


Abstract
Mammalian ribosomal RNA genes (rDNA) are transcribed by RNA polymerase I and at least two auxiliary factors, UBF and SL1/TFID/TIF-IB. It has also been reported that an additional factor(s) is required to reconstitute efficient initiation of rDNA transcription in vitro, depending upon the procedures of chromatographic separation. In an attempt to elucidate the molecular identity of such yet uncertain activities, we have developed agarose gel shift and UV cross-linking assays to detect proteins directly bound to the core promoter region of murine rDNA. With these techniques, we identified a 70 kDa protein (p70) in the flow-through fraction of a phosphocellulose column (TFIA-fraction). Interestingly, the binding of p70 to the rDNA core promoter was observed only in the presence of the SL1-containing fraction. The probable human orthologue of p70 was also detected in HeLa cells. Consistent with the observation that p70 bound to the core promoter only in the presence of the TFIA- and SL1-fractions, alteration of DNase I footprint pattern over the core promoter element was demonstrated by cooperative action of the TFIA- and SL1-fractions. A reconstituted in vitro transcription assay with further purified p70 indicated that p70 was required for accurate initiation of rDNA transcription. These results indicate that the p70 identified recently by the current DNA-binding experiments represents a novel transcription factor in rDNA transcription.
INTRODUCTION
The eukaryotic ribosomal RNA gene (rDNA) is transcribed by RNA polymerase I (Pol I) and its expression is regulated coordinately with cell growth and differentiation (1–4). In vertebrates, the core promoter element, located between –45 and +20 relative to the transcription initiation site, is necessary and sufficient for accurate initiation of rDNA transcription both in vitro and in vivo (5–7). The cis-acting upstream control element located between –160 and –80 augments the transcription (6,7). In human cells, an auxiliary transcription factor SL1, a multiprotein complex containing the TATA-binding protein (TBP) and three other polypeptides called TBP-associated factors for Pol I (TAFIs), primarily accounts for the promoter recognition (8,9). Cooperative interaction of SL1 with the upstream binding factor (UBF) leads to the formation of a stabilized ternary complex, resulting in the augmentation of transcription efficiency (10,11). In rodent cells, however, UBF is not absolutely required for basal transcription, and is suggested to counteract the transcriptional repression by an inhibitory activity (12,13). Instead, based on the requirement in reconstituted in vitro systems for rDNA transcription initiation, several other factors have been reported, including Factor C*, TFIA, TFIC, TIF-IA and TIF-IC (14–18). These factors, except for TFIA, have been demonstrated to exist in close proximity to Pol I, and shown to be involved in modulating the activity of the enzyme in response to growth conditions of the cells. These characteristics point to the possibility that some of the reported factors may represent the same biological entity. However, there are significant biochemical differences among these factors. For example, Factor C* is exhausted immediately after the initiation of rDNA transcription, whereas TIF-IA remains active to reinitiate transcription (14,19). Moreover, TIF-IA activity is purified with a single 75 kDa polypeptide, whereas TFIC activity co-purifies with three polypeptides with the molecular mass of 55, 50 and 42 kDa (19,20). The molecular size of Factor C* has not been determined. TIF-IC has been identified as a 65 kDa protein, which participates in both the initiation and the elongation of transcription by Pol I (18). TFIA has been found in a low salt flow-through fraction of phosphocellulose column chromatography of nuclear extracts, and shown to form a complex with TFID, a species-specific rDNA transcription factor and a probable equivalent to SL1, recruiting Pol I for efficient transcription (15). However, the molecular characteristics of this factor remain to be determined.
The molecular identities among these factors and their precise roles in rDNA transcription remain obscure, because neither complete purification nor molecular cloning of any factor has been accomplished. The difficulty in purifying these putative regulatory proteins is mainly attributable to their low abundance in a cell. To facilitate the purification of such proteins, one needs a rapid and sensitive assay system to identify the fractions containing the protein of interest from each column chromatography step during the factor purification. In an attempt to develop such an assay system, we have determined the conditions under which two related multiprotein complexes bound to the rDNA core promoter were separated in an agarose gel. One of the complexes, which migrates more slowly than the other, was shown to contain TBP as an integral part of the complex. This complex was preferentially formed with reconstituted protein fractions containing Pol I, SL1 and TFIA, but not UBF. In UV cross-linking experiments under the same binding conditions as the agarose gel shift assay, we identified a novel 70 kDa protein (p70) in the TFIA-containing protein fraction. Strikingly, the binding of p70 to the rDNA core promoter occurred only in the presence of the SL1-containing fractions. A similar 70 kDa protein is also present in human cells. Based on the core promoter-binding activity, mouse p70 was further purified from the TFIA-fraction. The partially purified p70 exhibited an essential role on the initiation of rDNA transcription in a reconstituted in vitro transcription assay. These results indicate that the present protein–DNA binding assay is useful to assess a novel transcriptional activity which is essential in mammalian rDNA transcription.
MATERIALS AND METHODS
Preparation and fractionation of whole-cell extracts
Whole-cell extracts were prepared from mouse ascites cell MH134 or HeLa cells as described previously (21). The extracts were dialyzed against buffer DB [20 mM Hepes–KOH (pH 7.9), 0.5 mM EDTA, 1 mM dithiothreitol (DTT), 0.2 mM phenylmethylsulfonyl fluoride (PMSF), 20% glycerol] containing 100 mM KCl (DB-0.1), and then applied to a phosphocellulose (P11, Whatman) column equilibrated with buffer DB-0.1. Typically, 30 ml of the dialyzed extract was loaded onto a 1.5 × 10 cm column of phosphocellulose at the flow rate of 0.3 ml/min. A flow-through fraction (fraction A) of the column was used for further purification (see below). Proteins retained on the phosphocellulose column were fractionated by stepwise elution with buffer DB containing 300 (fraction B), 600 (fraction C), 800 (fraction U) and 1200 mM KCl (fraction D), respectively. The fractions C, U and D contain RNA polymerase I, UBF and SL1, respectively. Each fraction was then dialyzed against buffer DB-0.1. Fraction A was applied onto a DEAE-Sepharose column (1.5 × 5 cm) equilibrated with buffer DB-0.1. The column was washed with buffer DB-0.1 and the bound proteins were eluted with buffer DB containing 300 mM KCl (DB-0.3). The eluate (TFIA-fraction) was dialyzed against buffer DB-0.1. The protein p70 was further purified from the TFIA-fraction by a linear gradient of KCl from 0.1 to 1.0 M on QAE-Sephadex A-50 (Pharmacia), or by eluting from the preparative scale of an SDS–polyacrylamide gel with the Disk Preparative Electrophoresis Apparatus NA-1800 (Nihon Eido, Tokyo) according to the manufacturer’s instructions.
Preparation of probes
Mouse rDNA promoter fragment containing from –168 to +30 was amplified by PCR with oligonucleotide primers 5′-CCCGGGTC-GACCAGTTGTTCCTTTGAGGTCCGGTTCTT-3′ and 5′-CC-CGGGTCGACGTGTTAATAGGGAAAGGACAGCGTG-3′, using either wild-type or mutant promoter having a G→A transition at –16 (22), and was subcloned into a plasmid pCRII (Invitrogen, San Diego, CA). The resultant plasmid (pCRIIMrSal or pCRII-16A, respectively) was digested with AvaI, and the DNA fragment containing the rDNA promoter sequence was purified from a gel. The fragment was then labeled with [α-32P]dCTP, dGTP and the Klenow fragment of DNA polymerase I.
The top-strand oligonucleotide covering from –56 to –3, and the bottom-strand oligonucleotide covering from –1 to –54 of either mouse or human rDNA promoter were synthesized and annealed. The double-stranded oligomers were then labeled by the filling-in reaction. For UV cross-linking experiments, the bottom-strand oligonucleotides were annealed by a primer complementary to 10 nucleotides from the 3′-end of the oligonucleotides followed by filling with [α-32P]dCTP, dATP, dGTP, dTTP and 5′-bromo-2′-deoxyuridine triphosphate. Unincorporated free nucleotides were removed by gel filtration.
Mouse rDNA promoter fragment containing from –330 to +66 was amplified by PCR with oligonucleotide primers 5′-AC-TAGTCTGGAGCTTTGGATCTTTTT-3′ and 5′-AGATCTG-AGCTCGGCGCCTTAAATCGAAAGGGTCTC-3′, and was subcloned into a plasmid pCRII (Invitrogen). The plasmid (pCRIIMr) was digested with ApaI and NarI. After the cohesive ends of the digest were converted to blunt ends with T4 DNA polymerase, the DNA fragment containing the rDNA promoter sequence was subcloned into the SmaI site of pBluescript II SK(+). The plasmid (pBSKIIMr10) was digested with HindIII and Bsp120I, and the DNA fragment containing the rDNA promoter sequence from –252 to +66 was purified from a gel. The fragment was then labeled with [α-32P]dCTP, dGTP and the Klenow fragment of DNA polymerase I, and used for DNase I footprint experiments.
DNA-binding experiments
Whole-cell extract or protein fractions were mixed as indicated in the individual reaction, supplemented with buffer DB-0.1 to 5 µl. Then, 5 µl of reaction mixture containing 600 µg/ml poly(dG)–poly(dC) and 10 mM MgCl2 was added. After incubation for 30 min at 30°C, 1 µl of probe solution (10 000 c.p.m., ∼5 ng/µl) was added, then the mixtures were incubated for 30 min at 30°C. In some cases, incubation was followed for 15 min with anti-TBP antiserum or preimmune serum (23). The reaction mixtures were loaded on an agarose gel cast in 0.5× TBE and run in 1× TBE at 100 V for 2 h [1× TBE: 0.045 M Tris–borate buffer (pH 8.3), 1 mM EDTA]. The gel was dried and autoradiographed.
DNA-binding reactions for UV cross-linking were performed as described above, except that the probes were replaced by internally labeled oligonucleotides (50 000–100 000 c.p.m./µl). After incubation for complex formation, the mixtures were directly irradiated by a UV lamp (Stratalinker; Stratagene, La Jolla, CA) for 10 min. Then, 4 µl of a nuclease mixture containing 1 µg each of DNase I and micrococcal nuclease in 18.75 mM MgCl2 and 3.75 mM CaCl2, was added followed by incubation for 30 min at 37°C. The mixtures were analyzed by SDS–PAGE.
For DNase I footprint, binding reactions were performed in 25 µl with 1–4 µl of TFIA-fraction and/or 8 µl of fraction D, and the probe (10 000 c.p.m.) for 30 min at 30°C followed by digestion with 0.1 U of DNase I for 1 min. The digestion reactions were terminated by the addition of 50 µl of stop mixture containing 50 mM EDTA, 0.1% SDS, 100 µg/ml yeast tRNA and 0.4 mg/ml proteinase K. After incubation at 37°C for 10 min, the digested DNAs were extracted with phenol and chloroform, precipitated twice with ethanol followed by loading onto a 6% sequencing gel.
In vitro transcription
MH134 nuclear extracts were fractionated by a phosphocellulose column. The 0.3–0.6 M KCl eluate was dialyzed against buffer DB-0.1 and was used as Pol I fraction. The 0.6–1.0 M KCl fraction of the column was further loaded onto a heparin–Sepharose column. The 0.4–0.6 M KCl eluate was dialyzed against buffer DB-0.1 and was used as SL1/UBF-fraction. A plasmid, pMrBKSP, which contains the mouse rDNA promoter region from –330 to +291 between the SacI and the BamHI site of pBluescript KS(+), was digested by EcoRV. An accurately initiated transcript with 320 nt is produced from this template.
A 25 µl transcription reaction containing 2 µl of the Pol I fraction, 4 µl of the UBF- and SL1-fractions and 0–6 µl of the TFIA-fraction, in 12 mM HEPES–KOH (pH 7.9), 4 mM MgCl2, 0.3 mM EDTA, 0.6 mM DTT, 0.12 mM PMSF, 12% glycerol, 600 µM ATP/CTP/GTP, 30 µM UTP, 10 µCi [α-32P]UTP, 100 µg/ml α-amanitin and 20 mg/ml template was incubated at 30°C for 60 min. The reaction was terminated by the addition of 200 µl of stop mixture containing 10 mM EDTA, 0.2% SDS, 200 µg/ml yeast tRNA and 0.1 mg/ml proteinase K. After incubation at 37°C for 15 min, the transcripts were extracted with phenol and chloroform, precipitated twice with ethanol followed by loading onto a 5% sequencing gel.
RESULTS
Detection of multiprotein complexes formed on the mouse rDNA gene promoter
To cover comprehensive interactions of factors involved in the initiation of rDNA transcription, we used a relatively large DNA fragment as a probe for electrophoretic mobility shift assay (EMSA), i.e. the mouse rDNA gene promoter from –168 to +30, which is sufficient for full promoter activity (5). Our early efforts to detect reproducible signals revealed that stability of protein complex on the rDNA gene promoter was varied with Mg2+ concentration and sorts of non-specific carrier DNA, and that the complex was too large to enter into polyacrylamide gel matrices (data not shown). We thus took advantage of an agarose gel with the binding reactions including 5 mM MgCl2, 300 µg/ml poly(dG)–poly(dC) and mouse whole-cell extracts. Under these conditions, two distinct protein–DNA complexes were separated, i.e. although rather smeary, complex A and B indicated in Figure 1A (see below). Unlabeled rDNA gene promoter fragment competed well with the probe (lanes 2–4), whereas a mutant promoter fragment containing a G→A transition at –16 did not (lanes 6–7). This mutation causes the rDNA promoter activity to decrease due to reduced binding affinity to factor(s) critical for the promoter recognition (22,24,25), indicating that the observed complex was formed in the DNA sequence-specific manner. We next examined whether the protein–DNA complexes detected in our agarose gel shift assay contain TBP, since the protein is an integral part of the promoter-selectivity factor SL1 (8). Addition of increasing amounts of anti-TBP antiserum resulted in the reduction of electrophoretic mobility and/or the dispersion of complex A, whereas complex B was not affected (Fig. 1B, lanes 2–6). Control preimmune antiserum had no effect on both the complexes (lanes 7–11). These results indicated that complex A contained TBP, but the faster migrating complex B lacked TBP. A DNA probe containing –56 to –1 of the mouse rDNA core promoter also formed a complex having the same sequence specificity and sensitivity to the antisera as observed in complex A (data not shown; see also below). Thus, we concluded that the present agarose gel shift assay firmly detected an rDNA promoter-specific protein complex.
Figure 1.
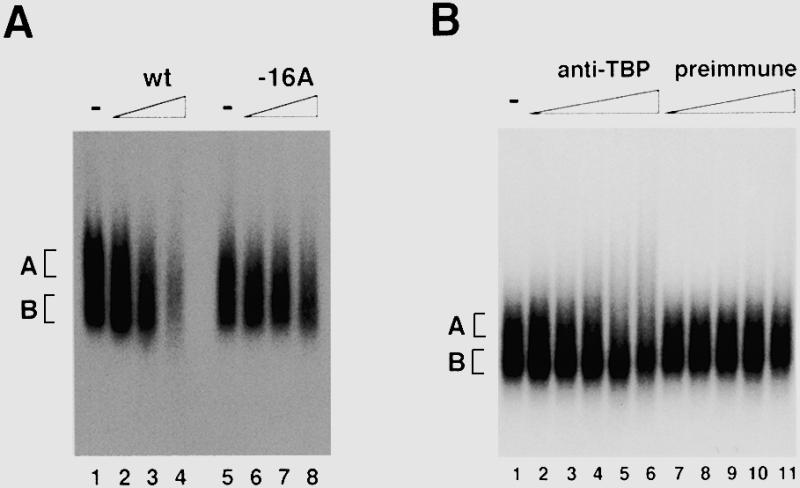
Detection of a specific complex formed with the rDNA promoter in mouse whole-cell extract. (A) Agarose gel shift assay was performed with mouse whole-cell extract and the mouse rDNA promoter fragment from –168 to +30 as probe in the absence (lanes 1 and 5) or the presence of 5- (lanes 2 and 6), 20- (lanes 3 and 7) and 80-fold (lanes 4 and 8) excess competitor of wild-type (lanes 2–4) and mutant (lanes 6–8) rDNA promoter fragments, respectively. Free probes run out of the gel. (B) Agarose gel shift assay was performed in the absence (lane 1) or presence of increasing amounts (lanes 2 and 7, 0.125 µl; lanes 3 and 8; 0.25 µl; lanes 4 and 9, 0.5 µl; lanes 5 and 10, 1.0 µl; lanes 6 and 11, 2.0 µl) of anti-mouse TBP antiserum (lanes 2–6) or preimmune antiserum (lanes 7–11). Positions of complexes A and B are indicated on the left.
Identification of a 70 kDa protein in the specific protein complex bound to the mammalian rDNA core promoters
As described in the above section, we have determined the condition to detect an rDNA promoter-specific complex which formed even in crude whole-cell extracts. We then fractionated the mouse whole-cell extract using column chromatography to clarify which factor(s) directly bind(s) to the mouse rDNA core promoter, Pol I, UBF, SL1 or yet unidentified factors. The KCl step-elution scheme on a phosphocellulose column was used to separate Pol I (0.3–0.6 M eluate), UBF (0.6–0.8 M eluate) and SL1 (0.8–1.2 M eluate) from whole-cell extracts (15,26). The flow-through (0.1 M KCl) fraction of phosphocellulose was further loaded onto a DEAE–Sepharose column. The 0.3 M KCl eluate of the column was also used for DNA-binding experiments with reconstituted column fractions, since this protein pool has been shown to contain a factor essential for rDNA transcription initiation, TFIA (15). Reconstitution of these fractions, when challenged to the core promoter region of mouse rDNA (–56 to –1), reproduced ever clearer promoter-bound complexes (Fig. 2A, lane 1 in the upper panel). Although the relative intensities of the separated complexes were varied in preparations of protein fractions, we reproducibly obtained two distinct complexes and confirmed that the slower migrating complex contained TBP (Fig. 2B). Thus, the complex produced by the reconstituted protein fractions is equivalent to complex A detected in cell extracts. Sequential combination of each fraction revealed that, although neither the TFIA-fraction nor the SL1-fraction alone gave any distinct signal (lanes 12 and 15), an intermediate complex was formed by simultaneous presence of both the fractions in binding reactions (lanes 3 and 8). This intermediate complex was further shifted and stabilized by the addition of the Pol I-fraction (lanes 1 and 4). The UBF-fraction had no effect on the formation of either the intermediate or stabilized complex.
Figure 2.
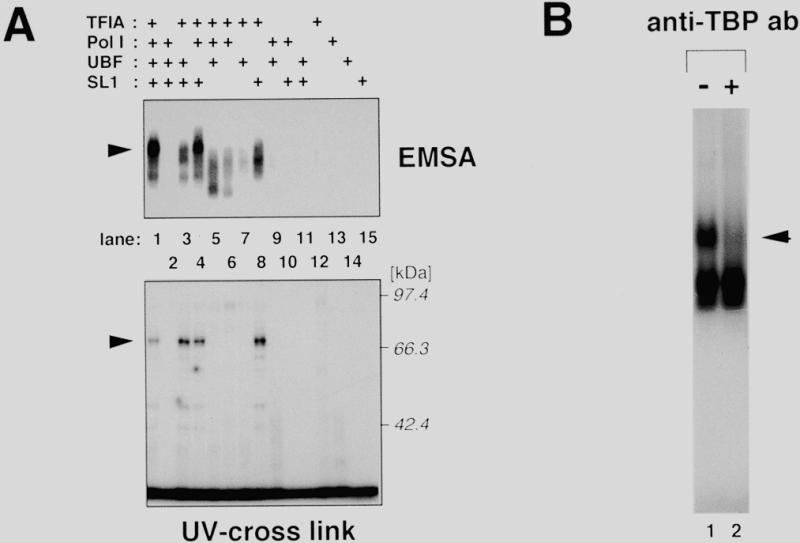
Reconstitution of column fractions derived from mouse whole-cell extract allows specific complex formation with the mouse rDNA core promoter. (A) Protein fractions containing TFIA, Pol I, UBF or SL1, were prepared from mouse whole-cell extract as described in Materials and Methods. The fractions, denoted as ‘+’ above each lane, were incubated with radiolabeled mouse rDNA core promoter fragment, and analyzed in an agarose gel. The amounts of protein added are as follows: TFIA-fraction, 14.8 µg; Pol I-fraction, 2.45 µg; UBF-fraction, 0.86 µg; SL1-fraction, 0.92 µg. The arrowhead indicates the specific complex reconstituted by the TFIA-, Pol I- and SL1-fractions. The lower panel is an autoradiogram of the UV cross-linking assay with the same combination of the fractions used in the agarose gel shift. Positions of a molecular weight marker are indicated on the right. The arrowhead indicates the position of p70. (B) TBP is involved in the reconstituted complex. An agarose gel shift assay was performed with the mouse core promoter probe and the mouse TFIA-, Pol I- and SL1-fraction. Following incubation for the DNA–protein complex formation, 1 µl of affinity-purified anti-TBP antibody (0.68 mg/ml) was added to the reaction mixture (lane 2). The mixtures were further incubated for 30 min at 30°C and then subjected to agarose gel electrophoresis. The arrowhead indicates the specific complex containing TBP.
Next, the same combination of the fractions was assayed by a UV cross-linking experiment with the same portion of DNA as in the agarose gel shift assay, in which cytosine residues are radiolabeled and thymine residues are partially replaced with bromodeoxyuridine. As shown in the lower panel of Figure 2A, a protein with an apparent molecular mass of 70 kDa (referred to as p70 hereafter) was efficiently cross-linked to the mouse core promoter. The DNA-binding activity of p70 was absolutely dependent on both the TFIA-fraction and the SL1-fraction (lanes 1, 3, 4 and 8), whereas no protein binding was detected with either fraction alone (lanes 12 and 15). Thus, the degree of p70 binding was well correlated with the appearance of the TBP-containing complex in the agarose gel shift assay. Neither the mouse TBP (35 kDa; 27) nor UBF1 and 2 (97 and 94 kDa, respectively; 28) was cross-linked to the core promoter region in this assay. We then investigated whether human cells contain a canonical orthologue of p70. To address this question, we fractionated HeLa cell extracts according to the same procedure for the preparation of the mouse fractions. As clearly demonstrated in Figure 3, a 70 kDa human protein binds to the human rDNA core promoter in the presence of human TFIA- and SL1-fractions (lanes 1, 3, 4 and 8). These results indicate that the protein complex bound to the rDNA core promoter contains at least two distinct molecules; TBP and a novel protein p70, binding of which is achieved by the cooperation of factors in TFIA- and SL1-fractions.
Figure 3.
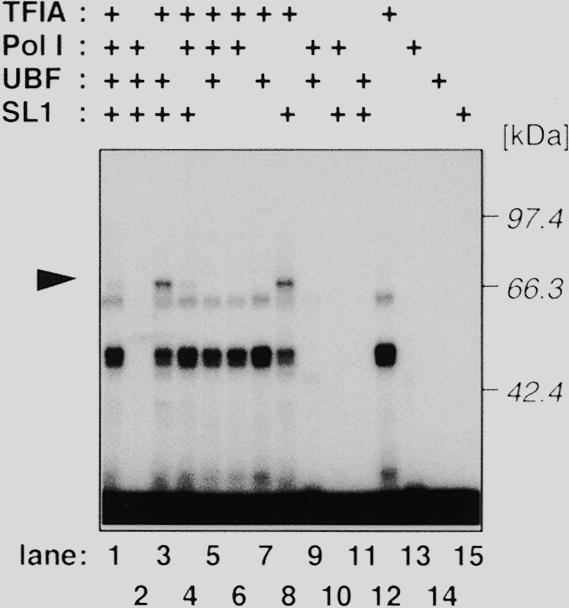
HeLa cells also contain the 70 kDa protein that binds to the human rDNA promoter in the presence of human SL1-fraction. Human TFIA-, Pol I-, UBF- and SL1-fractions were prepared from HeLa whole-cell extract as described. A UV-cross link experiment was performed essentially as described in Figure 2 with the photoreactive probe corresponding to the human rDNA promoter region from –56 to –1.
The TFIA-fraction contains p70
As the specific binding of p70 to the rDNA core promoter requires both TFIA- and SL1-fractions, the next obvious question is which fraction contains p70. To answer this, we further purified both fractions. Proteins contained in the TFIA-fraction were separated on the basis of electrophoretic mobility by a preparative polyacrylamide gel (Fig. 4, upper panel), and the fractions were analyzed by UV cross-linking in the presence of the SL1-fraction (Fig. 4, lower panel). Core promoter-binding activity was collected in the protein fractions with average molecular weight of 70 000 (Fig. 4, lanes 4–7). In a complementary experiment, proteins in the SL1-fraction were also fractionated and analyzed by cross-linking in the presence of the TFIA-fraction. However, none of the fractions showed DNA-binding activity (data not shown). Therefore, we conclude that the TFIA-fraction contains p70, and that its DNA-binding activity is dependent on factor(s) in the SL1-fraction.
Figure 4.
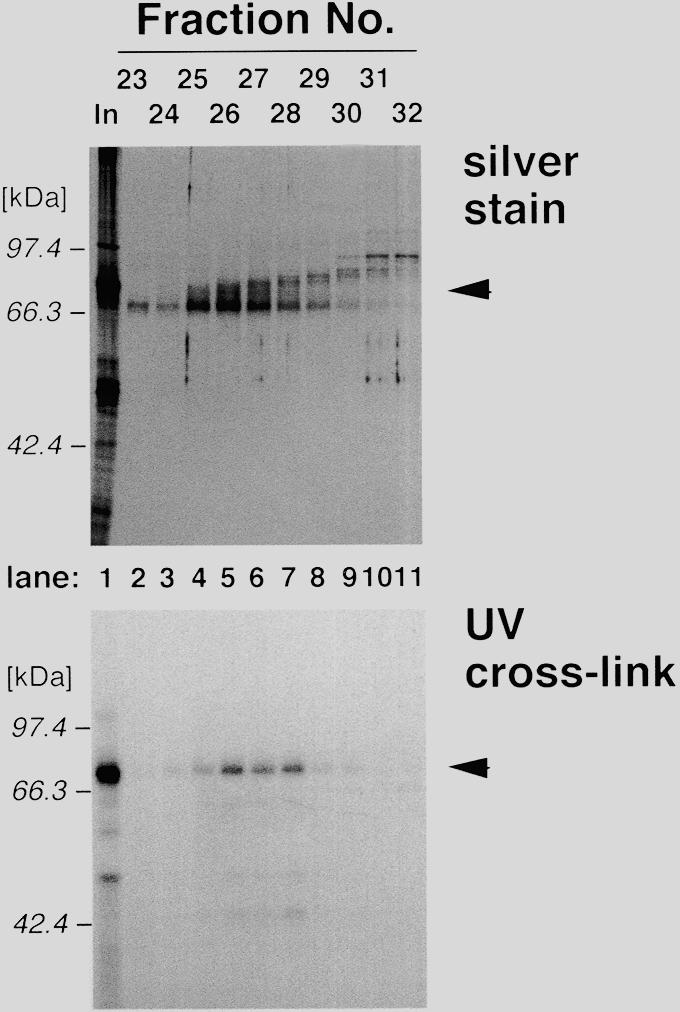
Size-fractionation of p70. Proteins in the TFIA-fraction (lane 1) were fractionated by size by a preparative scale of SDS–PAGE. Fractions containing 60–100 kDa proteins (fraction nos 23–32, lanes 2–11) were analyzed by silver stain (upper panel) and UV cross-linking in the presence of the SL1-fraction (lower panel). Positions of molecular weight markers are indicated on the left of each panel, and the positions of p70 are indicated by arrowheads.
The TFIA-fraction and SL1-fraction cooperate to protect the core promoter region from DNase I digestion
The data presented so far strongly suggest that the factors contained separately in the TFIA- and SL1-fractions cooperate to bind to the rDNA core promoter region. To detect such an interaction more directly, we performed a DNase I footprint experiment with the DNA fragment containing the mouse rDNA promoter from –252 to +66 as a probe. In the presence of the SL1-fraction alone, some of the bands showed relatively reduced intensity as compared with those of the free DNA (Fig. 5, lanes 2 and 3). Addition of increasing amounts of the TFIA fraction to the constant amount of the SL1-fraction caused local changes in the footprint. The presence of both the TFIA and SL1-fractions eliminated a specific set of bands in the regions from –10 to –30 and from –50 to –60, as compared with the SL1-fraction alone (lanes 4 and 5). Even at the highest amount, the TFIA-fraction alone showed essentially no obvious alterations, but general reduction, in the digestion ladder as compared with that observed in the absence of proteins (lanes 6 and 7). These results confirm that the factors present in the TFIA- and the SL1-fractions cooperate to bind to the rDNA core promoter region in the form of a specific conformation.
Figure 5.

Cooperative interaction between the factors in the TFIA- and SL1-fractions on the rDNA core promoter. The mouse rDNA promoter (–252 to +66) labeled at the 5′-end of the coding strand was used for the probe of DNase I footprint analysis. Reactions were performed with no proteins (lanes 2 and 7), and 0 (lane 3), 29.6 (lane 4) or 59.2 µg (lanes 5 and 6) of the TFIA-fraction in the presence (lanes 3–5) or absence (lane 6) of constant amounts (3.68 µg) of the SL1-fraction. The reaction product of a Maxam–Gilbert AG ladder was also loaded (lane 1). The nucleotide number with respect to the transcription initiation site as +1 is indicated on the left. Protected bases in the presence of both the fractions are indicated by arrowheads.
Partially purified p70 is essential for the rDNA transcription reconstituted in vitro
Finally we examined the effect of p70 on rDNA transcription. For this purpose, the TFIA-fraction was further purified by a gradient elution of a QAE-Sephadex column, and analyzed in an in vitro transcription system composed of partially purified Pol I and SL1/UBF-fractions. As demonstrated in the previous studies (15,26), no specific transcripts directed from the mouse rDNA promoter were detected in either combinations of the fractions lacking one or two (Fig. 6, lanes 3–8), confirming that each fraction was free from cross-contaminating activity from other fractions. In our reconstitution system, the Pol I-fraction, either alone or in combination with the SL1/UBF-fraction, produced variable lengths of transcripts (lanes 4 and 8). Addition of the peak fraction of p70 from QAE-Sephadex to the Pol I-fraction led to the remarkable reduction of the heterogeneity of the transcripts (lane 6). In the presence of both the Pol I- and the SL1/UBF-fractions, subsequent promoter-specific transcription occurred depending on the dose of p70 (lanes 9 and 10). Reduction of the transcription heterogeneity by p70 must result from the suppression of non-specifically initiated transcription by Pol I, but not from the inhibitory activity of potential ribonucleases in the p70-fraction, because ribonuclease inhibitors could not substitute for p70 (data not shown). These results strongly suggest that p70 not only participates in the initiation of rDNA transcription but also may have some effects to repress random transcription by RNA polymerase I.
Figure 6.

Transcription activity of partially purified p70 on the rDNA transcription in vitro. Transcription reactions were done in the presence or absence of 3.6 µg of Pol I-fraction (lanes 4, 6, 8, 9 and 10), 1.0 µg of SL1/UBF-fraction (lanes 5, 7, 8, 9 and 10) and 2.0 (lane 9) or 4.0 µg (lanes 3, 6, 7 and 10) of p70-fraction. In vitro transcribed RNA of 320 nt was produced as in the control reaction with the nuclear extract (lane 1).
DISCUSSION
We have demonstrated the specific interaction of a putative regulatory factor with the core promoter of mouse rDNA. The factor, tentatively referred to as p70 by its apparent molecular mass revealed by UV cross-linking, was found in the TFIA-fraction. The stable binding of p70 to the core promoter was absolutely dependent on the SL1-fraction. These two fractions were also required in EMSA for the formation of a core promoter-bound complex, which contained TBP as an integral component. Consistent with these observations, alterations of footprint over the core promoter region was detected by cooperative actions of the TFIA- and the SL1-fractions. A 70 kDa protein with similar DNA-binding characteristics was also identified in HeLa cells, suggesting that the same biological entity may be conserved at least in mammals.
TFIA is required for accurate initiation from both mouse and human rDNA promoter, and is suggested to function through interaction with SL1 to stabilize the pre-initiation complex on the rDNA promoter (15). Using the same chromatographic separation protocol by phosphocellulose to fractionate mouse S-100 extracts, Tower et al. (29) demonstrated that the TFIA-containing fraction can be replaced with an RNase inhibitor in the reconstituted in vitro transcription assay to reduce intrinsic RNase activity in the other fractions containing Pol I and SL1 activities. Both p70 and TFIA have the same chromatographic profile and the strict requirement of the SL1-containing fraction to act on the rDNA promoter, suggesting that p70 may represent the molecular entity of TFIA. In our reconstitution and transcription conditions, however, RNase inhibitor neither substituted the transcription stimulation activity of p70 nor reduced the background smearing detected in the absence of p70. Elucidation of the precise relationship between p70 and TFIA will be necessary to account for the discrepancy of the requirement of RNase inhibitor in the transcription systems.
In the current agarose gel shift and UV cross-linking assays, direct binding of UBF to the core promoter region was not detected. UBF derived from mouse and Xenopus laevis as well as human can bind to the upstream control element, and causes some structural changes in the core promoter region of the human rDNA promoter (30–33). In contrast, the mouse rDNA promoter interacts much more weakly with UBF of any origin (31,34). Thus, at least in the mouse system, UBF may not participate in the complex formation on the core promoter. Alternatively, the region of the core promoter element used in the UV cross-linking may be insufficient for the interaction with UBF. Site-specific photocross-linking experiments by Gong et al. (35) have shown that TBP and putative TAFIs of 145, 99, 96 and 91 kDa can be specifically cross-linked to different positions along the rDNA promoter of Acanthamoeba castellanii. In our system, however, there was no cross-linked adduct corresponding to the molecular mass of mouse TBP, while the core promoter-bound complex has been shown to definitely include TBP. Although TBP is involved in the transcription machinery for all three classes of the nuclear RNA polymerases (36,37), its function is likely to be different in each transcription system (38). Typically, in TATA box-containing promoters of Pol II and Pol III systems, TBP does bind to the TATA box. In TATA-less promoters for all classes, TBP seems only to associate with other proteins, such as TAFs, activators or polymerase subunits (39). In either case, TBP appears to function as the core molecule assembling components for promoter-commitment. In this regard, our data support the idea that TBP serves as a key protein to assemble the other proteins like TAFIs (see below; 8,9,40–43).
Recently, all the components of human SL1 have been cloned (43,44). Human SL1 is composed of TBP and three TAFIs with molecular masses of 48, 63 and 110 kDa, respectively (8,44). This composition is well conserved in mouse with slight differences in apparent the molecular weights of each TAFI (48, 68 and 95 kDa, respectively; 42,43). Close similarity in the apparent molecular weight of p70 with that of the 68 kDa component (TAFI68) raises the possibility that p70 is TAFI68 itself. Indeed, murine TAFI68 (mTAFI68), together with mTAFI48, contacts the mouse core promoter (42). DNA-binding activity of p70 was, however, detected only when the TFIA- and SL1-fractions were combined. The SL1-fraction is the high salt eluate of a phosphocellulose column (15,26). The TFIA-fraction, on the other hand, is the flow-through at low salt of the column and contains p70 (15 and this study). It means that p70 can easily dissociate from SL1 even if these factors are physically associated. This is quite different from the nature of mTAFI68 which tightly associates with the other component of SL1 throughout the phosphocellulose column chromatography even in the presence of 2 M urea (8). Consistent with this notion, immunoblots with antibodies against mTAFI68 revealed that mTAFI68 was found exclusively in the SL1-fraction, and that it was not detected in the TFIA-fraction (data not shown). Moreover, p70 is also separated from Pol I activity by the initial chromatography step, suggesting that it might be different from Pol I-associating factors, such as Factor C*, TFIC, TIF-IA, TIF-IC and PAF53 (14,18–20,45). It has been reported that several DNA-binding proteins interact with rat rDNA core promoter as well as with what seems an enhancer. These proteins, E1BF (46) and CPBF (47), can bind solely to the rDNA promoter, which remarkably contrasts to the fact that p70 requires the SL1-fraction to contact DNA. Although the precise relationship between p70 and these factors is still to be determined, the experimental system presented here will provide a powerful tool to complete the purification and characterization of important biological molecules involved in rDNA transcription.
Acknowledgments
ACKNOWLEDGEMENTS
We are indebted to Dr K. Hanada for helpful discussion, Dr A. Okuda for critical reading of the manuscript, and Dr K. Hisatake for valuable comments and discussion. We also thank Ms N. Hihara for excellent technical assistance. This work was supported in part by Grants-in-Aid from the Ministry of Education, Science, Sports and Culture of Japan and the grant provided by the Ichiro Kanehara Foundation.
REFERENCES
- 1.Reeder R.H. (1992) In McKnight,S.L. and Yamamoto,K.R. (eds), Transcriptional Regulation. Cold Spring Harbor Laboratory Press, New York, pp. 315–347.
- 2.Paule M.R. (1994) In Conaway,R.C. and Conaway,J.W. (eds), Transcription: Mechanisms and Regulation. Raven Press Ltd, New York, pp. 83–106.
- 3.Hannan K.M., Hannan,R.D. and Rothblum,L.I. (1998) Front. Biosci., 3, d376–d398. [DOI] [PubMed] [Google Scholar]
- 4.Grummt I. (1998) In Paule,M.R. (ed.), Transcription of Ribosomal RNA Genes by Eukaryotic RNA Polymerase I. Springer-Verlag, Berlin, pp. 135–154.
- 5.Yamamoto O., Takakusa,N., Mishima,Y., Kominami,R. and Muramatsu,M. (1984) Proc. Natl Acad. Sci. USA, 81, 299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haltiner M.M., Smale,S.T. and Tjian,R. (1986) Mol. Cell. Biol., 6, 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones M.H., Learned,R.M. and Tjian,R. (1988) Proc. Natl Acad. Sci. USA, 85, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Comai L., Tanese,N. and Tjian,R. (1992) Cell, 68, 965–976. [DOI] [PubMed] [Google Scholar]
- 9.Zomerdijk J.C.B.M. and Tjian,R. (1998) In Paule,M.R. (ed.), Transcription of Ribosomal RNA Genes by Eukaryotic RNA Polymerase I. Springer-Verlag, Berlin, pp. 67–73.
- 10.Learned R.M., Learned,T.M., Haltiner,M.M. and Tjian,R.T. (1986) Cell, 45, 847–857. [DOI] [PubMed] [Google Scholar]
- 11.Bell S.P., Learned,R.M., Jantzen,H.-M. and Tjian,R. (1988) Science, 241, 1192–1197. [DOI] [PubMed] [Google Scholar]
- 12.Kuhn A. and Grummt,I. (1992) Proc. Natl Acad. Sci. USA, 89, 7340–7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith S.D., O’Mahony,D.J., Kinsella,B.T. and Rothblum,L.I. (1993) Gene Expr., 3, 229–236. [PMC free article] [PubMed] [Google Scholar]
- 14.Brun R.P., Ryan,K. and Sollner-Webb,B. (1994) Mol. Cell. Biol., 14, 5010–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato H., Nagamine,M., Kominami,R. and Muramatsu,M. (1986) Mol. Cell. Biol., 6, 3418–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cavanaugh A.H. and Thompson,E.A.Jr (1985) Nucleic Acids Res., 13, 3357–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buttgereit D., Pflugfelder,G. and Grummt,I. (1985) Nucleic Acids Res., 13, 8165–8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schnapp G., Schnapp,A., Rosenbauer,H. and Grummt,I. (1994) EMBO J., 13, 4028–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schnapp A., Schnapp,G., Erny,B. and Grummt,I. (1993) Mol. Cell. Biol., 13, 6723–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahajan P.B. and Thompson,E.A. (1990) J. Biol. Chem., 265, 16225–16233. [PubMed] [Google Scholar]
- 21.Manley J.L., Fire,A., Cano,A., Sharp,P.A. and Gefter,M.L. (1980) Proc. Natl Acad. Sci. USA, 77, 3855–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagamine M., Kishimoto,T., Aono,J., Kato,H., Kominami,R. and Muramatsu,M. (1987) Mol. Cell. Biol., 7, 1486–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato K., Makino,Y., Kishimoto,T., Yamauchi,J., Kato,S., Muramatsu,M. and Tamura,T. (1994) Nucleic Acids Res., 22, 1179–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kishimoto T., Nagamine,M., Sasaki,T., Takakusa,N., Miwa,T., Kominami,R. and Muramatsu,M. (1985) Nucleic Acids Res., 13, 3515–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanaka N., Kato,H., Ishikawa,Y., Hisatake,K., Tashiro,K., Kominami,R. and Muramatsu,M. (1990) J. Biol. Chem., 265, 13836–13842. [PubMed] [Google Scholar]
- 26.Mishima Y., Financsek,I., Kominami,R. and Muramatsu,M. (1982) Nucleic Acids Res., 10, 6659–6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamura T., Sumita,K., Fujino,I., Aoyama,A., Horikoshi,M., Hoffmann,A., Roeder,R.G., Muramatsu,M. and Mikoshiba,K. (1991) Nucleic Acids Res., 19, 3861–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hisatake K., Nishimura,T., Maeda,Y., Hanada,K., Song,C.-Z. and Muramatsu,M. (1991) Nucleic Acids Res., 19, 4631–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tower J., Culotta,V.C. and Sollner-Webb,B. (1986) Mol. Cell. Biol., 6, 3451–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bell S.P., Pikaard,C.S., Reeder,R.H. and Tjian,R. (1989) Cell, 59, 489–497. [DOI] [PubMed] [Google Scholar]
- 31.Bell S.P., Jantzen,H.-M. and Tjian,R. (1990) Genes Dev., 4, 943–954. [DOI] [PubMed] [Google Scholar]
- 32.Pikaard C.S., McStay,B., Schultz,M.C., Bell,S.P. and Reeder,R.H. (1989) Genes Dev., 3, 1779–1788. [DOI] [PubMed] [Google Scholar]
- 33.Moss T., Stefanovsky,V.Y. and Pelletier,G. (1998) In Paule,M.R. (ed.), Transcription of Ribosomal RNA Genes by Eukaryotic RNA Polymerase I. Springer-Verlag, Berlin, pp. 75–94.
- 34.Kuhn A., Voit,R., Stefanovsky,V., Evers,R., Bianchi,M. and Grummt,I. (1994) EMBO J., 13, 416–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gong X., Radebaugh,C.A., Geiss,G.K., Simon,M.N. and Paule,M.R. (1995) Mol. Cell. Biol., 15, 4956–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cormack B.P. and Struhl,K. (1992) Cell, 69, 685–696. [DOI] [PubMed] [Google Scholar]
- 37.Schultz M.C., Reeder,R.H. and Hahn,S. (1992) Cell, 69, 697–702. [DOI] [PubMed] [Google Scholar]
- 38.Radebaugh C.A., Matthews,J.L., Geiss,G.K., Liu,F., Wong,J.-M., Bateman,E., Camier,S., Sentenac,A. and Paule,M.R. (1994) Mol. Cell. Biol., 14, 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rigby P.W.J. (1993) Cell, 72, 7–10. [DOI] [PubMed] [Google Scholar]
- 40.Eberhard D., Tora,L., Egly,J.-M. and Grummt,I. (1993) Nucleic Acids Res., 21, 4180–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zomerdijk J.C.B.M., Beckmann,H., Comai,L. and Tjian,R. (1994) Science, 266, 2015–2018. [DOI] [PubMed] [Google Scholar]
- 42.Rudloff U., Eberhard,D., Tora,L., Stunenberg,H. and Grummt,I. (1994) EMBO J., 13, 2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heix J., Zomerdijk,J.C.B.M., Ravanpay,A., Tjian,R. and Grummt,I. (1997) Proc. Natl Acad. Sci. USA, 94, 1733–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Comai L., Zomerdijk,J.C.B., Beckmann,M.H., Zhou,S., Admon,A. and Tjian,R. (1994) Science, 266, 1966–1972. [DOI] [PubMed] [Google Scholar]
- 45.Hanada K., Song,C.Z., Yamamoto,K., Yano,K., Maeda,K., Yamaguchi,K. and Muramatsu,M. (1996) EMBO J., 15, 2217–2226. [PMC free article] [PubMed] [Google Scholar]
- 46.Hoff C.M., Ghosh,A.K., Prabhakar,B.S. and Jacob,S.T. (1994) Proc. Natl Acad. Sci. USA, 91, 762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Z. and Jacob,S.T. (1994) J. Biol. Chem., 269, 16618–16626. [PubMed] [Google Scholar]