

Abstract
Rhabdomyosarcoma (RMS) is a pediatric soft tissue cancer with a lack of precision therapy options for patients. We hypothesized that with a general paucity of known mutations in RMS, chromatin structural driving mechanisms are essential for tumor proliferation. Thus, we carried out high-depth in situ Hi-C in representative cell lines and patient-derived xenografts (PDXs) to define chromatin architecture in each major RMS subtype. We report a comprehensive 3D chromatin structural analysis and characterization of fusion-positive (FP-RMS) and fusion-negative RMS (FN-RMS). We have generated spike-in in situ Hi-C chromatin interaction maps for the most common FP-RMS and FN-RMS cell lines and compared our data with PDX models. In our studies, we uncover common and distinct structural elements in large Mb-scale chromatin compartments, tumor-essential genes within variable topologically associating domains and unique patterns of structural variation. Our high-depth chromatin interactivity maps and comprehensive analyses provide context for gene regulatory events and reveal functional chromatin domains in RMS.
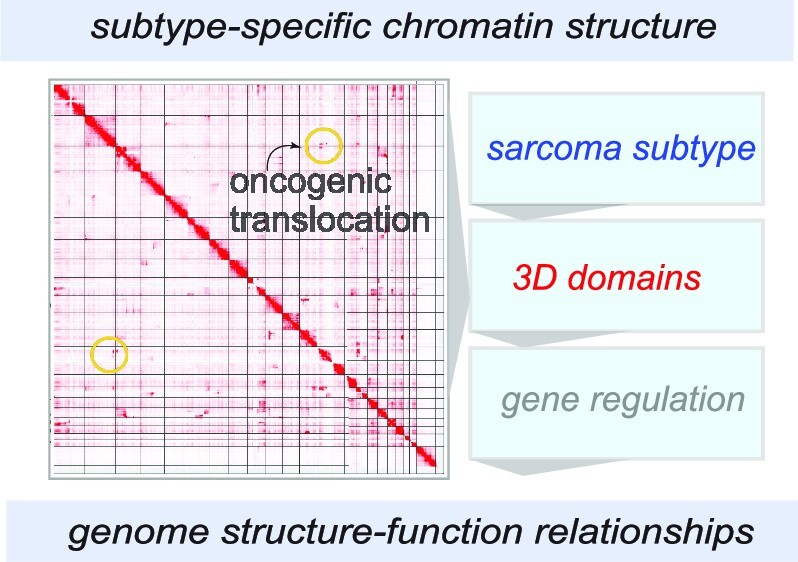
Graphical Abstract
Graphical Abstract.
INTRODUCTION
Rhabdomyosarcoma (RMS) is an aggressive pediatric solid tumor, with prognoses that are highly dependent on the subtype (1–4). The major RMS subtypes are defined by histological features, termed alveolar RMS (ARMS) and embryonal RMS (ERMS), while more rare forms also exist (2,4). The ARMS subtype is often driven by oncoproteins encoded by fusion events resulting in PAX3–FOXO1 or PAX7–FOXO1 expression (5–7). ERMS is driven by aberrantly active signaling pathways, including RAS (8), HIPPO (9,10) and NOTCH (11). It is of note that the myogenic master regulator MYOD is essential in RMS in its wild-type form (12–14), while mutations in MYOD cause a particularly aggressive and untreatable subtype of RMS (2,15,16). Thus, understanding the molecular and chromatin-recognition functions of MYOD is of high interest for defining altered RMS epigenetics and disease etiology/tumorigenicity. Clinical studies have revealed that fusion-positive RMS (FP-RMS) has poorer outcomes than fusion-negative RMS (FN-RMS) (17,18). However, key questions remain: To what extent can chromatin architecture itself be oncogenic, to stabilize aberrant epigenetic states? Are the genome structures of RMS subtypes conserved despite the divergent clinical risk associated with fusion status? These questions have motivated our comprehensive characterization and analysis of FN-RMS and FP-RMS, at the level of 3D chromatin structure and genome organization in cell lines and patient-derived tissues.
We sought to characterize genome structure–function relationships in RMS at several unique levels, including (i) larger (>Mb-scale) structural contact domains termed compartments (19–21), (ii) medium-range (<Mb-scale) contact domains, or topologically associating domains (TADs) (19,22,23), (iii) in the context of the cis-chromatin interactions at loci with copy number variation (CNV), or structural variations (SVs) (24–27), and (iv) at the level of the gene regulatory loci encoding the major fusion oncogenes in FP-RMS (28). In our comparative analyses, we discover substantial unique or subtype-specific regions at the compartment level (>1 Mb) amid convergence in epigenetic conservation of FP-RMS and FN-RMS genomes. We report a comprehensive set of subtype-specific TADs, often bracketing tumor-associated genes. Within the PAX3–FOXO1 locus in FP-RMS cells, we find evidence for unique structural events leading to the formation of the fusion alleles, which also has ramifications on local chromatin interactivity. Expanding upon this finding, in our structural analysis of the RMS landscape, we discover previously unknown SV events that are distinct within and across RMS subtypes. We also uncover recurrent SV and CNV patterns within the ERMS subtype (29) in cell lines and patient-derived xenografts (PDXs), suggesting that defects in genome integrity may be a common driver.
Our previous studies have focused on protein-centric chromatin conformation sequencing, modifying the HiChIP method in RMS for in situ spike-in (AQuA-HiChIP) of orthologous-species nuclei (14,30–32). Herein, we employed a modified version of AQuA-HiChIP allowing us to perform in situ Hi-C incorporating orthologous spike-in chromatin (Spice-C), to ensure that any absence of local chromatin interactions in our data was biological and not a product of experimental variation. Our 3D landscape presents a highly rigorous characterization of the initial high-resolution chromatin contact maps and an in-depth analysis of genome structure–function relationships in RMS. With our key definitions of conserved structural features in these tumors, we anticipate our studies will continue to catalyze impactful connections between primary genetic drivers and structural epigenetics.
MATERIALS AND METHODS
Cell lines
The human RMS cell lines RD (female), Rh30 (male), SMS-CTR (male) and Rh4 (female) were a gift from Dr Peter Houghton (University of Texas Health Science Center, San Antonio) and C2C12 myoblasts were obtained from ATCC. All lines were maintained in either DMEM or RPMI supplemented with 10% fetal bovine serum at 37°C with 5% CO2. Cell lines were authenticated by genotyping.
CRISPR/Cas9 knockout
CRISPR guides at the PAX3 promoter were designed using the Benchling® platform. The guides were cloned in pLentiCrisprV2-Puro or Hygro vectors. Lentivirus generated in 293T cells was used to infect RMS cells in the presence of 8 μg/ml protamine and selected for 5 days in puromycin (2 μg/ml) + hygromycin (100 ng/ml) supplemented growth media.
Cellular assays
Cell growth kinetics of PAX3 TAD boundary element knockout (KO) cells were monitored using the Incucyte® imaging platform. Colony assays were performed by plating serially diluted control and KO cells in growth media and stained with crystal violet after 15 days. Immunofluorescence staining was performed in cells after 3 days in differentiation medium (DMEM + 2% horse serum) and fixed with 4% paraformaldehyde, permeabilized in 0.5% Triton X-100/phosphate-buffered saline and incubated with MyHC and MEF2C antibodies followed by Alexa-488/563 secondary antibodies along with DAPI and imaged using an Olympus FL-2000 confocal microscope at 20× magnification.
Western blotting
Total cell lysate was obtained by lysing in 1× RIPA buffer supplemented with protease inhibitors (Roche) and MG213 (Calbiochem). Membranes (PVDF, Bio-Rad) were developed using ECL reagent (Immobilon, Millipore). Internal control (GAPDH, CST, #2118) and target antibodies PAX3 (R&D, #MAB2457), FOXO1 (CST, #2880S), MYOD1 (Santa Cruz, #sc760), MYOG (DSHB, #F5D), MEF2C (CST, #5030) and MyHC (DSHB, #MF20) were probed on different regions of the same blot and imaged (Li-Cor).
ChIP-seq library preparation
Formaldehyde (1%, 10 min) fixed cells were sheared to achieve chromatin fragmented to a range of 200–700 bp using an Active Motif EpiShear Sonicator. Chromatin samples were immunoprecipitated overnight at 4°C with antibodies targeting MYOD (CST, cat#13812S), H3K27ac (Active Motif, #39133), CTCF (Millipore, #07-729) and H3K9me3 (Active Motif, #39062). DNA purifications were performed with the modified ChIP protocol described previously (33). For sample normalization, 2 million C2C12 cells were added to 6 million RMS cells before sonication. ChIP-seq libraries were prepared using Illumina indexes and adaptors as described previously (31). Libraries were multiplexed and sequenced using the NextSeq500 (Illumina).
ChIP-seq data processing
ChIP-seq data were processed with the ENCODE ChIP-seq pipeline with chip.xcor_exclusion_range_max set at 25. Paired-end fastqs were aligned with bowtie2 to hg38, with parameters bowtie2 -X2000 --mm. Next, blacklisted region, unmapped, mate unmapped, not primary alignment, multi-mapped, low mapping quality (MAPQ < 30), duplicate reads, and PCR duplicates were removed. Peaks were called with MACS2, with parameters -p 1e-2 --nomodel -- shift 0 --extsize $[FRAGLEN] --keep-dup all -B --SPMR, where FRAGLEN is the estimated fragment length. IDR analyses were performed on peaks from replicate samples or pseudo-replicates for MYOD ChIP-seq with a threshold of 0.05. Motif analysis with HOMER (34) was then carried out on conservative IDR peaks. For visualization, bedGraph files were generated with MACS2 bdgcmp from the pile-up, and then converted to bigwig format with bedGraphToBigWig. Heatmaps were generated with deeptools. PAX3–FOXO1 ChIP-seq was analyzed as previously reported (33). Raw sequencing data and processed files are available at Gene Expression Omnibus (GSE215203).
Spike-in chromatin equalized Hi-C (Spice-C)
RD, SMS-CTR, Rh30 and Rh41 cells were fixed for 10 min at room temperature (23°C) and mouse myoblasts (C2C12) were spiked in at a 25% cell ratio. The cells were lysed gently with Hi-C buffer to release nuclei, permeabilized in 0.5% SDS for 10 min at 62°C, quenched with 10% Triton X-100 and digested with MboI (200 U, 2 h at 37°C), which was then heat inactivated (20 min, 62°C). Biotin incorporation was done with biotin-14-dATP (Thermo, cat#19524-016) and DNA Polymerase I, Large (Klenow) Fragment (NEB, cat#M0210) for 1 h at 37°C. We performed in situ ligation with T4 DNA ligase (4 h, room temperature). Nuclei were resuspended in TE and sonicated (five cycles with shearing ‘on’ time with 30 s on and 30 s off), using the Active Motif EpiShear Probe Sonicator, 30% power. The sheared DNA was reverse cross-linked with SDS and Proteinase K overnight and pulled down with M-280 Streptavidin Dynabeads (Thermo, cat#11205D), washed and eluted, followed by end repair, A-tailing, adapter ligation and library amplification all on-bead as previously reported (33). For a detailed protocol of the Spice-C method, refer to the Materials and Methods section in the Supplementary Data.
Hi-C sequencing
Libraries were indexed with Illumina barcodes and quantified using Qubit (Thermo Fisher), with concentrations measured between 1 and 10 ng/μl. Median fragment lengths for our libraries were measured with TapeStation (Agilent) and were between 200 and 500 bp with average size ∼350 bp for each indexed library. After validating indexed library concentrations and indexes with qPCR (IGM, Nationwide Children’s Hospital), we sequenced the libraries on the NovaSeq S1 platform (Illumina) with paired-end 150 bp reads and 300 sequencing cycles (∼1 billion reads) and demultiplexed for further analysis. Two independent biological replicates for each cell line were combined informatically to get contact maps with 30 million valid, long-range cis contacts.
Hi-C data processing
Hi-C data were processed in accordance with 4D Nucleosome processing pipeline recommendations. Briefly, fastq files were aligned to hg38 with bwa-mem (35), allowing for soft clipping. Mapped reads were then tagged for PCR duplications, parsed for ligation junction, merged and then sorted into binned matrix using pairtools (36). Matrix aggregation and normalization (ICE) were carried out with cooltools (Table 1).
Table 1.
Analytical tools for Hi-C analysis
| Software | Version | Reference |
|---|---|---|
| bwa | 0.7.17-r1188 | (35) |
| pairtools | v0.3.0 | (36) |
| cooltools | v0.4.0 | (36) |
| TopDom | v0.10.1 | (37) |
| HiCExplorer | v3.7 | (40) |
| neoloopfinder | 0.2.4.post2 | (39) |
| R | v4.0.0 | https://www.r-project.org/ |
| juicebox | 2.2.2 | (41) |
| diffHic | v1.22.0 | (38) |
| hicbreakfinder | NA | https://github.com/dixonlab/hic_breakfinder |
| ENCODE ChIP-seq | v2.1.2 | (42) |
| deeptools | v3.3.1 | (43) |
| HOMER | v4.11.1 | (34) |
| HiGlass | v1.11.7 | (44) |
Hi-C data analyses
Compartments were called at 100 kb using cooltools call-compartments, using H3K27ac as the reference track. TADs were called with R package TopDom (37). Differential TAD analysis was carried out with R package diffHic (38). We used hicbreakfinder (https://github.com/dixonlab/hic_breakfinder) to find structural break points, and then neoloopfinder (39) to estimate copy number and reconstruct SVs (Table 1).
RESULTS
In our previous studies, we developed an orthologous chromatin spike-in approach for short-range chromatin interaction domains (31). However, the resolution in these studies precluded our ability to define large-scale compartmental structures (19,20,45) and smaller-scale TADs. We were motivated to understand chromatin domains in RMS, and thus established our approach to perform in situ Hi-C for deep sequencing. With the same experimental workflow as in our previous studies (14,31), we developed a modified in situ Hi-C protocol Spice-C (see the Materials and Methods section in the Supplementary Data) (19). In our approach, we permeabilize nuclei with gentle SDS treatment and heat (32), perform restriction digests with MboI, biotinylate exposed DNA ends with a Klenow extension and perform proximity ligation to stitch together proximal chromatin ends associated with cis-interactions. We gently shear chromatin prior to reverse cross-linking and on-bead library preparation for next-generation sequencing. We noted that our early attempts to scale up the in situ Hi-C reactions resulted in quenching of the library amplification step with our streptavidin beads, which is a common caveat of on-bead library preparation. We circumvented this issue through preparation of individual amplification reactions with no more than 10 μl beads in sequencing library amplifications (see the Materials and Methods section in the Supplementary Data).
We established our modified in situ Hi-C method for FP-RMS cells (Rh4 and Rh30) and FN-RMS cells (RD and SMS-CTR) with ∼1 billion paired-end sequencing reads per cell line after combining replicate samples. We mapped the reads onto the human reference genome, tagged for PCR duplication removal and parsed into contact pairs. We binned valid contacts into interaction matrices and normalized interaction matrices using the ICE algorithm (46) for subsequent analyses.
Analyses of compartment structures in RMS
Compartments are Mb-scale, self-interacting chromatin structures (20). Generally, human genomes can be classified into ‘compartment A’ and ‘compartment B’ via principal component analysis of the interaction matrix (19–21). Regions in compartment A are gene-rich and are usually in an ‘active’ local epigenetic state. These regions usually exhibit higher GC content and contain more active chromatin markers such as H3K27ac (21,47,48). In contrast, regions in compartment B are relatively gene-poor and show higher levels of silent or repressive local epigenetic markers such as H3K9me3 (49,50). Based on these previous findings (19,20), we carried out compartment analysis with our in situ Hi-C at 100 kb resolution. Compartments were assigned based on the sign of the first principal component (PC1) value of the correlation matrices, using H3K27ac as the reference track (19,20). We generated compartment definitions for two FP-RMS cell lines (Rh4 and Rh30; Figure 1A and B) and two FN-RMS cell lines (RD and SMS-CTR; Figure 1C and D) and developed these maps for comparative analyses.
Figure 1.

Chromatin compartments in RMS. Correlation heatmaps along chromosome 2 in (A) Rh4 cells, (B) Rh30 cells, (C) RD cells and (D) SMS-CTR cells, with normalized eigenvectors shown below and correlation values shown on the right. The associations between compartment calls and GC richness, H3K27ac, H3K9me3, MYOD binding and PAX3–FOXO1 binding are shown for Rh4, Rh30, RD and SMS-CTR cells on (E) chromosome 16 and (F) chromosome 19 with A compartments shown in blue and B compartments shown in black. PC1 values for compartments are plotted against average intensity for chromatin marks and regulatory factors: H3K27ac (G) H3K9me3 (H), MYOD as scatter (I) and PAX3–FOXO1 (J). (K) A compartments and B compartments are plotted in blue and black, respectively, for Rh4, Rh30, RD and SMS-CTR on chromosome 16 (left) and chromosome 19 (right). (L) Discordant and concordant compartments’ association with deposition of H3K27ac, MYOD and PAX3–FOXO1. Discordant compartments were defined as A–B (compartment A in FP and compartment B in FN) and B–A (compartment B in FP and compartment A in FN).
The relationship of H3K9me3 with compartments A and B
To understand the roles of chromatin modifications and RMS drivers such as PAX–FOXO1 (6,7) as well as MYOD (14) in the compartmental structures of FP-RMS and FN-RMS, we established H3K27ac, H3K9me3, PAX3–FOXO1 and MYOD ChIP-seq in each cell model, using gentle sonication to avoid epitope shearing (33,51). We found that regions belonging to compartment A have a higher level of GC content and H3K27ac deposition compared to regions assigned to compartment B (Figure 1E and F) with exceptions within chromosomes 3, 12 and 18 (see Supplementary Figures S1–S4). Interestingly, H3K9me3, a repressive histone modification, is not necessarily depleted in compartment A across chromosomes (Figure 1E and F, and Supplementary Figures S1–S4). While the intensity of H3K9me3 is systematically higher in compartment B across many chromosomes, this histone mark is also present in compartment A, especially noticeable at chromosomes 5, 18, 21 and others (Supplementary Figures S1–S4). To our surprise, we also observed substantial signal of H3K27ac and H3K9me3 in both compartmental classes. This is consistent with histone modifications having unique roles in a context-dependent fashion, or with heterochromatin regions being punctuated with activating marks as has been shown by us and observed in other cell types (48,52). The relative enrichments of H3K9me3 in B compartments were more pronounced in the FN subtype (Figure 1E and F, and Supplementary Figures S1–S4). The greater degree of concordance between deposition of H3K9me3 and B compartments in FN-RMS relative to FP-RMS may be related to distinct large-scale compartment structure in each subtype, or alternative functions of H3K9me3 and heterochromatin formation in a subtype-specific context. As we used H3K27ac to assign compartmental identities, it follows that some chromosomes unexpectedly exhibit a positive correlation between levels of H3K27ac and H3K9me3. This unexpected concordance may have resulted from locally distinct epigenetic functions of H3K9me3, which can reside within both active and repressive chromatin regions (33,53). We noted that while GC richness is invariant in the context of local or global epigenetic state, deposition of histone marks is highly dependent upon the local chromatin context (Supplementary Figures S1–S4) (46,54).
In all four RMS cell lines, saddle plots (46,55,56) revealed a preference for self-interaction within compartment class (e.g. B–B interactions or A–A interactions), relative to cross-compartmental interactions (Supplementary Figure S5). Moreover, we found that for each RMS cell line, regardless of fusion status, B–B compartmental interactions were stronger than A–A homotypic interaction strengths. While these trends suggested epigenetic convergence across FP-RMS and FN-RMS, we did observe that Rh30 had a higher prevalence for intercompartmental interactions across compartment classes, relative to the other RMS cell lines that we investigated (Supplementary Figure S5). The unique compartmental architecture in Rh30, with increased tendency for chromatin interactions occurring across compartment classes, suggests that while there is a high degree of subtype-specific structure, variation also exists within subtypes.
PAX3–FOXO1 and MYOD have distinct binding profiles within compartments
We observed PAX3–FOXO1 and MYOD within both compartments A and B (Figure 1E and F, and Supplementary Figures S1–S4). MYOD deposition is more enriched in compartment A, and increases as the strength of compartment A increases: a trend reminiscent of H3K27ac (Figure 1G and I). On the other hand, PAX3–FOXO1 had a consistent signal intensity irrespective of the sign or magnitude of PC1 except for a slight increase in intensity in the strongest A compartments (Figure 1H). This profile is distinct from both H3K27ac (Figure 1I) and H3K9me3 (Figure 1J). Previous research has revealed that PAX3–FOXO1 has been shown to have pioneer transcription factor (TF) function, and MYOD may have pioneer factor-like properties that augment its association with compacted chromatin regions (Supplementary Figures S1–S4) (33,51). Our compartmental analyses are consistent with an ability of PAX3–FOXO1 to bind both active and inactive chromatin across a wide variety of chromatin structural domains (33,51). The different intensity profiles of MYOD and PAX3–FOXO1 in compartment A suggest that the master regulatory factors PAX3–FOXO1 and MYOD have distinct chromatin structural roles in RMS, as opposed to overlapping functions (Supplementary Figure S6).
Identification of subtype-specific compartments: PAX3–FOXO1 is associated with the opening of heterochromatin in FP-RMS, while H3K27ac is associated with maintaining compartment A status
We hypothesized that compartments that have distinct classifications between FP and FN cell lines would be salient when comparing across RMS subtypes. To investigate the similar (concordant) and dissimilar (discordant) compartments in each major subtype, we defined the subtype-specific compartments in FP-RMS and FN-RMS cell lines. Overall, we found that over 83% of the genome in each RMS cell model had been assigned a compartment, and 55% of all compartments are consistent in all four RMS cell lines. Seventy-two percent of FP-RMS compartments were consistent within the two cell lines (Rh4 and Rh30), while FN-RMS cell lines (RD and SMS-CTR) showed a higher level of consistency at 82% (Figure 1K and Supplementary Figure S7). In terms of the subtype-specific compartments, concordant A compartments (A–A) constituted 30% of all compartments, while concordant B compartments (B–B) accounted for another 25% of all compartments. Discordant compartments were defined as A–B (compartment A in FP and compartment B in FN) and B–A (compartment B in FP and compartment A in FN). Even though discordant compartments constituted a relatively small proportion of all compartments (6%), we did observe some stereotypic patterns of compartment discordance. Subtype-specific compartmentalization was especially prevalent on chromosomes 19 and 22 (18% and 17%, respectively), while chromosomes 17, 21 and 1 had more compartmental similarity across subtypes (74%, 73% and 72%, respectively; Figure 1K and Supplementary Figure S7, blue = compartment A, black = compartment B).
We next proceeded to examine whether chromatin regulatory factors were associated with discordant compartments. We examined the prevalence of histone acetylation at A–B, B–A, A–A and B–B regions. We found that H3K27ac has the highest concentration in A–A compartments. Sixty-six percent of FP and 76% of FN A–A regions have at least one H3K27ac site. In comparison, only 6% of FP and 9% of FN B–B regions have H3K27ac (Figure 1L). The stark contrast suggests that regions with consistent compartment assignments might share similar structures and functions between subtypes. In discordant compartments, H3K27ac deposition is higher in the A compartment, which is consistent with the definition. Overall, MYOD shows a similar profile to H3K27ac, both in percentage of compartments occupied and in the general trend of preferential binding within A compartments. This is consistent with MYOD signal intensity (Figure 1G) tracking closely to that of H3K27ac (Figure 1I). However, MYOD does occupy more B–B compartments and fewer A–A compartments than H3K27ac. Moreover, we find that at MYOD sites with B-to-A switching from FN to FP are associated with loci encoding WNT signaling molecules (Supplementary Table S1). PAX3–FOXO1 is less commonly observed in A–A and A–B compartments, mostly because PAX3–FOXO1 has much fewer FP-consensus binding events throughout the genome. While H3K27ac and MYOD are more common in A–A compartments, PAX3–FOXO1’s presence in both compartments is almost equal, showing a higher affinity to regions that changed from B to A. This is an extension of our findings that PAX3–FOXO1 has pioneer function in FP-RMS cells (33,51).
Analyses of TADs in RMS
Human genomes contain structures that are highly self-interacting and self-insulating (22,23). These structures, with early observations from Heard, Ren and co-workers, span tens of thousands of base pairs to a few Mb, and are known as TADs. To form TADs, CTCF and cohesin function together via a loop extrusion mechanism (46), with cohesin facilitating the loop extrusion and CTCF functioning as an insulator to block cohesin processivity (19,45,57,58). Interestingly, when CTCF is conditionally deleted, TADs are lost, while large-scale compartments persist, suggesting a decoupling of shorter-range and longer-range chromatin organization (57). There is evidence that TADs are generally conserved across mouse and human chromatin and conserved across tissues within human cell lines (19). However, we hypothesized that identification of subtype-specific TADs for RMS would allow us to conceptually and mechanistically connect chromatin structure and gene expression regulation in the tumor. Thus, we studied the chromatin domain structure of FP-RMS and FN-RMS, with a focus on the differential TADs between the subtypes.
We observed that TAD assignments are very sensitive to many parameters, among which include resolution of bins and size of the moving window, which has been seen by us and others, and remains a key area in bioinformatic research (59,60). For our analysis, we used 50 kb bins and a moving window of 10-fold on each side, resulting in median TAD size of ∼1 Mb (Figure 2A and B). We uncovered 1860, 1920, 2019 and 2059 TADs for Rh4, Rh30, RD and SMS-CTR cell lines, respectively. Most of the TADs are consistent within all four cell lines and have CTCF binding events at both TAD boundaries. For example, the TAD at chr2:216.75–217.75 Mb is present in all four RMS cell lines, demarcated with strong CTCF signal (Figure 2C). Even though the strengths of the TADs differ slightly across cell lines, we observe the signature ‘dot structures’ formed by the two TAD-defining CTCF binding sites at the boundaries in all four RMS cell lines (45,57,61). We also observe the distinct stripe pattern that may be evidence of cohesin extrusion in action (62). The sub-TAD strengths of CTCF-demarcated loop domains are relatively weaker in the RD cell line than in others, but sub-TAD boundaries at around 217.20 and 217.50 Mb are both visible, as exemplified in a highly representative TAD structure (Figure 2C).
Figure 2.

TADs in RMS. (A, B) Subtype-specific TADs are shown on chromosome 1 for Rh4, Rh30, RD and SMS-CTR with a colorimetric interaction intensity scale (shown, left) and subtypes (shown, right). (C) CTCF-bounded loop domains are shown with interaction intensity plotted in blue above CTCF ChIP-seq data for each cell line in green. Looping interactions are highlighted in yellow with arcs connecting the loop anchors on the ChIP-seq plots. (D) The most highly enriched motifs (top sequences, left; top TFs, right) at TAD boundaries are indicated for Rh4, Rh30, RD and SMS-CTR. (E) Tumor-specific genes associated with differential TADs in our RMS cell models are indicated.
PAX3–FOXO1 is not associated with defined TAD boundaries
We sought to determine whether there exists association between PAX3–FOXO1 and the genomic loci encoding boundaries. Based on recent studies in the muscle lineage (63) illustrating MYOD localization at loop anchors, we hypothesized that MYOD, a critical core regulatory factor essential for myogenic cell fate determination, would be localized to TAD boundaries. We found only moderate associations of the canonical E-box motif localized to these boundaries and there was no evidence of MYOD preferentially binding to TAD boundaries (permutation test; P-value ≤0.67). To our surprise, there was no evidence that PAX3–FOXO1 binding events were significantly enriched at the TAD boundaries (permutation test; P-value ≤0.99), compared to CTCF binding (P-value <10−5). The most highly enriched motif associated with TAD boundaries was CTCF (adjusted P-value <10−4). Both P53 and YY1 motifs were enriched within our datasets but not significantly (Figure 2D). Thus, we find evidence for CTCF as instructive for TAD maintenance in FN-RMS and FP-RMS, as well as the conservation of key motifs, including YY1 for TAD domain boundaries across subtypes. We attribute these findings to a fundamental distinction between loops and TADs. While some loop domains are synonymous with TADs, many can be differently defined based on the fundamental properties of the class of cis-chromatin interaction (19,45). In comparison, CTCF binding sites are highly concentrated at TAD boundaries (permutation test, P-value <10−5; Figure 2D). These results are in accordance with previous reports in other tissues where associations between CTCF/cohesin were found at boundaries (19,45,62,64). Understanding mechanisms of PAX3–FOXO1 and MYOD for shorter-range looping events will be of high interest in future studies. Mechanistically, it is interesting that the functions of these essential tumor driver TFs may exist at the larger compartment level, while being more context-specific or gene-specific for TAD structures.
To investigate whether TADs are associated with gene expression patterns in RMS, we analyzed 303 differentially called TADs between FP-RMS and FN-RMS cell lines (FDR < 0.01, FC > |log2(1.5)|). Some TADs that display distinct patterns between subtypes are located close to genes associated with tumorigenesis, such as RBFOXO1, ORC3, TCF4, PTN and NRG1 (Figure 2E and Supplementary Figure S8). We observed RBFOXO1 showing stronger gene body and downstream interactions in FP-RMS than in FN-RMS cell lines. For ORC3, there exist stronger interactions between upstream regions and the gene body in FP-RMS than in FN-RMS cell lines. In our compartment analysis, we also observed that the genes that are involved in A–B or B–A switching mostly encompassed a neurogenesis signature (128 genes) by GSEA analysis, while a small subset included genes involved in myogenesis (11 genes, Supplementary Table S2). Importantly, a majority of differential TADs between FP-RMS and FN-RMS were associated with genes involved in neurogenesis. Our findings provide a unique chromatin structural context for studies that find that FP-RMS tumors can express genes involved in neurogenesis (65,66).
Genome integrity and RMS: CNV, SV and classical fusions—pervasive copy number changes in RMS cell lines
SVs include genomic alterations such as somatic mutations, genome duplication, genome deletion and genomic translocation events. Recent studies have revealed that SVs and chromosomal imbalances are prevalent in cancer (25,67,68), and specifically in childhood tumors (69–71). For example, there is evidence that SVs occur in diffuse intrinsic pontine glioma, leukemias and lymphomas, and are relatively enriched in childhood sarcomas (29,69–74). In RMS, certain CNVs, including MYCN amplifications (2), have been associated with chemotherapy resistance and poor patient prognoses. Interestingly, it has been reported that FN-RMS genomes go through more structural alterations compared to FP-FMS genomes, while FP-RMS often undergoes genome duplication events in patients (27,29).
We applied neoloopfinder (39) to reconstruct copy number profiles from our Hi-C data for our FP-RMS (Rh4 and Rh30) and FN-RMS (RD and SMS-CTR) cell lines. We found pervasive copy number changes in all four RMS cell lines regardless of fusion status (Figure 3A–E and Supplementary Figure S9). The Rh30 genome has the highest percentage of altered copy number, as only 46.51% of the Rh30 genome remained copy number neutral. The SMS-CTR cell line has the next highest level of altered copy number, with 48.31% of the genome remaining copy number neutral. Rh4 and RD cell lines are slightly more stable, with 54.86% and 55.42% of the genome being copy number neutral, respectively. At the same time, Rh30 has slightly more single copy loss than the other cell lines, with 30.54% of the genome estimated to have CN = 1 (Figure 3F–I). Strikingly, PAX3–FOXO1 and MYOD both localize to CN-enriched loci in Rh4 cells, suggesting that primary and essential tumor drivers can interact with neo-scaffolding elements occurring through genome instability (Figure 3L and M). It is notable that CNV in RMS is associated with the PAX3–FOXO1 locus and also provides scaffolding for PAX3–FOXO1 binding (Figure 3F, G and M, and Supplementary Table S3). We hypothesize that this reveals a mechanism where CNV both enhances tumor-essential gene dosage and provides scaffolding for essential TFs to localize (Figure 3J–M). It was noteworthy that MYCN copy number is elevated in Rh30 cells, potentially connecting high MYCN expression with structural alterations (Figure 3G and Supplementary Table S3). We observed striking elevations in CNV at the loci encoding the cell cycle regulator CDK4 (Supplementary Table S3), which is associated with more severe patient outcomes in FP-RMS (2). Even though copy number profiles differ from cell line to cell line, there exist deletions that are common to both FN and FP cell lines, such as the deletion at the start of chr22 (Figure 3E) and amplification at the start of chr14 (Supplementary Figure S9). Our work has revealed key patterns of SV and CNV in each major subtype, which provides new evidence that events leading up to but not limited to the canonical fusion events in RMS may be critical for sarcomagenesis. We anticipate that CNV calls from our deep 3D genomic sequencing will continue to serve key roles in connecting chromatin architectural organization and structural alterations in human cancer.
Figure 3.

CNVs and SVs from Hi-C. CNV is indicated with colorimetric scale for Rh4, Rh30, RD and SMS-CTR for chromosome 1 (A), chromosome 5 (B), chromosome 12 (C), chromosome 19 (D) and chromosome 22 (E). On the scale, black = deletion/loss, blue = single copy, beige = two copies and red = three or more copies. The distributions of CNV gains and losses are shown for Rh4 (F), Rh30 (G), RD (H) and SMS-CTR (I) as a function of percent coverage across the genome on the y-axis. Fusion status for each cell line is indicated beneath the plot. The association of chromatin regulatory factors and histone marks with CNV in Rh4 cells is shown as a function of deposition of H3K27ac (J), H3K9me3 (K), MYOD binding (L) or PAX3–FOXO1 binding (M). Heatmaps detailing the translocation at PAX3 (chr2) and FOXO1 (chr13) loci in Rh4 FP-RMS cells (N). Recurrent patterns of SV along chromosome 8 are present with increased interaction frequencies between 71.8 and 127.6 Mb for FN-RMS PDXs, SJCRH13758_X2, SJCRHB0011_X and SJCRHB013_X (O), which is lacking in the FN-RMS cell lines RD and SMS-CTR (P).
The PAX3–FOXO1 fusion gene has different structures in different cell lines
We next asked whether SV identification from Hi-C could illuminate structural attributes in the locus encoding the major tumor driver in FP-RMS. Another type of SV is genome reconstruction resulting from a break in the linear chromosome fiber. A failure to repair the break could result in a translocation event, a deletion or an inversion [reviewed in (25)]. In FP-RMS, SVs lead to fusion genes such as PAX3–FOXO1 and PAX7–FOXO1, which are essential in FP-RMS but poorly understood at the molecular level. We asked the following questions: (i) whether the PAX3–FOXO1 fusion loci in Rh4 and Rh30 cell lines are structurally the same and (ii) whether there are structural breaks common to FN cell lines. Using bioinformatic tools, we were able to reconstruct and visualize PAX3–FOXO1 fusion locus along the altered genome (24,39). As one would expect, the chromosomal break is visible in Rh4 and Rh30 cell lines but absent in RD and SMS-CTR cell lines (Figure 3N and Supplementary Figure S10). The SV event in Rh30 cell line is a translocation between TTS of PAX3 and TSS of FOXO1. This is consistent with the interaction heatmap showing a symmetric radiating pattern originates at chr2:222.2 Mb and chr13:40.6 Mb (Supplementary Figure S11 and Figure 4A). The SV event resulting in PAX3–FOXO1 in the Rh4 cell line, however, is more complex. The SV involves an insertion of the PAX3 gene (chr2:222.2–222.3 Mb, anti-sense) between chr13:40.6 Mb and chr13:43.27 Mb, producing the two strip-like hotspots on the heatmap (Figure 3N). However, the square to the top left of the break site also suggests an inversion at chr2:222.2 Mb, just downstream of PAX3, and chr13:39.5 Mb. Furthermore, there are also long-range interactions of chr3 between 41.25 and 77.00 Mb, as well as interchromosomal interaction between chr2:222.0 Mb and chr13:77.50 Mb (Figure 3N and Supplementary Figure S11). The interactive architecture of chr2 and chr13 in Rh4 cell line is not well defined, as multiple SVs could exist from independent fusion or amplification events.
Figure 4.

Functional requirements for TADs. (A) Hi-C data corresponding to the reconstructed PAX3–FOXO1 TAD region is shown (top); the PAX3 TAD region is shown for Rh30 (middle) and SMS-CTR (bottom) at chromosome 2. The solid black lines delineate TADs in each case. (B) ChIP-seq for CTCF, SNAI2 and H3K27ac at the PAX3 locus in SMS-CTR cells. CRISPR guide RNAs targeting the TAD at this locus are indicated with Guides 1–4. (C) DepMap CRISPR score for RMS cell lines; values below −1 indicate essentiality of PAX3. (D) Genomic PCR of the CRISPR/Cas9 targeted region in control and PAX3 TAD boundary element deleted cells. (E) Expression of PAX3, FOXO1, PAX3–FOXO1 (P3F) and GAPDH control in Rh30 and SMS-CTR cells, with SNAI2–CTCF peak at PAX3 TAD boundary deleted (PAX3 TAD boundary element KO). (F) Clonal PCR analysis of isolated clones 1–7 with wild-type control, and primers spanning the CRISPR targeted region. (G) RT-qPCR RNA expression analysis of the CT75 gene, which is adjacent to the PAX3 gene (blue = control; red = CRISPR knockout of the PAX3 TAD boundary element). (H) Cell growth analysis of PAX3 TAD boundary element deleted compared to control Rh30 and SMS-CTR cells. (I) Colony assay of PAX3 TAD boundary element deleted cells compared to control Rh30 and SMS-CTR cells. (J) Expression of myogenic differentiation factors in PAX3 TAD deleted compared to control in Rh30 and SMS-CTR cells. (K) Confocal images of control and PAX3TAD boundary element deleted cells after 3 days of differentiation; cells were stained for DAPI (blue), differentiated myosin MyHC (green) and MEF2C (red). (L) Quantification of immunostaining from PAX3 TAD deleted cells compared to control cells grown in differentiation media.
We also studied TADs on the reconstructed Rh4 and Rh30 genomes (neo-TADs) by calculating directionality index (22) on the reconstructed genome. In both Rh4 and Rh30 cell lines, the PAX3–FOXO1 fusion gene is located at the inside of a neo-TAD (Figures 3N and 4A). In the Rh4 cell line, the TAD spans chr13:39.6–40.6 Mb, chr2:222.2–222.3 Mb and then chr13:43.27–43.9 Mb, with CTCF binding sites flanking both ends of the TAD and pointing to the interior of the TAD. We have discovered that convergent CTCF sites flanking the PAX3–FOXO1 neo-TAD are in agreement with the high degree of CTCF-motif convergence flanking TADs across mammalian chromatin architectural systems (19). In the Rh30 cell line, the neo-TAD spans 39.6–40.6 Mb on chr13, and then 222.2–222.7 Mb, with evidence suggesting a sub-TAD from chr13:40.0 Mb to chr2:222.7 Mb (Figure 4A). Thus, despite the common genetic drivers in FP-RMS, the events leading to the SV encoding the fusion alleles have occurred through distinct rearrangement mechanisms. We extended our analyses of SV into PDXs, obtained from the Childhood Solid Tumor Network (St Jude Children’s Research Hospital) (75), from the FN-RMS subtype to understand the context for frequent SV events reported in this subtype (29). We investigated the SVs in PDXs from FN-RMS patients obtained from St Jude Research Hospital (SJRHB013758_X2, SJRHB0011_X and SJRHB013_X). Using the same algorithms for SV detection, we found a structural break located on chr8 in all three PDX samples, near the MSC gene, while the same break is not conserved in RD or SMS-CTR cell lines (Figure 3O and P). Thus, high-depth Hi-C in RMS has revealed unique patterns of SV in each cell line or PDX, while some of these SV events produce convergent gene fusions, and others produce divergent structural and architectural features.
TAD structure upstream of PAX3 is essential to PAX3–FOXO1 expression
We identified chromatin contact domain structures near the PAX3 locus, which we hypothesized are essential to our FP-RMS cell lines (Figures 3N and 4A). PAX3 is an important regulator of stemness in the muscle (76,77). In FP-RMS the PAX3–FOXO1 oncogene is a driver of this disease (6,7), while a role for PAX3 in FN-RMS remains to be defined. We hypothesized that these conserved domain structures had intrinsic functional significance for RMS tumor maintenance. We noted that there are distinct contact domains at the PAX3 locus in Rh30 and SMS-CTR cells, while a neo-TAD is formed at a similar region at the PAX3–FOXO1 locus of Rh30 cells (Figure 4A). We also detected overlapping CTCF and SNAI2 binding peaks at the 5′ PAX3 TAD boundary region (Figure 4B). We found that a subset of SNAI2 binding peaks in FN-RMS, SMS-CTR and RD cells, overlapped with CTCF at predicted TAD boundaries. We hypothesized that this CTCF/SNAI2 overlapping site was important in defining the TAD boundary and may also be important for maintaining PAX3 expression in RMS cells (Figure 4B). We next asked whether PAX3 served essential roles in RMS and noted a differential requirement for expression of PAX3 in DepMap (78) in FP-RMS cells and not in FN-RMS cells (Figure 4C). However, it is challenging to distinguish whether the DepMap guide RNAs are able to target endogenous PAX3 versus the PAX3–FOXO1 fusion gene. We have previously observed that PAX3–FOXO1 is an essential gene in Rh4, while the loss of the fusion gene has lesser effects on Rh30 (79). Thus, to investigate the functional roles of chromatin domains for essentiality of PAX3 expression, we designed CRISPR/Cas9 guides to delete the CTCF/SNAI2 co-bound site in Rh30 and SMS-CTR cells, which is proximal to the TAD boundary (referred to herein as the PAX3 TAD boundary element). This regulatory element has a previously unknown function. We used a two-guide CRISPR/Cas9 strategy and deleted a 91 bp region upstream of the PAX3 gene that is proximal to the 5′ TAD boundary in both Rh30 and SMS-CTR cells. We confirmed the deletion by genomic PCR (Figure 4D) and performed western blot assays to assess PAX3 protein expression. We observed a >90% loss of PAX3 protein when the PAX3 TAD boundary was deleted in Rh30 and SMS-CTR cells, implying that this TAD boundary element is essential for endogenous PAX3 expression (Figure 4E). We next assessed expression of the PAX3–FOXO1 fusion protein using both the PAX3 and FOXO1 antibodies. We observed that FOXO1 and PAX3–FOXO1 expression was significantly increased in Rh30 cells upon deletion of the boundary element (Figure 4E). This is consistent with previous studies showing mutual exclusivity in expression of PAX3–FOXO1 relative to PAX3 (80,81). Interestingly, in SMS-CTR cells the endogenous FOXO1 protein was also increased (Figure 4E). Since our CRISPR/Cas9 approach may lead to mosaic deletion of the TAD, we isolated single clones from the PAX3 TAD boundary element deleted Rh30 population. We demonstrate that there exist clones with complete loss of the wild-type locus. However, other clones have indels, and many of these clones lack the wild-type locus (Figure 4F). We independently ablated the PAX3 TAD boundary element in RD cells and again show >90% loss of PAX3 expression (Supplementary Figure S12). We repeated the clonal analyses and can find clones with complete loss of the wild-type TAD boundary region. Since ablation of the PAX3 5′ TAD boundary element results in loss of PAX3 expression, we next assessed the expression of CT75, a long noncoding RNA of unknown function, which is located just outside the PAX3 TAD boundary. Deletion of the PAX3 TAD boundary element resulted in a slight but not significant increase in its expression (Figure 4G). Together, our experiments define the PAX3 5′ TAD boundary element, which is bound by CTCF and is required for PAX3 expression.
We next assessed the functional consequences of the loss of the PAX3 TAD boundary element. We performed growth assays and found that while Rh30 cells are viable, they have a deficit in growth compared to control cells, while in SMS-CTR cell growth was unaffected (Figure 4H). In colony forming assays, we saw a similar trend, where the PAX3 TAD boundary was essential for growth of Rh30 but not SMS-CTR cells (Figure 4I). Lastly, we assessed the effect of the loss of PAX3 on myogenic differentiation. In Rh30 cells, loss of PAX3 expression, through deletion of its TAD boundary element, resulted in an increase in MYOD, MYOG, MEF2C and differentiated myosin (MyHC expression, Figure 4J), while SMS-CTR cells did not show an induction of myogenic differentiation regulators. This effect was also observed in cells grown in myogenic differentiation media, where Rh30 cells with the PAX3 TAD boundary element deleted showed increased myogenic differentiation (Figure 4K and L), while SMS-CTR cells did not show any increase in myogenic differentiation. Together, we demonstrate that our identification of 3D chromatin structures at the PAX3 locus reveals essential components of its transcriptional and oncogenic regulation in RMS. Furthermore, by selectively targeting the PAX3 TAD we can selectively deplete PAX3 and not PAX3–FOXO1 expression, thus illuminating roles of PAX3 in the tumor.
DISCUSSION
We report the genome structure of RMS in each major subtype. In our Hi-C studies, we observe common and distinct architectural and epigenetic events across FN-RMS and FP-RMS subtypes. We anticipate further advances in this area as the epigenetics and genome structure associated with the infrequent PAX3–NCOA and PAX3–INO80 fusions in RMS are elucidated and compared with the PAX3–FOXO1 and PAX7–FOXO1 fusion drivers (4). In our studies, we identify unique structural elements underlying large-scale compartmental structures, TADs and SVs. In the cases of compartments, we find tumor-driving TFs highly associated with each class of compartment category (A and B). In our analyses of TADs, we find a surprising lack of co-occurrence of MYOD and PAX3–FOXO1 binding sites flanking these structural elements, while our evidence supports that the CTCF ‘rule’ of flanking convergent motifs applies strongly in each major subtype of the tumor. Considering CNV, we find evidence from our Hi-C data that gene loci encoding CDK4, MYCN and PAX3–FOXO1 are amplified suggesting that multiple pathways may reinforce the genetics driving tumorigenesis (2,27).
It is interesting to note that SVs are not shared between cell lines, even within a subtype. We did not detect any subtype-specific SVs that are common to both FN-RMS cell lines. In FP-RMS cell lines, the only common SV we detected is the PAX3–FOXO1 fusion locus. These results motivated the following question: What chromatin domains are functionally driving in FP-RMS? To further evaluate this question, and the structural elements within the PAX3–FOXO1 locus in Rh30, we performed CRISPR deletion with guide RNAs targeting CTCF and SNAI2 co-bound sites within the neo-TAD (Figure 4A and B). In these experiments, we found that PAX3 expression was highly dependent on the CTCF/SNAI2 sites at the TAD boundary (Figure 4C and D). The effects of PAX3 loss in these cells were minor decrease in proliferation and significant induction of myogenic differentiation in FP-RMS (Figure 4F–H). Thus, our chromatin structural studies have helped us define structure–function relationships in FP-RMS, within the PAX3 TAD. Further studies will be important to understand other ‘functional’ TADs in RMS, within each major subtype, as previous studies have shown a decoupling of TAD structure and RNA Pol II activity (45).
Interestingly, we and others (29) have identified many SV events in the FN subtype. Given the prevalence of SV in FN-RMS, and the new data indicating lack of clinical severity of RAS mutations in this subtype (2), we hypothesize that there may be fusion genes in FN-RMS cell lines. For example, our evidence suggests that there is excessive interaction between CGGBP1 on chr3 and ATP8A2 on chr13 in SMS-CTR cell line (Supplementary Figure S13). Long-read RNA sequencing and high content imaging (26,82) will be efficacious in further studies of potential fusion driver events of ERMS to expand the scope of known alterations in this tumor. Moreover, future studies will be impactful for domain-specific normalizations in the context of spike-in chromatin (Supplementary Figure S14).
To the best of our knowledge, our study defines chromatin architecture in RMS at high depth for the first time. Since there are no precision therapies yet reported for this tumor, more precise mechanistic and structural understanding of chromatin initiation and maintenance in RMS may reveal new possibilities for precision medicine. It is notable that the structural features we have identified are also functional features, reinforcing tumor growth at several key levels. Understanding the chromatin structural context for new driver genes as they emerge along with transcriptional mechanisms along the linear chromatin fiber will be impactful in the community of researchers and clinicians seeking to understand RMS etiology.
DATA AVAILABILITY
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO series accession number GSE215203 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE215203). Previously published SRA datasets used in the study were ChIP-seq and RNA-seq in RMS (GSE83728). A source data file accompanies this manuscript. The remaining data are available within the article and Supplementary Data, or available from the authors upon request. Source code for data analyses can be found at https://github.com/mxw010/RMS-HiC.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Amy Wetzel, Huachun Zhong, Shireen Woodiga and the Institute for Genomic Medicine sequencing facility at Nationwide Children’s Hospital for sequencing expertise and guidance. We would like to thank Dr Peter Houghton for deriving the RMS cell lines used in this study. We thank Dr Åsa Karlström and the Childhood Solid Tumor Network at St Jude Children’s Research Hospital for providing RMS PDX samples (data portal: cstn.stjude.cloud), and we thank Dr Ruoning Wang for providing mouse muscle tissue for methods development. We thank our colleagues at Nationwide Children’s Hospital and the OSU epigenetics community, UT Health, San Antonio, and NCI-CCR for conversations that were helpful in developing this work.
Contributor Information
Meng Wang, Nationwide Children’s Hospital, Center for Childhood Cancer, Columbus, OH 43205, USA.
Prethish Sreenivas, Greehey Children’s Cancer Research Institute, Department of Molecular Medicine, University of Texas Health Science Center, San Antonio, TX 78229, USA.
Benjamin D Sunkel, Nationwide Children’s Hospital, Center for Childhood Cancer, Columbus, OH 43205, USA.
Long Wang, Greehey Children’s Cancer Research Institute, Department of Molecular Medicine, University of Texas Health Science Center, San Antonio, TX 78229, USA.
Myron Ignatius, Greehey Children’s Cancer Research Institute, Department of Molecular Medicine, University of Texas Health Science Center, San Antonio, TX 78229, USA.
Benjamin Z Stanton, Nationwide Children’s Hospital, Center for Childhood Cancer, Columbus, OH 43205, USA; Department of Pediatrics, The Ohio State University College of Medicine, Columbus, OH 43205, USA; Department of Biological Chemistry and Pharmacology, The Ohio State University College of Medicine, Columbus, OH 43210, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
St. Baldrick’s Foundation (to B.Z.S.); CancerFree Kids Foundation (to B.Z.S.); Cancer Prevention and Research Institute of Texas (CPRIT) [RR160062 to M.I.]; Andrew McDonough B+ Foundation (to B.Z.S.); Mark Foundation for Cancer Research (to B.Z.S.); Nationwide Children’s Hospital (to B.Z.S.); National Cancer Institute [R00CA175184 to M.I.]; Max and Minnie Tomerlin Voelcker Fund (to M.I.).
Conflict of interest statement. The authors declare no competing interests.
REFERENCES
- 1. Dehner C.A., Armstrong A.E., Yohe M., Shern J.F., Hirbe A.C. Genetic characterization, current model systems and prognostic stratification in PAX fusion-negative vs. PAX fusion-positive rhabdomyosarcoma. Genes (Basel). 2021; 12:1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shern J.F., Selfe J., Izquierdo E., Patidar R., Chou H.C., Song Y.K., Yohe M.E., Sindiri S., Wei J., Wen X. et al. Genomic classification and clinical outcome in rhabdomyosarcoma: a report from an international consortium. J. Clin. Oncol. 2021; 39:2859–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shern J.F., Chen L., Chmielecki J., Wei J.S., Patidar R., Rosenberg M., Ambrogio L., Auclair D., Wang J., Song Y.K. et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014; 4:216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yohe M.E., Heske C.M., Stewart E., Adamson P.C., Ahmed N., Antonescu C.R., Chen E., Collins N., Ehrlich A., Galindo R.L. et al. Insights into pediatric rhabdomyosarcoma research: challenges and goals. Pediatr. Blood Cancer. 2019; 66:e27869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pandey P.R., Chatterjee B., Olanich M.E., Khan J., Miettinen M.M., Hewitt S.M., Barr F.G. PAX3–FOXO1 is essential for tumour initiation and maintenance but not recurrence in a human myoblast model of rhabdomyosarcoma. J. Pathol. 2017; 241:626–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Galili N., Davis R.J., Fredericks W.J., Mukhopadhyay S., Rauscher F.J. 3rd, Emanuel B.S., Rovera G., Barr F.G. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993; 5:230–235. [DOI] [PubMed] [Google Scholar]
- 7. Barr F.G., Galili N., Holick J., Biegel J.A., Rovera G., Emanuel B.S. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993; 3:113–117. [DOI] [PubMed] [Google Scholar]
- 8. Yohe M.E., Gryder B.E., Shern J.F., Song Y.K., Chou H.C., Sindiri S., Mendoza A., Patidar R., Zhang X., Guha R. et al. MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma. Sci. Transl. Med. 2018; 10:eaan4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oristian K.M., Crose L.E.S., Kuprasertkul N., Bentley R.C., Lin Y.T., Williams N., Kirsch D.G., Linardic C.M. Loss of MST/Hippo signaling in a genetically engineered mouse model of fusion-positive rhabdomyosarcoma accelerates tumorigenesis. Cancer Res. 2018; 78:5513–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tremblay A.M., Missiaglia E., Galli G.G., Hettmer S., Urcia R., Carrara M., Judson R.N., Thway K., Nadal G., Selfe J.L. et al. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell. 2014; 26:273–287. [DOI] [PubMed] [Google Scholar]
- 11. Ignatius M.S., Hayes M.N., Lobbardi R., Chen E.Y., McCarthy K.M., Sreenivas P., Motala Z., Durbin A.D., Molodtsov A., Reeder S. et al. The NOTCH1/SNAIL1/MEF2C pathway regulates growth and self-renewal in embryonal rhabdomyosarcoma. Cell Rep. 2017; 19:2304–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tapscott S.J., Thayer M.J., Weintraub H. Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science. 1993; 259:1450–1453. [DOI] [PubMed] [Google Scholar]
- 13. Pomella S., Sreenivas P., Gryder B.E., Wang L., Milewski D., Cassandri M., Baxi K., Hensch N.R., Carcarino E., Song Y. et al. Interaction between SNAI2 and MYOD enhances oncogenesis and suppresses differentiation in fusion negative rhabdomyosarcoma. Nat. Commun. 2021; 12:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gryder B.E., Pomella S., Sayers C., Wu X.S., Song Y., Chiarella A.M., Bagchi S., Chou H.C., Sinniah R.S., Walton A. et al. Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat. Genet. 2019; 51:1714–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Agaram N.P., Chen C.L., Zhang L., LaQuaglia M.P., Wexler L., Antonescu C.R. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for a common pathogenesis. Genes Chromosomes Cancer. 2014; 53:779–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kohsaka S., Shukla N., Ameur N., Ito T., Ng C.K., Wang L., Lim D., Marchetti A., Viale A., Pirun M. et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K–AKT pathway mutations. Nat. Genet. 2014; 46:595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Skapek S.X., Ferrari A., Gupta A.A., Lupo P.J., Butler E., Shipley J., Barr F.G., Hawkins D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Primers. 2019; 5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arndt C.A. Risk stratification of rhabdomyosarcoma: a moving target. Am Soc. Clin. Oncol. Educ. Book. 2013; 33:415–419. [DOI] [PubMed] [Google Scholar]
- 19. Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spracklin G., Abdennur N., Imakaev M., Chowdhury N., Pradhan S., Mirny L.A., Dekker J. Diverse silent chromatin states modulate genome compartmentalization and loop extrusion barriers. Nat. Struct. Mol. Biol. 2023; 30:38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N.L., Meisig J., Sedat J. et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012; 485:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dixon J.R., Xu J., Dileep V., Zhan Y., Song F., Le V.T., Yardimci G.G., Chakraborty A., Bann D.V., Wang Y. et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 2018; 50:1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Spielmann M., Lupianez D.G., Mundlos S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018; 19:453–467. [DOI] [PubMed] [Google Scholar]
- 26. Wang M., Sunkel B.D., Ray W.C., Stanton B.Z. Chromatin structure in cancer. BMC Mol. Cell Biol. 2022; 23:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen L., Shern J.F., Wei J.S., Yohe M.E., Song Y.K., Hurd L., Liao H., Catchpoole D., Skapek S.X., Barr F.G. et al. Clonality and evolutionary history of rhabdomyosarcoma. PLoS Genet. 2015; 11:e1005075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vicente-Garcia C., Villarejo-Balcells B., Irastorza-Azcarate I., Naranjo S., Acemel R.D., Tena J.J., Rigby P.W.J., Devos D.P., Gomez-Skarmeta J.L., Carvajal J.J. Regulatory landscape fusion in rhabdomyosarcoma through interactions between the PAX3 promoter and FOXO1 regulatory elements. Genome Biol. 2017; 18:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen X., Stewart E., Shelat A.A., Qu C., Bahrami A., Hatley M., Wu G., Bradley C., McEvoy J., Pappo A. et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell. 2013; 24:710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gryder B.E., Wachtel M., Chang K., El Demerdash O., Aboreden N.G., Mohammed W., Ewert W., Pomella S., Rota R., Wei J.S. et al. Miswired enhancer logic drives a cancer of the muscle lineage. iScience. 2020; 23:101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gryder B.E., Khan J., Stanton B.Z. Measurement of differential chromatin interactions with absolute quantification of architecture (AQuA-HiChIP). Nat. Protoc. 2020; 15:1209–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mumbach M.R., Rubin A.J., Flynn R.A., Dai C., Khavari P.A., Greenleaf W.J., Chang H.Y. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods. 2016; 13:919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sunkel B.D., Wang M., LaHaye S., Kelly B.J., Fitch J.R., Barr F.G., White P., Stanton B.Z. Evidence of pioneer factor activity of an oncogenic fusion transcription factor. iScience. 2021; 24:102867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Houtgast E.J., Sima V.M., Bertels K., Al-Ars Z. Hardware acceleration of BWA-MEM genomic short read mapping for longer read lengths. Comput. Biol. Chem. 2018; 75:54–64. [DOI] [PubMed] [Google Scholar]
- 36. Open2C Abdennur N., Fudenberg G., Flyamer I.M., Galitsyna A.A., Goloborodko A., Imakaev M., Venev S.V. Pairtools: from sequencing data to chromosome contacts. 2023; bioRxiv doi:15 February 2023, preprint: not peer reviewed 10.1101/2023.02.13.528389. [DOI] [PMC free article] [PubMed]
- 37. Shin H., Shi Y., Dai C., Tjong H., Gong K., Alber F., Zhou X.J. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 2016; 44:e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lun A.T., Smyth G.K. diffHic: a Bioconductor package to detect differential genomic interactions in Hi-C data. BMC Bioinformatics. 2015; 16:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang X., Xu J., Zhang B., Hou Y., Song F., Lyu H., Yue F. Genome-wide detection of enhancer-hijacking events from chromatin interaction data in rearranged genomes. Nat. Methods. 2021; 18:661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wolff J., Rabbani L., Gilsbach R., Richard G., Manke T., Backofen R., Gruning B.A. Galaxy HiCExplorer 3: a web server for reproducible Hi-C, capture Hi-C and single-cell Hi-C data analysis, quality control and visualization. Nucleic Acids Res. 2020; 48:W177–W184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Robinson J.T., Turner D., Durand N.C., Thorvaldsdottir H., Mesirov J.P., Aiden E.L. Juicebox.js provides a cloud-based visualization system for Hi-C data. Cell Syst. 2018; 6:256–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hitz B.C., Lee J.W., Jolanki O., Kagda M.S., Graham K., Sud P., Gabdank I., Seth Strattan J., Sloan C.A., Dreszer T. et al. The ENCODE uniform analysis pipelines. 2023; bioRxiv doi:06 April 2023, preprint: not peer reviewed 10.1101/2023.04.04.535623. [DOI]
- 43. Ramirez F., Dundar F., Diehl S., Gruning B.A., Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014; 42:W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kerpedjiev P., Abdennur N., Lekschas F., McCallum C., Dinkla K., Strobelt H., Luber J.M., Ouellette S.B., Azhir A., Kumar N. et al. HiGlass: web-based visual exploration and analysis of genome interaction maps. Genome Biol. 2018; 19:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rao S.S.P., Huang S.C., Glenn St Hilaire B., Engreitz J.M., Perez E.M., Kieffer-Kwon K.R., Sanborn A.L., Johnstone S.E., Bascom G.D., Bochkov I.D. et al. Cohesin loss eliminates all loop domains. Cell. 2017; 171:305–320.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R., Dekker J., Mirny L.A. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods. 2012; 9:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guelen L., Pagie L., Brasset E., Meuleman W., Faza M.B., Talhout W., Eussen B.H., de Klein A., Wessels L., de Laat W. et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008; 453:948–951. [DOI] [PubMed] [Google Scholar]
- 48. Popova L.V., Nagarajan P., Lovejoy C.M., Sunkel B.D., Gardner M.L., Wang M., Freitas M.A., Stanton B.Z., Parthun M.R. Epigenetic regulation of nuclear lamina-associated heterochromatin by HAT1 and the acetylation of newly synthesized histones. Nucleic Acids Res. 2021; 49:12136–12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nichols M.H., Corces V.G. Principles of 3D compartmentalization of the human genome. Cell Rep. 2021; 35:109330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dixon J.R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J.E., Lee A.Y., Ye Z., Kim A., Rajagopal N., Xie W. et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015; 518:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sunkel B.D., Stanton B.Z. Pioneer factors in development and cancer. iScience. 2021; 24:103132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Molina O., Vargiu G., Abad M.A., Zhiteneva A., Jeyaprakash A.A., Masumoto H., Kouprina N., Larionov V., Earnshaw W.C. Epigenetic engineering reveals a balance between histone modifications and transcription in kinetochore maintenance. Nat. Commun. 2016; 7:13334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Marques J.G., Gryder B.E., Pavlovic B., Chung Y., Ngo Q.A., Frommelt F., Gstaiger M., Song Y., Benischke K., Laubscher D. et al. NuRD subunit CHD4 regulates super-enhancer accessibility in rhabdomyosarcoma and represents a general tumor dependency. eLife. 2020; 9:e54993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Reinberg D., Vales L.D. Chromatin domains rich in inheritance. Science. 2018; 361:33–34. [DOI] [PubMed] [Google Scholar]
- 55. Schwarzer W., Abdennur N., Goloborodko A., Pekowska A., Fudenberg G., Loe-Mie Y., Fonseca N.A., Huber W., Haering C.H., Mirny L. et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature. 2017; 551:51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Akgol Oksuz B., Yang L., Abraham S., Venev S.V., Krietenstein N., Parsi K.M., Ozadam H., Oomen M.E., Nand A., Mao H. et al. Systematic evaluation of chromosome conformation capture assays. Nat. Methods. 2021; 18:1046–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nora E.P., Goloborodko A., Valton A.L., Gibcus J.H., Uebersohn A., Abdennur N., Dekker J., Mirny L.A., Bruneau B.G. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017; 169:930–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fudenberg G., Imakaev M., Lu C., Goloborodko A., Abdennur N., Mirny L.A. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016; 15:2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sefer E. A comparison of topologically associating domain callers over mammals at high resolution. BMC Bioinformatics. 2022; 23:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dali R., Blanchette M. A critical assessment of topologically associating domain prediction tools. Nucleic Acids Res. 2017; 45:2994–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rowley M.J., Nichols M.H., Lyu X., Ando-Kuri M., Rivera I.S.M., Hermetz K., Wang P., Ruan Y., Corces V.G. Evolutionarily conserved principles predict 3D chromatin organization. Mol. Cell. 2017; 67:837–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vian L., Pekowska A., Rao S.S.P., Kieffer-Kwon K.R., Jung S., Baranello L., Huang S.C., El Khattabi L., Dose M., Pruett N. et al. The energetics and physiological impact of cohesin extrusion. Cell. 2018; 173:1165–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang R., Chen F., Chen Q., Wan X., Shi M., Chen A.K., Ma Z., Li G., Wang M., Ying Y. et al. MyoD is a 3D genome structure organizer for muscle cell identity. Nat. Commun. 2022; 13:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bell A.C., Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000; 405:482–485. [DOI] [PubMed] [Google Scholar]
- 65. Wei Y., Qin Q., Yan C., Hayes M.N., Garcia S.P., Xi H., Do D., Jin A.H., Eng T.C., McCarthy K.M. et al. Single-cell analysis and functional characterization uncover the stem cell hierarchies and developmental origins of rhabdomyosarcoma. Nat. Cancer. 2022; 3:961–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Raghavan S.S., Mooney K.L., Folpe A.L., Charville G.W. OLIG2 is a marker of the fusion protein-driven neurodevelopmental transcriptional signature in alveolar rhabdomyosarcoma. Hum. Pathol. 2019; 91:77–85. [DOI] [PubMed] [Google Scholar]
- 67. Lupianez D.G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J.M., Laxova R. et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene–enhancer interactions. Cell. 2015; 161:1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vasudevan A., Schukken K.M., Sausville E.L., Girish V., Adebambo O.A., Sheltzer J.M. Aneuploidy as a promoter and suppressor of malignant growth. Nat. Rev. Cancer. 2021; 21:89–103. [DOI] [PubMed] [Google Scholar]
- 69. Dubois F.P.B., Shapira O., Greenwald N.F., Zack T., Wala J., Tsai J.W., Crane A., Baguette A., Hadjadj D., Harutyunyan A.S. et al. Structural variants shape driver combinations and outcomes in pediatric high-grade glioma. Nat. Cancer. 2022; 3:994–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Drews R.M., Hernando B., Tarabichi M., Haase K., Lesluyes T., Smith P.S., Morrill Gavarro L., Couturier D.L., Liu L., Schneider M. et al. A pan-cancer compendium of chromosomal instability. Nature. 2022; 606:976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Steele C.D., Abbasi A., Islam S.M.A., Bowes A.L., Khandekar A., Haase K., Hames-Fathi S., Ajayi D., Verfaillie A., Dhami P. et al. Signatures of copy number alterations in human cancer. Nature. 2022; 606:984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen X., Bahrami A., Pappo A., Easton J., Dalton J., Hedlund E., Ellison D., Shurtleff S., Wu G., Wei L. et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014; 7:104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Montefiori L.E., Bendig S., Gu Z., Chen X., Polonen P., Ma X., Murison A., Zeng A., Garcia-Prat L., Dickerson K. et al. Enhancer hijacking drives oncogenic BCL11B expression in lineage-ambiguous stem cell leukemia. Cancer Discov. 2021; 11:2846–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sungalee S., Liu Y., Lambuta R.A., Katanayeva N., Donaldson Collier M., Tavernari D., Roulland S., Ciriello G., Oricchio E. Histone acetylation dynamics modulates chromatin conformation and allele-specific interactions at oncogenic loci. Nat. Genet. 2021; 53:650–662. [DOI] [PubMed] [Google Scholar]
- 75. Stewart E., Federico S.M., Chen X., Shelat A.A., Bradley C., Gordon B., Karlstrom A., Twarog N.R., Clay M.R., Bahrami A. et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature. 2017; 549:96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Scaramozza A., Park D., Kollu S., Beerman I., Sun X., Rossi D.J., Lin C.P., Scadden D.T., Crist C., Brack A.S. Lineage tracing reveals a subset of reserve muscle stem cells capable of clonal expansion under stress. Cell Stem Cell. 2019; 24:944–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Der Vartanian A., Quetin M., Michineau S., Aurade F., Hayashi S., Dubois C., Rocancourt D., Drayton-Libotte B., Szegedi A., Buckingham M. et al. PAX3 confers functional heterogeneity in skeletal muscle stem cell responses to environmental stress. Cell Stem Cell. 2019; 24:958–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tsherniak A., Vazquez F., Montgomery P.G., Weir B.A., Kryukov G., Cowley G.S., Gill S., Harrington W.F., Pantel S., Krill-Burger J.M. et al. Defining a cancer dependency map. Cell. 2017; 170:564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Laubscher D., Gryder B.E., Sunkel B.D., Andresson T., Wachtel M., Das S., Roschitzki B., Wolski W., Wu X.S., Chou H.C. et al. BAF complexes drive proliferation and block myogenic differentiation in fusion-positive rhabdomyosarcoma. Nat. Commun. 2021; 12:6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Davis R.J., Barr F.G. Fusion genes resulting from alternative chromosomal translocations are overexpressed by gene-specific mechanisms in alveolar rhabdomyosarcoma. Proc. Natl Acad. Sci. U.S.A. 1997; 94:8047–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Keller C., Hansen M.S., Coffin C.M., Capecchi M.R. Pax3:Fkhr interferes with embryonic Pax3 and Pax7 function: implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev. 2004; 18:2608–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bintu B., Mateo L.J., Su J.H., Sinnott-Armstrong N.A., Parker M., Kinrot S., Yamaya K., Boettiger A.N., Zhuang X. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science. 2018; 362:eaau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO series accession number GSE215203 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE215203). Previously published SRA datasets used in the study were ChIP-seq and RNA-seq in RMS (GSE83728). A source data file accompanies this manuscript. The remaining data are available within the article and Supplementary Data, or available from the authors upon request. Source code for data analyses can be found at https://github.com/mxw010/RMS-HiC.