


Abstract
Puromycin, an analog of the 3′ end of aminoacyl-tRNA, causes premature termination of translation by being linked non-specifically to growing polypeptide chains. Here we report the interesting phenomenon that puromycin acting as a non-inhibitor at very low concentration (e.g. 0.04 µM) can bond only to full-length protein at the C-terminus. This was proved by using a carboxypeptidase digestion assay of the products obtained by Escherichia coli cell-free translation of human tau 4 repeat (tau4R) mRNA in the presence of low concentrations of puromycin or its derivatives. The tau4R mRNA was modified to code for three C-terminal methionines, which were radioactively labeled, followed by a stop codon. The translation products could not be digested by carboxy-peptidase if puromycin or a derivative was present at the C-terminus of full-length tau4R. Puromycin and its derivatives at 0.04–1.0 µM bonded to 7–21% of full-length tau4R, depending on the ability to act as acceptor substrates. Furthermore, the bonding efficiency of a puromycin derivative to tau4R was decreased by addition of release factors. These results suggest that puromycin and its derivatives at concentrations lower than those able to compete effectively with aminoacyl-tRNA can bond specifically to full-length protein at a stop codon. This specific bonding of puromycin to full-length protein should be useful for in vitro selection of proteins and for in vitro and in vivo C-terminal end protein labeling.
INTRODUCTION
The antibiotic puromycin (1), which is an analog of the 3′ end of Tyr-tRNATyr (2), acts in both prokaryotes and eukaryotes (3–5) as an inhibitor of peptidyl transferase (6,7). It has two modes of inhibitory action. The first is by acting as an acceptor substrate which attacks peptidyl-tRNA (donor substrate) in the P site to form a nascent peptide (6–9). The second is by competing with aminoacyl-tRNA for binding to the A′ site, defined as the binding site of the 3′ end of aminoacyl-tRNA within the peptidyl transferase center (10–12). It has been reported that the polypeptides released by puromycin are not full-length protein (6). Similarly, it has been shown that growing peptide chains on ribosomes are transferred to the α-amino group of puromycin, which interrupts the normal reaction of peptide bond formation (7). Therefore, these conventional studies suggest that puromycin is a non-specific inhibitor of protein synthesis as a result of competition with aminoacyl-tRNA. However, since most of the studies on puromycin have been performed at relatively high concentrations of puromycin to examine the non-specific inhibition of protein synthesis, the behavior of puromycin at lower concentrations, before non-specific inhibition occurs, is still open to question. Some studies have shown that full-length protein which fails to release from ribosomes at the final stage of protein folding requires treatment with puromycin or release factors (RFs) to be released (13,14). The results of these studies led us to hypothesize that puromycin might act as a non-inhibitor and bond specifically to full-length protein under certain conditions. Accordingly, the question we sought to answer here is whether puromycin has the ability to bond specifically to full-length protein in the process of normal translation, especially at low concentrations of puromycin, which do not effectively compete with aminoacyl-tRNA.
In order to obtain evidence of specific bonding of puromycin and its derivatives to full-length tau 4 repeats (tau4R) as non-inhibitors of protein synthesis, we have developed a carboxypeptidase digestion assay involving digestion with carboxypeptidase Y after Escherichia coli cell-free translation of modified tau4R mRNA. This approach provided evidence of the specific bonding of puromycin and its derivatives, as well as allowing a comparison of their efficiencies of specific bonding. We also discuss a possible model of this specific bonding of puromycin to full-length protein, and its potential applications to the establishment of mRNA and complete encoded protein fusions for the in vitro selection of proteins, as well as to in vitro and in vivo C-terminal end protein labeling for the analysis of various biological phenomena.
MATERIALS AND METHODS
Chemicals and enzymes
Puromycin dihydrochloride and DMT-deoxyuridine were obtained from Sigma Chemical Company. Bz-DMT-TBDMS-ribocytidine was a gift from Espec Oligo Service (Tsukuba Laboratory). Escherichia coli S30 Extract System for Linear Templates and T7 RNA polymerase were purchased from Promega. [35S]Methionine (1 µCi/pmol) and [γ-32P]ATP (3 µCi/pmol) were from Amersham. T4 polynucleotide kinase was from New England Biolabs. Taq DNA polymerase was from PE Applied Biosystems. Carboxypeptidase Y (sequencing grade) and endoproteinase Arg-C (Arg-C) from mouse submaxillary glands were from Boehringer Mannheim. Spleen phosphodiesterase (spleen PDase) was from Worthington Biochemical Company and nuclease P1 from Seikagaku Company. Purified E.coli release factor-1 (RF-1) and Salmonella release factor-2 (RF-2) (15,16) were gifts from Drs Y. Nakamura and K. Ito, Institute of Medical Science, University of Tokyo. Other chemicals were of reagent grade.
Preparation of puromycin derivatives
Puromycin derivatives, 2′-ribocytidylyl-(3′→5′)-puromycin (rCpPuro) and 2′-deoxyuridylyl-(3′→5′)-puromycin (dUpPuro), were synthesized mainly by the use of Harris and Hengesh’s coupling reaction (17,18). Here it was modified by using a solvent mixture of dry pyridine and 1,3-dimethyl-3,4,5,6,-tetrahydro-2(1H)-pyrimidinone (DMPU) to increase the yield. Twenty micromoles of 5′-O-phosphoryl-puromycin (19) and 40 µmol of fully protected deoxy- or ribonucleoside were treated with 0.1 mmol of dicyclohexyl carbodiimide (DCC) in a mixture of 2 ml of dry pyridine and DMPU (1:1, v/v) for 3 days at room temperature. The dimethoxytrityl (DMT) group and benzoyl group of puromycin derivatives were then deprotected. The derivatives were further purified by HPLC, analyzed by using two digestion reactions with spleen PDase and nuclease P1, and identified by MALDI/TOF/MS (PerSeptive Biosystems Voyager). [M+H]+ molecular ions at m/z 777 and 762 were seen for rCpPuro and dUpPuro, respectively. The modification of the coupling reaction caused an increase in the yield of the derivatives to 10% relative to starting puromycin, as determined using calculated molar extinction coefficients of 19 500 at 268 nm for puromycin, 28 900 at 265 nm for dUpPuro and 30 200 at 271 nm for rCpPuro in 0.1 M HCl (17).
Preparation of mRNA
The tau4R region (Fig. 1A) corresponding to amino acids 192–318 of human tau protein was amplified by PCR from a plasmid (pAR3040) (20,21). The tau4R (N-terminal Met) mRNA (Fig. 1B) was amplified by PCR with primer 1, the product generated by PCR with primer 1-1 (5′-GAGCATAGATCTCGATCCCG-CGAAATTAATACG-3′) containing a part of the T7 promoter and primer 1-2 (5′-GGACATGACATTCATCATGTCTGGCATATGTAT-3′) containing an initiation codon, and primer 2 (5′-GTTCTCGGATCCTTACAGCTTGTGGGTTTCAATCTTTTTA-3′) containing an ochre stop codon. The tau4R (C-terminal Met) mRNA (Fig. 1C) was amplified by PCR with primer 1-1 and primer 3 (5′-GCAGCCCATCATCATCTACTTGTGGGTTTCAAT-3′) containing three methionine triplets followed by an ochre stop codon. Thus, the natural CUG leucine coding triplet of tau4R was substituted by a UAA ochre stop codon, and the natural CTG leucine, AAG lysine and CAC histidine were substituted by three AUG methionine triplets (Fig. 1A). PCR was carried out with Taq DNA polymerase, using 30 cycles each consisting of 30 s at 92°C, 30 s at 65°C and 1 min at 73°C. PCR-generated fragments were purified with QIA quick PCR purification kit (Qiagen). Transcription was done by using T7 RNA polymerase with PCR-generated fragments for 2 h at 37°C, followed by DNase I treatment for 15 min at 37°C. Both mRNAs were purified by 8 M urea/4% PAGE.
Figure 1.

Amino acid sequence (A) and DNA templates (B and C) for modification of tau4R. (A) The amino acid sequence of tau4R is from the literature (21). The last asterisked amino acid was replaced by an ochre stop codon. Three underlined and three double underlined amino acids for construction of tau4R (N-terminal Met) and tau4R (C-terminal Met) mRNA were replaced by methionine, respectively. Tau4R was digested at the C-terminal side of square-boxed arginine by treatment with Arg-C. (B) DNA template of tau4R (N-terminal Met) mRNA. Three bold black bars indicate positions at which the fourth, fifth and eighth amino acids of tau4R were replaced by methionine, respectively, as described in (A). (C) DNA template of tau4R (C-terminal Met) mRNA. Three bold black bars indicate positions at which the 125th, 126th and 127th amino acids of tau4R were replaced by methionine, respectively, as described in (A). SD, the Shine–Dalgarno sequence; square-boxed Met, the starting methionine residue.
Cell-free translation
Cell-free translation was performed as described by Lesley et al. (22) using an E.coli S30 Extract System for Linear Templates (Promega) with minor modifications. The reaction volume was 25 µl and the translation was carried out at 37°C for 10 min with 10 pmol of tau4R (N-terminal) mRNA and 15 pmol of [35S]methionine (1 µCi/pmol) or 2 pmol of [5′-32P]puromycin derivative labeled with [γ-32P]ATP (3 µCi/pmol) using T4 polynucleotide kinase. Electrophoresis was performed by 8 M urea/1% SDS–PAGE with 18% acrylamide and 0.5% bisacrylamide for the separating gel in 1.5 M Tris–HCl/0.4% SDS, pH 8.8 and 5% acrylamide and 0.5% bisacrylamide for the stacking gel in 0.5 mM Tris–HCl/0.4% SDS, pH 6.8, after a pre-run for 30 min at room temperature. The gels were exposed to an imaging plate for 30 min for analysis with an imaging analyzer (Fuji Film BAS2000).
Carboxypeptidase digestion assay
Cell-free translation was performed with 10 pmol of tau4R (C-terminal Met) mRNA and 15 pmol of [35S]methionine (1 µCi/pmol) in a 25 µl reaction mixture, and then an aliquot of 6 µl of each 25 µl reaction mixture in the absence or presence of puromycin or a derivative was digested with 0.8 µg of Arg-C for 1 h at 37°C, followed by 0.6 µg of carboxypeptidase Y for 1 h at 25°C. Electrophoresis was carried out by 1% SDS–PAGE with 18% acrylamide and 0.5% bisacrylamide for the separating gel in 1.5 M Tris–HCl/0.4% SDS, pH 8.8 and 5% acrylamide and 0.5% bisacrylamide for the stacking gel in 0.5 mM Tris–HCl/0.4% SDS, pH 6.8, after a pre-run for 30 min at room temperature. The gels were analyzed with the imaging analyzer, as described in the previous section.
RESULTS
Inhibitory activity of puromycin and its derivatives at low concentrations
The cell-free system containing non-radioactive puromycin, rCpPuro or dUpPuro with [35S]methionine and the tau4R (N-terminal Met) mRNA (Fig. 1B) was employed to examine the degree of inhibition of protein synthesis by puromycin and its derivatives at low concentrations. Instead of using a conventional peptidyl transferase assay (23,24), we calculated the degree of inhibition by scanning the gels with an imaging analyzer. Fifty percent inhibition corresponds to the formation of one-half of the products of cell-free translation of tau4R mRNA (N-terminal Met) as compared with the amount obtained in the absence of puromycin and its derivatives. The concentrations of puromycin, rCpPuro and dUpPuro affording 50% inhibition were ~6, 1 and 20 µM, respectively (Fig. 2). rCpPuro was a stronger inhibitor than puromycin and dUpPuro was the weakest inhibitor. The results are consistent with studies of the function of CCA sequences required for recognition of aminoacyl-tRNA during peptide bond formation (17,23–25). The inhibition of protein synthesis by puromycin and its analogs was difficult to detect at 0.04 µM and the inhibition by dUpPuro was difficult to detect even at 1.0 µM (Fig. 2).
Figure 2.

Inhibition of protein synthesis in an E.coli cell-free system by puromycin and its derivatives. Tau4R (N-terminal Met) mRNA was used for the cell-free translation with non-radioactive puromycin, rCpPuro or dUpPuro. Open circles, filled circles and filled squares represent puromycin, rCpPuro and dUpPuro, respectively. Fifty percent inhibition corresponds to the formation of one-half of the products of cell-free translation of tau4R mRNA (N-terminal Met) compared to the amount obtained in the absence of puromycin and its derivatives. Degree of inhibition was evaluated by scanning the gels with an imaging analyzer (Fuji Film BAS2000).
Bonding of 32P-labeled puromycin derivatives to full-length tau4R at low concentration
Synthesized and directly 32P-labeled puromycin derivatives, rCpPuro and dUpPuro, were added to an E.coli cell-free translation system with tau4R (N-terminal) mRNA (Fig. 1B). Surprisingly, we found that 32P-labeled rCpPuro and dUpPuro at 0.04 µM seemed to be bonded to full-length tau4R (Fig. 3A, lanes 3 and 5). These bands disappeared completely upon addition of the non-radioactive derivatives. The bonding efficiency of 32P-labeled rCpPuro and dUpPuro was 17 and 10% of total tau4R, respectively (Fig. 3B). The total amount of tau4R corresponding to the product of cell-free translation of tau4R mRNA (N-terminal Met) in the absence of puromycin and its derivatives showed no difference between the absence (Fig. 3A, lane 1) and presence (Fig. 3A, lane 2) of non-radioactive puromycin as a control, since the concentration (0.04 µM) was lower than that capable of inhibiting protein synthesis effectively (Fig. 2). This result strongly supports the view that 32P-labeled puromycin derivatives bonded only to full-length tau4R at 0.04 µM (Fig. 3A, lanes 3 and 5).
Figure 3.
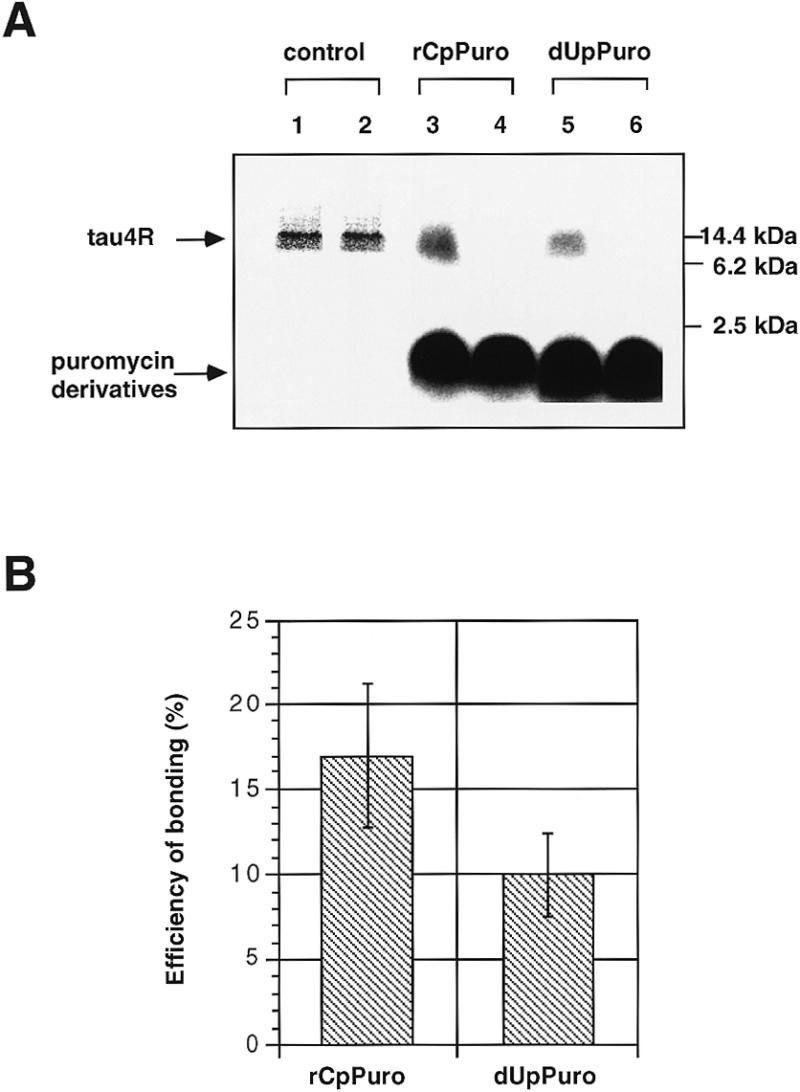
Specific bonding of 32P-labeled puromycin derivatives to full-length tau4R. (A) Autoradiograms of SDS–PAGE of the cell-free translation products obtained with tau4R (N-terminal Met) mRNA. Lanes 1 and 2, [35S]methionine-labeled products in the absence and presence of 0.04 µM puromycin, respectively. Lanes 3 and 5, translation products in the presence of 0.04 µM 32P-labeled rCpPuro and 32P-labeled dUpPuro, respectively. Lanes 4 and 6, translation products without mRNA in the presence of 0.04 µM 32P-labeled rCpPuro and 32P-labeled dUpPuro, respectively. Molecular weight bars on the right side of the gel show the positions of non-radioactive tau4R and molecular markers (molecular weight range 2512–16 949, Code no. 80-1129-83, Pharmacia LKB Biotechnology). (B) Efficiency of bonding (%) of 32P-labeled rCpPuro and dUpPuro to the C-terminal end of full-length tau4R. The value of 100% corresponds to the total products of cell-free translation of tau4R mRNA (N-terminal Met) in the absence of puromycin and its derivatives. Bonding efficiency was evaluated by scanning the gels with an imaging analyzer (Fuji Film BAS2000). Data represent the mean ±SD of two separate experiments.
A system to detect specific bonding of puromycin or its derivatives to the 35S-labeled C-terminal of full-length tau4R
To confirm that puromycin or its derivatives bonded to full-length protein, we have developed a new carboxypeptidase digestion assay (Fig. 4). The cell-free translation was performed using [35S]methionine and the tau4R (C-terminal Met) mRNA (Fig. 1C) constructed with three methionine triplets followed by an ochre stop codon to detect full-length tau4R. Tau4R was observed at a slightly higher position (14 kDa) on the gel (Fig. 5A, lane 1) than the expected position (12 kDa) based on the molecular size markers because it is a basic protein (Fig. 1A). However, the fragment of tau4R (4 kDa) digested with Arg-C at the carboxyl end of the arginine residue (Figs 1A and 4) was detected at the expected position (Fig. 5A, lane 2). This assay is based on the idea that, if the 4 kDa fragment of tau4R produced by Arg-C treatment is bonded to puromycin or its derivatives at the C-terminus, it will not be digested by carboxypeptidase Y (Fig. 4). It was confirmed that treatment with carboxypeptidase Y for 60 min was sufficient to digest completely the 4 kDa fragment of control tau4R (Fig. 5A, lane 3), whereas a part of the 4 kDa fragment from tau4R synthesized in the presence of 0.04 µM rCpPuro, puromycin or dUpPuro remained undigested (Fig. 5A, lanes 5, 7 and 9). Hence this assay can determine the amount of full-length tau4R that is bonded to puromycin or its derivatives on the basis of the protection by puromycin or its derivatives of the 35S-labeled C-terminal end of the 4 kDa tau4R fragment from digestion by carboxypeptidase Y (Fig. 4).
Figure 4.

The carboxypeptidase digestion assay to detect specific bonding of puromycin and its derivatives to full-length tau4R. A shaded circle indicates puromycin or a derivative. M, bold black bars and R indicate the starting methionine residue, three methionine residues at the C-terminal end and an arginine residue, respectively. Tau4R and tau4R bonded to puromycin or a derivative can be digested by Arg-C, producing a 4 kDa fragment of tau4R and a 4 kDa fragment of tau4R bonded to puromycin or a derivative. The 4 kDa fragment of tau4R can be completely digested by carboxypeptidase Y, producing amino acids and oligopeptides, but the 4 kDa fragment of tau4R bonded to puromycin or a derivative cannot be digested by carboxypeptidase Y.
Figure 5.
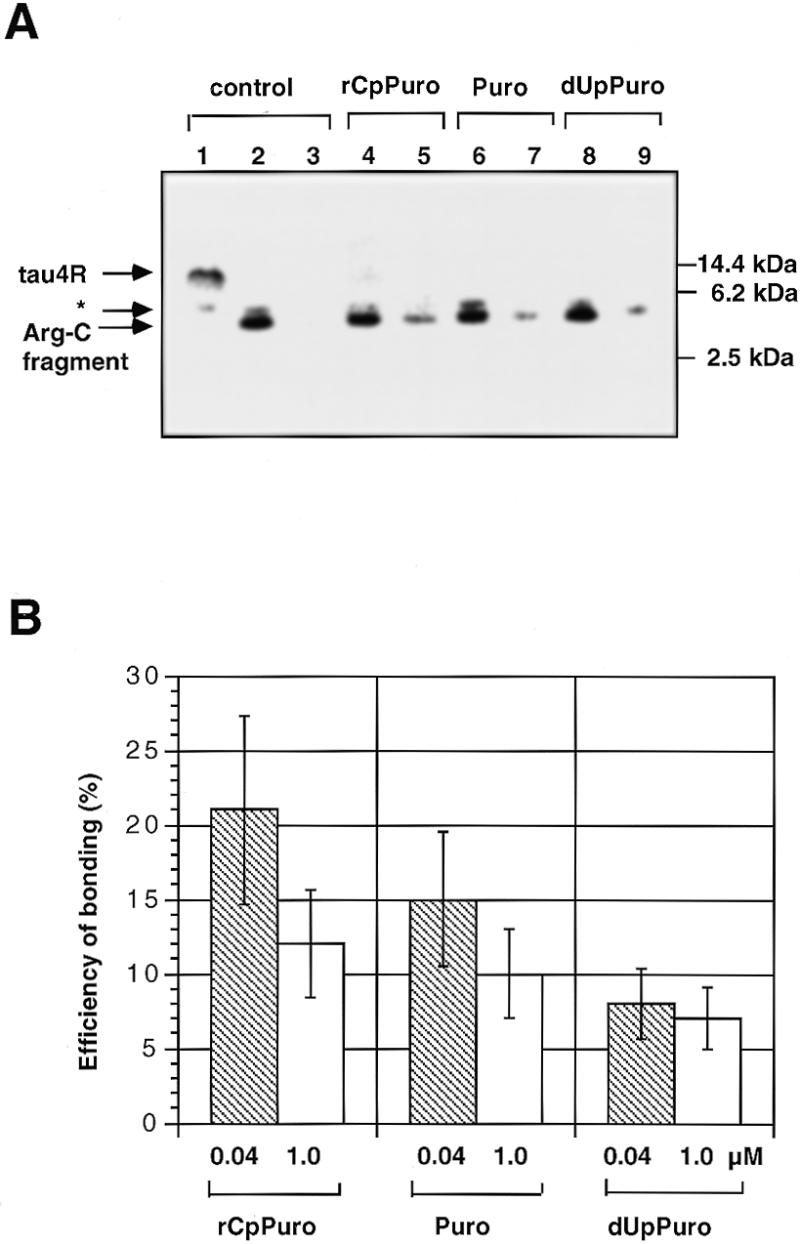
Specific bonding of puromycin or its derivatives to the 35S-labeled C-terminal of full-length tau4R. (A) Autoradiograms of SDS–PAGE of the carboxypeptidase digestion products following the cell-free translation with tau4R (C-terminal Met) mRNA. Lane 1, 35S-labeled full-length tau4R product in the absence of puromycin or its derivatives. Lane 2, product of lane 1 digested with Arg-C. Lane 3, product of lane 2 digested with carboxypeptidase Y for 60 min. Lanes 4, 6 and 8, full-length tau4R product synthesized in the presence of 0.04 µM rCpPuro, puromycin and dUpPuro after digestion with Arg-C. Lanes 5, 7 and 9, products of lanes 4, 6 and 8 digested with carboxypeptidase Y for 60 min. Molecular weight bars on the right side of the gel show non-radioactive tau4R and molecular markers (molecular weight range 2512–16 949, Code no. 80-1129-83, Pharmacia LKB Biotechnology). The asterisked band appearing just above the band corresponding to the Arg-C fragment was independent of tau4R mRNA and therefore was not studied further. (B) Specific bonding efficiency (%) of puromycin, rCpPuro and dUpPuro at 0.04 and 1.0 µM to the 35S-labeled C-terminal end of full-length tau4R. The value of 100% corresponds to the total products of cell-free translation of tau4R mRNA (C-terminal Met) in the absence of puromycin and its derivatives. The result of the carboxypeptidase assay was evaluated by scanning the gels with an imaging analyzer (Fuji Film BAS2000). Data represent the mean ±SD of two or more separate experiments.
Demonstration and comparison of specific bonding of puromycin and its derivatives to full-length tau4R
Using the carboxypeptidase digestion assay, we found that not only rCpPuro and dUpPuro, but also puromycin itself showed a capacity for specific bonding to the 35S-labeled C-terminal end of full-length tau4R at a concentration of 0.04 µM (Fig. 5A). The 4 kDa fragments of tau4R bonded to puromycin, rCpPuro and dUpPuro all remained on the gel after the carboxypeptidase Y treatment for 60 min (Fig. 5A, lanes 5, 7 and 9), in contrast to the control (Fig. 5A, lane 3). Analysis of the efficiency of specific bonding showed rCpPuro (21%) > puromycin (15%) > dUpPuro (8%) at a concentration of 0.04 µM (Fig. 5B). Thus, rCpPuro gave the highest efficiency of specific bonding and one in every five producing full-length protein molecules seems to have rCpPuro at its C-terminus. A comparison of the results at 0.04 and 1.0 µM indicated that the efficiencies of specific bonding of rCpPuro and puromycin were distinctly decreased and that of dUpPuro was insignificantly decreased with the increase in concentration (Fig. 5B). In principle, the efficiencies of specific bonding of stronger inhibitors seemed to show sharper falls with increasing concentration (Fig. 5B). In other words, the efficiency of specific bonding decreased with increasing degree of inhibition due to the decrease of the total produced full-length protein molecules. The efficiencies of specific bonding (Fig. 5B) were in the same order as the degree of inhibition of protein synthesis (Fig. 2). This suggests that this specific bonding to full-length protein depends on the ability of these compounds to act as acceptor substrates for peptide bond formation in the A site of the ribosome. Moreover, the efficiencies of bonding (Fig. 5B) were in good agreement with those of 32P-labeled puromycin derivatives (Fig. 3B).
Inhibition by RFs of 32P-labeled rCpPuro bonding to tau4R
Since puromycin and its derivatives bonded to full-length protein, as judged from the results of the bonding experiments of 32P-labeled puromycin derivatives and the carboxypeptidase digestion assay, the specific bonding of puromycin and its derivatives might occur at a stop codon. To test this hypothesis, E.coli RF-1 or Salmonella RF-2 (15,16) was added to the E.coli cell-free translation system in the presence of 0.04 µM 32P-labeled rCpPuro (Fig. 6). The efficiency of specific bonding of 32P-labeled rCpPuro to tau4R was reduced to 80% by addition of 150 pmol of RF-1 and to 60% by addition of 375 pmol of RF-2, respectively (Fig. 6 lanes 2 and 3), based on the bonding efficiency of 0.04 µM 32P-labeled rCpPuro to tau4R in the absence of RFs, taken as 100% (Fig. 6, lane 1). These results strongly suggest that specific bonding of puromycin to full-length protein occurs at a stop codon in the process of termination of protein synthesis.
Figure 6.

Inhibition of specific bonding of 32P-labeled rCpPuro to tau4R by RF-1 and RF-2. Cell-free translation using mRNA of tau4R (N-terminal Met) was performed as described in Materials and Methods. Lane 1, [32P]rCpPuro-labeled tau4R produced in the presence of 0.04 µM 32P-labeled rCpPuro with 5 µl of blank buffer [50 mM Tris–HCl (pH 7.9), 50 mM KCl, 3 mM DTT]. Lane 2, [32P]rCpPuro-labeled tau4R produced in the presence of 0.04 µM 32P-labeled rCpPuro with 5 µl of RF-1 (30 pmol/µl). Lane 3, [32P]rCpPuro-labeled tau4R produced in the presence of 0.04 µM 32P-labeled rCpPuro with 5 µl of RF-2 (75 pmol/µl).
DISCUSSION
This is the first report to present evidence that puromycin and its derivatives can bond only to full-length protein molecules at very low concentrations (e.g. 0.04 µM) where they act as non-inhibitors of protein synthesis. First, we confirmed that the inhibition of protein synthesis by puromycin and its derivatives at a concentration of 0.04 µM was almost undetectable (Fig. 2). Secondly, we found that 32P-labeled rCpPuro and dUpPuro at 0.04 µM seemed to be bonded to full-length tau4R (Fig. 3). Thirdly, we proved by means of the carboxypeptidase digestion assay that puromycin and its derivatives (rCpPuro and dUpPuro) at 0.04 µM showed specific bonding to the 35S-labeled C-terminal end of full-length tau4R (Fig. 5A) to an extent that depends on the ability of each compound to act as an acceptor substrate for peptide bond formation in the A site (Figs 2 and 5B). We confirmed the agreement of the bonding efficiencies obtained by using 32P-labeled rCpPuro and dUpPuro (Fig. 3B) and those examined using 35S-labeled tau4R (Fig. 5B). Finally, we showed that the efficiency of specific bonding of 32P-labeled rCpPuro to tau4R was decreased by RF-1 and RF-2. Taken together, these results support the idea that puromycin and its derivatives can bond specifically to full-length protein.
We propose a possible model to explain the specific bonding of puromycin to full-length protein. It is well known that puromycin at high concentrations competes with aminoacyl-tRNA, causing premature termination of protein synthesis and forming peptidyl puromycin by being linked to the growing peptide chain (Fig. 7A) (6–9). On the other hand, puromycin at lower concentrations (e.g. 0.04 µM) hardly inhibits protein synthesis (Fig. 2), so that it could bond specifically to full-length protein molecules (Figs 3A and 5A). In other words, puromycin at sufficiently low concentrations could have a chance to be bonded to proteins only at a stop codon, where puromycin does not need to compete with aminoacyl-tRNA. Since termination is a relatively slow step involving a translational pause in eukaryotes (26,27) and E.coli (28), it is possible that puromycin even at very low concentrations binds to the A′ site, defined as the binding site of the 3′ end of aminoacyl-tRNA within the peptidyl transferase center (10–12), and competes with RFs to release full-length protein from ribosomes (Fig. 6). This concept is supported by the findings that complex formation of ribosomes with RF was completely blocked by the presence of the ternary complex (aminoacyl-tRNA with elongation factor-Tu) and decreased by the presence of aminoacyl-tRNA at the A site, but was unimpaired by the presence of deacylated tRNA at the A site (29). Therefore, we propose a model in which puromycin can bond to the full-length protein at the stop codon in the process of normal termination of protein synthesis (Fig. 7B). This phenomenon should be useful for studying aspects of the mechanism of translational termination, especially in relation to RFs (Fig. 6), such as the determination of efficiency of RFs independently of the influence of termination codon context (30). At low concentrations such as 0.04 µM, specific bonding of puromycin occurs efficiently because sufficient full-length protein molecules are produced (Fig. 7B). However, at intermediate concentrations such as 1.0 µM, both specific bonding of puromycin to full-length protein molecules (Fig. 7B) and non-specific bonding of puromycin to nascent protein molecules (Fig. 7A) could occur at the same time. This was because the efficiency of specific bonding of puromycin would decrease with increasing degree of inhibition, due to decreasing formation of full-length protein molecules and increasing amounts of nascent protein molecules (Figs 2 and 5B).
Figure 7.

A possible model of specific bonding of puromycin to full-length protein. (A) Non-specific bonding to nascent protein in competition with aminoacyl-tRNA at higher concentrations of puromycin. (B) Specific bonding to full-length protein at the stop codon, not in competition with aminoacyl-tRNA, at lower concentrations of puromycin. Puro stands for puromycin.
If this model (Fig. 7B) is correct, the specific bonding of puromycin should be applicable to the in vitro selection of proteins by methods such as in vitro virus (31) and RNA–peptide fusion (32), based on the use of puromycin as a physical linkage between mRNA and its encoded peptide or protein, because the establishment of fusions of mRNA and its complete encoded protein is preferable for the accurate assignment of genotype (mRNA) and phenotype (its encoded protein). The above methods [in vitro virus (31) and RNA–peptide fusion (32)] required the use of mRNA without a stop codon to establish the fusions of mRNA and its encoded peptide or protein, so that another method independent of the stop codon was developed (33), which could be applied to the screening of natural mRNA or cDNA libraries. However, in the present work, we have shown that puromycin, under certain conditions, bonds to complete encoded tau4R obtained by using mRNA with a stop codon (Figs 3, 5 and 6). In other words, the requirement of no stop codon may no longer be essential for the in vitro virus (31) and RNA–peptide fusion (32) approaches.
There are a number of other interesting potential applications of this specific bonding of puromycin to full-length protein at the C-terminus for the analysis of various biological phenomena. For example, puromycin and its derivatives should be useful as new protein labeling reagents for the rapid and convenient analysis of protein–protein interaction and protein folding, not only in vitro but also in vivo, since the specific bonding of puromycin and its derivatives can occur using natural mRNAs with a stop codon (Figs 3, 5 and 6). Here we should point out that C-terminal end labeling of full-length protein with puromycin and its derivatives should not impair the function of the proteins (13,14). Furthermore, the fact that the efficiency of specific bonding seems to depend on the ability to act as an acceptor substrate (Figs 3 and 5) could be a useful hint in the design of new protein labeling reagents. We are currently working on an application of specific bonding of a fluorescent puromycin as a C-terminal end protein labeling reagent to analyze protein–protein interactions by means of fluorescence polarization measurement.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs Y. Nakamura and K. Ito for the gift of purified release factors, Dr S. Inoue for useful advice on the synthesis of puromycin derivatives, M. Nakamura and Y. Ogawa for help in the synthesis of puromycin derivatives, Dr T. Tsuji for the mass spectrometric measurement of chemically synthesized puromycin derivatives and Dr N. Doi for useful suggestions about the manuscript.
REFERENCES
- 1.Cooperman B.S., Weitzmann,C.J. and Fernandez,C.L. (1990) In Hill,W.E., Dahlberg,A., Garrett,R.A.G., Moore,P.B., Schlessinger,D. and Warner,J.R. (eds), The Ribosome: Structure, Function, and Evolution. American Society for Microbiology, Washington, DC, pp. 491–501.
- 2.Yarmolinsky M.B. and De La Hara,G.L. (1959) Proc. Natl Acad. Sci. USA, 45, 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeda Y., Hayashi,S., Nakagawa,H. and Suzuki,F. (1960) J. Biochem., 48, 169–177. [Google Scholar]
- 4.Ferguson J.J. (1962) Biochim. Biophys. Acta, 57, 616–617. [Google Scholar]
- 5.Nemeth A.M. and De La Haba,G.L. (1962) J. Biol. Chem., 237, 1190–1193. [PubMed] [Google Scholar]
- 6.Allen D.W. and Zamecnik,P.C. (1962) Biochim. Biophys. Acta, 55, 865–874. [DOI] [PubMed] [Google Scholar]
- 7.Nathans D. (1964) Proc. Natl Acad. Sci. USA, 51, 585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Traut R.R. and Monro,R.E. (1964) J. Mol. Biol., 10, 63–72. [DOI] [PubMed] [Google Scholar]
- 9.Zamir A., Leder,P. and Elson,D. (1966) Proc. Natl Acad. Sci. USA, 56, 1794–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nathans D. and Neidle,A. (1963) Nature, 197, 1076–1077. [DOI] [PubMed] [Google Scholar]
- 11.Steiner G., Kuechler,E. and Barta,A. (1988) EMBO J., 7, 3949–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirillov S., Porse,B.T., Vester,B., Woolley,P. and Garrett,R.A. (1997) FEBS Lett., 406, 223–233. [DOI] [PubMed] [Google Scholar]
- 13.Kudlicki W., Odom,O.W., Kramer,G. and Hardesty,B. (1994) J. Biol. Chem., 269, 16549–16553. [PubMed] [Google Scholar]
- 14.Hardesty B., Kudlicki,W., Odom,O.W., Zhang,T., McCarthy,D. and Kramer,G. (1995) Biochem. Cell Biol., 73, 1199–1207. [DOI] [PubMed] [Google Scholar]
- 15.Ito K., Uno,M. and Nakamura,Y. (1998) Proc. Natl Acad. Sci. USA, 95, 8165–8169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito K. and Nakamura,Y. (1997) Biochimie, 79, 287–292. [DOI] [PubMed] [Google Scholar]
- 17.Hengesh E.J. and Morris,A.J. (1973) Biochim. Biophys. Acta, 299, 654–661. [DOI] [PubMed] [Google Scholar]
- 18.Harris R.J., Mercer,J.F.B., Skingle,D.C. and Symons,R.H. (1972) Can. J. Biochem., 50, 918–926. [DOI] [PubMed] [Google Scholar]
- 19.Yoshikawa M., Kato,T. and Takenishi,T. (1969) Bull. Chem. Soc. Jpn, 42, 3505–3508. [Google Scholar]
- 20.Yanagawa H., Chung,S-H., Ogawa,Y., Sato,K., Shibata-Seki,T., Masai,J. and Ishiguro,K. (1998) Biochemistry, 37, 1979–1988. [DOI] [PubMed] [Google Scholar]
- 21.Goedert M., Spillantini,M.G., Potier,M.C., Ulrich,J. and Crowther,R.A. (1989) EMBO J., 8, 392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lesley S.A., Brow,M.A.D. and Burgess,R.R. (1991) J. Biol. Chem., 266, 2632–2638. [PubMed] [Google Scholar]
- 23.Harris R.J., Hanlon,E. and Symons,R.H. (1971) Biochim. Biophys. Acta, 240, 244–261. [DOI] [PubMed] [Google Scholar]
- 24.Symons R.H., Harris,R.J., Clarke,L.P., Wheldrake,J.F. and Elliott,W.H. (1969) Biochim. Biophys. Acta, 179, 248–250. [DOI] [PubMed] [Google Scholar]
- 25.Tezuka M. and Chladek,S. (1990) Biochemistry, 29, 667–670. [DOI] [PubMed] [Google Scholar]
- 26.Wolin S.L. and Walter,P. (1988) EMBO J., 7, 3559–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lodish H.F. and Jacobsen,M. (1972) J. Biol. Chem., 247, 3622–3629. [PubMed] [Google Scholar]
- 28.Bjornsson A. and Isaksson,L.A. (1996) Nucleic Acids Res., 24, 1753–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tate W.P., Horning,H. and Luhrman,R. (1983) J. Biol. Chem., 258, 10360–10365. [PubMed] [Google Scholar]
- 30.Tate W.P. and Brown,C.M. (1992) Biochemistry, 31, 2443–2450. [DOI] [PubMed] [Google Scholar]
- 31.Nemoto N., Miyamoto-Sato,E., Husimi,Y. and Yanagawa,H. (1997) FEBS Lett., 414, 405–408. [DOI] [PubMed] [Google Scholar]
- 32.Roberts R.W. and Szostak,J.W. (1997) Proc. Natl Acad. Sci. USA, 94, 12297–12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doi N. and Yanagawa,H. (1999) FEBS Lett., 457, 227–230. [DOI] [PubMed] [Google Scholar]