


Abstract
Polymerases from the Pol-I family which are able to efficiently use ddNTPs have demonstrated a much improved performance when used to sequence DNA. A number of mutations have been made to the gene coding for the Pol-II family DNA polymerase from the archaeon Pyrococcus furiosus with the aim of improving ddNTP utilisation. ‘Rational’ alterations to amino acids likely to be near the dNTP binding site (based on sequence homologies and structural information) did not yield the desired level of selectivity for ddNTPs. However, alteration at four positions (Q472, A486, L490 and Y497) gave rise to variants which incorporated ddNTPs better than the wild type, allowing sequencing reactions to be carried out at lowered ddNTP:dNTP ratios. Wild-type Pfu–Pol required a ddNTP:dNTP ratio of 30:1; values of 5:1 (Q472H), 1:3 (L490W), 1:5 (A486Y) and 5:1 (Y497A) were found with the four mutants; A486Y representing a 150-fold improvement over the wild type. A486, L490 and Y497 are on an α-helix that lines the dNTP binding groove, but the side chains of the three amino acids point away from this groove; Q472 is in a loop that connects this α-helix to a second long helix. None of the four amino acids can contact the dNTP directly. Therefore, the increased selectivity for ddNTPs is likely to arise from two factors: (i) small overall changes in conformation that subtly alter the nucleotide triphosphate binding site such that ddNTPs become favoured; (ii) interference with a conformational change that may be critical both for the polymerisation step and discrimination between different nucleotide triphosphates.
INTRODUCTION
DNA polymerases constitute a core component of DNA sequencing methods, a widespread and important bio-technology, based on chain-termination by dideoxynucleotide-triphosphates (ddNTPs), either the ddNTPs themselves (1,2) or fluorescent derivatives (3). Discrimination between chain-terminating ddNTPs and dNTPs plays a key role in DNA sequencing performance. Effective ddNTP incorporation is associated with a high uniformity of signal intensity in sequencing ladders (4). Furthermore, efficient usage of ddNTPs requires lower concentrations, an advantage when fluorescent terminators (3) are used, as large excesses give rise to high backgrounds. Bacteriophage T7 DNA polymerase incorporates ddNTPs much more efficiently than the enzymes from Escherichia coli and Taq and, as a consequence, gives superior sequencing ladders (4). The molecular basis for discrimination between dNTPs and ddNTPs resides in a single amino acid, at an equivalent location: Y526 (T7); F762 (E.coli); F667 (Taq) (4). The T7 mutant, Y526F, shows a much reduced ability to use ddNTPs and, consequently, gives poor sequencing ladders. The F762Y and F667Y variants of E.coli and Taq use ddNTPs effectively and show much improved sequencing properties. The important role of F762, in the E.coli polymerase, for deoxynucleotide-triphosphate recognition has been confirmed by a more complete kinetic analysis (5). Thermo-SequenaseTM, a Taq polymerase mutant that lacks exonuclease activity, and contains tyrosine at position 667, has excellent sequencing properties and is perhaps the most widely used enzyme for DNA sequencing (6,7). All three polymerases belong to the Pol-I family (also called family A polymerases) and the critical aromatic amino acid is found in a highly conserved stretch, the B-motif (8–10) (also called region III) (Fig. 1). Structural data has shown that the B-motif amino acids form an α-helix (the O-helix in the case of E.coli) with the most conserved amino acids on one side of the helix, forming part of the dNTP binding site (Fig. 1) (11–17). The tyrosine/phenylalanine is near the sugar of the dNTP, rationalising its critical role in dNTP/ddNTP selection.
Figure 1.
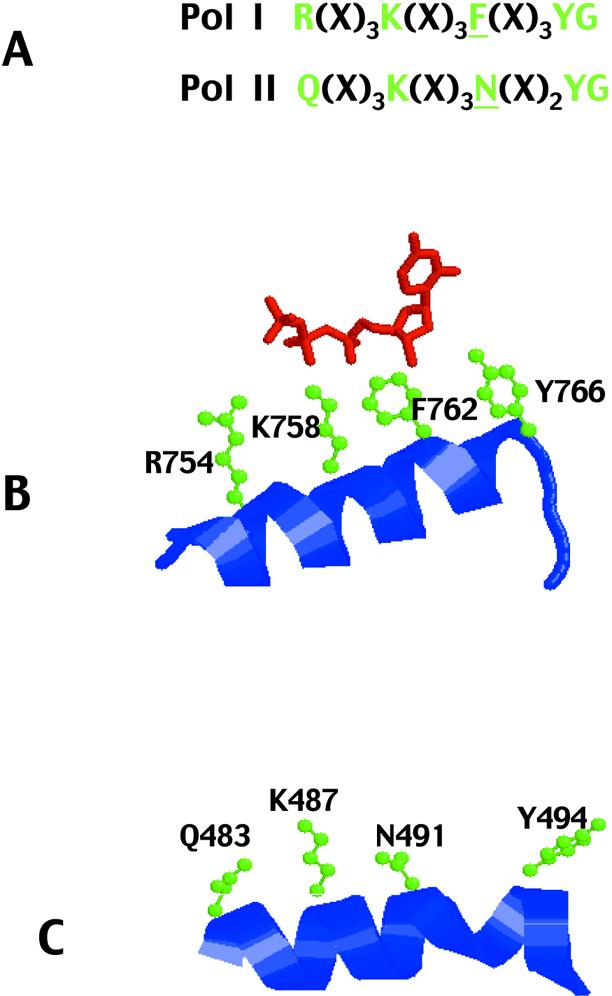
(A) The B-motif (region III) of polymerases from the Pol-I and Pol-II families. With Pol-I the R, K, F and YG indicated in green, and the spacing between them, are highly conserved (8–10). The F, shown in green and underlined, is critical in discrimination between dNTPs and ddNTPs (4). In the case of Pol-II the Q, K, N and YG shown in green, and their spacing, are conserved. It is difficult to deduce the optimal line up, between the two families, due to variation in both the conserved amino acids and their spacing (8–10). The alignment shown (others are possible by including gaps) has the critical F in Pol-I replaced by an N (underlined) in Pol-II. (B) Structure of the B-motif of a Pol-I enzyme; the E.coli Klenow fragment (11). The conserved amino acids (side chains shown in green) lie on one side of an α-helix and interact with dNTP (shown in red). F762 is near the sugar ring explaining its role in dNTP/ddNTP selection. (C) Structure of the B-motif of the Pol-II enzyme from T.gorgonarius (28). Although the conserved amino acids (green) are on one side of an α-helix, as for Pol-I, the side chains, and their relative disposition, differ between the two families and are unlikely to interact with dNTPs identically. The α-helix of Tgo–Pol is also distorted by the presence of a short stretch of 310-helix between N491 and Y494, resulting in a further difference between the two families. All structures were generated using RasMol (36).
Polymerases with thermal stability are routinely used for DNA sequencing. Not only are they generally more robust than mesophilic enzymes but are essential for cycle-sequencing protocols, which involve heat–cool cycles (18,19). The extreme thermostability of polymerases purified from hyper-thermophilic archaea (20–23) suggests that these enzymes have potential use in DNA sequencing. Unfortunately, archaeal polymerases often use ddNTPs poorly and, as a result, are generally not as useful in DNA sequencing as, for example, Thermo SequenaseTM. The archaeal polymerase from Pyrococcus furiosus, studied in this publication, belongs to the Pol-II family (8–9) (also called the α-family, or the B-family), a different group to the better characterised Pol-I enzymes. However, sequence alignment shows that the Pol-II family also has a B-motif, even though it cannot be exactly aligned with that of Pol-I (8,9) (Fig. 1), and there is no exact counterpart to the aromatic amino acid critical for ddNTP/dNTP selection in the Pol-I family. Nevertheless, mutations in the B-motif of Pol-II polymerases influence dNTP binding (24–26), suggesting a role in deoxynucleoside-triphosphate recognition. Recently, crystal structures of two Pol-II members, bacteriophage RB69 gp43 (27) and Thermococcus gorgonarius (Tgo) (28), have been published. The B-motif amino acids form an α-helix (the P-helix with both enzymes) which, as suggested by sequence alignments, is similar, but not structurally identical to the corresponding α-helix in the Pol-I family (Fig. 1). Although both structures lack bound nucleic acid it was possible to model primer-template and dNTP into RB69. The B-region was located near both primer/template and dNTP but did not appear to provide an amino acid that binds near the sugar of the dNTP. Rather, this might be supplied by tyrosine 416, an amino acid from another part of the polymerase, which packs under the sugar ring of the dNTP (28).
Sequence and structural comparisons of Pol-I and Pol-II members indicate that homologous regions are most likely used for dNTP binding and recognition. However, the exact details of the interaction with dNTPs, and hence discrimination between dNTPs and ddNTPs, differs between the two classes. Therefore, the simple tyrosine/phenylalanine switch, so successful in converting Taq to a good sequencing polymerase, is unlikely to be possible with archaeal polymerases. Nevertheless, there seems no reason why other mutations should not alter dNTP/ddNTP discrimination and so give improved sequencing variants. This publication reports on the preparation of P.furiosus polymerase mutants that recognise ddNTPS a factor of up to 150-fold better than the wild-type enzyme and are superior for cycle-sequencing protocols.
MATERIALS AND METHODS
Construction of a Pfu–Pol expression vector
A gene coding for Pfu–Pol (29, accession number D12983) was digested from the plasmid pTM121 (prepared in house by Amersham Pharmacia Biotech Inc.) as a NdeI–SmaI fragment and ligated into NdeI–EcoRV cut pET-17b (Novagen) to give pET-17b(Pfu–Pol). This manipulation destroys the unique EcoRV restriction site present in pET-17b. The Pfu–Pol gene used contains the mutation D215A, which abolishes the 3′ to 5′ exonuclease activity of the enzyme. All experiments were performed with the exo– form of the enzyme.
Expression and purification of Pfu–Pol
Escherichia coli BL21(DE3) (Novagen) containing pET-17b(Pfu–Pol) was grown at 37°C until an A600 of 0.5 was reached. Protein expression was induced by adding IPTG, to a final concentration of 1 mM, and continuing growth for another 4 h. Cells were harvested by centrifugation at 4°C (5000 r.p.m. for 20 min) and resuspended in 10 mM Tris pH 8.0, 100 mM NaCl, 1 mM phenylmethanesulphonyl-fluoride, 1 mM benzamidine. After sonication on ice (10 × 15 s pulses) samples were centrifuged at 10 000 r.p.m. for 20 min. The supernatant was incubated with ~20 U of DNase I (Boehringer-Mannheim) for 30 min to hydrolyse DNA. Next the supernatant was heated at 75°C for 20 min to denature most of the E.coli proteins and inactivate the DNase I. Precipitated proteins were removed by centrifugation (10 000 r.p.m. for 20 min) and the supernatant was loaded onto a 20 ml DEAE-Sephacel column, equilibrated and eluted with Tris pH 8.0, 100 mM NaCl. The flow through was collected and immediately applied to a 20 ml heparin–Sepharose column, equilibrated with Tris pH 8.0, 100 mM NaCl. The column was developed with a 100–700 mM NaCl gradient in Tris pH 8.0. Fractions were analysed by SDS–PAGE using Coomassie Blue staining and those containing a protein running at ~90 kDa were pooled and concentrated using Centriprep 50 spin concentrators (Amicon). Protein samples were estimated to be >95% pure as judged by SDS–PAGE. All purified protein samples were stored at –20°C as 50% glycerol stocks.
Random mutagenesis of Pfu–Pol and selection of mutants by colony screening
Random mutagenesis was carried out on a section of the Pfu–Pol gene (in pET-17b) flanked by unique EcoRV and SacI restriction sites; a region comprising bases 1293–1596 and amino acid 431–532 (i.e. the P-helix and surrounding amino acids). A PCR-based method in conjunction with the mutagenic base analogues 8-oxo-dGTP and dPTP was used (30). The mutagenic primers used were GAACTATGATATCGCTCC (EcoRV primer) and CTTTTCTTCGAGCTCCTTCCATACT (SacI primer). Initially 30 rounds of PCR were carried out under the following conditions: 10 mM Tris–HCl, pH 8.8, 50 mM KCl, 1.5 mM MgCl2, 0.08% Nonidet P-40, 500 µM each dNTP (four normal and two mutagenic), 0.5 U of Taq DNA polymerase; cycle 94°C for 1 min, 55°C for 3 min, 72°C for 2 min. The amplified products were used in a second PCR reaction (conditions identical to the first but with only the four normal dNTPs at 250 µM) to generate a library of mutated DNA fragments. The library was digested with EcoRV and SacI and ligated into pET-17b(Pfu–Pol) from which the EcoRV–SacI fragment of the wild-type Pfu–Pol gene had been excised. The resulting plasmids were used to transform E.coli BL21(DE3) to ampicillin resistance. Transformants containing Pfu–Pol variants better able to incorporate ddNTPs were selected by modifying a ‘colony screening rapid filter assay’ usually used to detect DNA polymerase activity (31). Bacterial colonies were gridded onto duplicate LB/ampicillin agar plates and allowed to grow overnight at 37°C. The colonies from one plate were replica-plated onto a nitrocellulose filter containing activated calf-thymus DNA (31), which was then overlaid onto an LB/ampicillin plate containing 1 mM IPTG. After a further 4 h, at 37°C, during which Pfu–Pol expression was induced, the nitrocellulose filters were removed and cells lysed using toluene/chloroform (31). The nitrocellulose filters were soaked in 20 mM Tris–HCl, pH 8.5, 10 mM KCl, 20 mM (NH4)2SO4, 2 mM MgSO4, 0.1 mg/ml bovine serum albumin and 0.1% Nonidet P-40 and baked at 70°C for 30 min to destroy E.coli DNA polymerases. The filters were re-soaked with this buffer but also containing 12 µM each of dTTP, dCTP and dGTP plus 1 µl of [α-32P]ddATP (3000 Ci/mmol, Nycomed-Amersham) and incubated at 70°C for 30 min to allow 32P incorporation into polymeric material. The filters were washed with trichloroacetic acid and pyrophosphate (31), dried and any radioactivity retained on the filters (i.e. incorporated into polymeric material) determined using autoradiography (31). Under these conditions wild-type Pfu–Pol results in minimal retention of 32P on the filters. Clones that were associated with 32P retention were rescued from the duplicate LB/ampicillin plate and used for both the preparation of mutant Pfu–Pol (exactly as for the wild- type enzyme) and plasmid preparation for DNA sequencing.
Site-directed mutagenesis of Pfu–Pol
Site-directed mutagenesis of the Pfu–Pol gene was carried out using the PCR-based ‘overlap extension’ method (32). Most of the directed mutants were made to the P-helix i.e. within the EcoRV–SacI fragment described above. Therefore the EcoRV and SacI oligonucleotides were used as the common outer primers, along with appropriate primers containing the required mutation, to produce two overlapping DNA fragments. These two fragments, containing the desired mutation, were used as a template in a subsequent PCR reaction, together with the EcoRV and SacI primers to generate the ‘full-length’ mutated EcoRV–SacI fragment. The protocol for PCR and the subsequent cloning of the mutated fragment were as described above. The amino acid Y410 is not in the P-helix and so does not lie between the EcoRV and SacI restriction sites. The mutations Y410F/A were produced analogously using an EcoRI flanking primer (GGGAAAGAATTCCTTCC) and the SacI primer. The Y410 codon is located between the unique EcoRI and SacI restriction sites.
Activity assay for Pfu–Pol
The activity of Pfu–Pol (wild-type and mutants) was determined using the ‘activated calf thymus DNA’ assay (33). Assays were carried out in volumes of 50 µl containing 20 mM Tris, pH 8.5, 10 mM (NH4)2SO4, 10 mM KCl, 2 mM MgSO4, 0.1% Triton and 5 µg (100 µg/ml) bovine serum albumin. The concentration of the four dNTPs was 200 µM (including ~1 µCi of [α-32P]dATP) and 0.4 U of activated calf-thymus DNA (Amersham-Pharmacia Biotech) was used. The reaction was initiated by addition of Pfu–Pol (amount added depended on the activity of the variant under study) and incubation was for 30 min at 72°C (reactions were found to be linear over this time range). After this time the amount of radioactivity incorporated into acid insoluble material was determined by scintillation counting as described (33).
DNA sequencing using Pfu–Pol mutants
DNA sequencing reactions were performed using cycle-sequencing (18,19). A kit purchased from Stratagene (Exo-‘Cyclist’ DNA sequencing kit) together with M13 mp18 single- stranded template DNA and a universal primer was used. Reactions were carried out exactly as defined in the user manual of the kit. Approximately 1–2 U of mutant polymerase was added per sequencing reaction, corresponding to the units of wild-type Pfu–Pol normally used. Reactions were initially performed at a 30:1 ddNTP:dNTP ratio, the optimal nucleotide ratio for the wild-type enzyme. This ratio was progressively lowered for mutant enzymes that showed an increased preference for dideoxynucleotides.
RESULTS
Although the B-region in polymerases belonging to the Pol-I and Pol-II families are similar, rather than identical, it is clear that both play a role in dNTP binding. Therefore, mutagenesis to amino acids that constitute this region is a logical place to begin if one is attempting to produce Pol-II variants able to efficiently use ddNTPs. The amino acid sequence in the vicinity of the B-region for P.furiosus polymerase (Pfu–Pol), the enzyme investigated in this study, is shown in Figure 2. For comparison the homologous sequence of the structurally characterised T.gorgonarius enzyme (Tgo–Pol) is also shown. With Tgo–Pol the B-motif consists of an α-helix (The P-helix) flanked by loop regions (Fig. 2). Tgo–Pol and Pfu–Pol have 79% sequence identity and variant amino acids mostly involve conservative changes. We have used Swiss-Model/SwissPdb viewer (34) to deduce a structure for Pfu–Pol based on Tgo–Pol. As expected the two structures are almost identical and the derived P-helix from Pfu–Pol is shown in Figure 2. A variety of mutations has been made to these amino acids in Pfu–Pol (Table 1), using either random mutagenesis (30) or back-to-back PCR site-directed mutagenesis (32). The mutants were expressed in E.coli, using pET-17b, and purified using a heat step, followed by two chromatography columns. All proteins appeared pure by heavily-loaded SDS–PAGE, stained with Coomassie Blue.
Figure 2.
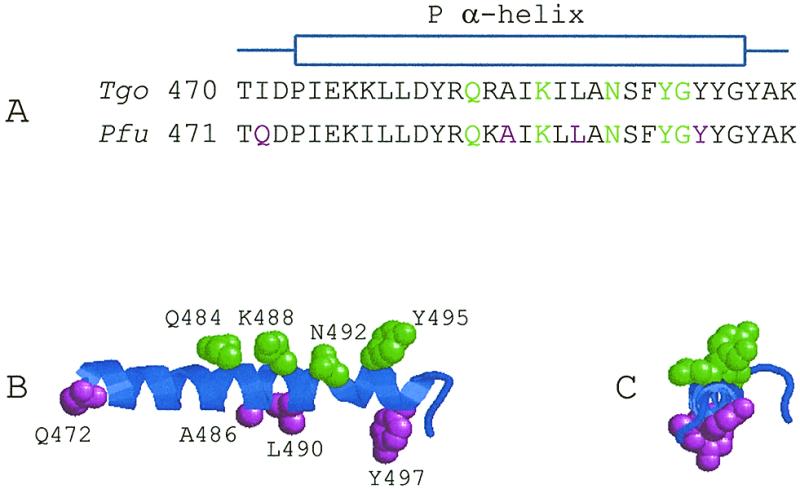
(A) The amino acids in, and surrounding, the B-region of T.gorgonarius (Tgo) and P.furiosus (Pfu) polymerases. The amino acids shown in green are highly conserved and correspond to the amino acids also illustrated in green in Figure 1. Amino acids shown in magenta, when mutated in Pfu–Pol, give better sequencing performance. Note: Pfu–Pol contains a single amino acid insertion, upstream of this region, when compared to Tgo–Pol. This accounts for the numbering of corresponding amino acids differing by one. (B) The structure formed by the Pfu–Pol amino acids shown in (A), which comprises an α-helix (the P-helix) flanked by loop regions (shown in blue). The conserved, green, amino acids (Q484, K488, N492 and Y495) lie on one side of the helix and form part of the dNTP binding site. Three of the ‘improving’, magenta, amino acids (A486, L490 and Y497) lie in the helical region but on the opposite side to the conserved amino acids. The fourth (Q472) is in a stretch of loop that precedes the α-helix. The side chains of these amino acids are shown in space-filling mode. (C) End-on view of the P-helix region of Pfu–Pol clearly showing that the side chains (shown in space-filling mode) of the conserved (green) and ‘improving’ (magenta) amino acids protrude on opposite sides of the helix. The structures given for Pfu–Pol were modelled from Tgo–Pol using Swiss-Model/SwissPdb viewer (34). The structures shown were generated using RasMol (36).
Table 1. DNA sequencing properties of Pfu–Pol mutants.
| Category of enzyme | Mutant | Activity (%) | ddNTP:dNTP ratio optimised for DNA sequencing ladders | Fold improvement |
|---|---|---|---|---|
| Wild-type | Wild-type | 100 | 30:1 | |
| Alterations to highly | Q484A | <1 | Low activity | |
| conserved amino acids | K488A | 27 | Worse than wild-type | |
| in P-helix (shown in | N492Y | <1 | Low activity | |
| green in Figs 1 and 2) | N492H/K/G | ~40 (all) | Worse than wild-type | |
| Y495/I/D/C/S | n.d. | Similar to wild-type | ||
| Insertions into P-helixa | A(Y)N and TIN(Y)GVL | <1 (both) | Low activity | |
| Alterations to other | L479Y/W/P | 33/25/28 | Similar to wild-type | |
| amino acids in | A486Y | 26 | 1:5 | 150 |
| P-helix (see Fig. 2) | A486W | 20 | Worse than wild-type | |
| L490W | 123 | 1:3 | 90 | |
| L490Y | 78 | 5:1 | 6 | |
| S493Y | 100 | Similar to wild-type | ||
| F494Y/C/S/T/V | ~50 (all) | Similar to wild-type | ||
| G496P/S/A | 80/45/47 | Similar to wild-type | ||
| Y497F | 51 | Worse than wild-type | ||
| Y497W | 46 | 10:1 | 3 | |
| Y497A | 40 | 5:1 | 6 | |
| Alterations to Y410 | Y410A/F | 33/47 | Worse than wild-type | |
| Alterations to loop | Q472H | 82 | 5:1 | 6 |
| preceding the P-helix | ||||
| (see Fig. 2) | ||||
| Multiple mutations | Q472H-N492H | 100 | 5:1 | 6 |
| A486Y-L490W | 40 | 1:5 | 150 | |
| Q472H-A486Y-N492H | 25 | Worse than A486Y | ||
| Q472H-L490W-N492H | 43 | Worse than L490W |
N492H/K/G/Y indicates that N492 was changed to H, K, G and Y etc.
aA(Y)N has a Y inserted between A491 and N492 of the wild-type enzyme. In TIN(Y)GVL this sequence replaces the amino acids between 489 and 494 (LLANSF) in the wild-type enzyme. Activity was determined using the ‘activated calf thymus’ method (33) and the activity seen with the wild-type set to 100. The wild-type enzyme gives readable DNA sequencing gels at a 30:1 ddNTP:dNTP ratio, but unreadable gels at 5:1. Mutants described as similar to wild type have this property; mutants noted as worse give unreadable gels at the 30:1 ratio. For mutants with improved discrimination the number reported is the lowest ratio of ddNTP:dNTP at which readable sequencing gels were obtained. In these cases the ‘fold improvement’ (defined as [ddNTP/dNTP]wild type/[ddNTP:dNTP]mutant) is also given.
The DNA-sequencing performance of the mutants was assessed using a standard cycle-sequencing protocol with the four [α-32P]ddNTPs as chain terminators (18,19). In our hands wild-type Pfu–Pol gave good sequencing ladders (clearly defined bands, lack of non-specific termination, sequencing information spread over the entire gel) at a ddNTP:dNTP ratio of 30:1 (Fig. 3). However, when a 5:1 ratio was used much of the radioactivity was found towards the top of the gel (due to lower than optimal levels of termination giving rise to long products) and excessive non-specific termination was also seen, resulting in shadow bands across all four lanes. Both effects make it difficult to deduce the DNA sequences from the sequencing ladders. As improved DNA-sequencing performance correlates with increased selectivity for ddNTPs, we looked for mutants that give a readable sequencing ladder at ddNTP:dNTP ratios of <30:1. Therefore, mutants were initially tested at the wild-type 30:1 ratio. Enzymes which gave readable sequencing gels or were associated with increased radioactivity towards the bottom of the gel (indicative of better ddNTP incorporation resulting in shorter products) were then evaluated at a 5:1 ratio of ddNTP:dNTP and, if warranted, at progressively decreasing ratios of the two triphosphates.
Figure 3.
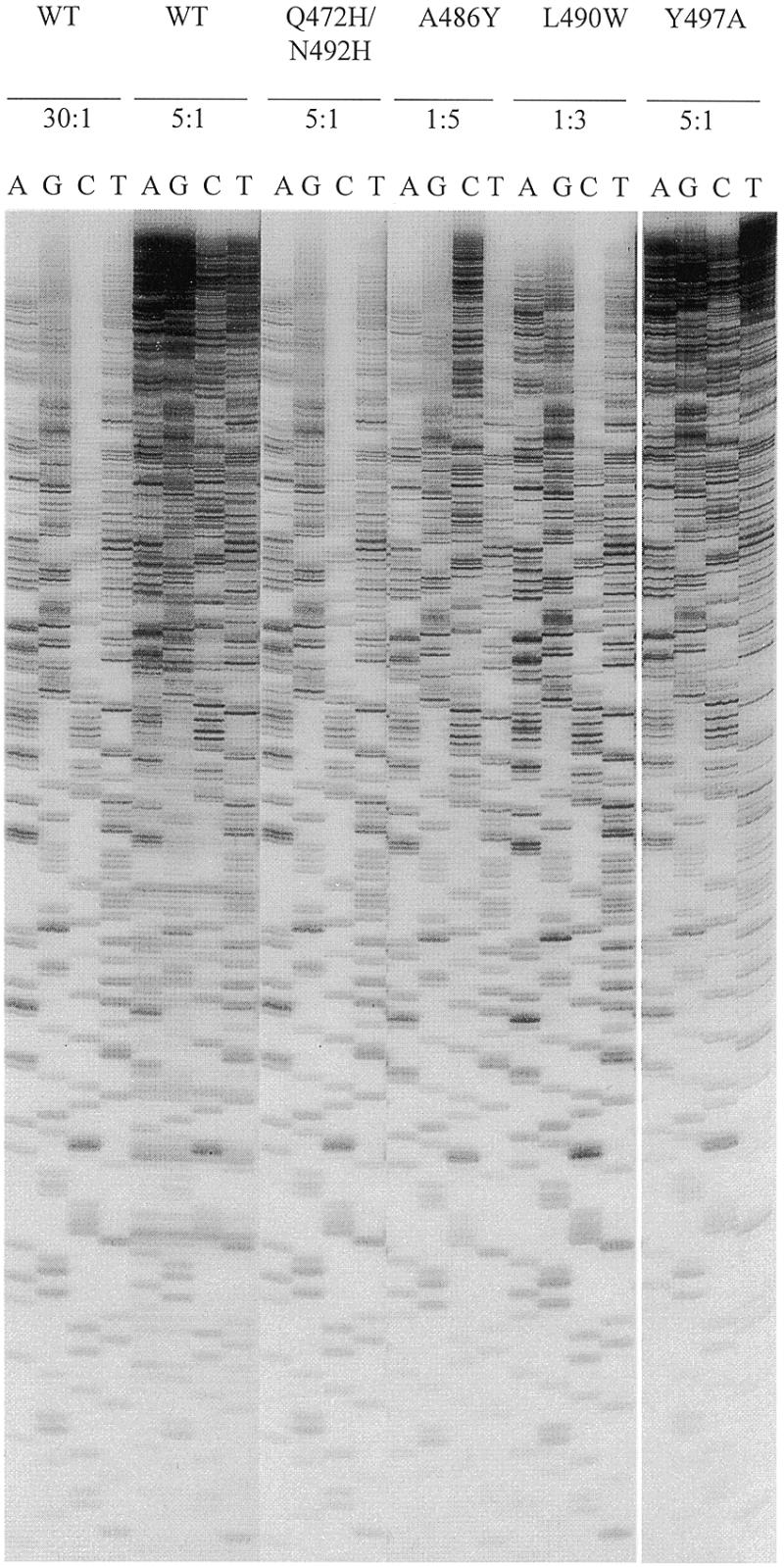
Sequencing ladders found with wild-type Pfu–Pol and selected mutants at various ddNTP:dNTP ratios. For the wild-type enzyme (WT) sequencing information is readable from the ladders produced at the 30:1 ddNTP:dNTP but not at the 5:1 ratio. For the four mutants illustrated the sequencing ladders produced at the ddNTP:dNTP ratios given are of similar quality to that seen with the wild-type at 30:1. Therefore all four mutants give comparable sequencing ladders to the wild type enzyme at reduced ratios of ddNTP:dNTP. A, G, C and T represent the individual tracks for these deoxynucleosides.
Initially, the four most highly conserved amino acids (Q484, K488, N492, Y495), on the side of the P-helix that faces the ddNTP binding site (Figs 1 and 2), were investigated. Most of the mutants showed inferior sequencing ladders at the 30:1 ddNTP:dNTP ratio used for initial screening, when compared to the wild-type enzyme (Table 1). The few mutants that appeared equivalent to the wild type under these conditions all gave hard to read sequencing gels, characterised by radioactivity towards the top of the gel and non-specific termination, at a 5:1 ratio. Both sequence and structural comparisons (Fig. 1) show that the Pol-II family does not have an exact counterpart to the critical phenylalanine/tyrosine found with Pol-I enzymes. The best guess as to which amino acid, if any, would play this role is N492: a residue that is highly conserved (8,9) and structurally at least in a similar location to the phenylalanine/tyrosine (Fig. 1). However, N492Y (i.e. an attempt to introduce a tyrosine near a location shown to give good sequencing performance with the Pol-I family) gave an inactive enzyme. Other mutations to this amino acid, e.g. N492H/K/G, while active resulted in difficult to read ladders at the initial 30:1 ddNTP:dNTP ratio. Changes to Q484, K488 and Y495 gave sequencing phenotypes at best equivalent to, but more commonly worse than, the wild-type enzyme (Table 1).
As shown in Figure 1, not only do the conserved amino acids on the same side of the O- or P-helix vary in the nature of their side chains but also in the spacing between them. The B-motif in the Pol-II family is missing an amino acid when compared to Pol-I. Alternative alignments to that shown in Figure 1, in which a gap in Pol-II is placed opposite the key phenylalanine/tyrosine in Pol-I, have been proposed (8,9). Therefore, insertion mutations consisting of: addition of single tyrosine immediately before N492 [A(Y)N]; replacement of several Pfu–Pol P-helix amino acids with the corresponding regions of the Taq O-helix [TIN(Y)GVL] (Table 1), have been prepared. In these Pfu–Pol variants the ‘missing’ amino acid is replaced with a tyrosine either in the Pfu–Pol or the Taq polymerase context, allowing a more exact sequence alignment of the mutated B-motif with that of the Pol-I family. These insertion mutations also represent another approach to placing a tyrosine near the important phenylalanine/tyrosine in the Pol-I family. Unfortunately, all insertion mutations were inactive (Table 1).
Following alterations to the conserved amino acids and variation in their spacing a number of mutations have also been made in most of the other P-helix amino acids. As can be seen in Table 1 the majority of the changes lead to a sequence performance roughly equivalent to the wild-type polymerase i.e. readable ladders at ddNTP:dNTP ratios of 30:1, no useful sequencing data, with most radioactivity at the top of the gel and non-specific termination at the 5:1 ratio. Therefore, most changes offered no improvement in DNA sequencing performance over that of the wild type. Three mutations, A486Y, L490W and Y497A, were found which gave readable sequencing ladders at the 5:1 ratio. Progressively decreasing the amount of chain terminator showed that A486Y could be used at a ddNTP:dNTP ratio of 1:5, L490W at 1:3 and Y497A at 5:1; representing an improvement over wild type (in terms of ddNTP usage) of 150-, 90- and 6-fold respectively (Table 1, Fig. 3). With both A486Y and L490W there was little accumulation of radioactivity at the top of the gels at the final ddNTP:dNTP ratios used. Although counts are seen at the top of the gel for Y497A at the 5:1 ratio of ddNTP:dNTP, the ladder produced is superior to that seen for the wild type at the same levels of nucleoside triphosphates; little non-specific termination is seen with the mutant, whereas this is considerable with the wild type (Fig. 3). Other mutations to these amino acids e.g. L490Y and Y497W also resulted in preferential usage of ddNTPs when compared to the wild type, although the ddNTP:dNTP ratios useable with these mutants were not as good as with L490W and Y497A respectively. We also found certain mutations to these amino acids e.g. A486W and Y497F (Table 1) that resulted in poorer ddNTP utilisation as compared to the wild- type enzyme.
As well as changing the amino acids in the P-helix in a directed manner, random mutagenesis (30) has been used between amino acids 431 and 532. This stretch of 101 amino acids encompasses the P-helix as well as amino acids flanking this structural element (Fig. 2). Preparing large numbers of random mutants is quick and straightforward; screening for improved phenotype is often more tedious and difficult. We have adapted a ‘colony screening rapid filter assay’ (31), originally used to identify recombinants containing wild-type Taq DNA polymerase, to directly assess improved incorporation of ddNTPs. Essentially E.coli colonies, on nitrocellulose filters containing activated calf-thymus DNA, were incubated with [α-32P]ddATP and dTTP/dCTP/dGTP. Colonies expressing a Pfu–Pol mutant that effectively uses ddNTPs transfer radioactivity to polymeric DNA immobilised on the nitrocellulose filter, allowing subsequent detection by autoradiography. The method includes a heat step, prior to the assay, to destroy host polymerases and so automatically scores for thermostability in the mutant Pfu–Pol. Only one mutant, that appeared to incorporate ddNTPs particularly well, was found and revealed by sequencing to be a double mutant, Q472H-N492H. This double mutant gave good sequencing data at a ddNTP:dNTP ratio of 5:1 (Fig. 3) i.e. a 6-fold improvement over wild type. However, the amount of ddNTP could not be further decreased and so the double mutant does not appear to be as good as either A486Y or L490W. The amino acid at 492, normally asparagine, is one of the conserved P-helix amino acids, described above (Figs 1 and 2). We therefore thought that the improvement in phenotype was due to the N492H change, a combination not tested above. Remarkably single directed mutants showed that the key change was to Q472H, which gave good sequencing ladders, equivalent to those produced by the double mutant, at 5:1 ddNTP:dNTP ratios. The single mutant N492H showed non-specific termination at the 5:1 ratio and in fact gave slightly inferior performance to the wild type when tested at 30:1 ddNTP:dNTP. The amino acid Q472 is located in a loop that connects the P-helix to another long α-helix (Fig. 2) and its location is such that it is unlikely to interact directly with dNTPs. Not only does the mutation Q472H improve ddNTP incorporation but it fully rescues a mutant, N492H, with sequencing performance worse than the wild type. Changes have also been made to Y410, an amino acid that is not in the P-helix, but has been shown by crystallography (27,28) to have an important role in binding dNTPs, forming the bottom of the nucleotide binding cleft. In our hands both Y410F and Y410A offered no improvement over the wild type (Table 1).
Attempts to further reduce the amount of ddNTP needed for sequencing, by combining the improved mutants A486Y, L490W and Q472H-N492H were unsuccessful. One double mutant A486Y-L490W behaved in an identical manner to the single mutant A486Y i.e. gave readable sequencing gels at 1:5 ddNTP:dNTP (Table 1). The other two, Q472H-A486Y-N492H and Q472H-L490W-N492H, actually gave worse sequencing performance, characterised by unreadable gels at ddNTP:dNTP ratios of 1:5 and 1:3, than was seen with A486Y and L490W alone.
DISCUSSION
The aim of this work was to produce a variant of Pfu–Pol capable of efficient incorporation of chain-terminating ddNTPs, a property associated with good sequencing performance, at least in the Pol-I family (4). With Pol-I enzymes a change of a single phenylalanine to tyrosine converts ddNTPs from poor to good substrates (4). It was suggested that the 3′-OH of the natural dNTP substrate was one of the ligands for Mg2+, essential for polymerisation. With ddNTPs, which lack a 3′-OH, this interaction cannot take place, accounting for their poor substrate properties. Most Pol-I enzymes contain a phenylalanine, near the sugar of the dNTP, which is sometimes (either in a few natural Pol-I enzymes e.g. from T7 or by site-directed mutagenesis) replaced by tyrosine. It has been proposed that the phenolic-OH of this tyrosine can replace the Mg2+ ligand lost when ddNTPs are substituted for dNTPs, accounting for the superior sequencing performance of Pol-I enzymes containing a tyrosine at this location. However, sequence alignments (available at the outset of the project; 8,9) and structural information (which became known during the course of the project) show that Pfu–Pol, as a family-II enzyme, does not have an equivalent of the critical phenylalanine/tyrosine.
This publication demonstrates that changes to the amino acids likely to interact directly with the dNTP do not improve discrimination for ddNTPs over dNTPs. This includes the conserved amino acids on one side of the P-helix and Y410 (Figs 1 and 2, Table 1). Structural data (27,28) shows that all these residues are near the dNTP and several are located in the vicinity of the sugar ring. None of the mutations improve ddNTP incorporation and sequencing performance and an effect as profound as that caused by the single phenylalanine/tyrosine switch with the Pol-I family was never observed. Additionally, inserting a single amino acid into the P-helix of Pfu–Pol (to produce a better alignment with the O-helix of Pol-I) or helix swaps between the two categories gave inactive enzyme. This probably arises because of a large overall disruption to Pol-II structure when its P-helix is replaced with the equivalent of an O-helix. Recently a study with the archaeal polymerase from Thermococcus literalis (VentTM DNA polymerase) reached the same conclusion (35) i.e. changes to amino acids in direct contact with the dNTP do not increase the enzyme’s preference for ddNTPs over dNTPs.
Fortunately, changes at three positions within the P-helix, amino acids A486, L490 and Y497 gives rise to a higher selectivity for ddNTPs. As shown in Figure 2, the side chains of these amino acids protrude from the P-helix on the side facing away from the DNA-binding cleft. The three amino acids are unlikely, therefore, to interact directly with the dNTP, as postulated for the four highly conserved amino acids, Q484, K488, N492 and Y495. Mutagenesis of VentTM polymerase also concluded that changes to the equivalent of the Pfu–Pol A486 (A488 in VentTM) lead to an increase in the efficiency of ddNTP usage (35). This study showed that the bigger the side chain used to replace alanine, the better the utilisation of ddNTP. The largest side chain used was phenylalanine and this led to 15-fold better usage of ddNTPs. The change we made, A486Y, allows readable sequencing gels at a ddNTP:dNTP ratio of 1:5, a 150-fold improvement over the wild type. The determination of ‘sequencing performance’ by visual inspection of gels is somewhat subjective; therefore, deciding the ddNTP:dNTP ratio which gives clearly interpretable patterns is difficult. However, we routinely use the Pfu–Pol A486Y mutant for sequencing purposes at the 1:5 ddNTP:dNTP ratio and find it gives reliable sequencing information. Using the largest available side chain in the mutation A486W resulted in poorer ddNTP usage as compared to both A486Y and the wild- type enzyme. What is clear, though, is that this amino acid represents a residue important for ddNTP usage, even though any improvement is far less than the phenylalanine/tyrosine switch in the Pol-I family. L490 is one turn along the helix from A486 and the side chains of these two amino acids point in almost identical directions (Fig. 2). This amino acid was not studied with VentTM but we have found that the mutation L490W improves ddNTP usage, by a factor only slightly less than the A486Y change. L490Y, while slightly improved over wild type, is far less effective than L490W. This may, like the changes to the alanine described above, result from a correlation between side chain size and ddNTP utilisation. However, as there are not many larger side chains than the wild-type leucine, it is difficult to test this experimentally with a series of mutations as carried out with VentTM polymerase and A488 (35). Two other P-helix amino acids, L479 and Y497, have side chains that point in almost the same direction as those of A486 and L490. Changes to L479 give a phenotype similar to wild type (Table 1). However, some changes to Y497 (Y497W and Y497A) lead to a slightly improved preference for ddNTPs (Table 1) whereas others Y497F decrease ddNTP utilisation. A similar effect was seen with VentTM with the corresponding amino acid Y499 (35).
Not only are A486 and L490 unlikely to interact with dNTPs directly, but they are not highly conserved in the Pol-II family. Although the equivalent position to A486 is most usually alanine, other amino acids e.g. asparagine, serine, isoleucine, leucine and phenylalanine are found (8,9). Similarly L490 is commonly replaced by either another hydrophobic amino acid or threonine/serine. This non-conservation emphasises that A486 and L490 are unlikely to have a direct critical function e.g. binding dNTP, primer/template or in catalysis. How then do they influence ddNTP:dNTP selectivity? The mutations A486Y and L490W (and also Y497A) might cause small movements throughout the polymerase, resulting in a slight change in the arrangement of the amino acids that constitute the dNTP binding site. This could, fortuitously favour ddNTP incorporation over dNTP. Alternatively, as suggested in the studies with VentTM (35), the discrimination on the kinetic level between nucleoside triphosphates may be controlled by a protein conformational change. The successful mutations with both Pfu–Pol and VentTM may manipulate this conformational change such that ddNTPs become better substrates.
The indirect action of the mutants A486Y, L490W and Y497A was confirmed by the detection of a fourth mutant, associated with better ddNTP usage. Random mutagenesis to a region including both the P-helix and its flanking sequences revealed the double mutant Q472H-N492H. The improved properties do not result from the change to N492 (the conserved asparagine in the P-helix, Figs 1 and 2) but, as shown by subsequent single mutants (Table 1), to alteration to Q472. The residue Q472 is in a loop that lies between the P-helix and a second long α-helix. These two anti-parallel helices comprise the bulk of the fingers domain of the RB69 (27) and Tgo (28) polymerases and presumably also of the Pfu–Pol. Q472 is highly variable between Pol-II enzymes and even differs within the more closely related archaeal sub-set of this class (8,9) e.g. this residue is isoleucine with Tgo–Pol and VentTM (Fig. 2). Q472H, or the double mutant Q472H-N492H, only improve ddNTP incorporation, as assessed by sequencing gels, by a factor of 6-fold. However, this effect has proved to be reproducible in very many DNA sequence analyses. The Q472H mutation could improve performance by bringing about slight conformational changes that alter the active site to suit ddNTPs, as discussed above. However, we prefer the interpretation that the alteration facilitates a conformational change, critical for both polymerisation and nucleotide triphosphate selectivity.
Although both this study and a similar one with VentTM polymerase (35) have managed to isolate Pol-II mutants that preferentially use ddNTPs and give better DNA sequencing gels, the derivatives so far obtained are a long way short of being competitive with current state of the art sequencing enzymes derived from the Pol I family, like Thermo-SequenaseTM (6,7). Converting an archaeal Pol-II enzyme to a good DNA sequencer is likely to be a much more subtle and difficult task than in the Pol-I case.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Pauline Heslop for expert technical assistance. This work was supported by a LINK grant (industrial partner Amersham Pharmacia Biotech Inc.) from the UK MRC.
REFERENCES
- 1.Sanger F., Nicklen,S. and Coulson,A.R. (1977) Proc. Natl Acad. Sci. USA, 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bankier A.T., Weston,K.M. and Barrell,B.G. (1987) In Wu,R. (ed.), Methods in Enzymology, Vol. 155. Academic Press Inc., San Diego, CA, pp. 51–93. [DOI] [PubMed]
- 3.Prober J.M., Trainer,G.L., Dam,R.J., Hobbs,F.W., Robertson,C.W., Zagursky,R.J., Cocuzza,A.J., Jensen,M.A. and Baumeister,K. (1987) Science, 238, 336–341. [DOI] [PubMed] [Google Scholar]
- 4.Tabor S. and Richardson,C.C. (1995) Proc. Natl Acad. Sci. USA, 92, 6339–6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Astatke M., Grindley,N.D.F. and Joyce,C.A. (1988) J. Mol. Biol., 278, 147–165. [DOI] [PubMed] [Google Scholar]
- 6.Reeve M.A. and Fuller,C.W. (1995) Nature, 376, 796–797. [DOI] [PubMed] [Google Scholar]
- 7.Vander Horn P.B., Davis,M.C., Cunniff,J.J., Ruan,C., McCardle,B.F., Samols,S.B., Szasz,J.G., Hujer,K.M., Domke,S.T., Brummet,S.R., Moffett,R.B. and Fuller,C.W. (1997) Biotechniques, 22, 758–765. [DOI] [PubMed] [Google Scholar]
- 8.Delarue M., Poch,O., Tordo,N., Moras,D. and Argos,P. (1990) Protein Eng., 3, 461–467. [DOI] [PubMed] [Google Scholar]
- 9.Braithwaite D.K. and Ito,J. (1993) Nucleic Acids Res., 21, 787–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joyce C.M. and Steitz,T.A. (1994) Annu. Rev. Biochem., 63, 777–782. [DOI] [PubMed] [Google Scholar]
- 11.Ollis D.L., Brick,P., Hamlin,R., Xuong,N.G. and Steitz,T.A. (1985) Nature, 313, 762–766. [DOI] [PubMed] [Google Scholar]
- 12.Freemont P.S., Friedman,J.M., Beese,L.S., Sanderson,M.R. and Steitz,T.A. (1998) Proc. Natl Acad. Sci. USA, 85, 8924–8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beese L.S., Derbyshire,V. and Steitz,T.A. (1993) Science, 260, 352–355. [DOI] [PubMed] [Google Scholar]
- 14.Kim Y., Eom,S.H., Wang,J., Lee,D.S., Suh,S.W. and Steitz,T.A. (1995) Nature, 376, 612–616. [DOI] [PubMed] [Google Scholar]
- 15.Kiefer J.R., Mao,C., Hansen,C.J., Basehore,S.L., Hogrefe,H.H., Bramman,J.C. and Beese,L.S. (1997) Structure, 5, 95–108. [DOI] [PubMed] [Google Scholar]
- 16.Kiefer J.R., Mao,C., Bramman,J.C. and Beese,L.S. (1998) Nature, 391, 304–307. [DOI] [PubMed] [Google Scholar]
- 17.Doublie S., Tabor,S., Long,A.M., Richardson,C.C. and Ellenberger,T. (1998) Nature, 391, 251–258. [DOI] [PubMed] [Google Scholar]
- 18.Murray V. (1989) Nucleic Acids Res., 17, 8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koop B.F., Rowan,L., Chen,W.-Q., Deshpande,P., Lee,H. and Hood,L. (1993) Biotechniques, 14, 442–447. [PubMed] [Google Scholar]
- 20.Perler F.B., Comb,D.G., Jack,W.E., Moran,L.S., Qiang,B., Kucera,R.B., Benner,J., Slatko,B.E., Nwankwo,D.O., Hempstead,S.K., Carlow,C.K.S. and Jannasch,H. (1992) Proc. Natl Acad. Sci. USA, 89, 5577–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uemori T., Ishino,Y., Toh,H., Asada,K. and Kato,I. (1993) Nucleic Acids Res., 21, 259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Southworth M.W., Kong,H., Kucera,R.B., Ware,J., Jannasch,H.W. and Perler,F.B. (1996) Proc. Natl Acad. Sci. USA, 93, 5281–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perler F.B., Kumar,S. and Kong,H. (1996) Adv. Protein Chem., 63, 377–435. [DOI] [PubMed] [Google Scholar]
- 24.Blasco M.A., Lazaro,J.M., Bernad,A., Blanco,L. and Salas,L. (1992) J. Biol. Chem., 267, 19427–19434. [PubMed] [Google Scholar]
- 25.Dong Q., Copeland,W.C. and Wang,T.S. (1993) J. Biol. Chem., 268, 24163–24174. [PubMed] [Google Scholar]
- 26.Dong Q. and Wang,T.S. (1995) J. Biol. Chem., 270, 21563–21570. [DOI] [PubMed] [Google Scholar]
- 27.Wang J., Sattar,A.K.M.A., Wang,C.C., Karam,J.D., Konigsberg,W.H. and Steitz,T.A. (1997) Cell, 89, 1087–1099. [DOI] [PubMed] [Google Scholar]
- 28.Hopfner K.-P., Eichinger,A., Engh,R.A., Laue,F., Ankenbauer,W., Huber,R. and Angerer,B. (1999) Proc. Natl Acad. Sci. USA, 96, 3600–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uemori T., Ishino,Y., Toh,H., Asada,K. and Kato,I. (1993) Nucleic Acids Res., 21, 259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zacolo M., Williams,D.M., Brown,D.M. and Gherardi,E. (1996) J. Mol. Biol., 255, 589–603. [DOI] [PubMed] [Google Scholar]
- 31.Sagner G., Rüger,R. and Kessler,C. (1991) Gene, 97, 119–123. [DOI] [PubMed] [Google Scholar]
- 32.Ho S.N., Hunt,H.D., Horton,R.M., Pullen,J.K. and Pease,L.R. (1989) Gene, 77, 51–59. [DOI] [PubMed] [Google Scholar]
- 33.Richardson C.C. (1996) In Cantini,G.L. and Davies,D.R. (eds), Procedures in Nucleic Acids Research. Harper and Row, New York, NY, pp. 263–273.
- 34.Guex N. and Peitsch,M.C. (1997) Electrophoresis, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- 35.Gardner A.F. and Jack,W.E. (1999) Nucleic Acids Res., 27, 2545–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sayle R. and Milner-White,J.E. (1995) Trends Biochem. Sci., 20, 374–376. [DOI] [PubMed] [Google Scholar]