot analysis
Oocyte RNA was extracted using a slight modification of the procedure described by Chomczynski and Sacchi (16) and detailed in Weeks et al. (17). RNA was extracted from 10–15 oocytes, previously frozen in dry ice and stored at –70°C. Gel electrophoresis and RNA transfer were performed similarly to the procedure of Fourney et al. (18) using formaldehyde–agarose gels. Initial estimates of equal loading were determined by ethidium bromide staining and visualization of rRNA under UV light. The RNA was transferred to a Nytran membrane (Schleicher & Schuell) with a PosiBlot Pressure Blotter (Stratagene). The RNA was crosslinked to the membrane by 120 mJ of UV light for 10 s in a Stratalinker (Stratagene) and pre-hybridized for at least 2 h at 65°C in 10% polyethylene glycol (MW8000), 7% SDS and 1.5× SSPE (0.27 M NaCl, 15 mM NaPO4, pH 7.7, 1.5 mM EDTA). The CAT probe was made from a 551 bp fragment generated by cleavage of pCAT-control (Promega) with HindIII and NcoI, the cyclin B probe was made from a 1.6 kb fragment generated by cleavage of Xlcyc1 (19) with EcoRI and HindIII. Isolated fragments were labeled by random primed synthesis with [α-32P]dATP (Amersham, Piscataway, NJ). After denaturing the CAT probe by boiling at 100°C for 5 min, it was added to the membrane in fresh hybridization solution and incubated overnight at 65°C. Unbound probe was washed from the membrane using 0.1× SSC (15 mM NaCl, 1.5 mM sodium citrate, pH 7.0) and 1% SDS for 10 min at room temperature, followed by washes at 65°C. The membrane was then placed in plastic wrap and exposed to X-ray film (Kodak, Rochester, NY) for 1–3 days at –70°C, after which the film was developed. After removing the CAT probe by boiling the membrane in 0.1× SSPE/0.5% SDS for 5 min, the blot was prehybridized and probed (by the same procedure as for the CAT probe) with the cyclin B1 probe to control for variations in RNA extraction and loading.
Southern blot analysis
DNA was extracted from oocytes injected with pCAT-target and pCAT-control in the presence and absence of TFO by an alkaline lysis procedure. Oocytes were homogenized in 100 µl oocyte homogenization buffer (0.1 M NaCl, 1% Triton X, 20 mM Tris, pH 7.6, 1 mM PMSF) and 100 µl of 0.2 N NaOH, 1% SDS was added by gentle mixing. After 10 min incubation on ice, 150 µl 5 M potassium acetate, pH 4.8, was added by gentle mixing and incubated for 10 min on ice. The preparation was centrifuged for 10 min at 15 000 g at 4°C. The pellet was discarded and the supernatant was extracted with phenol:chloroform (1:1) and then chloroform:isoamyl alcohol (24:1). Isopropyl alcohol (200 µl) was added to the aqueous phase and incubated at –20°C for several hours. The DNA was pelleted by centrifugation at 15 000 r.p.m. for 15 min and the pellet was resuspended in 100 µl water and precipitated with 250 µl 100% ethanol at –20°C for several hours. After centrifugation for 15 min at 15 000 r.p.m., the pellet was washed twice with 70% ethanol and resuspended in sterile water. The DNA was linearized by restriction enzyme digestion (EcoRI) for 2 h at 37°C and subjected to electrophoresis on a 1% agarose gel in 1× TAE (40 mM Tris base, 20 mM sodium acetate, 1 mM EDTA, adjusted to pH 7.2). The DNA was transferred from the gel on a Posiblot Pressure Blotter (Stratagene) as described for the transfer of RNA in the northern blot analysis and the same probe and procedure was used to identify the CAT plasmids. After hybridization and washing to remove non-specific interactions, the filter was exposed to X-ray film (Kodak) for 1–3 days at –70°C, after which the film was developed.
Restriction analysis of plasmids after injection into oocytes
Xenopus oocytes were collected and microinjected with 10 ng of circularized labeled pCAT-Eco72I mixed with dextran-conjugated rhodamine (70 kDa dextran; Molecular Probes, Eugene, OR). Dextran-conjugated rhodamine was added to easily visualize the nuclei that were successfully injected. pCAT-Eco72I was prepared as described in Gargiulo and Worcel (20). Briefly, pCAT-Eco72I was digested with XbaI and the linearized plasmid was separated on a 1% agarose gel and purified with Geneclean (Bio101). The 5′-phosphate groups of the linearized plasmid were removed by calf intestinal phosphatase and replaced using [γ-32P]ATP and T4 polynucleotide kinase. T4 DNA polynucleotide kinase (Promega) was inactivated by heating to 70°C for 5 min. The end-labeled DNA was diluted to a concentration of 1 µg/ml and ligated overnight at 18°C with T4 DNA ligase (Promega). The DNA was sequentially extracted with phenol:chloroform (1:1) and chloroform:isoamyl alcohol (24:1) and then was precipitated with a 2.5× vol of 100% ethanol. The DNA was pelleted by centrifugation for 10 min at 15 000 r.p.m. and washed twice with 75% ethanol. After resuspending in a small volume, the concentration of the labeled, circularized plasmid was determined by absorbance at 260 nm. A small amount of the DNA was separated on a 1% agarose gel and compared to linearized DNA to determine the success of circularization.
Twelve hours after injection of labeled plasmid, nuclei were gently removed from oocytes by manually tearing open the animal pole of the oocyte with watchmaker’s forceps. The nuclei were suspended in 85 mM KCl, 5 mM PIPES, pH 7.0, and 5.5% (w/v) sucrose at a ratio of 10 µl/oocyte and 100, 200 and 400 U Eco72-I (Stratagene) were added and the samples were incubated at room temperature for 2 h. The digestions were terminated by adding an equal volume of 1% SDS, 20 mM EDTA, 30 mM Tris–HCl, pH 7.9. Proteinase K (500 µg/ml) was added and the samples were incubated for 2 h at 37°C. The DNA was extracted twice with phenol:chloroform (1:1) and once with chloroform:isoamyl alcohol (24:1) and precipitated with 2.5 vol 100% ethanol. After resuspension in a small volume of sterile water, the samples were digested with PstI (Promega) at 37°C for 2 h. As a control an equal number of non-injected oocyte nuclei were submitted to the same procedure, except that 10 ng of circularized radiolabeled plasmid was added (not injected). This serves as a control for evaluating enzyme digestion with Eco72-I. The DNA was separated by electrophoresis on a 1% agarose gel and the gel was dried and exposed to X-ray film (Kodak).
RESULTS
The formation of a triplex within the CAT gene inhibits expression while one placed outside the reporter gene does not
Our previous studies using the reporter plasmid pCAT-target examined TFO-mediated inhibition when the triplex target site was positioned 30 nt after the transcription initiation site. We showed that at this position triplex formation resulted in loss of CAT activity regardless of the orientation of the target site. It is possible that the placement of the target site in close proximity to the assembly site of the transcription complex may have disrupted initiation rather than elongation. An endogenous triplex-forming site is present in pCAT-control, a purine-rich sequence 578 bp after the transcription initiation site (Figs 2 and 3). A DEED-modified oligonucleotide (TFO-578) capable of forming a purine motif triplex at this site was synthesized. If sequence-specific association of TFO within the transcribed portion of a gene stops mRNA production, then pCAT-control should be regulated by TFO-578. Figure 3 shows representative CAT assays with pCAT-control injected with and without TFO-578. TFO-578 was able to significantly inhibit gene expression of pCAT-control but had no effect on expression of the β-galactosidase control plasmid.
Figure 3.
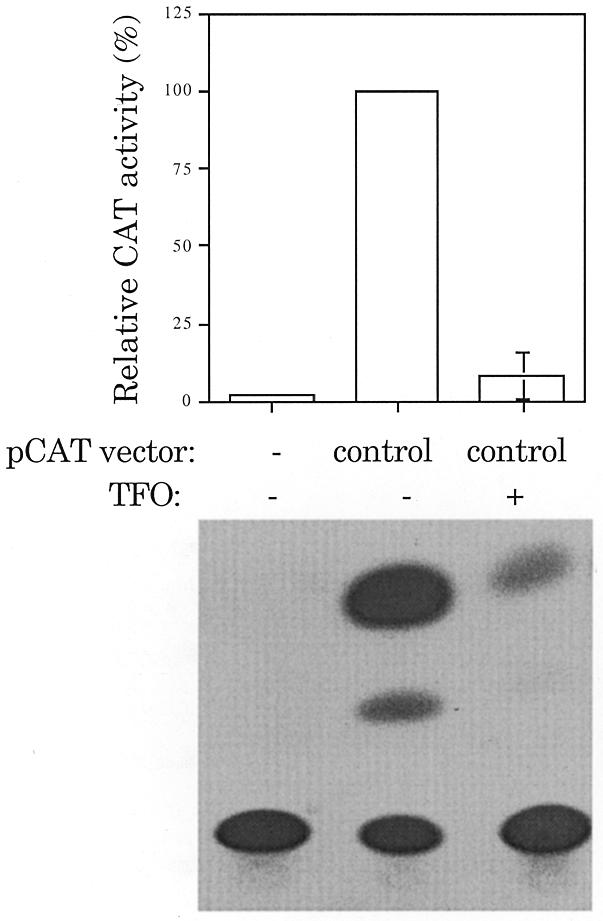
TFO inhibition of elongation. CAT activity present in oocytes injected with pGL3-luciferase and pCAT-control in the presence or absence of +578 (5′-GAAGAAGTGGGGGTAAAAG) with a DEED-modified backbone were compared. pCAT-control contains a purine rich sequence, 5′-GAAAACGGGGGCGAAGAAG. The percent CAT activity was adjusted for luciferase expression and is given as the mean ± SE of two independent trials. pCAT-control was arbitrarily set to 100% to allow for interassay comparison.
However, what if TFO binding to the plasmid at any site leads to reduced expression from the plasmid? To discriminate between specific inhibition of the CAT gene and a general inhibition of expression, the same target site sequence used in pCAT-target was positioned outside the CAT gene (pCAT-AatII, Fig. 2). Figure 4 compares the CAT activity of pCAT-AatII in the presence and absence of TFO. There was no detectable decrease in CAT activity in the presence of TFO. TFO binding to the target site outside the CAT gene does not alter reporter plasmid activity.
Figure 4.
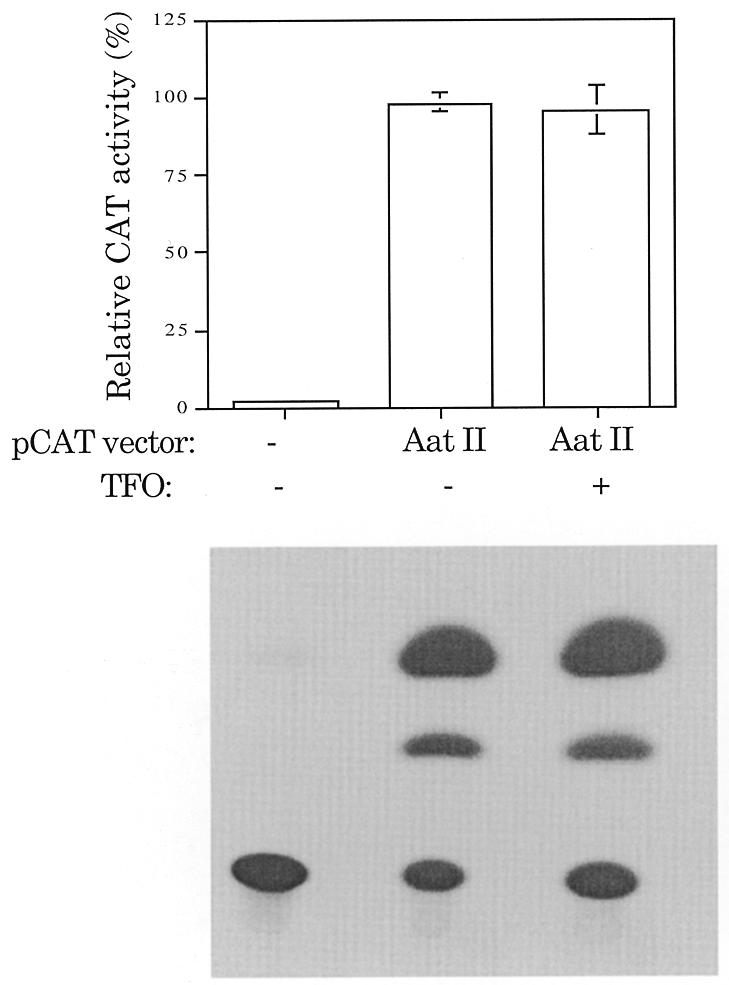
Target site outside the CAT gene. No significant TFO inhibition of gene expression was seen with the target site positioned outside the CAT gene. CAT activities present in oocytes injected with pCAT-AatII(–679) and pSV-β-galactosidase in the presence or absence of TFO (5′-AAAATATAGGGGGAGAG) with a DEED-modified backbone were compared. The percent CAT activity was adjusted for pSV-β-galactosidase expression and is given as the mean ± SE of two independent trials. On this scale, the error bars are sometimes not visible because they are smaller than the width of the bar line. pCAT-control was arbitrarily set to 100% to allow for interassay comparison.
The presence of an oligonucleotide-mediated triplex does not lead to large scale degradation or mutation of reported plasmids in oocytes
We have shown previously that the CAT activity encoded by a reporter plasmid in oocytes can be dramatically reduced using TFOs. We have examined whether the formation of a triplex leads to the degradation or a sequence alteration of the plasmid that may account for the overall reduction in CAT activity. In Figure 5, circular DNA isolated from oocytes injected with pCAT-control or pCAT-target, in the presence or absence of TFO, was compared by Southern blot analysis. The oocytes were injected and incubated for 24 h, conditions previously shown to lead to oligonucleotide-mediated inhibition of gene activity (11). Neither pCAT-target nor pCAT-control were detectably degraded during the 24 h incubation of the oocytes and similar amounts of plasmid were recovered in the presence or absence of TFO. The re-isolated plasmid was subjected to sequence analysis in the region of the triplex target. Although it is possible that mutations occurred in a small portion of the total plasmid, there was no discernable sequence difference between the control plasmid and the plasmid injected in the presence of oligonucleotide. Thus, the inhibitory effect of the TFO on gene expression is unlikely to be due to either degradation or sequence changes in pCAT-target.
Figure 5.
Southern blot analysis of injected oocytes. Comparison of DNA isolated from oocytes injected with pCAT-target in the presence and absence of TFO. Digestion of pCAT-target with EcoRI produced 4293 and 459 bp fragments. Non-injected (NI) oocytes served as a negative control.
TFOs lead to a reduction in CAT mRNA level
The simplest explanation for TFO inhibition of pCAT-target production of CAT activity is that there is a direct effect on transcription. CAT activity depends on the synthesis of CAT protein and therefore does not distinguish between a reduction in transcription versus translation. In Figure 6 the production of RNA from the reporter plasmid was directly assayed by northern blot analysis of 30 oocytes injected with pCAT-control or pCAT-target in the presence or absence of TFO. Injected oocytes were incubated for 24 h prior to RNA extraction. The endogenous maternal mRNA cyclin B1 served as a control for RNA recovery and loading. The CAT mRNA was present in all the plasmid-injected samples, except for when TFO was present with the pCAT-target plasmid. These data suggest that the inhibition of gene expression in the presence of TFO was due to inhibition of transcription and that TFO did not affect transcription when the target site was not present (pCAT-control and TFO). We note that the TFO is not complementary to the transcript and thus could not serve to degrade the mRNA in a matter similar to that seen for antisense oligonucleotides.
Figure 6.
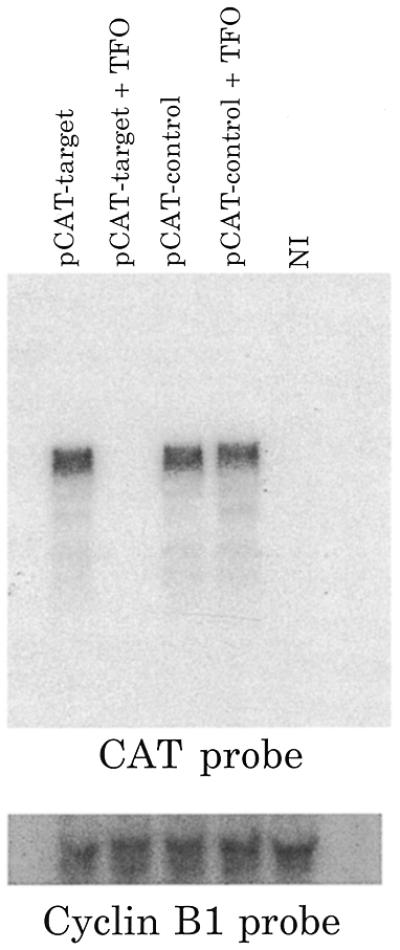
Northern blot analysis of injected oocytes. Comparison of total RNA isolated from oocytes injected with pCAT-control and pCAT-target injected with and without TFO. Non-injected (NI) oocytes served as a negative control. Cyclin B1 probe and ethidium bromide staining served to control for even extraction and loading of RNA.
Target site accessibility after reporter plasmid injection
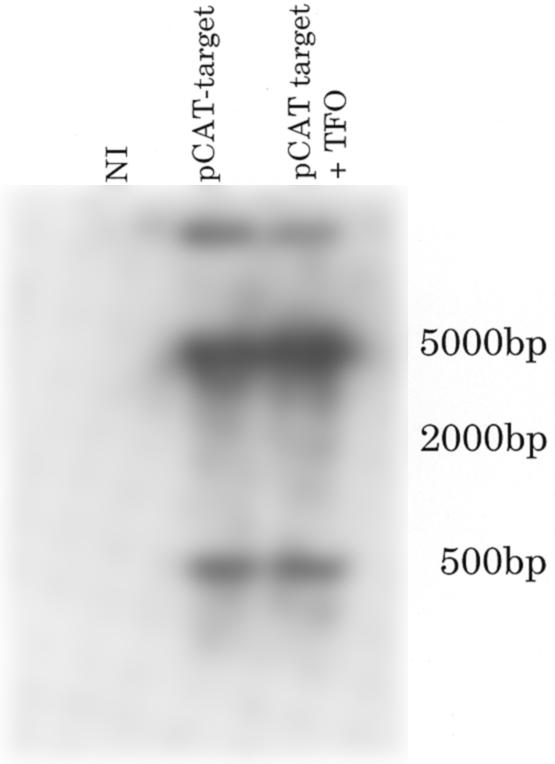
In our previous studies, preformed triplex inhibited gene expression of pCAT-target, however, when plasmid was injected 30 min prior to oligonucleotide injection, CAT activity remained high (11). We determined if the major groove of the target site was accessible after plasmid injection into oocytes by probing chromatinized plasmid by restriction enzyme digestion. The target site was changed to generate an Eco72I restriction enzyme recognition site. The sequence of the target site including an Eco72-I site can be found in Materials and Methods. This target site was inserted 30 nt after the start site of transcription into the pCAT-control plasmid to generate pCAT-Eco72. After determining that pCAT-Eco72 had the same expression characteristics as pCAT-target in order of addition experiments, we injected and then re-isolated plasmids from oocyte nuclei. The re-isolation method was based on experiments that map nucleosomes on plasmid DNA injected into oocyte nuclei and was amenable to restriction enzyme analysis of injected plasmid DNA (20–24). Injected plasmids were resistant to cleavage with Eco72-I (6–8%, Fig. 7). These results suggest that the major groove of the target site in pCAT-Eco72-I is predominately inaccessible to TFO binding, due to occupancy by either transcription factors or nucleosomes.
Figure 7.
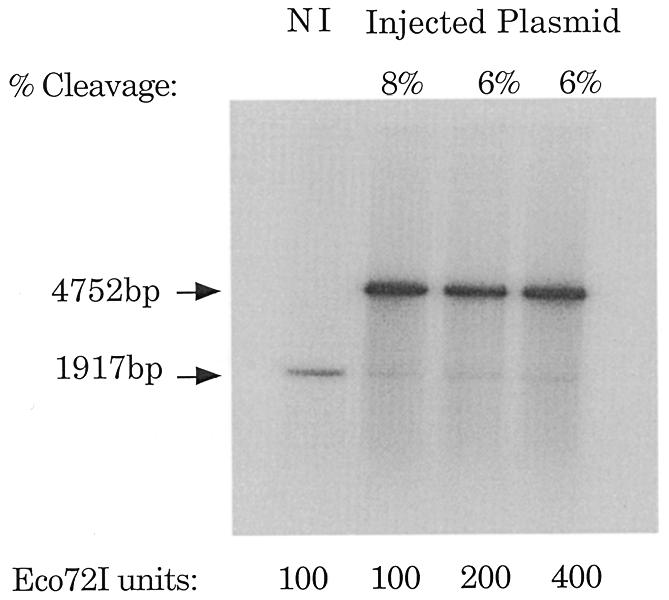
Restriction digest analysis of injected pCAT-Eco72I. Eco72I digestion of pCAT-Eco72I injected into Xenopus oocytes was evaluated in manually separated nuclei. Labeled, circular pCAT-Eco72I was added to non-injected (NI) samples (whole cell extract or nuclei) as a control for the Eco72I activity. Nuclei were subjected to increasing concentrations of Eco72I to ensure that the enzyme was not limiting. The fragments digested with both Eco72I and PstI were separated at 1917 bp, while the single digested fragment appeared at 4782 bp. The data were quantitated by electronic autoradiography (InstantImager; Packard Instrument Co., Meriden, CT).
To a reasonable first approximation, in oocyte nuclei a nucleosome consists of ~130 bp of DNA wrapped around a histone octamer core with a linker region of 40–50 bp of DNA (25). We sought to determine whether a TFO would be able to bind and inhibit gene expression if target sites were placed in the linker region. To ensure that a target site was in the linker region, a reporter plasmid with six target sites was constructed (pCAT-6target). The repeated target sites span 216 bp and thus at least one of the triplex targets should be in the linker region between core particles. In contrast to our previous studies, pCAT-6target plasmid expression was inhibited by the TFO regardless of injection order (Fig. 8), presumably due to the availability of a target site in a nucleosome spacer region.
Figure 8.
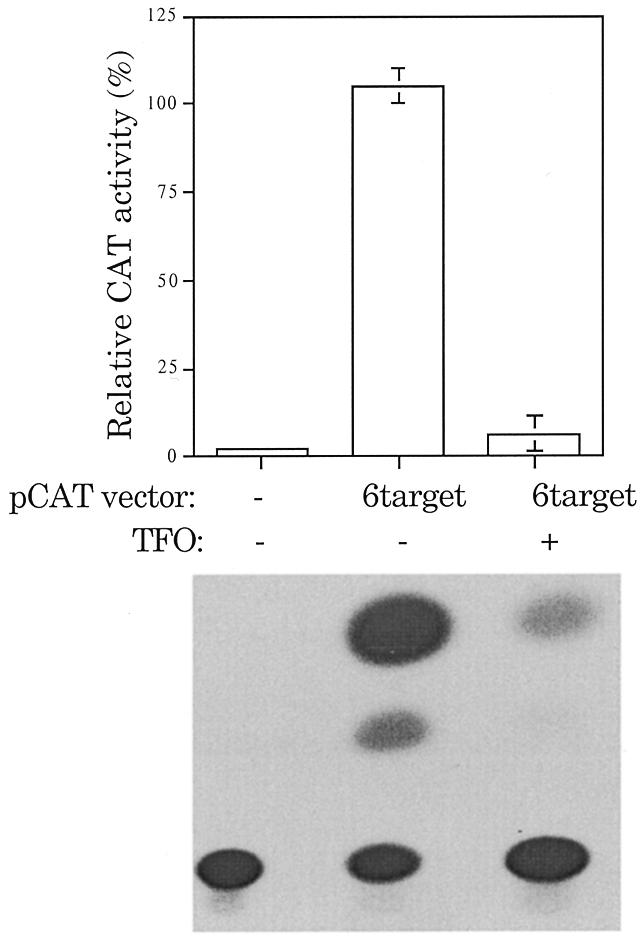
Availability of target affects ability to inhibit gene expression. Oocytes were injected with pCAT-6target 30 min prior to injection with TFO. CAT activity was monitored 24 h after plasmid injection. pCAT-6target contains six consecutive repeated target sites at the +30 position, relative to the start of transcription. The TFO (5′-AAAATATAGGGGGAGAG) used has a DEED-modified backbone. The percent CAT activity was adjusted for luciferase expression and is given as the mean ± SE of at least three independent trials. pCAT-target was arbitrarily set to 100% to allow for interassay comparison.
DISCUSSION
The strong and specific binding of oligonucleotides to appropriate target sites in the major groove of a DNA duplex under in vitro conditions provides the impetus to seek ways in which a similar interaction could be used to regulate gene activity in vivo. However, the use of TFOs to inhibit gene expression in vivo requires the design of oligonucleotides of sufficient nuclease resistance, specificity and binding strength under cellular conditions. With these constraints in mind, we have pursued the potential of DEED-modified purine TFOs for in vivo use. The DEED modification alters the charge of the oligonucleotide at neutral pH, making it cationic. The modification changes the phosphodiester linkages between sugars to a phosphoramidate linkage. Such a change increases the resistance of oligonucleotides to cellular nucleases (26). In addition, in vitro band shift analysis of the triplex-forming characteristics of DEED-modified TFOs indicate that they bind strongly and specifically to their target site at neutral pH in the presence of physiological concentrations of potassium and magnesium (10). These in vitro studies encouraged us to continue the examination of DEED-modified oligonucleotides in vivo, using reporter plasmids injected into Xenopus oocyte nuclei (11). In summary, we found that DEED-modified TFO triplex formation within the transcribed portion of a CAT reporter plasmid reduced the production of active CAT protein by more than 50-fold. We also showed that the interaction was sequence specific and target sequence orientation independent. These studies used a target site that contained two pyrimidine interruptions in a 17 nt stretch of purines, thus increasing the number of potentially useful target sequences that might exist in a gene. However, several important issues relating to the in vivo action of DEED-modified TFOs remained unresolved, and were addressed in the current study.
Two questions arise concerning the location of the triplex target site. First, would the formation of a triplex at any site within the reporter plasmid be sufficient to inhibit gene expression and, second, was the position of the triplex target site in our previous studies (30 bp downstream of the transcription start site) uniquely suited to inhibit transcription or would the formation of a triplex in another position within the transcribed region of the reporter similarly inhibit gene expression? With regard to the first question, placement of the triplex target site outside the transcribed portion of the CAT gene (pCAT-AatII) did not support TFO-mediated reduction in CAT activity. In contrast, an endogenous triplex target site within the CAT gene and 578 bp downstream of the transcription start site could be successfully used for TFO-mediated inhibition of CAT expression. This finding suggests that any potential triplex-forming region within the transcribed portion of a gene may be a sufficient target for oligonucleotide-mediated inhibition of transcription. This observation may not hold true for other types of modified oligonucleotides. For example, there has been a recent report that the formation of triplexes using phosphorothioate-modified TFOs would not inhibit gene expression unless they were crosslinked to the DNA duplex (27). The DEED-modified oligonucleotides do not seem to require covalent attachment to the duplex to function.
In our prior studies we did not determine if the TFO effect was based upon destabilization or alteration of the reporter plasmid. Here we show that both pCAT-target and pCAT-control were as stable in the presence of TFO as in its absence. This suggests that the inhibition of gene expression was not due to a cellular response to the triplex structure causing degradation of the plasmid. Others have shown that under some circumstances the introduction of triplex DNA into cells can lead to sequence changes in the duplex (1,28–30). We did not see any changes in plasmid sequence in these assays, although a very infrequent alteration would not have been detected. We conclude that the inhibition that we have reported using TFOs is neither due to plasmid degradation nor sequence alteration.
The inhibition of gene expression appears to be due to inhibition of transcription. The CAT transcript as assayed by northern blot analysis was undetectable in the presence of pCAT-target and triplex-forming oligonucleotide, in contrast to pCAT-target alone. The CAT mRNA appears as a slightly broad band, possibly due to a cluster of transcripts of different lengths arising from transcription beginning or ending at slightly different sites. It has been suggested that the pCAT targets have three start sites of transcription, –402, –5 and +1 bp, in cell culture, although initiation at +1 bp is predominant (communication with Promega, Madison, WI).
In our previous studies, preformed triplex inhibited gene expression of pCAT-target, however, when plasmid was injected first, followed by oligonucleotide, CAT activity was unaffected (11). Although full chromatinization may take several hours, others have shown that plasmid DNA injected into Xenopus oocyte nuclei are rapidly bound by cellular DNA-binding proteins (20,21). We showed that soon after injection the triplex target site in our reporter plasmid was not accessible to restriction enzyme digestion, indicating that nucleosomes or other DNA-binding proteins occlude the triplex target. We conclude that for our reporter plasmid pCAT-target, the TFO was unable to displace nucleosomes or DNA-binding proteins that masked the target site. In vitro studies of triplex formation on reconstituted nucleosomes demonstrated that nucleosome core particles inhibit DNA triplex formation (31,32). In vivo studies of transient transfection assays required electroporation of preformed triplex to inhibit the promoter of murine c-pim-1, a protooncogene; target DNA was inaccessible to triplex formation if electroporated first into the cell (33,34). Because the single site we were targeting appeared to be blocked, we explored whether a triplex target site that was not blocked by nucleosome formation would restore oligonucleotide-mediated control of gene expression when the rest of the reporter plasmid was assembled into nucleosomes. We created a reporter plasmid pCAT-6target that spaced target sites in a pattern that prevented all of the sites from being covered by a single nucleosome. We found that even after the establishment of protein–plasmid complexes pCAT-6target was inhibited by subsequent injection of TFO. Therefore, the prior establishment of protein–DNA associations, such as the presence of nucleosomes, need not prevent the effective use of TFOs as modulators of gene expression. We propose that if several potential triplex-binding regions were identified in a gene, spaced so that they could not be simultaneously covered by nucleosomes, then a mix of TFOs to those target sites would inhibit gene expression. We are currently testing this hypothesis.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Dr John Dagle and Vera Ogniewski for helpful discussions and suggestions. They also thank Chip Greaves for technical support and Drs Andy Russo and Marc Wold for sharing equipment. This work was supported by grants from the NIH to D.L.W.
REFERENCES
- 1.Vasquez K.M. and Wilson,J.H. (1998) Trends Biochem. Sci., 23, 4–9. [DOI] [PubMed] [Google Scholar]
- 2.Kim H.G. and Miller,D.M. (1995) Biochemistry, 34, 8165–8171. [DOI] [PubMed] [Google Scholar]
- 3.Kovacs A., Kandala,J.C., Weber,K.T. and Guntaka,R.V. (1996) J. Biol. Chem., 271, 1805–1812. [DOI] [PubMed] [Google Scholar]
- 4.Maher L.J.,III (1992) Biochemistry, 31, 7587–7594. [DOI] [PubMed] [Google Scholar]
- 5.Escude C., Giovannangeli,C., Sun,J.S., Lloyd,D.H., Chen,J.K., Gryaznov,S.M., Garestier,T. and Helene,C. (1996) Proc. Natl Acad. Sci. USA, 93, 4365–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim H.G. and Miller,D.M. (1998) Biochemistry, 37, 2666–2672. [DOI] [PubMed] [Google Scholar]
- 7.Kim H.G., Reddoch,J.F., Mayfield,C., Ebbinghaus,S., Vigneswaran,N., Thomas,S., Jones,D.E.,Jr and Miller,D.M. (1998) Biochemistry, 37, 2299–2304. [DOI] [PubMed] [Google Scholar]
- 8.Xodo L., Alunni-Fabbroni,M., Manzini,G. and Quadrifoglio,F. (1994) Nucleic Acids Res., 22, 3322–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Durland R.H., Rao,T.S., Revankar,G.R., Tinsley,J.H., Myrick,M.A., Seth,D.M., Rayford,J., Singh,P. and Jayaraman,K. (1994) Nucleic Acids Res., 22, 3233–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dagle J.M. and Weeks,D.L. (1996) Nucleic Acids Res., 24, 2143–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bailey C.P., Dagle,J.M. and Weeks,D.L. (1998) Nucleic Acids Res., 26, 4860–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Froehler B.C., Ng,P.G. and Matteucci,M.D. (1986) Nucleic Acids Res., 14, 5399–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumont J.N. (1972) J. Morphol., 136, 153–179. [DOI] [PubMed] [Google Scholar]
- 14.Colman A. (1984) In Hames,D. and Higgins,S. (eds), Transcription and Translation—A Practical Approach. IRL Press, Oxford, UK.
- 15.Jones N.C., Richter,J.D., Weeks,D.L. and Smith,L.D. (1983) Mol. Cell. Biol., 3, 2131–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chomczynski P. and Sacchi,N. (1987) Anal. Biochem., 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 17.Weeks D.L., Walder,J.A. and Dagle,J.N. (1991) Development, 111, 1173–1178. [DOI] [PubMed] [Google Scholar]
- 18.Fourney R.M., Miyakoshi,J., Day,R.S. and Paterson,M.C. (1988) Focus, 10, 5–7. [Google Scholar]
- 19.Minshull J., Blow,J.J. and Hunt,T. (1989) Cell, 56, 947–956. [DOI] [PubMed] [Google Scholar]
- 20.Gargiulo G. and Worcel,A. (1983) J. Mol. Biol., 170, 699–722. [DOI] [PubMed] [Google Scholar]
- 21.Gargiulo G., Razvi,F., Ruberti,I., Mohr,I. and Worcel,A. (1985) J. Mol. Biol., 181, 333–349. [DOI] [PubMed] [Google Scholar]
- 22.Kroll K.L. and Amaya,E. (1996) Development, 122, 3173–3183. [DOI] [PubMed] [Google Scholar]
- 23.Fragoso G., Pennie,W.D., John,S. and Hager,G.L. (1998) Mol. Cell. Biol., 18, 3633–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mymryk J.S., Berard,D., Hager,G.L. and Archer,T.K. (1995) Mol. Cell. Biol., 15, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolffe A. (1995) Chromatin Structure and Function, 2nd Edn. Academic Press, San Diego, CA.
- 26.Dagle J.M., Weeks,D.L. and Walder,J.A. (1991) Antisense Res. Dev., 1, 11–20. [DOI] [PubMed] [Google Scholar]
- 27.Ebbinghaus S.W., Fortinberry,H. and Gamper,H.B.,Jr (1999) Biochemistry, 38, 619–628. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z. and Rana,T.M. (1997) Proc. Natl Acad. Sci. USA, 94, 6688–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang G., Levy,D.D., Seidman,M.M. and Glazer,P.M. (1995) Mol. Cell. Biol., 15, 1759–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang G., Seidman,M.M. and Glazer,P.M. (1996) Science, 271, 802–805. [DOI] [PubMed] [Google Scholar]
- 31.Brown P.M. and Fox,K.R. (1996) Biochem. J., 319, 607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westin L., Blomquist,P., Milligan,J.F. and Wrange,O. (1995) Nucleic Acids Res., 23, 2184–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svinarchuk F., Cherny,D., Debin,A., Delain,E. and Malvy,C. (1996) Nucleic Acids Res., 24, 3858–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Svinarchuk F., Debin,A., Bertrand,J.R. and Malvy,C. (1996) Nucleic Acids Res., 24, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]