


Abstract
The newt hammerhead ribozyme is transcribed from Satellite 2 DNA, which consists of tandemly repeated units of 330 bp. However, different transcripts are synthesized in different tissues. In all somatic tissues and in testes, dimeric and multimeric RNA transcripts are generated which, to some extent, self-cleave into monomers at the hammerhead domain. In ovaries, primarily a distinct monomeric unit is formed by transcription, which retains an intact hammerhead self-cleavage site. The ovarian monomeric RNA associates to form a 12S complex with proteins that are poorly characterised so far. In this work we identified NORA, a protein that binds the ovarian form of the newt ribozyme. We show that the newt ribozyme binds to the Escherichia coli-expressed protein, as well as to a protein of identical size that is found exclusively in newt ovaries. Also NORA mRNA was detectable only in ovary, but in neither somatic tissues nor testes. The tissue-specific expression of NORA is analogous to the ovary-specific transcription of the newt ribozyme. Although NORA was identified by its ability to bind to the newt ribozyme in the presence of a vast excess of carrier RNA, it was able to interact with certain other RNA probes. This novel RNA-binding protein does not contain any motif characteristic for RNA-binding proteins or any other known protein domain, but it shares a striking similarity with a rat resiniferatoxin-binding protein.
INTRODUCTION
The hammerhead ribozyme is characterised by a short sequence motif composed of three helices that are connected by three short conserved regions. RNA cis-cleavage is induced at a defined position of the catalytic domain (reviewed in 1). Hammerhead ribozymes have been found naturally amongst some subviral plant pathogens (some satellite RNAs and three exceptional viroids), which undergo self-cleavage during their replication cycle [reviewed by Symons (2)]. Besides these small RNA replicons, the hammerhead domain is also found in some RNA molecules in the newt (3,4) where it serves an unknown function. Recently, hammerhead-containing transcripts of a satellite DNA have also been found in the trematode Schistosoma (5).
The newt ribozyme is coded by highly repetitive DNA sequences known as Satellite 2 (Sat2). Transcription is done by RNA polymerase II and controlled by a proximal sequence element and a distal sequence element, similarly to snRNAs (6,7). In every newt tissue examined, a monomeric strand-specific Sat2 transcript of ∼330 nt could be detected as well as multiple forms of it (3). However, there is a different distribution of monomers to multimers among various tissues. The ovary contains mostly monomers, whereas somatic tissues and testes accumulate predominantly dimers and larger multimers. Dimer-sized Sat2 synthetic transcripts undergo site-specific self-cleavage in vitro (4). The termini of the non-ovarian monomeric RNA transcripts coincide with those produced by the in vitro self-cleavage reaction. In contrast to this, the monomeric RNAs found in the oocytes map 47 nt upstream of the in vitro self-cleavage site (8). As a result, each monomeric ovarian transcript retains an intact catalytic domain that could trans-cleave an unknown RNA target. It was indeed shown that the ovarian newt ribozyme can cleave an artificial RNA substrate in trans under some experimental conditions (6,9). This finding suggested that the ovarian newt hammerhead ribozyme may be involved in RNA processing events and that it could be part of a ribonucleoprotein (RNP) complex (6). Along this line, it has been recently shown that the newt ribozyme is in fact present as an RNP particle of about 12S in the oocytes of the newt Triturus (10). Reconstitution experiments and gel-shift analyses have shown that a complex is formed in vitro on the monomeric ribozyme molecules. UV cross-linking studies have identified in the newt ovary a 40 kDa protein associated to the newt ribozyme with high affinity. A similarly sized protein with high affinity for the newt ribozyme was also found in Xenopus ovary extracts. The observation that an appropriate oligoribonucleotide substrate could be specifically cleaved in newt ovary extracts (10) supports the hypothesis of involvement of an RNP containing the newt ribozyme in a tissue-specific RNA processing event. It appears that the ribonucleolytic activity of the ribozyme–protein complex is much higher than the in vitro activity of the synthetic ribozyme. This suggested that the protein components of the ribozyme RNP might help the endoribonuclease activity, perhaps by chaperoning the newt ribozyme into a functional conformation. Examples of protein-associated ribozymes whose activity is enhanced by the presence of the cognate protein are common among other classes of ribozymes (RNAse P, introns of group I and II, HDV ribozyme) (11–13), but so far the newt hammerhead ribozyme is the only ribozyme of its class that is known to require association with proteins.
Such observations make it likely that the newt ribozyme is biologically significant but shed little light on the process(es) in which it is involved. To address the possible biological function(s) of the newt ribozyme we sought to identify and characterise the ovarian protein components that are part of the newt ribozyme RNP. Since the starting material (newt ovaries) was scarce, we could not apply classical biochemical purification schemes for the identification of proteins. Instead, we decided to use the recently developed RNA-ligand screening procedure of Sägesser et al. (14) to identify cDNA clones from an expression library that express such proteins. By using this methodology we detected a protein that binds to the newt ribozyme. This novel RNA-binding protein, which we termed NORA (for newt ovary ribozyme-associated protein), was in vitro expressed, its exclusive presence in the ovary was assessed by northern and western assays and its RNA-binding properties were characterised by northwestern and gel shift analyses.
MATERIALS AND METHODS
The sequence of NORA cDNA has been submitted to the DDBJ/EMBL/GenBank database under accession no. AJ010793.
RNA-ligand screening
The cDNA library used in this work had been constructed in λZAPII (Stratagene) using poly(A)+ RNA isolated from ovary of the newt Triturus carnifex. For preparation of the 32P-labelled RNA probe, the plasmid pGEM-AluI (6) was linearised with HindIII and used as template for in vitro transcription with T7 RNA polymerase, as described recently (15). This delivered a 330 nt RNA called ‘Sat2’, which is similar to the monomeric newt ribozyme found in ovary tissue. The screening procedure was performed according to Sägesser et al. (14) with slight modifications. Unlike the original protocol, the screening was performed with a buffer (SB) containing 25 mM HEPES, pH 7.9, and lacking Mn2+ and Zn2+. The amount of 32P-labelled RNA probe used was 0.1–0.4 × 106 c.p.m./ml. Yeast RNA (RNA type VI, torula yeast; Sigma-Aldrich, Germany) was added to SB buffer at a final concentration of 0.1 mg/ml for blocking of non-specific RNA-binding proteins during pre-hybridisation and hybridisation. The same conditions were applied for plaque lift assays during secondary and tertiary screening.
The phage clone λZ-NORA, which could be identified by screening, was subjected to in vivo excision procedure according to the protocol supplied by Stratagene. This delivered plasmid pNORA, which is based on the pBluescript SK(–) vector, containing an insert between the EcoRI and XhoI sites. For in vitro synthesis of sense- and antisense-specific RNAs, pNORA was linearised with XhoI and transcribed with T3 RNA polymerase or cut with EcoRI and transcribed with T7 RNA polymerase, respectively.
DNA sequence analysis
The DNA sequence was obtained by the dideoxy sequencing method modified for a double-stranded DNA template using a Sequenase® 2.0 kit (Amersham) or by MediGene GmbH (Martinsried, Germany). DNA sequence analysis was performed with the programs of the Wisconsin Package. Comparisons of two nucleotide or amino acid sequences were performed with the program PILEUP. Comparisons of a query sequence and either a protein or a DNA database were performed with the programs FASTA, TFASTA, BLASTP, BLASTX and TBLASTN (16,17).
Animals, RNA isolation and tissue extract preparation
Triturus carnifex newts were collected near Pisa (Italy) and were anaesthetised in 0.1% tricane-methane sulfonate (MS222; Sigma-Aldrich) before removing the tissues. RNAs were extracted from ovary and several other tissues by homogenisation in guanidinium isothiocyanate, followed by iso-propanol precipitation, as described by Chomczynski and Sacchi (18). Poly(A)+ RNA purification was carried out with a DYNABEADS® mRNA purification kit (DYNAL) according to the manufacturer’s instructions. Total tissue extracts were prepared as described (10).
Rapid amplification of cDNA ends (RACE) cloning of the 5′ end of NORA and construction of a full-length cDNA clone
RACE was used to identify the 5′ end of the mRNA (19) employing a 5′ RACE System kit (Gibco BRL) according to the instructions provided by the manufacturer. Briefly, T.carnifex ovary poly(A)+ RNA was prepared as described above and was reverse transcribed using SuperScript II RNAse H-RT and a specific 20mer primer (5′-CCAATATTCCAAGCTCTGAT-3′) located 190 bp from the 5′ end of NORA. The resulting single-stranded cDNA was purified trough a GlassMax DNA Isolation Spin Cartridge (Gibco BRL) and tailed at its 3′ end with cytidine nucleotides by terminal deoxynucleotidyltransferase. A specific 22mer primer (5′-CAGAGCCCCAGATATTGTTCAT-3′) located 138 bp from the 5′ end of NORA was used for subsequent rounds of amplification by PCR, together with the AAP primer (Gibco BRL). The DNA was extended with Taq polymerase (Gibco BRL) in a MiniCycler (MJ Research) using 35 cycles of amplification (30 s at 94°C; 30 s at 55°C; 1 min at 72°C) followed by a final extension of 7 min at 72°C. A subsequent nested amplification was performed using AUAP primer (Gibco BRL) and the 22mer primer. PCR products were cut with PvuII and SpeI restriction enzymes and cloned into the EcoRV/SpeI sites of pBluescript SK(+) (Stratagene). The clone pNORAfull was constructed by joining the EcoRI/PvuII fragment of the longest clone obtained by RACE, containing the first 340 nt of NORA cDNA, to the PvuII/XhoI fragment of pNORA, and by cloning the fragment into EcoRI/XhoI-cut pBluescript SK (+) (Stratagene).
Northern blotting
RNA samples were separated on 1% agarose/formaldehyde gels (20). The RNA was blotted from the gel on a GeneScreen Membrane (DuPont NEN®) by capillary transfer overnight at room temperature with 20× SSC. The membrane was cross-linked in a Stratalinker® UV cross-linker (Stratagene). Pre-hybridisation and hybridisation with pNORA antisense probe were carried out at 65°C as described (20).
In vitro translation
For in vitro translation pHisNORAfull DNA was linearised with BamHI and transcribed with T7 RNA polymerase. The capped RNA was incubated in Flexi Rabbit Reticulocyte Lysate (Promega, 50 µl reaction, as described by the manufacturer) for 90 min, together with 20 mCi [35S]methionine (Amersham). To visualise the protein product, the reactions were analysed by SDS–PAGE followed by autoradiography.
Protein expression and purification
The pHis vector used for the production of fusion proteins is a modification of vector pET3a (21) that includes six histidine residues following the initiator methionine. To produce pHis-NORA, a 1464 bp SmaI/XhoI fragment from pNORA was gel-isolated, treated with Klenow fragment to blunt XhoI protruding ends and sub-cloned into the HpaI site of the pHis vector.
To create pHisNORAfull, pNORAfull was cut with RsaI and the 1380 bp fragment containing NORA open reading frame (ORF) was cloned into SmaI-cut pHis.
The clones were checked by DNA sequencing for the correct frame and orientation and were then transformed into Escherichia coli BL21(DE3), which is the appropriate host strain for expression of pET vectors (21). Transformed E.coli BL21(DE3) cells were grown from overnight pre-cultures in LB medium supplemented with ampicillin (100 µg/ml) at 37°C to OD600 = 0.8. Cells were induced with 0.4 mM IPTG and grown for an additional 2 h.
His-tagged proteins were purified with Ni2+-NTA–agarose resin (Qiagen) under native conditions using batch methods. Briefly, cell pellets from 2 l of cells were thawed at room temperature and resuspended in 25 ml of lysis buffer (50 mM Tris pH 8.0, 100 mM NaCl, 5 mM β-mercaptoethanol, 5% glycerol) containing 1.0 mM imidazole. Cells were lysed by sonication and cell debris were removed from the cell lysate by centrifugation at 18 000 g for 30 min at 4°C. Cleared lysate was bound to 5 ml of Ni2+-NTA–agarose resin in a total volume of 35 ml by gentle rocking for 30 min at 4°C. The resin was washed twice with lysis buffer containing 1.0 mM imidazole and twice with lysis buffer containing 10.0 mM imidazole. Elution was carried out by two rounds of rocking the washed resin at 4°C for 20 min in 20 ml of lysis buffer containing 100 mM imidazole. Eluted proteins were dialysed against 10 mM Tris–Cl, 100 mM NaCl pH 8.0 and concentrated to ∼3 mg/ml in Centriprep 30 concentrators (Amicon). Purified His-NORA protein (1 mg) was injected in rabbits for the production of NORA-specific antibodies.
Western blot analysis
Proteins were fractionated by SDS–PAGE and transferred to Protran BA nitrocellulose membrane (Schleicher and Schuell) in 25 mM Tris, 200 mM glycine, 20% methanol at 30 V overnight at 4°C. For the analysis with Penta-His antibody (Qiagen), the membrane was blocked in buffer 1 (10 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% Tween-20) containing 3% BSA at room temperature for 1 h and incubated with the antibody diluted (1:2000) in buffer 1 containing 1% BSA. Following extensive washings in buffer 1, the membrane was incubated with goat anti-mouse IgG peroxidase-conjugate (Jackson ImmunoResearch Laboratories; 1:5000 in buffer 1 containing 1% BSA) washed again and detected with DAB chromogen (Sigma). Western analysis with the NORA-specific antibody was performed by blocking the membrane in buffer 2 (10 mM Tris–HCl pH 8.0, 250 mM NaCl, 0.1% Tween-20, 5% fat-free milk), and incubating with NORA-antibody (1:5000 in buffer 2). After washes in buffer 2 lacking milk, goat anti-rabbit IgG alkaline peroxidase-conjugate (Promega) diluted (1:20 000) in buffer 2 was added. After incubation for 1 h and extensive washes, detection was performed with BCIP and NBT chromogens (Promega) in 100 mM Tris–HCl pH 8.8, 5 mM MgCl2, 100 mM NaCl.
Northwestern blot analysis
Labelled RNA transcripts were produced by inclusion of [α-32P]UTP (Amersham) in the reaction, as described (15). The Sat2Δ probe is a 145 nt transcript synthesised from NdeI-linearised pGEM-AluI (6) by T7 RNA polymerase. BS probe is a T3-polymerase 98 nt transcript of XbaI-linearised pBluescript II SK (+). U1 corresponds to the T7-polymerase 197 nt transcript of HindIII-linearised plasmid PU1 (22). Gem3 probe (269 nt) was transcribed from a PvuII-cut pGEM3Z vector (Promega) by T7 RNA polymerase.
Bacterial pellets were treated as described (14). Protein samples were resolved on 12% SDS–polyacrylamide gels and transferred to Immobilon nitrocellulose membranes (Millipore). Pre-blocking and probing were carried out according to Sägesser et al. (14) using 0.1 × 106 c.p.m./ml 32P-labelled riboprobe and SB buffer containing yeast blocking RNA at 0.1 mg/ml (described above). Washes were performed with SB buffer devoid of yeast RNA.
Gel mobility shift assays
For analysis of complex formation, His-NORA protein was incubated in binding buffer (10 mM Hepes–KOH pH 7.9, 50 mM KCl, 0.1 mM EDTA, 4% glycerol) for 1 h at 25°C (final volume 10 µl), together with 32P-labelled RNA. The RNA probes were heated at 90°C and snap cooled prior to the reaction, to allow proper folding. The amounts of protein and RNA used varied in the different experiments and are indicated in the text and in the legends to the figures. In supershift experiments His-NORA protein was incubated together with 32P-labelled Sat2Δ RNA in binding buffer containing RNAse inhibitor (1 U/µl), to allow complex formation. After 15 min, NORA-specific immune serum was added to a final dilution 1:500 and the samples were incubated for a further 1 h. Samples were separated on 5 or 6% polyacrylamide gels (acrylamide:bisacrylamide ratio 29:1) containing 0.5× Tris–borate–EDTA run for 3 h at 25°C with constant voltage of 200 V. The dried gels were scannned and quantified using a PhosphorImager (Molecular Dynamics) and the program ImageQuant.
RESULTS
Identification of a newt ribozyme-binding protein
For the current study we used a cDNA expression library that had been prepared from newt ovary mRNA and that had been cloned in bacteriophage λZAPII (Stratagene). This library was subjected to RNA-ligand screening (14) with an in vitro synthesised 32P-labelled RNA probe (Sat2) corresponding to the ovarian monomeric newt ribozyme. A total of about 2 × 105 plaques were tested for binding of the RNA probe in a primary screening. In this way, we identified an original plaque with phages that expressed a protein with the potential to bind to the newt ribozyme. Since the plaques were confluent during the original screening, phage progeny of this plaque was used to single out the positive phage clone in a second screening round. A secondary screening is shown in Figure 1A. To confirm the homogeneity of this phage clone, progeny of the secondary screening was subjected to a further plaque lift assay, either alone or mixed with phages of the cDNA library (Fig. 1B–D). All plaques originating from phages of the secondary screening resulted in a uniquely intense hybridisation signal. The phage clone was termed λZ-NORA.
Figure 1.
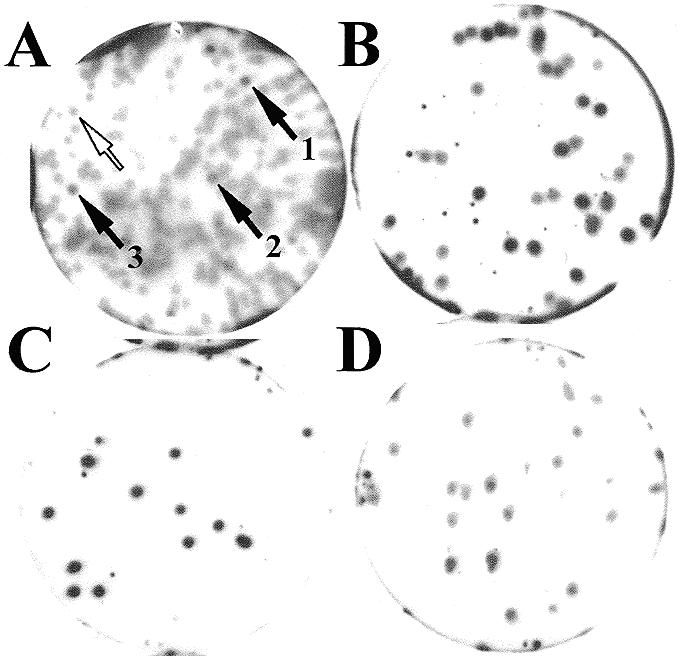
Secondary (A) and tertiary screening (B–D) of λZ-NORA clone. (A) Clones picked from the primary screening were plated at ∼200 p.f.u./plate. The black arrows show the plaques corresponding to three clones of λZ-NORA, which were subsequently found to be identical. The empty arrow shows an example of non-ribozyme-binding (negative) plaque. Clone λZ-NORA picked from secondary screening was plated pure (∼15 p.f.u./plate) (C) or mixed with ∼20 p.f.u. from the λ ZAPII cDNA library (B), as a negative control. The same amount of phages from λ ZAPII library was also plated alone (D). Plaques were induced with IPTG and transferred to filters, as described in Materials and Methods. Binding was performed with 32P-labelled Sat2 probe (2 × 105 c.p.m./ml) in SB buffer containing 0.1 mg/ml yeast RNA. Where a mixture had been plated, the ratio of positive:negative plaques, which could be discriminated by their signal intensity, was as expected.
Sequence analysis of NORA
λZ-NORA was rescued into pBluescript SK(–), yielding plasmid pNORA whose sequencing revealed a 1440 bp cDNA insert. Since neither a start codon nor a 5′-untranslated region (5′-UTR) was detectable, it was clear that pNORA did not contain the full-length cDNA. In accordance with this, northern blot analysis also suggested a longer mRNA of ∼1.7 kb (Fig. 2).
Figure 2.
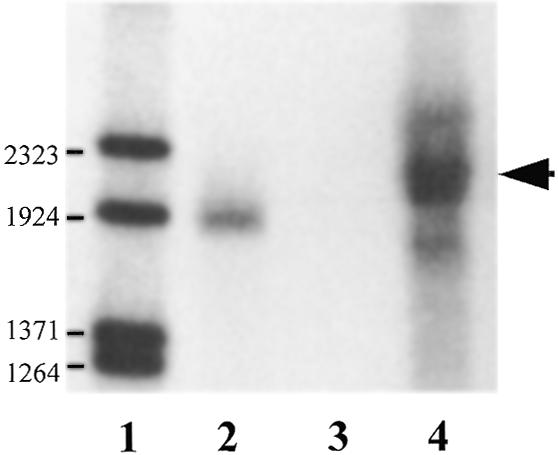
Northern blot analysis of NORA mRNA in T.carnifex ovary. RNA samples were fractionated in a 0.8% agarose/formaldehyde gel. The blot was hybridised with 32P-labelled NORA antisense probe. Lane 1, lambda DNA/BstEII end-labelled molecular weight marker; lane 2, NORA sense cold transcript (1530 nt); lane 3, NORA antisense cold transcript; lane 4, 15 µg total RNA from T.carnifex ovary. Numbers on the left refer to the size of the DNA fragments. The arrow on the right points to the signal corresponding to NORA mRNA.
In order to obtain a full-length cDNA clone of NORA we used a RACE protocol. Several clones of different lengths were obtained and sequenced. This allowed deduction the entire 1767 nt sequence of NORA cDNA. By joining the longest of the clones obtained by 5′ RACE to the insert in pNORA, a plasmid containing the full-length cDNA was obtained and named pNORAfull.
NORA sequence contains a 5′-UTR of 131 bp, an ORF of 1197 bp and a 3′-UTR of 428 bp, followed by a poly(A) stretch. The 3′-UTR sequence has a single polyadenylation signal, AATAAA, which is located 15 nt upstream of the 3′ poly(A) stretch (23). The ORF translates into 399 amino acids, starting at position 132 and ending with a stop codon at position 1329. The predicted molecular weight of the NORA protein is 45 kDa and its pI is 5.12.
No known consensus protein motif, including RNA-binding domains, was found in NORA amino acid sequence by any of the standard computer programs tested. However, the protein contains a variety of consensus phosphorylation sites (Table 1) and a bipartite nuclear localisation signal in position 248–265 (KKDLISKVVQTIGKKKA) (24).
Table 1. Consensus phosphorylation sites in NORA protein.
| Phosphorylation sites | First residue number |
|---|---|
| cAMP- and cGMP-dependent protein kinase | 287 |
| Casein kinase II | 10, 12, 64, 68, 70 |
| Protein kinase C | 39, 55, 58, 286, 324 |
| Tyrosine kinase | 145 |
Comparison of amino acid sequence: similarity with a previously described rat protein
A database search revealed significant similarity with a 26 kDa rat protein, called resiniferatoxin-binding protein (RBP-26). The 1526 bp cDNA clone encoding RBP-26 had been isolated from a rat neonatal dorsal root ganglion library (25) (EMBL nucleotide sequence database accession no. X67877). The function of RBP-26 is not known but the similarity to NORA is high, both at the nucleic acid level (68% on a 1052 bp region) as well as at the amino acid level (73% similarity, 56% identity over a 249 amino acid region). In addition to the similarity with RBP-26, homologous short cDNA sequences were also found in cDNA libraries from human liver and breast tumour cells, from mouse cells (embryonic carcinoma, 9.5 days embryo posterior region, melanoma and T cells) and zebrafish 0 day fin regenerates.
A main difference between NORA and its homologous protein in rat lies in their size. While the ORF of RBP-26 stops already after 230 amino acids, the ORF in NORA continues for 154 additional amino acids. However, the DNA sequences of NORA and RBP-26 show a high degree of similarity also in the putative 3′-UTR of RBP-26, so we assume that the attributed difference is most likely due to a sequence inaccuracy in the RBP-26 cDNA. In line with this assumption, the 3′-UTR of RBP-26 contains an ORF (from nucleotide 719 to 1182) with a very high degree of similarity with the region of the NORA protein from amino acid 175 to 333 (85% similarity, 68% identity). Thus, a single extra nucleotide in the RBP-26 sequence would cause a frame shift so that the coding region of RBP-26 would be extended for an additional 444 nt. The putative new RBP-protein of 385 amino acids would then have 79% similarity and 63% identity with the NORA protein (Fig. 3).
Figure 3.

Multiple alignment of deduced amino acid sequence of NORA protein and of a RBP protein of 385 amino acids obtained by joining ORF 31–733 with ORF 700–1182 of RBP-26 cDNA. Numbers on the right refer to the position of the last residue of a line. The alignment was performed with the program PILEUP. This algorithm introduces gaps to maximise the similarities and these are denoted by dots. Amino acids identical to NORA sequence are denoted by dashes and those amino acids which differ from NORA are indicated by single lowercase letters.
NORA protein expression
In order to confirm the size of the encoded translation product, a plasmid was constructed which encoded NORAfull protein carrying six histidines at the N-terminus, under the control of T7 RNA polymerase promoter. The recombinant protein His-NORAfull was expressed in E.coli. Following electrophoresis of total E.coli protein extracts, the expected protein with an apparent molecular weight of 49 kDa could be detected in western blot experiments performed with an antibody recognising the histidine tag (Fig. 4A).
Figure 4.
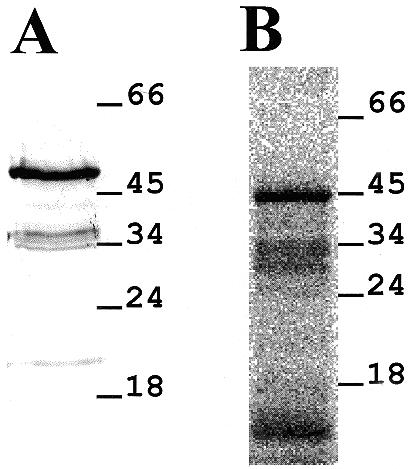
NORA protein expression. (A) Western blot analysis. pHis-NORAfull was expressed in E.coli and the total protein extract was electrophoresed on a 12% SDS–polyacrylamide gel, transferred to a nitrocellulose membrane and detected by the Penta-His antibody. Relative migration of size standards is shown on the right side of the panel. (B) In vitro translation. Rabbit reticulocyte lysates were used to translate His-NORAfull cRNA in [35S]methionine labelled proteins. A radioautograph of a 12% polyacrylamide gel is shown with molecular weights of markers in kDa shown on the right.
In addition, rabbit reticulocyte lysates were used to translate a T7 RNA polymerase transcript of pHis-NORAfull. Polyacrylamide gel electrophoresis revealed one main [35S]methionine labelled translation product of 45 kDa (Fig. 4B). Western blot assays have shown that this in vitro-translated protein does not have an N-terminal 6-His tag (data not shown), as expected if the translation initiates at the start codon proper of the NORA cDNA sequence.
This observation contrasts with the report of Ninkina et al. (25), who detected three small translation products of 23, 25 and 26 kDa in analogous experiments performed with RBP-26 mRNA, and confirms the correctness of the NORA predicted ORF.
NORA mRNA and protein are found exclusively in the ovary
To determine the expression pattern and the size of NORA mRNA, total RNA and poly(A)+ RNA were extracted from various newt tissues (ovary, testes, liver, stomach, intestine, heart, lungs, oviduct) and subjected to northern analysis. No hybridisation signal could be detected in RNA samples isolated from somatic tissues, nor from testes. However, in the RNA prepared from ovary tissue a single and abundant RNA of ∼1.7 kb could be detected (Fig. 5).
Figure 5.
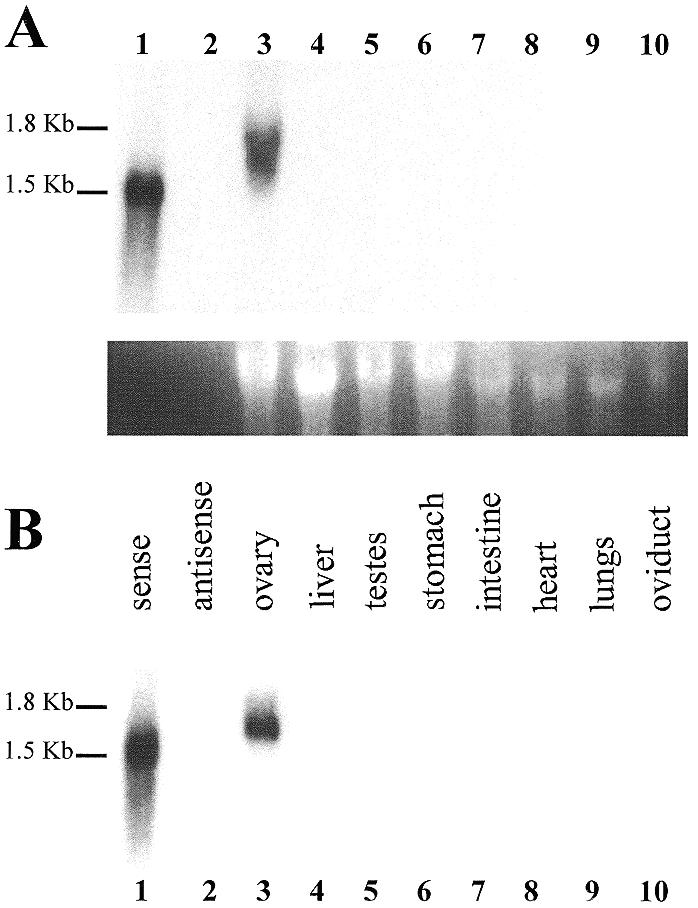
Northern blot analysis of NORA mRNA expression pattern in various newt tissues. Total RNA (A) and poly(A)+ RNA (B) samples from T.carnifex ovary, testes and somatic tissues were fractionated on 1% agarose/formaldehyde gels. The blots were hybridised with 32P-labelled NORA antisense probe. (A) Lane 1, NORA sense cold transcript (1530 nt); lane 2, NORA antisense cold transcript; lanes 3–10, total RNA (10 µg) from T.carnifex tissues. Exposure time was 3 days. Ethidium bromide staining of the 18S rRNA was used as a loading control. (B) Same as (A), except that poly(A)+ RNA extracted from 10 µg of total RNA was loaded in lanes 3–10.
To assay for the presence of NORA protein in newt tissues, immunoblot analyses were performed with a rabbit polyclonal antibody raised against purified His-NORAfull protein. Total protein extracts from eight different tissues of adult newts (ovary, testes, liver, stomach, intestine, heart, lungs, kidney) were resolved on an SDS–polyacrylamide gel and stained with Coomassie (Fig. 6A). As expected, different tissues contained different protein patterns. A gel run in parallel and subjected to immunoblot analysis revealed a band of size ∼45 kDa exclusively in the ovary extract (Fig. 6B). The size of the signal corresponds to the size of NORA protein.
Figure 6.
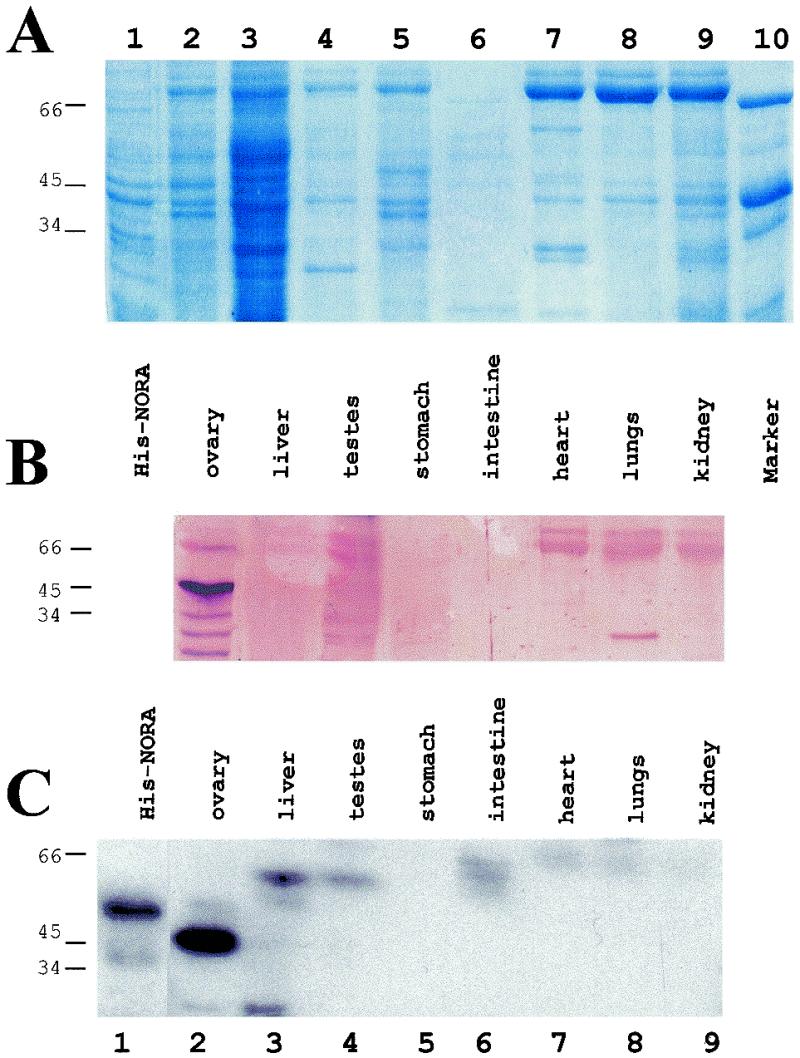
Analysis of NORA protein presence in various newt tissues. (A) Total extracts (corresponding to ∼2 mg of tissue) were separated by electrophoresis on a 12% SDS–polyacrylamide gel, which was Coomassie stained. Total protein extract from His-NORAfull expressing E.coli was loaded as positive control (lane 1). Lane 10, protein size marker. (B) A duplicate gel was run in parallel, transferred to nitrocellulose membrane and subjected to immunoblot analysis with a NORA-specific rabbit polyclonal antibody. (C) A third identical gel was transferred to nitrocellulose membrane and probed with 32P-labelled Sat2 in SB buffer containing 0.1 mg/ml yeast RNA. Exposure time was 4 h. Molecular weights of markers in kDa are shown on the left. Only in ovarian extracts could a strong signal be detected. The size of the signal was identical regardlessof whether the antibody or the RNA probe was used.
In order to test whether NORA protein was also detectable by virtue of its RNA binding, extracts from various newt tissues, including testes and ovaries, were resolved on an SDS–polyacrylamide gel as in Figure 6A and subjected to northwestern blotting analysis. Probing with a 32P-labelled Sat2 transcript revealed only a single strong signal in the ovary, while no signal could be detected in any of the other seven newt tissues examined (Fig. 6C). The signal corresponded to an ovarian protein of ∼45 kDa, exactly as NORA. As a control a crude extract from imidazole-induced E.coli cells expressing His-tagged NORAfull was loaded (Fig. 6C, lane 1). In this total extract only a protein of the approximate size of His-NORAfull (49 kDa) showed binding to the probe. When extracts from induced E.coli cells not carrying the pHis-NORAfull plasmid were run on an SDS–polyacrylamide gel and probed with labelled Sat2 RNA, no signal was detected (data not shown).
Thus, while a single band of identical size could be detected in ovary extracts, by two independent methodologies using two different probes (antibody and RNA), no such signal could be found in the residual seven other newt tissues. This is in accordance with the absence of NORA mRNA in these tissues (Fig. 5).
RNA-binding activity of NORA protein: northwestern and gel mobility shift assays
We intended to confirm and to characterise the RNA-binding capacity of the in vitro-expressed NORA protein in a northwestern assay. His-tagged NORA (40 kDa) and NORAfull (49 kDa) proteins were overexpressed in E.coli and purified. The proteins were separated on an SDS–polyacrylamide gel and transferred to a filter. After probing with 32P-labelled Sat2 RNA, a strong signal could be detected (Fig. 7A) indicating that both full-length and shorter His-tagged NORA proteins are able to bind Sat2 RNA, once immobilised on nitrocellulose filters.
Figure 7.
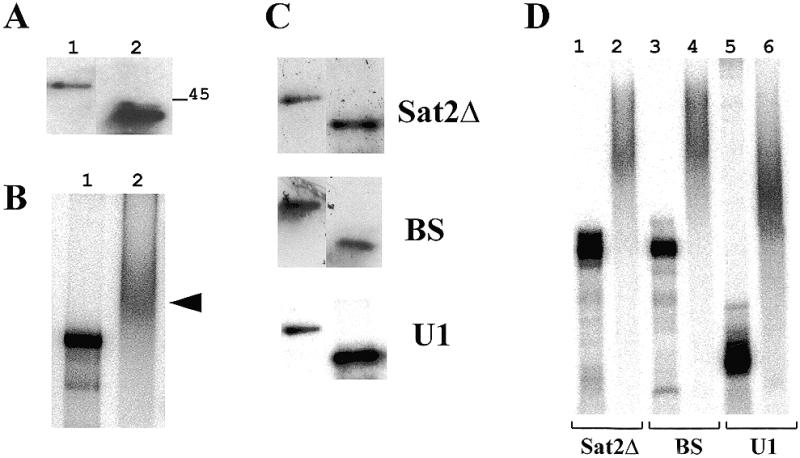
Binding of purified NORA protein to Sat2 RNA and other diverse RNA probes. (A) Northwestern blot analysis. Purified His-tagged NORAfull (lane 1) and His-NORA (lane 2) proteins (1 µg) were subjected to electrophoresis on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose membrane and probed with 32P-labelled Sat2 in SB buffer containing 0.1 mg/ml yeast RNA. Relative migration of the size standard is shown on the right of the panel. (B) Gel mobility shift analysis was performed with the radiolabelled Sat2 RNA (10 pM; 3000 c.p.m.) in the absence (lane 1) or presence (lane 2) of recombinant His-NORA protein (6 µM). The reaction was performed in SB buffer (see Materials and Methods) containing 500 ng/µl yeast RNA. Samples were separated on a 6% polyacrylamide gel. The arrow on the right indicates the shifted band. (C) Northwestern blot analysis performed as in (A), using the probes indicated on the right of the panels. (D) Gel mobility shift analysis performed with diverse RNA probes in the absence (lanes 1, 3 and 5) or presence (lanes 2, 4 and 6) of recombinant His-NORA protein, as described in (B). Lanes 1 and 2, Sat2Δ RNA; lanes 3 and 4, BS RNA; lanes 5 and 6, potato U1 RNA.
We also tested by gel mobility shift experiments the ability of the purified short His-tagged NORA protein to bind Sat2 RNA in solution. The binding of NORA protein (6 µM) to Sat2 RNA probe (10 pM; ∼3000 c.p.m.) was tested in the presence of 500 ng/µl competitor yeast RNA (estimating an average size of ∼100 nt after gel-electrophoretic analysis, this amount of RNA corresponds to a concentration of ∼15 µM). As shown in Figure 7B, also under these experimental conditions NORA was able to bind to Sat2 RNA. However, binding was also detected in northwestern experiments (Fig. 7C) and gel mobility shift assays (Fig. 7D) when various other RNA probes were used: a shorter transcript of Sat2 sequence (Sat2Δ), a transcript of the polylinker of pBluescript (BS) and a probe corresponding to potato U1 snRNA sequence (U1).
Since under the experimental conditions used for gel mobility shift assays the amount of input protein is high and the probe concentration is relatively low, the shift observed might be due to a non-specific association of proteins to the RNA. To address this issue, we performed gel mobility shift experiments in which a range of various protein concentrations was tested (1 µM–100 pM), the yeast RNA was omitted and the concentration of the RNA probe was increased to 100 pM (3000 c.p.m./sample). These experiments showed that a protein concentration of 100 nM was sufficient to observe complex formation (data not shown). In subsequent experiments, therefore, the concentration of protein was held constant at 100 nM.
Also, the binding of NORA to Sat2Δ was tested in a different set of gel mobility shift experiments, in which the probe concentration was varied between 100 pM and 500 nM (Fig. 8A). However, the total amount of labelled RNA was kept constant for all the binding reactions so that the specific activity of the RNA was varied. As shown in Figure 8A, there was a progressive decrease in the formation of the shifted complex as the molar ratio of NORA protein to Sat2Δ RNA decreased. After quantification of the signals the data were subjected to Scatchard analysis. The plot was not consistent with the results expected for a single binding site, but rather indicates a more complex situation. Therefore it was not possible to easily determine a proper Kd value for NORA.
Figure 8.
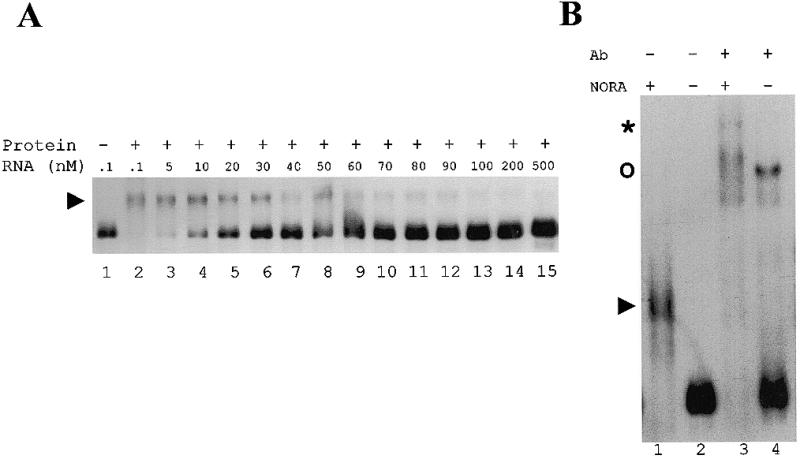
Analysis of NORA binding to Sat2Δ RNA. (A) Gel mobility shift assay. The His-NORA protein concentration was held constant at 100 nM and the RNA concentration was varied between 100 pM and 500 nM (lanes 2–15). The total amount of labelled RNA was kept constant in the binding reaction (1000 c.p.m.), but the specific activity of the RNA was varied. In lane 1 no protein was added. The complexes were separated from the unbound RNAs on a 5% polyacrylamide gel. The arrowhead on the left shows the shifted complex. (B) Antibody supershift assay. Sat2Δ RNA (100 pM) was incubated in the presence (lanes 1 and 3) or absence (lanes 2 and 4) of His-NORA protein (100 nM). After 15 min incubation at 25°C, a rabbit polyclonal NORA-specific antibody was added at a final concentration 1:500 (lanes 3 and 4). The arrowhead on the left shows the shifted complex in lane 1. The asterisk indicates the supershift in lane 3. The upper band in lane 4 (indicated by a circle) is due to a non-specific interaction of the immune serum with the RNA probe.
The presence of NORA protein in the shifted complex was confirmed by supershift experiments performed with a NORA-specific antibody (Fig. 8B).
In competition binding experiments labelled Sat2Δ RNA (100 pM) was competed against either unlabelled Sat2Δ RNA or unlabelled Gem3 RNA. In these experiments, NORA showed no difference in its affinity for the two competitor RNA sequences (not shown). In both cases the D (i.e. the concentration of competitor required to reduce binding by 50%) was measured to be ∼10 nM. When yeast RNA was used as a competitor RNA in the same conditions, however, its D was measured to be ∼1 µM (33 ng/ml). No shifted complex was observed in these conditions when 3 µM yeast RNA was present (not shown).
DISCUSSION
A cDNA expression library from newt ovaries was used to identify, via functional RNA-ligand screening (14), proteins that interact with the monomeric ovarian form of the newt hammerhead ribozyme. A cDNA clone that encoded an RNA-binding protein (subsequently called NORA) was characterised. Northern blot analysis showed that NORA mRNA is present at a high level in newt ovary, while it was not detectable in all other newt tissues examined, including testes. In accordance with these data, we could show the presence of a protein of the expected size in newt ovary tissues that was able to interact with the ovarian monomer of the newt ribozyme as well as with a NORA-specific antibody. Neither testes, nor any somatic tissues contain this protein in detectable quantities. The ovary specificity of NORA coincides with the occurrence of its RNA partner, the particular monomeric form of the newt ribozyme that still contains the functional hammerhead, which is likewise exclusively found in the ovary tissue (compare with Introduction). Moreover, the ovarian 45 kDa protein was the only strong signal detected with the Sat2 RNA probe and the antibody. In view of the tissue specificity, NORA is a promising candidate for a constituent of the RNP particle identified by Luzi et al. (10). In the same study, Luzi and colleagues identified by UV cross-linking a protein with high affinity for the ovarian-like RNA monomer. Its estimated molecular weight of ∼40 kDa is in reasonable agreement with the size of NORA.
Inspection of the NORA sequence showed that none of the consensus RNA-binding motifs identified so far (26) is present. However, it is noteworthy that the protein is rich in lysine (11.5%), which could contribute to the interaction with RNA.
Database searches showed a striking degree of similarity of the NORA protein with RBP-26 described by Ninkina et al. (25). RBP-26 had been isolated from a rat neonatal dorsal root ganglion library for its ability to bind resiniferatoxin (RTX), a naturally occurring diterpene, structurally related to phorbol esters (27). Ninkina et al. (25) isolated the RBP-26 protein while searching for an intra-membrane RTX receptor. They found that RBP-26 did indeed bind RTX, but its cytoplasmic localisation and other considerations prompted them to conclude that RBP-26 was not the RTX receptor, and that the functional role of RBP-26 remained to be established. While the significance of the dual binding activity of NORA/RBP-26 towards RNA and RTX is difficult to interpret, we think that the function of NORA could be connected to its RNA-binding capacity.
Moreover, while, in the newt, NORA mRNA was found solely in the ovary, a 1.6 kb RBP-26 mRNA was present in different tissues of newborn rats (25), in agreement with the isolation of cDNAs homologous to NORA cDNA from a variety of human and mouse tissues. Therefore, despite a very high degree of sequence conservation between the mammalian and amphibian protein, the tissue specificity seems to be completely different. However, since the tissue specificity was so far examined only for adult newt and in newly born rats, a developmental regulation of gene expression cannot be ruled out.
We analysed the RNA-binding activity of purified NORA fusion protein by northwestern assay and gel mobility shift analysis. As evident from Figure 7, in the conditions that we used NORA protein binds RNA in a non-sequence-specific manner. However, we cannot exclude a sequence-specific interaction in vivo perhaps due to the assistance of other proteins. Along this line, the size of the 12S ribonucleoprotein complex observed by Luzi et al. (10) argues in favour of a multiprotein complex assembled in vivo on Sat2 monomeric transcripts. Moreover, NORA could be identified in plaque lift assays and was able to bind Sat2 RNA in northwestern and gel shift assays, despite the excess of competitor yeast RNA. Most likely NORA recognises a structural motif that occurs in all the RNA probes tested, but less frequently in the yeast competitor RNA used. In fact, the affinity of NORA for Sat2Δ RNA measured in the competition binding assays was approximately equal to the affinity for Gem3 RNA but was 100-fold higher than for yeast RNA. Also, the tissue-specific expression argues that despite the lack of sequence specificity NORA may interact in vivo with Sat2 RNA. However, the protein could also interact with other RNAs present in the newt ovary. The availability of a NORA-specific antibody allows us to investigate this issue in greater detail in the future.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank Christian Hammann (University of Dundee, Scotland, UK), Kriton Kalantidis (IMBB-FORTH, Heraklion, Greece) and Ettore Luzi (MPI, Göttingen, Germany), who critically read the manuscript. We thank Ana M. Aransay (Faculty of Medicine, University of Crete, Heraklion, Greece) for precious advice with RACE procedure and for help with sequencing. We are grateful to Silvia Marracci (University of Pisa, Italy) for kindly providing the cDNA expression library. M.A.D. acknowledges receipt of Short Term Fellowships from the European Molecular Biology Organisation (EMBO) and from the Federation of European Biochemical Societies (FEBS).
DDBJ/EMBL/GenBank accession no. AJ010793
REFERENCES
- 1.Birikh K.R., Heaton,P.A. and Eckstein,F. (1997) Eur. J. Biochem., 245, 1–16. [DOI] [PubMed] [Google Scholar]
- 2.Symons R.H. (1997) Nucleic Acids Res., 25, 2683–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Epstein L.M., Mahon,K.A. and Gall,J.G. (1986) J. Cell Biol., 103, 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Epstein L.M. and Gall,J.G. (1987) Cell, 48, 535–543. [DOI] [PubMed] [Google Scholar]
- 5.Ferbeyre G., Smith,J.M. and Cedergren,R. (1998) Mol. Cell. Biol., 18, 3880–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cremisi F., Scarabino,D., Carluccio,M.A., Salvadori,P. and Barsacchi,G. (1992) Proc. Natl Acad. Sci. USA, 89, 1651–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coats S.R., Zhang,Y. and Epstein,L.M. (1994) Nucleic Acids Res., 22, 4697–4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Epstein L.M. and Coats,S.R. (1991) Gene, 107, 213–218. [DOI] [PubMed] [Google Scholar]
- 9.Marusic L., Luzi,E., Barsacchi,G. and Eckstein,F. (1997) Eur. J. Biochem., 247, 397–401. [DOI] [PubMed] [Google Scholar]
- 10.Luzi E., Eckstein,F. and Barsacchi,G. (1997) Proc. Natl Acad. Sci. USA., 94, 9711–9716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altman S., Kirsebom,L. and Talbot,S. (1993) FASEB, 7, 7–14. [DOI] [PubMed] [Google Scholar]
- 12.Jeng K.-S., Su,P.Y. and Lai,M.M.C. (1996) J. Virol., 70, 4205–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lambowitz A.M. and Perlman,P.S. (1990) Trends Biochem. Sci., 15, 440–444. [DOI] [PubMed] [Google Scholar]
- 14.Sägesser R., Martinez,E., Tsagris,M. and Tabler,M. (1997) Nucleic Acids Res., 25, 3816–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hammann C., Hormes,R., Sczakiel,G. and Tabler,M. (1997) Nucleic Acids Res., 25, 4715–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pearson W.R. and Lipman,D.J. (1988) Proc. Natl Acad. Sci. USA, 85, 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 18.Chomczynski P. and Sacchi,N. (1987) Anal. Biochem., 162, 156–164. [DOI] [PubMed] [Google Scholar]
- 19.Frohman M.A., Dush,M.K. and Martin,G.R. (1988) Proc. Natl Acad. Sci. USA, 85, 8998–9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, NY.
- 21.Studier F.W., Rosenberg,A.H., Dunn,J.J. and Dubendorff,J.W. (1990) Methods Enzymol., 185, 60–89. [DOI] [PubMed] [Google Scholar]
- 22.Beven A.F., Simpson,G.G., Brown,J.W.S. and Shaw,P.J. (1995) J. Cell Sci., 108, 509–518. [DOI] [PubMed] [Google Scholar]
- 23.Wilusz J., Pettine,S.M. and Shenk,Th. (1989) Nucleic Acids Res., 17, 3899–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robbins J., Dilworth,S.M., Laskey,R.A. and Dingwall,C. (1991) Cell, 64, 615–623. [DOI] [PubMed] [Google Scholar]
- 25.Ninkina N.N., Willoughby,J.J., Beech,M.M., Coote,P.R. and Wood,J.N. (1994) Mol. Brain Res., 22, 39–48. [DOI] [PubMed] [Google Scholar]
- 26.Burd C.G. and Dreyfuss,G. (1994) Science, 265, 615–621. [DOI] [PubMed] [Google Scholar]
- 27.Appendino G. and Szallasi,A. (1997) Life Sci., 60, 681–696. [DOI] [PubMed] [Google Scholar]