

Abstract
Individual nucleotide resolution UV cross-linking and immunoprecipitation followed by high-throughput sequencing (iCLIP-seq) is a powerful technique that is used to identify RNA-binding proteins’ (RBP) binding sites on target RNAs and to characterize the molecular basis of posttranscriptional regulatory pathways. Several variants of CLIP have been developed to improve its efficiency and simplify the protocol [e.g., iCLIP2 and enhanced CLIP (eCLIP)]. We have recently reported that transcription factor SP1 functions in the regulation of alternative cleavage and polyadenylation through direct RNA binding. We utilized a modified iCLIP method to identify RNA-binding sites for SP1 and several of the cleavage and polyadenylation complex subunits, including CFIm25, CPSF7, CPSF100, CPSF2, and Fip1. Our revised protocol takes advantage of several features of the eCLIP procedure and also improves on certain steps of the original iCLIP method, including optimization of circularization of cDNA. Herein, we describe a step-by-step procedure for our revised iCLIP-seq protocol, that we designate as iCLIP-1.5, and provide alternative approaches for certain difficult-to-CLIP proteins.
Key features
Identification of RNA-binding sites of RNA-binding proteins (RBPs) at nucleotide resolution.
iCLIP-seq provides precise positional and quantitative information on the RNA-binding sites of RBPs in living cells.
iCLIP facilitates the identification of sequence motifs recognized by RBPs.
Allows quantitative analysis of genome-wide changes in protein-RNA interactions.
Revised iCLIP-1.5 protocol is more efficient and highly robust; it provides higher coverage even for low-input samples.
Graphical overview
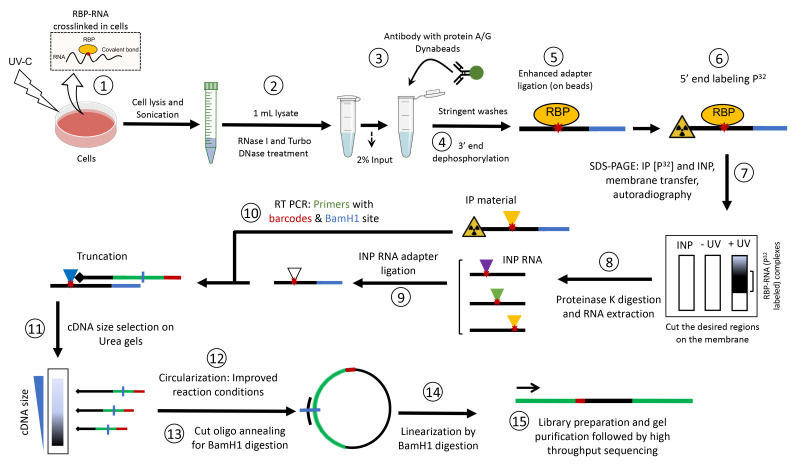
Keywords: iCLIP, eCLIP, Posttranscriptional regulation, RNA-binding proteins, UV crosslinking
Background
RNA-binding proteins (RBPs) are key players in posttranscriptional regulation, as they regulate essentially all aspects of mRNA metabolism, including 5′ cap formation, pre-mRNA splicing, 3′ end formation, mRNA export, and mRNA stability (Hentze et al., 2018). RBPs that decorate an RNA molecule during its life can dynamically change in space and time while they tightly co-ordinate posttranscriptional regulation (Hafner et al., 2021). Increasingly, links are being detected between defects in RBPs’ functions and human diseases, including neurological disorders and cancer (Pereira et al., 2017). Thus, understanding the role of RBPs in posttranscriptional regulation, and identifying their full repertoire of RNA targets in cells, is of fundamental importance for both better characterizing gene expression regulatory programs and drug development.
Although numerous methods exist to characterize RBPs’ target RNAs in cells (e.g., RIP, TRIBE, and APEX-seq), the cross-linking and immunoprecipitation (CLIP)-based methods are considered the gold standard for precisely identifying endogenous RNA-binding sites of RBPs (Hafner et al., 2021). Several variants of CLIP have been developed, including HITS-CLIP, individual-nucleotide resolution CLIP (iCLIP), ir-CLIP, enhanced CLIP (eCLIP), and iCLIP2, aiming to simplify the protocol and improve its efficiency (Buchbender et al., 2020; Konig et al., 2011; Lee et al., 2021; Van Nostrand et al., 2016; Zarnegar et al., 2016). These methods all rely on exposure of live cells to UV-C light, which covalently crosslinks RBPs to their target RNAs. The CLIP method can be carried out so as to provide precise positional and quantitative information on the crosslink sites. In particular, iCLIP aims to detect RNA fragments that truncate at the crosslink site during cDNA preparation, and thus reveals RBP binding sites on target RNA at individual nucleotide resolution. Since the original iCLIP method is laborious, and the library preparation efficiency can become limiting for low input samples (Huppertz et al., 2014), eCLIP was developed to omit the inefficient cDNA circularization step, improve the adapter ligation conditions, and implement a size-matched input (SMI) to control for background signal (Van Nostrand et al., 2016). Although eCLIP maintains the individual nucleotide resolution of iCLIP, it does not include direct visualization of protein-associated RNA. Hence, the quality of immunoprecipitated RNA, as well as the possibility of co-purifying background RBPs, cannot be directly monitored.
We have recently modified the original iCLIP method to interrogate the posttranscriptional regulatory functions of the ubiquitously expressed transcription factor SP1 (Song et al., 2022). By identifying its RNA-binding sites, we found that SP1 preferentially binds to A/G-rich sequence motifs on target RNAs. We further utilized our revised iCLIP procedure to examine the binding profiles of several cleavage and polyadenylation factors on target mRNAs (Song et al., 2022). Our data yielded high-resolution RNA-binding maps for SP1, as well as the cleavage and polyadenylation complex subunits, and unraveled a previously unknown function of SP1 in alternative cleavage and polyadenylation regulation. We have used this protocol with several cell lines (e.g., mouse neuroblastoma N2a cells, cgr8 embryonic stem cells, HeLa cells, glioblastoma, and HEK293 cells) to identify RNA targets of RBPs involved in diverse functions, including splicing regulation, mRNA stability, mRNA export, 5′ capping, and mRNA modifications (Nitoiu et al., 2021; Han et al., 2017 and 2022; Nabeel-Shah et al., 2022 ). Our revised iCLIP protocol combines the improvements previously implemented in eCLIP (e.g., enhanced adapter ligation and SMI), with the optimization of several steps in the original iCLIP method, including improved circularization of the cDNA by increasing the incubation time and adding Betain in the reaction mixture, which helps the circularization of difficult-to-ligate substrates. Moreover, we utilize two-step dephosphorylation of RNA fragments to ensure the complete removal of the 3′-cyclic phosphate group left behind by RNase I cleavage. Since iCLIP2 is more similar to eCLIP (i.e., omission of circularization and linearization steps), whereas our protocol essentially follows the workflow of the original iCLIP procedure, we designate our revised protocol as iCLIP-1.5.
Materials and reagents
Protein G Dynabeads (Life Technologies, catalog number: 10004D)
Protein A Dynabeads (Life Technologies, catalog number: 10002D)
GFP recombinant rabbit monoclonal antibody (Thermo Fisher Scientific, catalog number: G10362)
Rabbit polyclonal anti-GFP (Abcam, catalog number: ab290)
SP1 antibody (Santa Cruz, catalog number: sc-17824)
Protease Inhibitor Cocktail Set III (Calbiochem/Merck, catalog number: 539134-1SET)
RNase I (Life Technologies, catalog number: AM2295)
Turbo DNase (Life Technologies, catalog number: AM2238)
T4 PNK plus 10× PNK buffer (NEB, catalog number: M0201L)
Murine RNase inhibitor 40 U/µL (NEB, catalog number: M0314L)
T4 RNA ligase 1 high conc 30 U/µL (NEB, catalog number: M0437M)
10× T4 RNA ligase reaction buffer (NEB, catalog number: B0216SVIAL)
FastAP 1 U/µL (Life Technologies, catalog number: EF0652)
Proteinase K 0.8 U/µL (NEB, catalog number: P8107S)
MyONE Silane beads (Life Technologies, catalog number: 37002D)
Pre-adenylated adapter L3-App IDT (rAppAGATCGGAAGAGCGGTTCAG/ddC/)
ATP [γ-32P] (PerkinElmer, catalog number: NEG502A250UC)
4%–12% NuPage gels (Life Technologies, catalog number: NP0322BOX)
LDS-4× sample buffer (NuPage loading buffer) (Life Technologies, catalog number: NP0007)
Pre-stained protein size marker (Fermentas, catalog number: SM1811)
Nitrocellulose membrane protran BA85 (VWR, catalog number: 732-4174)
20× transfer buffer (Life Technologies, catalog number: NP0006-1)
20× MOPS-SDS running buffer (Life Technologies, catalog number: NP0001)
Whatman filter paper (GE Healthcare, catalog number: 3030917)
Film (Fuji, catalog number: 4741019236)
Phenol/chloroform (Sigma, catalog number: P3803)
Phase lock gel heavy tube (VWR, catalog number: 713-2536)
GlycoBlue (Ambion, catalog number: 9510)
3 M sodium acetate pH 5.5 (Life Technologies, catalog number: AM9740)
2× TBE-urea loading buffer (Life Technologies, catalog number: LC6876)
6% TBE-urea pre-cast gels (Life Technologies, catalog number: EC68652B)
Low molecular weight marker (NEB, catalog number: N3233L)
TBE running buffer (Life Technologies, catalog number: LC6675)
SYBR green II (Life Technologies, catalog number: S-7564)
19 G syringe needle (BD, Microlance, catalog number: 300637)
Glass pre-filters (Whatman, catalog number: 1823010)
Costar SpinX column (Corning, catalog number: 8161)
CircLigaseTM (Lucigen, catalog number: CL4115K)
10× Circligase buffer (Lucigen, catalog number: CL4115K)
Betain solution 5 M (Sigma, catalog number: B0300-1VL)
Cut_oligo ‘GTTCAGGATCCACGACGCTCTTCaaaa’ (HPLC-purified, IDT)
BamHI (Fermentas, catalog number: FD0055)
Fast digest buffer (Fermentas, catalog number: FD0055)
2× Phusion High-Fidelity PCR Master Mix (NEB, catalog number: M0531L)
NaOH (Merck/Sigma, catalog number: S8045-500G)
RLT buffer (Qiagen, catalog number: 79216)
PBS (Sigma-Aldrich, catalog number: D8537)
UltraPureTM agarose (Invitrogen, catalog number: 16500-500)
QIAquick Gel Extraction kit (Qiagen, catalog number: 28706)
Tris (VWR, catalog number: 0826)
Sodium chloride (NaCl) (VWR, catalog number: 27810.364)
Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M2670)
Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: C1016)
Potassium chloride (KCl) (Thermo Fisher Scientific, catalog number: AM9640G)
Urea (Thermo Fisher Scientific, catalog number: 15505035)
Sodium deoxycholate (Thermo Fisher Scientific, catalog number: 89904)
Tween-20 (Thermo Fisher Scientific, catalog number: 28320)
Sodium dodecyl sulfate (SDS) (Thermo Fisher Scientific, catalog number: 28364)
Triton X-100 (Thermo Fisher Scientific, catalog number: 28314)
-
Primers (HPLC-purified from IDT):
P5_Solexa: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT
P3_Solexa: CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT
Rt1clip/5Phos/NNAACCNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt2clip/5Phos/NNACAANNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt3clip/5Phos/NNATTGNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt4clip/5Phos/NNAGGTNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt5clip/5Phos/NNCGCCNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt6clip/5Phos/NNCCGGNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt7clip/5Phos/NNCTAANNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt8clip/5Phos/NNCATTNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt9clip/5Phos/NNGCCANNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt10clip/5Phos/NNGACCNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt11clip/5Phos/NNGGTTNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt12clip/5Phos/NNGTGGNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt13clip/5Phos/NNTCCGNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt14clip/5Phos/NNTGCCNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt15clip/5Phos/NNTATTNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Rt16clip/5Phos/NNTTAANNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
Solutions
Lysis buffer (see Recipes)
High-salt wash (see Recipes)
PNK buffer (see Recipes)
PK buffer (see Recipes)
PK buffer + 7 M urea (see Recipes)
1× RNA ligase buffer (see Recipes)
10× RNA ligase buffer (see Recipes)
1× FastAP buffer (see Recipes)
Recipes
-
Lysis buffer
50 mM Tris-HCl, pH 7.4
100 mM NaCl
1% Igepal CA-630
0.1% SDS
0.5% sodium deoxycholate
On the day: 1/100 volume of Protease Inhibitor Cocktail Set III, for tissues: 1/1,000 volume of ANTI-RNase
-
High-salt wash
50 mM Tris-HCl, pH 7.4
1 M NaCl
1 mM EDTA
1% Igepal CA-630
0.1% SDS
0.5% sodium deoxycholate
-
PNK buffer
20 mM Tris-HCl, pH 7.4
10 mM MgCl2
0.2% Tween-20
-
PK buffer
100 mM Tris-HCl, pH 7.4
50 mM NaCl
10 mM EDTA
-
PK buffer + 7 M urea
100 mM Tris-HCl, pH 7.4
50 mM NaCl
10 mM EDTA
7 M urea
-
1× RNA ligase buffer (no DTT)
50 mM Tris-HCl pH 7.5
10 mM MgCl2
-
10× RNA ligase buffer (no DTT)
500 mM Tris-HCl pH 7.5
100 mM MgCl2
-
1× FastAP buffer
10 mM Tris pH 7.5
5 mM MgCl2
100 mM KCl
0.02% Triton X-100
Equipment
Refrigerated microcentrifuge
Picofuge (Thermo Fisher Scientific, catalog number: 75004061)
Stratalinker UV crosslinker 2400 Stratagene
Denaturing agarose gel apparatus (Bio-Rad Laboratories)
Magnetic stand (Thermo Fisher Scientific, Invitrogen)
Electrophoresis chamber (Life Technologies, catalog number: EI0002)
Transfer apparatus (Life Technologies, catalog number: EI0002)
Sponge pads for XCell II blotting (Life Technologies, catalog number: EI9052)
Thermoblock (Thermo Fisher Scientific, Invitrogen)
Thermomixer (Boekel Scientific)
Sonicator (Branson Digital Sonifier)
Geiger counter sensitive to beta particles (Ludlum 3 survey meter Geiger counter radiometer 44-9 pancake probe)
3/8" or 1/2" Plexiglas benchtop shield (P32 protective shield)
Cell scraper
Liquid nitrogen (N2)
Saran wrap
Blue light transilluminator
UV transilluminator
X-ray film cassette
Software
iMAPs (Jernej Ule’s lab, https://imaps.goodwright.com/)
PureCLIP (Sabrina Krakau, https://pureclip.readthedocs.io/en/latest/index.html)
CTK package (Chaolin Zhang’s lab, https://zhanglab.c2b2.columbia.edu/index.php/CTK_Documentation)
Procedure
We describe below the step-by-step procedure for performing an iCLIP-seq experiment using GFP-tagged proteins with modifications/improvements implemented at various steps. This procedure (designated as iCLIP-1.5) has been applied to both epitope-tagged proteins (i.e., GFP- or FLAG-tagged proteins) and endogenous proteins (e.g., SP1) using validated antibodies. For doxycycline-inducible epitope-tagged cell lines, induce the expression of protein 24 h prior to harvesting. Since this protocol follows the workflow of the original iCLIP-seq method (Huppertz et al., 2014), the data generated using this protocol can be subjected to the standard iCLIP analysis pipeline.
Store all buffers at 4 °C and perform the procedure on ice. Room temperature is defined as 22 °C throughout this protocol. All washing steps throughout the protocol are performed with a volume of 900µL unless stated differently.
-
UV crosslinking of cells
Grow HEK293 cells in 15 cm diameter tissue culture plates (~80% confluent) (Note 1).
Remove the media and wash cells once with ice-cold 1× PBS. Gently add 6 mL of ice-cold PBS and place the plate on a small ice tray that fits inside the Stratalinker (Note 2).
Remove the lid and irradiate the cells once with 150 mJ/cm2 at 254 nm in a Stratalinker 2400 (or equivalent UV-C crosslinker) (Notes 3, 4, 5).
Scrape off the cells and transfer to a 15 mL Falcon tube. Centrifuge at 1,000× g for 5 min at 4 °C, remove the supernatant, and snap freeze the cells for later use.
-
Lysis, partial RNA digestion, and immunoprecipitation
Prepare protein G Dynabeads for conjugation with anti-GFP antibody (Note 6). Wash 100 μL of protein G Dynabeads with 900 μL of lysis buffer (with protease inhibitors added) by gently pipetting up and down a few times. Place the tubes on a magnetic rack to separate the bead and remove the supernatant. Repeat the washing step three times and, after the last wash, resuspend beads in 100 μL of lysis buffer.
Add 10 μg of anti-GFP antibody. Rotate tubes for 1 h at room temperature (i.e., ~22 °C) (Note 7).
While beads are rotating, resuspend the cell pellets in 2.2 mL of iCLIP lysis buffer supplemented with protease inhibitors (1:100 Protease Inhibitor Cocktail Set III). Cells expressing free FLAG/GFP can be used as negative controls (Note 9).
Sonicate the samples with a probe sonicator: 15 cycles, 0.3 s on, 0.7 s off (three rounds) (Note 10).
Transfer 1 mL of cell lysate to a 1.5 mL Eppendorf tube.
Prepare at least two RNase I dilutions in PBS: 1:50 dilution for high RNase I and 1:250 dilution for low RNase I (Notes 11, 12, 13).
-
Add 20 µL of RNase I (from the dilution stocks) and 2 µL of Turbo DNase to 1 mL of lysate and place the tubes on a thermomixer. Shake the samples in a thermomixer (1,200 rpm) for exactly 5 min at 37 °C, and immediately put back on ice for 5 min.
Critical: Optimization of RNase I dilution is extremely important for the success of the experiment (see Note 11). Incubate the samples for exactly 5 min at 37 °C.
Centrifuge the lysate at 16,100× g in a microcentrifuge for 30 min at 4 °C. Transfer the clarified supernatant to a new 1.5 mL Eppendorf tube and keep on ice.
Transfer 20 µL of clarified lysate to an Eppendorf tube as input. Snap freeze in dry ice/liquid nitrogen and store at -80 °C until further use.
After 1 h, wash the antibody-conjugated beads (from Step B1) twice with 900 µL of ice-cold lysis buffer.
After the last wash, add the clarified cell lysate to the beads. Rotate in a cold room (4 °C) for 2 h or overnight (Note 14).
Place the tubes on a magnetic rack to separate the beads, remove the supernatant, and add 900 µL of high-salt buffer (Note 15).
Rotate for 5 min in the cold room. Repeat three times.
Wash three times with PNK buffer (no need to rotate in the cold room). Add 900 µL of PNK buffer and gently pipette up and down. Place the tubes back on a magnetic rack and discard the supernatant (Note 16).
Wash once with 1× FastAP buffer (no need to rotate in the cold room). Add 900 µL of 1× FastAP buffer and gently pipette up and down. Place the tubes back on a magnetic rack and discard the supernatant.
-
RNA 3′ end dephosphorylation, L3 adapter ligation, and 5′ end labeling
To prime RNA fragments for 3′ adapter ligation, RNA 3′ end dephosphorylation is carried out in two steps. This ensures the complete removal of the 3′-cyclic phosphate group left behind by RNase I cleavage.
-
Resuspend the beads in 100 µL of the following mix and shake at 37 °C for 15 min in a thermomixer (1,200 rpm).
H2O 79 μL
10× FastAP buffer 10 μL
RNase inhibitor 2 μL
Turbo DNase 1 μL
FastAP enzyme 8 μL
-
While tubes are shaking, prepare 300 µL of the following mix for each sample. Add the mix to each tube and incubate for another 20 min while shaking.
H2O 230 µL
5× PNK (pH 6.5) 60 µL
0.1 M DTT 3 µL
Turbo DNase 1 µL
RNase inhibitor (murine) 2 µL
T4 PNK enzyme 4 µL
Separate the beads and wash once with 900 µL PNK buffer (no need to rotate in the cold room).
Wash three times with high salt buffer. Rotate each time for 5 min in the cold room.
Wash three times with PNK buffer (no need to rotate in the cold room).
Wash once with 1× ligase buffer (no DTT) by gently pipetting up and down (Note 16).
-
Remove the buffer and resuspend the beads in 30 μL of the ligation mixture.
H2O 12 µL
10× ligase buffer (no DTT) 3 µL
100% DMSO 0.6 µL
50% PEG 8000 9 µL
RNase inhibitor (murine) 0.4 µL
T4 RNA ligase 1 (high conc 30 U/µL) 2.5 µL
Pre-adenylated adapter L3-App (20 μM) 2.5 µL
Incubate at room temperature for 90 min. Flick the tube every 10–15 min. Alternatively, the tubes can be shaken in a thermomixer at room temperature.
Add 500 μL of PNK buffer and place on the magnetic rack to separate the beads. Discard the supernatant.
Wash three times with high-salt buffer. Rotate each time in the cold room for 5 min.
Wash three times with PNK buffer without rotation in the cold room. Gently pipette up and down a few times. Place the tubes back on a magnetic rack and discard the supernatant.
Resuspend the beads in 1 mL of PNK buffer.
-
Transfer 200 μL of beads (20%) (hot beads) to a new tube. Keep the remaining 800 μL of beads in PNK buffer on ice (cold beads). Remove the supernatant from the hot beads and resuspend in the 5′ end labeling mixture.
T4 PNK 0.2 μL
32P-γ-ATP 0.4 μL
10× PNK buffer 0.4 μL
H2O 3.0 μL
Caution: All 32P-related work must be performed in a designated radioisotope area. Always work behind the protective shield when handling radioactive material.
Incubate for 5 min at 37 °C while shaking at 1,200 rpm in a thermomixer.
Separate the hot beads using the magnetic rack and discard the supernatant as radioactive waste.
Resuspend hot beads in 20 μL of 1× NuPage loading buffer (LDS sample buffer). Optional: add reducing agent (DTT) in the 1× NuPage loading buffer to break the antibody chains.
Remove supernatant from cold beads and mix them with hot beads.
Take out input tubes from -80 °C. Thaw on ice and add LDS sample loading buffer.
Heat the beads and input tubes at 70 °C for 5 min. Magnetically separate the beads and transfer the supernatant into a new tube.
-
Load the samples (20 μL) onto a 4%–12% NuPAGE Bis-Tris gel (Note 17). Use 1× MOPS-SDS running buffer (diluted from 20× MOPS-SDS running buffer). Also, load 5 μL of a pre-stained protein size marker.
1 2 3 4 5 6 7 8 Ladder Input Ladder Sample1 Empty Sample2 Empty Control Run the gel for 1 h at 180V. For high molecular weight proteins, run the gel for 72 min.
Cut the ATP-containing dye front from the gel bottom and discard as solid radioactive waste.
Set up the transfer using a pure nitrocellulose membrane and Whatman filter papers according to the manufacturer's instructions. Use 1× transfer buffer (diluted from 20× transfer buffer) (Note 17).
Transfer in the cold room at 14 V overnight.
-
-
RNA isolation
Rinse the membrane in ice-cold PBS and wrap it with saran wrap. Expose to Fuji film at -80 °C for 30 min in a dark room (Notes 18, 19, 20).
-
Isolate the RNA-protein complexes using the autoradiograph as a mask for cutting the desired regions on the membrane (Figure 1). Cut the membrane into small pieces (~1–2 mm slices) using a new sterile razor blade for each sample and place them in Eppendorf tubes. Keep the tubes on ice until all samples have been excised.
Note: Representative autoradiographs shown in A and B are for two unrelated RBPs.
For inputs, cut the membrane in parallel with the IP samples, matching the sizes of the excised areas (Figure 1B).
Add 20 μL of Proteinase K into 180 μL of PK buffer. Add this Proteinase K mixture to tubes containing the membrane pieces.
Shake the tubes in a thermomixer at 37 °C for 20 min.
Add 200 μL of PK-7M urea buffer and shake for another 20 min at 37 °C. For making PK-7M urea buffer, first dissolve 420 mg of urea in 500 µL of PK buffer, then bring to a final volume of 1 mL by slowly adding PK buffer.
Centrifuge the tubes briefly, collect the solution, and add it together with 400µL phenol/chloroform to a 2mL Phase Lock Gel Heavy tube.
Shake at 37 °C for 5 min in a thermomixer.
Separate the phases by centrifuging the tubes for 5 min at full speed in a tabletop centrifuge at room temperature.
Transfer the aqueous layer into a new tube (be careful not to touch the gel matrix). Centrifuge the supernatant again for 1min and transfer into a new tube.
Add 0.75µL of GlycoBlue and 40µL of 3M sodium acetate (pH 5.5), mix, and add 1mL of ice-cold 100% ethanol (EtOH). Mix by inverting the tubes several times and place overnight at -20 °C.
The next day, centrifuge the samples at 16,100× g for 20 min at 4 °C.
Remove the supernatant and wash the pellet with 900 µL of ice-cold 80% EtOH and centrifuge again for 10 min.
Resuspend the pellet in 5 μL of H2O and transfer to a PCR tube. Keep on ice (or -80 °C) until the input samples are ready for cDNA synthesis.
-
Input adapter ligation
Prior to converting RNA samples into cDNA, input RNA must undergo L3 adapter ligation. Of note, for certain spliceosome subunits (e.g., U2AF65 and PUF60), L3 adapter ligation to the immunoprecipitated protein-RNA complexes does not occur efficiently, presumably due to steric hinderance (Shao et al., 2014; Han et al., 2022). In such cases, we recommend that the IP samples should be processed along with inputs so that, after immunoprecipitation and three high-salt buffer washes (Step B12), you proceed directly to radiolabeling (Step C11) and omit the dephosphorylation and adapter ligation steps described above in section C. Isolate the RNA from the membrane as detailed above and continue with the steps noted below for input samples.
-
To 5 μL input samples, add:
H2O 15 μL
10× FastAP buffer 2.5 μL
RNase inhibitor 0.5 μL
FastAP enzyme 2.5 μL
-
Incubate at 37 °C for 15 min while shaking at 1,200 rpm. Then, add 75 μL of the following master mix:
H2O 48 µL
5× PNK (pH 6.5) 20 µL
0.1M DTT 1 µL
RNase inhibitor (murine) 1 µL
Turbo DNase 1 µL
T4 PNK enzyme 4 µL
Incubate for another 20 min at 37 °C while shaking at 1,200 rpm.
Using the magnetic rack, separate 20 µL of MyONE Silane beads per sample.
Wash once with 900 µL RLT buffer. Pipette up and down a few times and then remove the buffer.
Resuspend beads in 300 µL of RLT buffer. Then, add these beads in RLT buffer to the samples.
Add 10 µL of 5 M NaCl and 615 µL of 100% EtOH.
Mix and rotate samples at room temperature for 15 min.
After 15 min, place tubes on the magnetic rack and remove supernatant.
Resuspend beads in 1 mL of 75% EtOH and move the suspension to a new tube.
Let it sit for 30 s and place back on the magnetic rack.
Remove the liquid and resuspend again in 1 mL of 75% EtOH. Let sit for 30 s. Remove the liquid and repeat the process once more.
After the final wash, spin down the tubes using a picofuge and place them back on the magnetic rack. Remove all residual liquid.
Let air dry for 3 min.
Resuspend in 10 µL of H2O. Let it sit for 5 min at room temperature.
Using the magnetic rack, separate the beads and collect RNA in 10 µL of H2O. Freeze 5 µL at -80 °C as the backup input sample (Note 21).
-
To the remaining 5 µL input samples, add:
H2O 1.2 µL
10× T4 RNA ligase reaction buffer 2 µL
100% DMSO 0.3 µL
50% PEG 8000 8 µL
RNase inhibitor (murine) 0.2 µL
T4 RNA ligase 1 (high conc 30 U/µL) 1.3 µL
Pre-adenylated adapter L3-App (20 μM) 2 µL
Incubate at room temperature for 90 min with shaking at 1,200 rpm.
Take 20 µL of MyONE Silane beads per sample.
Wash once with 900 µL of RLT buffer.
Resuspend beads in 61.6 µL of RLT buffer.
Add beads in 61.6 µL of RLT buffer to the sample.
Add 61.6 µL of 100% EtOH and incubate at room temperature for 15 min with the pipette tip left in the tube. Pipette up and down every 3-5 min to mix the samples.
After 15 min, pipette resuspend in 1 mL 75% EtOH and move to a new tube. Let it sit for 30 s at room temperature and then remove the liquid.
Add 1 mL of 75% EtOH, pipette resuspend, and after 30 s remove the liquid.
Wash once again with 1 mL of 75% EtOH.
After the third wash, briefly spin down the samples using a picofuge and remove residual liquid.
Air dry samples for 3 min at room temperature. Resuspend in 5 µL of H2O.
-
-
Reverse transcription and gel extraction of cDNA
Now that the Input and CLIP samples are synchronized, remove the CLIP samples from -80°C and perform reverse transcription (RT) for all samples (Figure 2).
Note: RT primer sequence corresponds to Rt1clip, as listed in the Materials and Reagents section.
-
Transfer RNA (5 µL) to PCR tubes and add the following reagents (Note 22):
Rt#CLIP primer (0.5 pmol/μL) 1 μL
dNTP mix (10 mM) 1 μL
-
Place tubes in PCR machine and use the following thermal program:
70 °C for 5 min.
Then, hold at 25 °C until RT mix is added.
-
In the meantime, prepare RT mix:
H2O 7 µL
5× first strand buffer 4 µL
0.1 M DTT 1 µL
RNase inhibitor (murine) 0.5 µL
Superscript III 0.5 µL
-
Add 13 µL of RT mix to each tube and use following thermal program:
25 °C 5 min
42 °C 20 min
50 °C 40 min
80 °C 5 min
4 °C hold
Add 1.65 μL of 1 M NaOH to each sample and incubate at 98 °C for 20 min.
Add 20 μL of 1 M HEPES–NaOH pH 7.3 to each sample. This will eliminate radioactivity from strongly labeled samples and prevent RNA from interfering with subsequent reactions (Note 23).
Add 350 μL of TE buffer, 0.75 μL of GlycoBlue, and 40 μL of 3 M sodium acetate (pH 5.5), mix, then add 1 mL of ice-cold 100% EtOH. Mix again and precipitate at -20 °C overnight.
The next day, centrifuge at 16,100× g for 15 min at 4 °C. Remove the supernatant and wash the pellet with 500 μL of ice-cold 80% EtOH. Centrifuge again, remove supernatant, and resuspend the pellet in 6 μL of H2O.
Add 6 μL of 2× TBE-urea loading buffer to the cDNA and heat samples to 80 °C for 5 min immediately before loading. Also, when loading the gel, leave one lane empty between each sample to avoid cross-contamination.
Set up the gel running apparatus with 6% TBE-urea gel in 1× TBE running buffer according to the manufacturer’s instructions. Make sure to flush out urea from the wells using a P1000 pipette tip prior to loading samples.
Run 6% TBE-urea gel for 40 min at 180 V until the lower (dark blue) dye is close to the bottom (Figure 3, Note 24).
Cut off the gel lane containing the size marker. Incubate it for 10 min in 20 mL of TBE buffer with 2 μL of SYBR green II added while gently shaking. Wash once with TBE and visualize by UV transillumination. Print the result using a 100% scale and use the printout as a mask to guide the excision of the cDNA bands from the rest of the gel (Note 25).
Together with the full L3-App sequence, the primer sequence accounts for 52 nt of the cDNA. Cut three bands, at 70–85 nt, 85–120 nt, and 120–200 nt with a sterile razor (Figure 3) (Note 26).
-
Use one of the following methods to crush the gel pieces:
-
Add 400 μL of TE to each gel piece and crush it with a 1 mL syringe plunger.
or
Prepare 0.5 mL tubes by piercing a hole in the bottom using a sterile 19G needle. Needle can be heated with a flame to assist in piercing a hole. Place a gel fragment inside and then place the tubes into 2 mL collection tubes. Centrifuge at room temperature for 2 min at full speed. Remove the 0.5 mL tubes and add 400 μL of TE to each collection tube.
-
Incubate samples for 1 h at 37 °C while shaking in a thermomixer. Then, place on dry ice for 5 min and put back for another hour at 37 °C while shaking.
Place two 1 cm glass pre-filters into a Costar SpinX column and transfer the liquid portion of the supernatant to these columns for each sample.
Centrifuge at full speed for 1 min in a tabletop centrifuge at room temperature. Collect the solution and add it together with 400 μL of RNA phenol/chloroform to a 2 mL phase lock gel heavy tube.
Shake at 37 °C for 5 min in a thermomixer.
Separate the phases by centrifuging the tubes for 5 min at full speed in a tabletop centrifuge at room temperature.
Transfer the aqueous layer into a new tube (be sure not to touch the gel matrix). Optionally, centrifuge the supernatant again for 1min and transfer the liquid into a new tube.
Add 1.5µL of GlycoBlue and 40µL of 3M sodium acetate (pH 5.5). Mix and add 1mL of ice-cold 100% EtOH. Mix by inverting the tubes several times and place overnight at -20°C.
-
-
Circularization of cDNA
The next day, centrifuge the samples at 16,100× g for 15 min at 4 °C. Remove the supernatants and wash once with 500 µL of ice-cold 80% EtOH. Centrifuge samples at 16,100× g for 10 min at 4 °C and remove supernatant.
-
Resuspend pellets in 8 μL of ligation mix:
H2O 4.9 μL
10× Circligase buffer 0.8 μL
50 mM MnCl2 0.4 μL
Circligase II 0.3 μL
Betain (5 M) 1.6 μL
Transfer to PCR tubes and incubate samples at 60 °C for 2 h (Note 27).
-
Then, add 30 μL of oligo annealing mix to each tube:
H2O 26 μL
FAST digest buffer 3 μL
10 μM Cut_oligo 1 μL
Anneal the oligonucleotide with the following program: 95 °C for 2 min, followed by successive incubations of 20 s, starting at 95 °C and decreasing the temperature by 1 °C each time until reaching 25 °C. Then, hold at 25 °C.
Add 2 μL of BamHI and incubate for 30 min at 37 °C; then, incubate for 5 min at 80 °C (Figure 2).
Transfer samples to 1.5 mL Eppendorf tubes. Add 350 μL of TE, 0.75 μL of GlycoBlue, and 40 μL of 3 M sodium acetate (pH 5.5) and mix. Then, add 1 mL of ice-cold 100% EtOH. Mix again and precipitate at -20 °C overnight.
-
Library preparation and gel extraction
The next day, centrifuge samples at 16,100× g for 15 min at 4 °C. Remove the supernatant and wash with 500 μL of ice-cold 80% EtOH as described above.
Resuspend the samples in 23 μL of H2O.
-
To determine the minimum number of PCR cycles required for library preparation, we recommend performing at least two initial PCR reactions using 17 and 20 cycles. Additional cycle numbers can also be tested. We usually do not sequence iCLIP libraries that require more than 22 cycles (Notes 28, 29).
Set up the following reaction mixture for each sample:
cDNA 1 μL
P3/P5 Solexa primer mix (10 μM each) 0.25 μL
2× Phusion HF PCR Master Mix 5 μL
H2O 3.75 μL
-
Run the PCR with the following conditions:
98 °C 30 s 98 °C 10 s Test 16–22 cycles. In our hands, libraries for Input and strong RBPs, such as PTBP1 and SRSF1, often need less than 16 cycles without over-amplification 65 °C 30 s 72 °C 30 s 72 °C 3 min 10 °C hold Mix 8 μL of PCR product with 2 μL of 5× TBE loading buffer, load on a 6% TBE gel, run at 180 V and 120 mA for 25–30 min, and stain the gel with SYBR Gold for 15 min.
Image the gel and estimate the minimum number of cycles needed for library preparation (Note 30). Overamplification during PCR often results in secondary bands that migrate at higher sizes, as detailed previously (Huppertz et al., 2014) (Figure 4). We amplify libraries in two halves using 10 μL of cDNA template in each half. Since 2.5-fold more concentrated cDNA is used for the first half (and second half), the number of cycles needed for library preparation is one less than the number of cycles used in the initial test PCR.
-
Prepare the following reaction mix:
cDNA 10 μL
P3/P5 Solexa primer mix (10 μM each) 1 μL
2× Phusion HF PCR Master Mix 20 μL
H2O 9 μL
Run the same PCR program with the determined optimal cycle number.
Mix 8 μL of PCR product with 2 μL of 5× TBE loading buffer, load on a 6% TBE gel, run at 180 V and 120 mA for 25–30 min, stain, and visualize with SYBR Gold (Figure 4).
If the library appears under-amplified (i.e., the band is not clearly visible on the gel), immediately place back into the PCR machine for two or more cycles and re-run the gel.
Amplify the second half of the cDNA using the appropriate number of cycles, then combine both halves and proceed to agarose gel extraction steps.
Prepare 2% agarose gel in TBE. Let it cool down, add 1:10,000 SYBR Safe, mix, and pour gel.
Estimate the volume of each sample and accordingly add 6× loading dye into each.
Load samples on gel along with a low molecular weight DNA marker. Leave one well empty between samples.
Run at 90 V for 1 h (Note 31).
Use blue light illumination to visualize and then cut gel slices of appropriate sizes (see library amplification image for sizes) and place them into 15 mL Falcon tubes. Use a fresh razor blade for each sample.
Perform gel extraction using the Qiagen Gel Extraction kit according to the manufacturer’s instructions, with the following modifications: melt the gel pieces at room temperature. Elute in 20 μL of elution buffer. For improved yield, elute twice by taking the flowthrough from first elute and adding it back to the column.
Quantify the iCLIP libraries using Bioanalyzer and qPCR, as detailed previously (Huppertz et al., 2014). Mix High, Medium, and Low fractions into a 5:5:1 ratio for sequencing (Note 32).
iCLIP libraries can be sequenced using standard Illumina protocols. 50-nucleotide single-end runs are recommended.
Figure 1. Optimization of RNase 1 dilutions and CLIP-autoradiography.

A. CLIP autoradiography of 32P-labeled RBP-RNA complexes was performed after RNase I treatment (1:25–1:250 dilution). Under high RNase 1 condition (1:25), the radiolabeled RNA signal diminished. Optimal RNase 1 concentration (1:250 here) results in radiolabeled RNA that runs as a smear above the predicted size of the RBP (~72 kDa here). Note: Antibody chains are often observed on autoradiographs. Bottom panel shows western blotting results to indicate the recovery of GFP-tagged bait protein. Black bar on the right indicates region on the membrane to be excised. B. Autoradiograph of input (INP) and 32P-labeled immunopurified (IP) RBP-RNA complexes after partial RNase I digestion (RNase I dilution, 1:250). Since the input sample was not radiolabeled, no radioactive signal was observed. Red boxes indicate the regions on the membrane that were excised to extract RNA. RBP (SP1) size is ~82 kDa here. GFP-only sample is shown as a negative control.
Figure 2. Schematic representation of cDNA synthesis and iCLIP library preparation.

Adapter-ligated RNA fragments are reverse transcribed using barcoded primers. Adapter and RT primer sequences are shown. cDNA is size selected, circularized, and linearized prior to performing PCR for library preparation.
Figure 3. Gel purification and size selection of cDNA.

Cartoon illustration of 6% TBE-urea gel electrophoresis to guide the excision of iCLIP cDNA products. The pattern of migration for cDNA and two dyes (indicated) is highly reproducible when the gel is run for 40 min at 180 V (make sure to note the time required to obtain the indicated pattern of migration of the bands on the gel when using your own powerpack in initial experiments). Gel slices that are excised to purify cDNA corresponding to high (H), medium (M), and low (L) molecular size cDNA fractions are indicated. This figure was made after Konig et al. (2011).
Figure 4. Representative PCR analysis of iCLIP libraries.

Left: Preliminary PCR of iCLIP and input samples to determine the optimal cycle number. The iCLIP sample (labeled as IPs) was amplified using two different cycle numbers, i.e., 17 and 19, whereas the input (INP) was amplified using 17 cycles. While 19 and 17 cycles for IPs and input, respectively, lead to overamplification resulting in secondary products of higher sizes, 17 was chosen as the optimal cycle number to prepare the final libraries for iCLIP samples (labeled as IPs). Right: Gel image showing the final libraries for iCLIP and input samples prepared using the indicated cycle numbers. Note: Since the template cDNA was 2.5-fold more concentrated for the final PCR in comparison with the preliminary PCR, the iCLIP library (labeled as IPs) was prepared using 16 rather than 17 cycles. The final input library was prepared using 15 cycles. PCR products were resolved on 6% TBE gels and stained with SYBR gold.
Data analysis
Since this protocol essentially follows the workflow of the original iCLIP-seq method (Huppertz et al., 2014), the data can be subjected to the standard iCLIP analysis pipeline. Data analysis involves several steps, including de-multiplexing if multiple samples were run within one sequencing lane, mapping reads to the genome, collapsing of random barcodes and removing PCR duplicates, and identification of binding sites. Briefly, 51 nt raw reads that consist of three random positions, a 4 nt multiplexing barcode, and another two random positions, followed by the cDNA sequence, are initially de-duplicated based on the first 45 nt. Reads are de-multiplexed. The random positions, barcodes, and any 3′-bases matching Illumina adaptors are removed, and reads shorter than 25 nt are filtered out. The remaining reads are trimmed to 35 nt. These steps can be carried out using Trimmomatic (Bolger et al., 2014). The remaining reads are mapped to the human genome/transcriptome. To prevent false assignments of reads from repetitive regions, we generally remove any reads with a mapping quality <3 from further analysis. Several freely available pipelines can be utilized (e.g., PureCLIP, CTK, and iMAPs) (Krakau et al., 2017; Shah et al., 2017; Kuret et al., 2022). Moreover, the MEME package can be used to identify enriched sequence motifs (Bailey et al., 2015).
Validation of protocol
The number of PCR cycles required to amplify the cDNA during library preparation can directly impact the quality of the resulting data (i.e., libraries amplified with higher numbers of cycles contain higher amounts of PCR duplicates). The required number of cycles depends on several factors, including the protein abundance, quality of the antibody, IP efficiency, and technical limitations of the experimental procedure (Buchbender et al., 2020). In the original iCLIP method, 27–30 cycles were often needed to generate libraries (Huppertz et al., 2014). We have observed that, for our revised iCLIP-1.5 protocol, the number of cycles required for the amplification of cDNA libraries often lies between 16 and 20 cycles. Moreover, input material and abundant RBPs, such as PTBP1 and U2AF35, require even fewer PCR cycles, ranging from 14 to 16.
This revised protocol has been used to perform iCLIP-seq for a number of RBPs, including both well-characterized and relatively poorly studied RBPs, in several recent publications (Han et al., 2022; Nabeel-Shah et al., 2022; Song et al., 2022). In our most recent study, we performed iCLIP-seq for SP1 using our revised protocol (Song et al., 2022). SP1 iCLIP required 17 PCR cycles to obtain a DNA concentration suitable for high-throughput sequencing. After PCR duplicate removal, we obtained >18 million uniquely mapped reads per replicate (Figure 5A) (Song et al., 2022). In parallel to these experiments, we also performed one replicate of SP1 iCLIP-seq using the original protocol [data not published in Song et al. (2022)]. That iCLIP library required 28 cycles of PCR to obtain a DNA concentration sufficient for high-throughput sequencing. After discarding PCR duplicates, we obtained only ~1.6 million uniquely mapped reads (Figure 5A) (data unpublished), indicative of a low-complexity library and high PCR duplication rate. Thus, our revised iCLIP-1.5 protocol represents a substantial improvement to the original iCLIP-seq method, including enhanced adapter ligation, incorporation of SMI, and improved circularization of the cDNA.
Figure 5. Sample results for iCLIP-seq data analysis.

A. Comparison of number of uniquely mapping reads for SP1 iCLIP-seq data generated using either the revised protocol (reported here) or the original protocol. iCLIP-seq data for SP1 replicates 1–3 was reported as SP1.S2, SP1.S3, and SP1-E, respectively, in Song et al. (2022) (GSE165739). B. Left: Standardized metaplot profile showing the signal for CPSF5 along mRNA transcripts. iCLIP-seq was performed using GFP-tagged CPSF5 as reported in Song et al. (2022). Raw iCLIP data was acquired from the GEO database using the accession number GSE165739. Crosslink-induced truncation sites were identified using the CTK package (FDR ≤ 0.01). Right: Top RNA-binding motif identified in CPSF5 iCLIP-seq. MEME software was used for de novo motif discovery. U in the target RNA sequences was replaced by T for MEME analysis. Statistical significance was calculated against randomly assorted sequences.
In Figure 5B, we showcase the analysis of our published iCLIP-seq data generated using this revised protocol for a well-characterized RBP, CPSF5 (CFIm25) (Song et al., 2022). The analysis was performed using the CTK package to identify crosslink-induced truncation sites (Shah et al., 2017). We found that CPSF5 preferentially binds in the 3′ UTRs of target transcripts, consistent with its known function as a subunit of the cleavage and polyadenylation complex. Moreover, we identified the UGUA motif as the most enriched 4 mer around the cross-link sites of CPSF5, as reported in previous studies (Martin et al., 2012). These results show that our revised iCLIP-1.5-seq protocol faithfully captures known RNA-binding preferences of RBPs.
Notes
For a standard experiment, ∼5–7 million HeLa cells or ∼10 million HEK293 cells are sufficient. We usually grow cells in two independent batches to represent biological replicates.
Make sure that tray/plate does not touch the UV bulbs or cover the energy detector. This can lead to errors in UV exposure readings.
The length of UV exposure can be optimized for new RBPs (range 150–400 mJ/cm2). However, increasing the UV dose could distort library preparation due to damaged RNA and could trigger DNA damage response pathways in the cells. Therefore, care should be taken when increasing the UV dose.
Cells grown in a monolayer are equally exposed to the UV light and hence only require a single round of irradiation to crosslink equally.
For suspension cells, centrifuge the cells, wash three times with 1× PBS (6 mL), leave cells in 1× PBS, and transfer to the 100 mm dishes. Irradiate and harvest cells as described.
Although Proteins A and G are structurally similar, they have different affinities for IgG subclasses across different species. For example, Protein A has greater affinity for rabbit, pig, dog, and cat IgG, whereas protein G has greater affinity for mouse and human IgG. Therefore, host species of the RBP-specific primary antibody should match the target species of the Dynabeads used. Protein A Dynabeads should be used for rabbit antibodies.
The quantity of required antibody and time of incubation depend on antibody quality and purity. This should be optimized in preliminary experiments. For GFP-tagged cell lines, we routinely use GFP recombinant rabbit monoclonal antibody from Thermo Fisher Scientific (G10362) or rabbit polyclonal anti-GFP from Abcam (ab 290). For SP1 iCLIP-seq reported in Song et al. (2022), we used 10 μg of SP1 antibody (Santa Cruz sc-17824).
RNA or protein concentration should be determined using Nanodrop or the Bradford assay, respectively. Normalize all the samples to the lowest concentration. 2 mg/mL is recommended as the optimal protein concentration. We take 1 mL of cell lysate per purification for each sample. Therefore, 2.2 mL of cell lysate is usually sufficient for two iCLIP experiments.
We employ several kinds of controls as needed, including samples where the RBP is absent from the original material (such as knockout or knockdown cells), a control where no crosslinking is done, and a control where no antibody is used during IP. In the case of epitope-tagged proteins (e.g., GFP and FLAG), use of cells expressing free FLAG/GFP should be used. It should be noted that FLAG/GFP-only cells usually yield iCLIP libraries with extremely low complexity, which are dominated by PCR duplicates, and hence not ideal for downstream computational analysis.
Sonication is strongly recommended for nuclear proteins. It shears the genomic DNA and helps to release proteins from the chromatin. Keep the probe ~0.5 cm above the bottom and avoid touching the tube sides. Repeat sonication (15 cycles, 0.3 s on, 0.7 s off) three times on ice. Leave the tubes on ice for ~3 min between rounds. Wash the probe with 70% EtOH and then with dH2O before sonication and in between samples.
Optimization of RNA fragmentation is extremely important for the success of the experiment. Enzymatic RNA digestion depends on the cell type and quantity of RNA present in the sample. Each new batch of RNase I should be tested for the optimal dilution. Treatment with high RNase I concentration (1:5 or 1:50) results in sharp bands on SDS-PAGE, corresponding to ~5 kDa above the predicted size of the protein. The optimal RNA fragments should range between 50 nt and 200 nt. The optimal RNase I dilution should yield RBP-RNA complexes that run in a diffused fashion ~20–80 kDa above the predicted size of RBP. Optimal RNase I dilution must be determined in the initial experiments prior to performing full iCLIP-seq experiment for a given RBP. For initial experiments, dephosphorylation and L3 adapter ligation steps should be omitted (see also Note 16 below). The presence of multiple sharp bands under high RNase I conditions generally indicates the co-purification of strongly interacting non-specific RBPs. To prevent the co-purification of a non-specific RBP, we recommend performing dual immunoprecipitation with urea denaturation, as detailed in Huppertz et al. (2014). For a detailed description of the RNase I optimization step, see Huppertz et al. (2014).
Although the optimal RNase I dilution should be determined for each new RBP, in our experience 1:250 dilution works for most RBPs in HEK293. For GFP-only, no-UV, and/or no-antibody control samples, RNase I dilution of 1:250 should suffice, and there is no need to test several dilutions since these controls do not yield any appreciable radioactive signal.
iCLIP lysis buffer contains SDS, which disables RNase I after prolonged incubation. Additionally, SUPERase•In RNase Inhibitor can be used to inhibit RNase I.
In our experience, overnight IP at 4 °C does not cause any appreciable RNA degradation. Optional: Cell lysate can be pre-incubated with 25 µL of protein A/G Dynabeads for 2 h at 4 °C to reduce background binding. Take 25 µL of protein A/G Dynabeads, wash three times with iCLIP lysis buffer, and incubate with the clarified lysate (before taking input samples) for 2 h at 4 °C with end-to-end rotation. After 2 h, place tubes on a magnetic rack and transfer the supernatant to a new tube. Continue with the procedure from Step 9.
If antibody depletion efficiency needs to be monitored, collect 20 μL at this step (flowthrough) for western blot comparison of the lysate before and after IPs. Additionally, it is possible to take input from the flowthrough rather than whole-cell lysates. Input from the clarified whole-cell lysate (recommended) retains the target RBP-RNA complexes, whereas flowthrough is specifically depleted of the bait.
For experiments aimed only at optimization of the RNase I concentration, and not for library preparation, steps involving washing with FastAP buffer, RNA 3′ end dephosphorylation, and L3 adapter ligation should be omitted. Proceed straight to radiolabeling after washing with high-salt and PNK buffers. Moreover, no-UV and no-antibody controls do not need to be subjected to RNA 3′ end dephosphorylation and L3 adapter ligation steps, since these controls often fail at the library preparation steps. In rare instances when we were able to successfully prepare iCLIP libraries for controls, sequencing data were almost always dominated by PCR duplicates. Since DTT can decrease the IP efficiency, it is removed from the ligation buffer used for L3 adapter ligation steps for IPs.
The Novex NuPAGE gels are important for the success of iCLIP. A pour-your-own SDS-PAGE gel (Laemmli) changes its pH during the run, which can get to pH ∼9.5 and lead to alkaline hydrolysis of the RNA. The Novex NuPAGE buffer system maintains the pH around 7 throughout the whole run. MOPS NuPAGE running buffer is recommended. Do not forget to add methanol when diluting 20× transfer buffer.
If the experiment is aimed only at optimizing RNase I conditions, then also perform western blotting to assess the recovery of the bait.
Different exposure times might be needed for autoradiography depending upon the RBP and IP efficiency. Expose for 30 min, 1 h, or even overnight at -80 °C. Although iCLIP buffer conditions eliminate physical protein–protein interactions, it is possible that undesired interacting proteins might co-purify with your protein of interest. If contaminating bands of other protein–RNA complexes are observed above your protein–RNA complex, only cut up to, but not including, these bands. Alternatively, IPs can be performed under denaturing urea conditions, as detailed in Huppertz et al. (2014). We do not use 32[P] that is older than two weeks. If radioactive smearing in +UV samples is not observed even after prolonged exposure, it might be due to failed IP, your protein binding very weakly to RNA, or cells not being properly exposed to UV during the crosslinking step. We do not recommend continuing the experiment for library preparations in such cases.
When comparing different RNase I conditions, the size of the radiolabeled band must change. Under high-RNase conditions, the radioactive smearing will diminish in comparison to the low-RNase, confirming that the band corresponds to a protein–RNA complex. Furthermore, the high-RNase condition helps to determine the size of the immunoprecipitated RBP, as the RBP will be bound to short RNAs and thus will migrate as a less diffuse band ∼5 kDa above the expected molecular weight of the protein. The radioactive band should become more diffuse in the low-RNase condition. On this basis, proceed to RNA isolation using the following guidelines: the average molecular weight of 70 nt RNA is ∼20 kDa. To isolate a broad range of RNAs between 40 and 300 nt in size (including the adapter), we recommend cutting a wide band of ∼15–80 kDa above the expected molecular weight of the protein. For further details refer to Huppertz et al. (2014). Note: At extremely low-RNase condition (e.g., 1:1,200), RBP–RNA complexes might not enter the wells. Control samples should not show any bands.
The remaining 5 µL can be used later in case the input cDNA libraries fail to amplify. When using antibody against endogenous proteins, the same input can be used for RBPs of similar molecular weights.
iCLIP primers are barcoded, as reported in Huppertz et al. (2014). Use distinct primers (Rt1clip–Rt16clip) for the control and the experimental replicates. Since primers contain distinct 4 nt barcode sequences, samples can be multiplexed. Distinct barcodes also allow us to control for cross-contamination between samples during library preparation. Moreover, RT polymerases other than Superscript III can also be used. For example, irCLIP utilized f TIGRT-III reverse transcriptase (Ingex, #TGIRT50).
It is possible to multiplex up to three samples with different barcodes at this stage. Alternatively, cDNA libraries can be made separately for each sample and mixed after the PCR for sequencing.
We have observed that the gel running time can vary depending on the power pack that is used. For initial experiments, it is important to note the time it takes for the lower (dark blue) dye to nearly reach the bottom. At least in the initial experiments, it is recommended to include DNA size markers.
This step is not needed if you are not using DNA size markers.
We recommend avoiding cutting gel slices higher than 200 nt or lower than 70 nt. cDNAs fragments below 70 are often too short to accurately map to the genome. cDNA fragments that are too long are not suitable to obtain desired resolution. If binding to miRNAs or other short RNAs is not of interest, it is not necessary to isolate low band.
We have increased the incubation time and included betain in the reaction mixture. Increasing the incubation time and the addition of betain to a final concentration of 1M in the reaction mixture have been found to improve the circularization of difficult-to-ligate substrates.
cDNA libraries must never be brought to the area where iCLIP RNA or cDNA work is done, in order to avoid cross-contaminating samples. Library preparation should be carried out in a designated area, ideally far away from general lab workplaces.
Additional cycle numbers can also be tested. It is important to optimize PCR cycle numbers in order to avoid overamplification. In our experience, 17–20 cycles produce a sufficient cDNA concentration for sequencing for most RBPs. Strong RBPs and input libraries often require fewer cycle numbers, usually ranging from 14 to 16.
The size of the cDNA will be the size of the product minus the combined length of the P3/P5Solexa primers and the barcode (128 nt). Therefore, a band cut at 70–85 nt on the cDNA gel (with 20–30 nt cDNA + 52 nt primer) is expected to generate 145–160 nt PCR products. For a detailed description, see Huppertz et al. (2014).
Longer runs may give better resolution but larger cut sizes. We incubate the gel running apparatus in 20% bleach in between different runs. Rinse thoroughly with water to clean the apparatus before using it for the next run.
The ratio of the bands can be adjusted. For example, if miRNAs are important, use more of the lower bands. Also, take the quality of each PCR sample into account; if one of the ½ PCR reactions was overamplified, use more of the reaction that is in the right range of amplification.
Acknowledgments
This research was funded by the Canadian Institutes of Health Research Foundation Grant FDN-154338 to JFG. We thank Dr. Benjamin J Blencowe and his laboratory members including Drs. Thomas Gonatopoulos-Pournatzis, Ulrich Braunschweig, Andrew Best, and Esha Sharma for helpful discussions and technical advice. We also thank all members of the Greenblatt laboratory, particularly Nujhat Ahmed for helpful discussions and her critical reading of the manuscript, and Shuye Pu for his help with iCLIP data analysis. This protocol was used in Song et al. (2022).
Competing interests
The authors declare no competing interests.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
Q&A
Post your question about this protocol in Q&A and get help from the authors of the protocol and some of its users.
References
- 1.Bailey T. L., Johnson J., Grant C. E. and Noble W. S.(2015). The MEME Suite. Nucleic Acids Res 43(W1): W39-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolger A. M., Lohse M. and Usadel B.(2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15): 2114-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buchbender A., Mutter H., Sutandy F. X. R., Kortel N., Hanel H., Busch A., Ebersberger S. and Konig J.(2020). Improved library preparation with the new iCLIP2 protocol. Methods 178: 33-48. [DOI] [PubMed] [Google Scholar]
- 4.Hafner M., Katsantoni M., Köster T., Marks J., Mukherjee J., Staiger D., Ule J. and Zavolan M.(2021). CLIP and complementary methods. Nat Rev Dis Primers 1(1): 1-23. [Google Scholar]
- 5.Han H., Best A. J., Braunschweig U., Mikolajewicz N., Li J. D., Roth J., Chowdhury F., Mantica F., Nabeel-Shah S., Parada G., et al.(2022). Systematic exploration of dynamic splicing networks reveals conserved multistage regulators of neurogenesis. Mol Cell 82(16): 2982-2999 e2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han H., Braunschweig U., Gonatopoulos-Pournatzis T., Weatheritt R. J., Hirsch C. L., Ha K. C. H., Radovani E., Nabeel-Shah S., Sterne-Weiler T., Wang J., et al.(2017). Multilayered Control of Alternative Splicing Regulatory Networks by Transcription Factors. Mol Cell 65(3): 539-553 e537. [DOI] [PubMed] [Google Scholar]
- 7.Hentze M. W., Castello A., Schwarzl T. and Preiss T.(2018). A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol 19(5): 327-341. [DOI] [PubMed] [Google Scholar]
- 8.Huppertz I., Attig J., A. D’Ambrogio, Easton L. E., Sibley C. R., Sugimoto Y., Tajnik M., König J. and Ule J.(2014). iCLIP: Protein–RNA interactions at nucleotide resolution. Methods 65(3): 274-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Konig J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B., Turner D. J., Luscombe N. M. and Ule J.(2011). iCLIP--transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. J Vis Exp(50): 2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krakau S., Richard H. and Marsico A.(2017). PureCLIP: capturing target-specific protein-RNA interaction footprints from single-nucleotide CLIP-seq data. Genome Biol 18(1): 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuret K., Amalietti A. G., Jones D. M., Capitanchik C. and Ule J.(2022). Positional motif analysis reveals the extent of specificity of protein-RNA interactions observed by CLIP. Genome Biol 23(1): 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee F. C. Y., Chakrabarti A. M., Hänel H., Monzón-Casanova E., Hallegger M., Militti C., Capraro F., Sadée C., Toolan-Kerr P., Wilkins O., et al.(2021). An improved iCLIP protocol. bioRxiv, 2021.08.27.457890. 10.1101/2021.08.27.457890 [DOI] [Google Scholar]
- 13.Martin G., Gruber A. R., Keller W. and Zavolan M.(2012). Genome-wide analysis of pre-mRNA 3' end processing reveals a decisive role of human cleavage factor I in the regulation of 3' UTR length. Cell Rep 1(6): 753-763. [DOI] [PubMed] [Google Scholar]
- 14.Nabeel-Shah S., Lee H., Ahmed N., Burke G. L., Farhangmehr S., Ashraf K., Pu S., Braunschweig U., Zhong G., Wei H., et al.(2022). SARS-CoV-2 nucleocapsid protein binds host mRNAs and attenuates stress granules to impair host stress response. iScience 25(1): 103562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nitoiu A., Nabeel-Shah S., Farhangmehr S., Pu S., Braunschweig U., Blencowe B. J. and Greenblatt J. F.(2021). KRAB Zinc Finger protein Znf684 interacts with Nxf1 to regulate mRNA export. bioRxiv, 2021.09.29.462476. 10.1101/2021.09.29.462476 [DOI] [Google Scholar]
- 16.Van Nostrand E. L., Pratt G. A., Shishkin A. A., Gelboin-Burkhart C., Fang M. Y., Sundararaman B., Blue S. M., Nguyen T. B., Surka C., Elkins K., et al.(2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP(eCLIP). Nat Methods 13(6): 508-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pereira B., Billaud M. and Almeida R.(2017). RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer 3(7): 506-528. [DOI] [PubMed] [Google Scholar]
- 18.Shah A., Qian Y., Weyn-Vanhentenryck S. M. and Zhang C.(2017). CLIP Tool Kit(CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics 33(4): 566-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao C., Yang B., Wu T., Huang J., Tang P., Zhou Y., Zhou J., Qiu J., Jiang L., Li H., et al.(2014). Mechanisms for U2AF to define 3' splice sites and regulate alternative splicing in the human genome. Nat Struct Mol Biol 21(11): 997-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song J., Nabeel-Shah S., Pu S., Lee H., Braunschweig U., Ni Z., Ahmed N., Marcon E., Zhong G., Ray D., et al.(2022). Regulation of alternative polyadenylation by the C2H2-zinc-finger protein Sp1. Mol Cell 82(17): 3135-3150 e3139. [DOI] [PubMed] [Google Scholar]
- 21.Zarnegar B. J., Flynn R. A., Shen Y., Do B. T., Chang H. Y. and Khavari P. A.(2016). irCLIP platform for efficient characterization of protein-RNA interactions. Nat Methods 13(6): 489-492. [DOI] [PMC free article] [PubMed] [Google Scholar]