


Abstract
Regulation of gene expression in trypanosomatid parasites is predominantly post-transcriptional. Primary transcripts are trans-spliced and polyadenylated to generate mature mRNAs and transcript stability is a major factor controlling stage-specific gene expression. Degenerate PCR has been used to clone the gene encoding the Leishmania homologue of poly(A)-binding protein (LmPAB1), as an approach to the identification of trans-acting factors involved in this atypical mode of eukaryotic gene expression. lmpab1 is a single copy gene encoding a 63 kDa protein which shares major structural features but only 35–40% amino acid identity with other PAB1 sequences, including those of other trypanosomatids. LmPAB1 is expressed at constant levels during parasite differentiation and is phosphorylated in vivo. It is localised predominantly in the cytoplasm but inhibition of transcription with actinomycin D also reveals diffuse localisation in the nucleus. LmPAB1 binds poly(A) with high specificity and affinity but fails to complement a null mutation in Saccharomyces cerevisiae. These properties are indicative of functional divergence in vivo.
INTRODUCTION
Gene regulatory mechanisms in trypanosomatid parasites are atypical of eukaryotes in general. Constitutive transcription of protein coding genes (by RNA polymerases I and II) generates polycistronic RNAs which are extensively processed to produce mature monocistronic mRNAs. As a consequence, regulation of gene expression is predominantly post-transcriptional (reviewed in 1). Fundamental differences in mRNA maturation include the uncoupling of 5′ capping from early transcriptional elongation as a consequence of the trans-splicing of an independently transcribed, capped spliced-leader (SL) sequence to the 5′ end of mRNAs (reviewed in 2). Trans-splicing and polyadenylation are coupled processes (3) apparently regulated by hierarchical signals typically occurring within intergenic polypyrimidine tracts (4–7). Trypanosomatids lack the AAUAA polyadenylation consensus found in higher eukaryotes and display both micro-heterogeneity in polyadenylation site usage and multiplicity of sites within a single transcriptional unit (3 and references therein). This observation suggests that transcriptional termination is not coupled to cleavage and polyadenylation. In addition, trypanosome RNA polymerase II lacks the characteristic C-terminal heptad repeats found in other organisms although this region is still phosphorylated in vivo (8 and references therein). The implications of these features for the formation and function of a complex analogous to the postulated transcription/processing complex found in other eukaryotes (9) have yet to be extensively investigated.
Multiple points of regulation have been identified in the expression of specific trypanosomatid transcripts. Factors affecting mRNA stability have been most extensively studied to date and signals that mediate stability within the 3′-UTR, intergenic and coding regions of specific transcripts have been identified (10,11). Examples of translational and post-translational regulation have also been characterised (12) but little progress has been made in identifying factors involved in stage-specific regulation of gene expression.
The poly(A)-binding protein I (PAB1) of eukaryotes is the major cytoplasmic mRNA binding protein and also plays a role in polyadenylation of nuclear transcripts (13–15). In the cytoplasm, PAB1 has been implicated in translational initiation (16) and termination (17) and in mRNA turnover. When complexed to the poly(A) tail, PAB1 circularises mRNA molecules via its interaction with translational initiation factors at the cap, enhancing translational initiation and stabilising mRNA (16). In addition, PAB1 is important in mRNA decay: dissociation of PAB1 from the poly(A) tail is necessary for progressive deadenylation, the first step in the major decay pathway for many transcripts (18–23).
The central role of PAB1 in the metabolism of mRNA provides the rationale for characterising the functional homologue in Leishmania. These trypanosomatid parasites, which cause a spectrum of human diseases, cycle between extracellular and intracellular habitats (24) and are assumed to be dependent on regulated expression of stage-specific genes to survive extreme environmental changes. Given the unusual gene regulatory mechanisms described above, it is likely that Leishmania PAB1 will have both conserved and divergent functions in comparison to other eukaryotic homologues. Here, we describe cloning of the Leishmania gene using a degenerate PCR approach, expression of recombinant protein and in vitro analysis. We demonstrate that LmPAB1 is constitutively expressed in at least two phosphorylated isoforms in the cytoplasm but is also present in the parasite nucleus, where it accumulates upon transcriptional inhibition. Divergence in one of the RNA binding domains, coupled with the inability of LmPAB1 to complement function in Saccharomyces cerevisiae, suggest that the Leishmania protein may have distinct functional interactions in vivo.
MATERIALS AND METHODS
Parasite strains and culture
Promastigotes of Leishmania major Friedlin (MHOM/IL/81/Friedlin) were maintained in vitro at 26°C as previously described (25).
PCR and plasmid construction
Degenerate PCR. Primers 286F (CTAGTCTAGACTGGGCT-ACGGCTATCGTGCAACTTT) and 1013R (GCCGGAATTCG-TCGAGAAGTTCTTCACGTACGAGGATT), designed with reference to the most conserved motifs of Trypanosoma cruzi PAB1 and adjusted for L.major codon bias (26), were used with 200 ng target genomic DNA. Amplification with Taq DNA polymerase (Promega) was optimised to 59°C using temperature gradient PCR. The 745 bp product was cloned into pBS SK+ to generate the plasmid LmpabH3.
Constructs for bacterial expression and yeast complementation studies. The open reading frame of lmpab1 was amplified using Pfu polymerase (Stratagene) from subclone pL7152.23 (derived from cosmid L7152; 27) using primers PABPF1c (TGGGCCCGGGATGGCTGCTGCTGTCCAGGAAG) and PABPR3b (CCATCGATACTGCTGGCTTCTCGCTTAC-GCCGT) for yeast constructs, or PABPF1b (CTACATATGGCTGCTGCTGTCCAGGAAG) and PABPR2c (GGATCCCTGCTGGCTTCTCGCTTACGCCGT) for bacterial constructs. The 1.7 kb products were A-tailed with Taq polymerase, cloned into pGEM-T (Promega) and sequenced. For bacterial expression of recombinant protein, the cloned product was digested with NdeI and BamHI and cloned into pET15b (Novagene) to obtain pET15b PABPA7. For yeast complementation studies, the cloned product was digested with XmaI and ClaI and cloned into p415 GAL1 and p416 GAL1 (28), to obtain clones p415A and p416A respectively.
RNA and DNA analysis
High molecular weight genomic DNA was extracted, digested, blotted and hybridised at low stringency (50% formamide, 2× SSC, 2% SDS, 5× Denhardt’s solution at 37°C; washes in 2× SSC at 20°C and 2× SSC, 1% SDS at 42°C), as described (29). Total RNA was extracted (30), denatured with glyoxal and DMSO, blotted and hybridised as described (31). Blots were washed twice at room temperature in 2× SSPE, 2% SDS and twice at 42°C in 0.5× SSPE, 1% SDS before exposure to BioMAX MR™ film (Kodak). Autoradiographs were scanned and density-analysed using IP Lab Spectrum Software (Scanalytics, Vienna, VA). DNA sequencing was carried out by the dideoxy chain termination method using T7 DNA polymerase (Sequenase; USB). The nucleotide sequence of lmpab1 has been submitted to the EMBL database and assigned the accession number AF093062.
Recombinant protein purification for antibody production
Expression of His6-tagged LmPAB1 from pET15b PABA7 (in Escherichia coli BL21 DE3) was induced by IPTG. Cells were then lysed under denaturing conditions and His6-tagged protein bound to and eluted from Ni–agarose (Qiagen). The protein was further purified by 6% SDS–PAGE and used for immunisation (Eurogentec). Antibodies specific to LmPAB1 (abSK375) were purified by incubation with recombinant His6-LmPAB1 blotted on to PVDF membrane, followed by elution with 100 mM glycine pH 2.5 (32).
Parasite protein analysis
Promastigote proteins were extracted and analysed by SDS–PAGE and immunoblotting as previously described (33), using the following antibodies: abSK375 (described above); ab336 (anti-HASPB, affinity-purified polyclonal antiserum; 25,29); ab415 [anti-Sw3 (histone H1) (34)]. Immune complexes were detected by enhanced chemiluminescence using horseradish peroxidase-conjugated goat anti-rabbit IgG (Sigma) or by alkaline phosphatase detection (33). PhD secondary structure prediction software was used to analyse the deduced sequence of LmPAB1 (35).
Indirect immunofluorescence microscopy
Leishmania major promastigotes were cultured in the presence of actinomycin D for 10 h. Parasites were harvested, washed three times in PBS and allowed to settle onto polylysine-coated slides at 4°C for 1 h. Slides were washed in PBS and the cells fixed in 4% paraformaldehyde, followed by 10% Nonidet P-40 (NP-40) as described (38). Parasites were then incubated with purified abSK375 (1:20 dilution in 5% milk powder in PBS) or preimmune serum for 40 min at 37°C. After three washes in PBS, slides were further incubated with FITC-conjugated goat anti-rabbit IgG (Sigma; 1:2000 in 5% milk powder in PBS) for 40 min at 37°C, prior to washing. After DAPI staining (0.5 µg/ml in PBS) and mounting (Vector Shield, Vector Labs), slides were viewed using a Nikon Microphot-FX epifluorescent microscope. No fluorescence was obtained when preimmune serum was used as the first antibody (data not shown). Images were captured with a Photometrics CH350 CCD camera and analysed using IPLab Spectrum software (Scanalytics Inc.).
Nuclear fractionation
Cellular fractionation was performed as described (34). Washed parasites resuspended in 1/100 volume of lysis buffer (140 mM NaCl, 1.5 mM MgCl2, 10 mM Tris pH 8.6, 0.5% NP-40) containing protease inhibitors (2 µg/ml leupeptin, 0.4 mM PMSF) were vigorously mixed and centrifuged (6000 g) at 4°C for 3 min. The supernatant (cytoplasmic fraction) was removed and the pellet washed once in lysis buffer. After further centrifugation, the pellet obtained was the nuclear enriched fraction. All fractions were snap frozen and stored at –70°C.
RNA binding analysis
Poly(A) (Sigma) was hydrolysed with 0.2 M NaOH at 50°C for 10 min, precipitated and 5′-end labelled with polynucleotide kinase and [γ-32P]dATP. Radiolabelled poly(A) was purified on a G50 Sephadex™ spin column (Pharmacia). Promastigotes were harvested, washed twice in cold PBS, lysed in low-salt lysis buffer (10 mM Tris pH 8.0, 1 mM MgCl2, 4 mM KCl, 1 mM DTT, 1% Triton X-100) and debris removed by centrifugation (6000 g). Ten microlitre extracts (1 × 107 cell equivalents) were incubated in a total volume of 30 µl with 500 ng radiolabelled poly(A) and 15 µg yeast tRNA (Sigma) in binding buffer (20 mM Tris pH 8.0, 150 mM NaCl, 5 mM EDTA, 1 mM DTT). The concentration of NaCl was adjusted and unlabelled competitor poly(A) added where indicated. After incubation for 5 min at room temperature, samples were UV cross-linked (Stratalinker™) prior to SDS–PAGE. Gels were dried and exposed to BioMAX MR™ film (Kodak) using a Hyper-cassette™ (Amersham).
Protein complexes were immunoprecipitated from 100 µl binding reactions by incubation for 30 min at 4°C with a 50% slurry of protein A–Sepharose coupled to abSK375 or preimmune serum, diluted to a total volume of 200 µl with IPP150 buffer (10 mM Tris–HCl pH 8.0, 0.15 mM NaCl, 0.1% NP-40) (37). After four washes in IPP150 buffer, resin samples were boiled in 2× SDS loading buffer and analysed by 10% SDS–PAGE.
In vivo phosphorylation analysis
Eight day cultured promastigotes (1 × 108) were washed in phosphate-free DMEM, incubated for 2 h at 26°C in the same medium containing 0.2% bovine serum albumin and then incubated for a further 2 h in the presence of 200 µCi [32P]orthophosphate, before extensive washing with cold medium. After pelleting, parasites were lysed (20 mM Tris pH 7.4, 10 mM sodium pyrophosphate, 50 mM sodium fluoride, 2 mM sodium-o-vanadate, 10 µg/ml leupeptin, 10 µg/ml aprotinin, 1 mM PMSF, 1% Triton X-100) for 30 min on ice and the lysate precleared by centrifugation at 12 000 g for 10 min. Supernatant aliquots were incubated for 3 h at 4°C with 10 µl specific antiserum, 30 µl protein A–Sepharose added and incubation continued for a further 2 h. Immune complexes were washed, recovered from the beads and analysed as described above, using 8% SDS–PAGE.
Yeast strains and transformation
All strains were grown in supplemented minimal media with the addition of a carbon source and complete supplement mixture minus leucine and uracil (BIO 101, Inc.) to maintain selection. Transformation was carried out using the lithium acetate method (36). Strain YAS2031 [MAT a pab1::HIS3, ade2-1, his3-11, leu2-3.112, trp 1-1, ura 3-1, can1-100; (38)] was transformed with the LEU2CEN vector p415 (28) containing lmpab1; vector alone was used as a control. The control strain yDM146 [MAT a pbp 1::LEU2, ade2-1, his3-11, leu2-3.112, trp 1-1, ura 3-1, can1-100; (15)] was transformed with the URA3CEN plasmid p416 (28) containing lmpab1; vector alone was used as a control. Transformants competent for the expression of LmPAB1 were then streaked onto plates lacking leucine in the presence of 5′-fluoro-orotic acid and uracil, with galactose as the sole source of carbon (36), grown for 1 week at 30°C and examined for growth.
RESULTS
LmPAB1 is highly conserved but displays several unusual features
All poly(A)-binding proteins to date are characterised by the presence of four highly conserved RNA binding domains (RBD) containing ribonucleoprotein (RNP) motifs. To facilitate cloning from L.major, degenerate primers 296F and 1013R were used to amplify the region between the RNP1 motif of RBD1 and the RNP2 motif of RBD4 from genomic DNA. The single 745 bp product was cloned (LmpabH3) and sequenced. Analysis of the deduced amino acid sequence showed high identity with RBD2 and RBD3 of known PAB1 sequences (data not shown). LmpabH3 was then used to screen a metacyclic cDNA library and a genomic cosmid library. A single cDNA clone (p8c1) and 11 contiguous cosmids were isolated; the cDNA and regions of the cosmid clones were sequenced on both strands to generate the lmpab1 gene sequence.
The 2.7 kb cDNA, p8c1, is complete at the 5′ end as indicated by the presence of the 39 nt spliced leader sequence found on all mature L.major transcripts. A 262 bp 5′-UTR precedes the 1680 bp open reading frame encoding the 560 amino acid poly(A)-binding protein (LmPAB1). The 3′-UTR is incomplete, as judged by the length of the single 3.2 kb transcript identified by RNA blotting (see Fig. 3).
Figure 3.
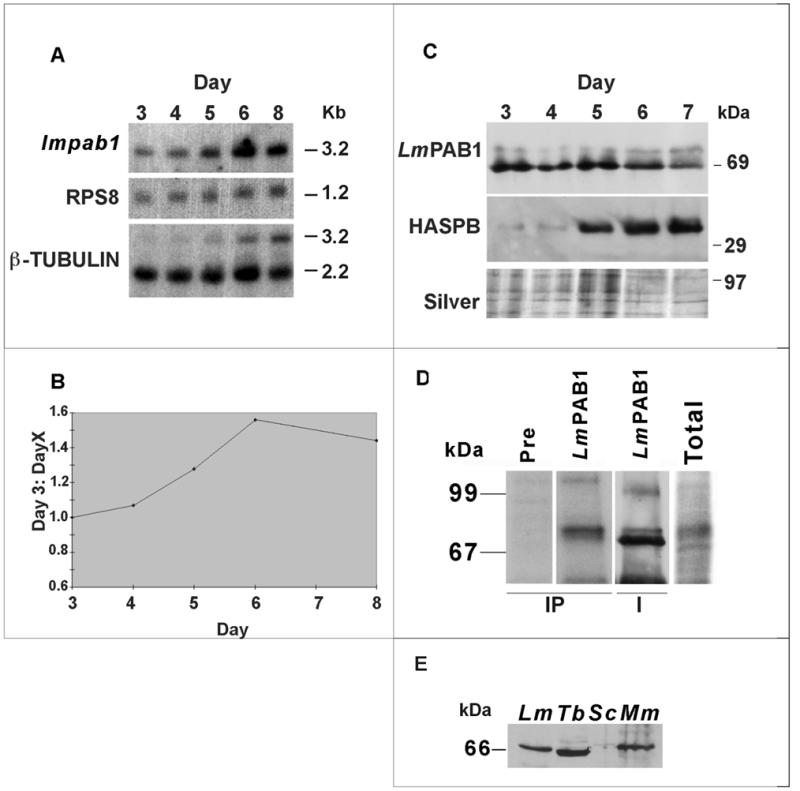
Analysis of the expression pattern of LmPAB1. (A) Total RNA, isolated from differentiating parasites over a time course (3–8 days), was size-separated, blotted and probed with the open reading frame of lmpab1. The same blot was re-probed with RPS8 (constitutively expressed, S8 ribosomal protein gene) and β-tubulin (which recognises differentially expressed transcripts of 2.2 and 3.2 kb; 45). (B) Expression of the lmpab1 transcript normalised against RPS8 expression and plotted as a ratio of day 3 (logarithmic parasite) expression levels. (C) Expression of LmPAB1 in differentiating parasites. Total proteins were extracted at daily intervals (3–7 days), separated by 10% SDS–PAGE, electroblotted and identical blots probed with abSK375 (anti-LmPAB1, top panel) and ab366 (anti-HASPB, middle panel). Silver staining was used to visualise protein loading (bottom panel). (D) In vivo phosphorylation of LmPAB1. Total lysate from 32P-radiolabelled parasites was immunoprecipitated (IP tracks) with anti-LmPAB1 or pre-immune serum, prior to analysis by 8% SDS–PAGE. The IP/LmPAB1 track was then immunoblotted against anti-LmPAB1 (I track). Relative exposure times at –70°C: total, 1 × 107 parasites labelled with 100 µCi 32P, 14 h; IP, 1 × 108 parasites labelled with 200 µCi 32P, 96 h. (E) Cross-reactivity of anti-LmPAB1with PAB1 from other species. Cell extracts from L.major (Lm, 5.0 × 106), T.brucei procyclics (Tb, 2.0 × 107), S.cerevisiae (Sc, 2.0 × 107) and mouse macrophages (Mm, 2.0 × 106) were separated by 10% SDS–PAGE and immunoblotted with anti-LmPAB1.
The 5′-UTR of lmpab1 (EMBL accession no. AF093062) contains two short A-rich tracts rather than the more extended regions of A residues present in the 5′-UTRs of all other pab1 transcripts identified. These stretches of up to 18 consecutive As within A-rich regions of 50–70 nt have been implicated in the auto-regulation of pab1 translation (39,40). In contrast, the longest consecutive stretch of As within the 5′-UTR of lmpab1 is four residues.
The 3′-UTR of lmpab1 is long (∼1.2 kb), a characteristic of pab1 transcripts from evolutionary diverse eukaryotes (41) but also typical of many trypanosomatid mRNAs. It displays at least one 90 bp A-rich tract containing regions of up to nine consecutive A residues, again typical of other pab1 transcripts, causing frequent internal oligo(dT) priming during cDNA cloning and subsequent underestimation of transcript length (41).
LmPAB1 itself shows strong identity with the PAB1s of other eukaryotes (Fig. 1). The sequence includes the four characteristic RBDs, although the generally well-conserved RBD4 (42) is more divergent in LmPAB1. Within the RNP1 motifs, the charged and aromatic residues important in RNA binding specificity are conserved (38). There is also strong identity with other functional homologues at the RBD1/RBD2 junction and at the extreme C-terminus.
Figure 1.

Alignment of LmPAB1 with PAB1 homologues. The open reading frame of LmPAB1 is aligned (CLUSTALV; 61) against PAB1 homologues from Human (H.s) [genpept accession no. g129617], S.cerevisae (S.c) [genpept accession no. P04147] and T.cruzi (T.c) [GenBank accession no. U06070]. Coloured boxes delineate the four N-terminal RNA binding domains (RBD1, blue, amino acids 22–105; RBD2, red, amino acids 106–193; RBD3, green, amino acids 194–288; RBD4, orange, amino acids 303–386) and the conserved C-terminal domain (purple, amino acids 498–560). RNP1 motifs are highlighted (asterisks) and potential sites of phosphorylation indicated (circles) (35).
More unexpectedly, LmPAB1 shows no more overall conservation with other trypanosomatid PAB1s than with a range of other eukaryotic PAB1s. Thus, the Trypanosoma brucei and T.cruzi PAB1 sequences share 86% identity with each other (43) but only 35% identity to the L.major protein (Fig. 1). Similarly, LmPAB1 is 36 and 38% identical to the Homo sapiens and S.cerevisiae sequences respectively, although identity is concentrated within the RBDs (44% identity across RBD1–4) rather than throughout the protein.
In all PAB1s characterised to date, the sequence between the N-terminal RBDs and the highly conserved C-terminal domain is the least conserved region of the protein and usually rich in proline and methionine. In LmPAB1, this ‘linker’ region (amino acids 386–495) contains 13.2% proline (compared to 15–19% in equivalent regions of other PAB1s) but is rich in basic residues (13.7%; Fig. 1). In S.cerevisiae, a protein which regulates polyadenylation has been identified by its interaction with the PAB1 linker region: the PAB1-binding protein 1 (Pbp1p) has a region of sequence similarity with PAB1 and mutations in this region prevent interaction of the two proteins (15). Secondary structure predictions indicate that this region of interaction is predominantly looped, the precise structure presumably varying between species, dependent on sequence. In addition a short helical domain, immediately N-terminal to the looped region, is necessary for the PAB1:Pbp1p interaction. Neither of these regions is conserved in LmPAB1, suggesting that any interacting proteins may also be divergent in the parasite.
LmPAB1 is expressed from a single copy gene during parasite differentiation
DNA blotting was used to determine the copy number and chromosomal location of the lmpab1 gene (Fig. 2). Probes covering both the N-terminal region of the deduced protein (including RBD 1–4; probe B) and the C-terminus (incorporating the linker region and the last few residues of RBD4; probe C) hybridised to a simple pattern of bands on blots of DNA digested with enzymes cutting both outside and within the gene sequence. These data, obtained from blots hybridised and washed at low stringency, indicate that there are no other sequences within the genome that are closely related to lmpab1. The original LmpabH3 probe, together with the 11 contiguous cosmids isolated by library screening, localised to a single locus on chromosome 35 (data not shown; 27). This analysis is consistent with the presence of a single lmpab1 gene copy in the Leishmania genome, rather than a closely-linked tandem repeat, as observed in T.brucei and T.cruzi (43,44).
Figure 2.
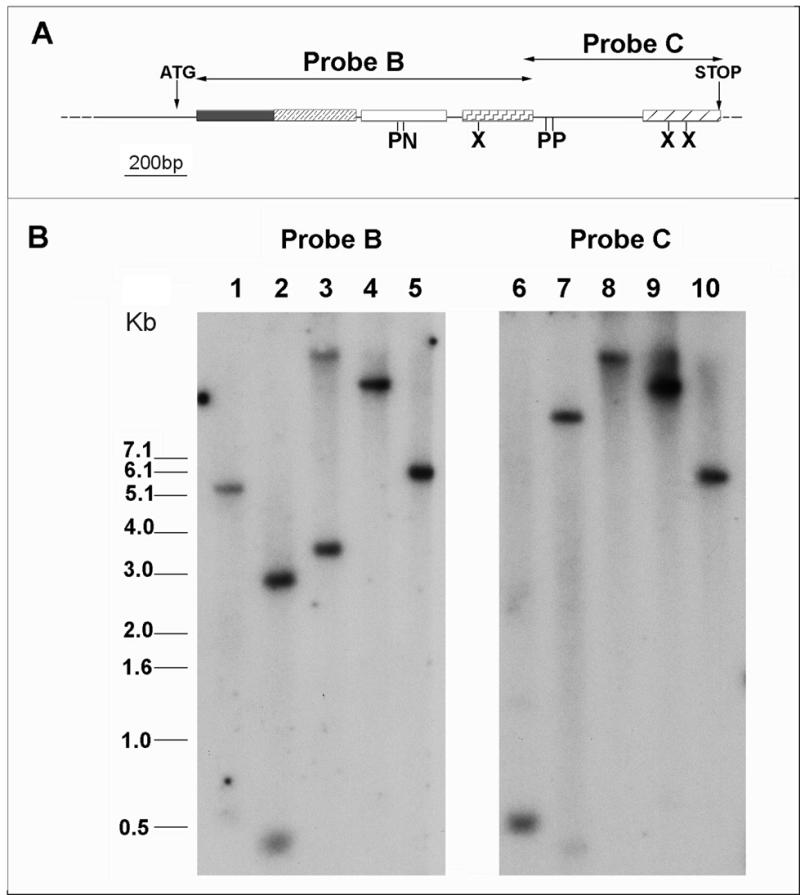
Southern analysis of L.major genomic DNA. (A) Partial restriction map of the lmpab1 locus. Regions encoding the RNA binding domains and the C-terminal domain are boxed and highlighted differentially. Positions of the initiator and stop codons and extent of probes B and C are shown. Internal restriction sites are indicated: N, NotI; P, PstI; X, XhoI. (B) Southern hybridisation of probes B and C to L.major genomic DNA digested with XhoI (lanes 1 and 6); PstI (lanes 2 and 7); NotI (lanes 3 and 8); EcoRI (lanes 4 and 9); ClaI (lanes 5 and 10). Blots were hybridised and washed at low stringency, as described.
The expression pattern of lmpab1 during in vitro differentiation of L.major promastigotes was determined by both RNA and immunoblotting (Fig. 3). Leishmania major grows logarithmically in culture over a period of several days before differentiating into infective metacyclic cells, which are metabolically quiescent and non-dividing. Total RNA extracted from parasites over a 7–8 day growth period was analysed by hybridisation with probes derived from the lmpab1 gene, the ribosomal protein S8 gene, RPS8 (constitutively expressed to normalise loading) and the β-tubulin locus (differentially expressed during metacyclogenesis; 45). Analysis of the relative band densities of lmpab1 and RPS8 transcripts demonstrates maximum expression of lmpab1 mRNA at day 6, when it is 1.5 times more abundant than in day 3 parasites (Fig. 3B). This correlates with parasite differentiation, monitored by hybridisation of β-tubulin transcripts of 2.2 kb (expressed throughout the growth cycle) and 3.2 kb (stabilised in metacyclic parasites).
To analyse protein expression during parasite differentiation, polyclonal antibodies were raised against recombinant LmPAB1. This antiserum recognises wild-type LmPAB1 as a 69 kDa protein, together with a second, low-abundance molecule of ∼75 kDa (Fig. 3C). The deduced size of LmPAB1 is 62.6 kDa; the aberrant migration of this protein is presumably due to its high proportion (15.4%) of positively charged amino acids (as observed elsewhere; 44). Extensive antibody purification failed to reduce the intensity of the 75 kDa band, and serum purification against the 75 kDa antigen did not reduce recognition of LmPAB1 (data not shown), suggesting that these proteins share a number of epitopes and may be isoforms. Indeed, use of high antibody titres on immunoblots reveals the presence of multiple faint bands in the 69–75 kDa range (data not shown).
PAB1 isoforms have been described in yeast, sea urchin and wheat (46,47) and are presumed to be differentially processed forms of the protein. It has been postulated that this modification is due to phosphorylation (46). To address this issue in Leishmania, parasites were radiolabelled with inorganic phosphate in vivo and the phosphorylated proteins subsequently immunoprecipitated with anti-LmPAB1 (Fig. 3D). By this analysis, LmPAB1 is clearly one of several proteins within the 60–70 kDa size range that are phosphorylated in stationary phase Leishmania and this modification is predominantly targetted to the 75 kDa form of the protein (compare IP and I tracks).
Immunoblotting protein extracts taken from parasites grown over 7 days in vitro reveals expression of the two LmPAB1isoforms throughout the growth cycle (Fig. 3C). Differentiation of the parasites is monitored by expression of HASPB, a surface marker for metacyclic cells, from day 5 onwards (25). This correlates with a modest reduction in the level of the 69 kDa LmPAB1 band; the abundance of yeast PAB1 is similarly reduced in stationary phase yeast cells (15). Thus, the 50% increase in mRNA abundance observed in stationary phase is not concomitant with an increase in the level of protein.
The LmPAB1 antibodies cross-react with T.brucei and mouse macrophage protein extracts but display very weak cross reactivity with S.cerevisiae proteins, suggesting that the epitopes recognised are poorly conserved in the yeast protein (Fig. 3E).
LmPAB1 is a major cytoplasmic poly(A) binding protein
In vitro binding assays were performed to confirm the function of LmPAB1. Poly(A) binding analysis with cytoplasmic fractions of L.major parasites identifies a single poly(A) binding complex of ∼60 kDa (Fig. 4A). The retention of ~50% of the binding activity in high salt (1 M NaCl) and complete abrogation of binding in the presence of a 10× concentration of specific competitor [poly(A)] indicates that binding is of high specificity and relatively high affinity. High affinity binding to poly(A) is a defining characteristic of PAB1; reduced affinity of LmPAB1 for poly(A) would indicate significant functional divergence of this protein. Direct confirmation of the binding activity of LmPAB1 was obtained by specific immunoprecipitation of the binding complexes (Fig. 4B). This was achieved with similar efficiency at both high and low salt concentration. The equal intensity of the bands obtained with anti-LmPAB1 is indicative of binding with equivalent affinity under both conditions. Thus, it can be estimated that LmPAB1 accounts for ∼50% of the cytoplasmic poly(A) binding activity observed. This suggests that there is at least one binding activity in the Leishmania cytoplasm, immunologically unrelated to LmPAB1, which binds poly(A) with low affinity. Such an activity could result from an abundant but relatively non-specific RNA binding protein; this has not been investigated further to date.
Figure 4.
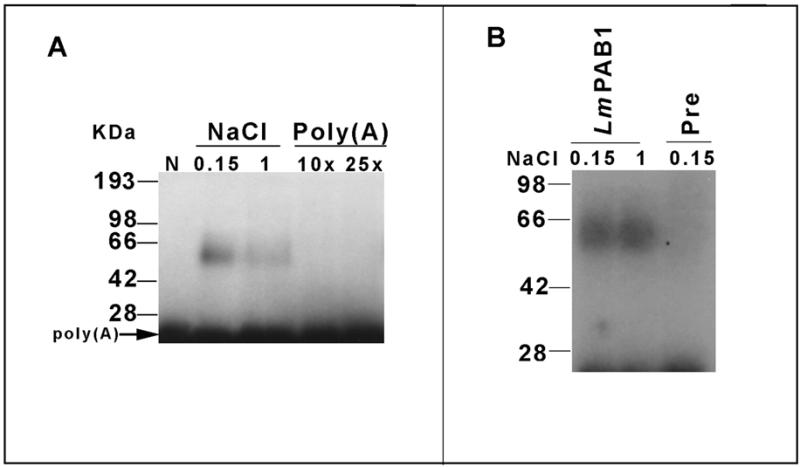
RNA binding analysis. (A) Binding of cytoplasmic parasite proteins to end-labelled poly(A). Reactions were performed at low (0.15 M) or high salt (1 M NaCl); unlabelled poly(A) competitor was added as indicated to reactions containing low salt. Complexes were UV cross-linked and separated by 10% SDS–PAGE followed by autoradiography. N, no lysate added. (B) Immunoprecipitation of LmPAB1:poly(A) complexes bound at low (0.15 M) or high (1 M) salt, with anti-LmPAB1 or with pre-immune serum (Pre), followed by 10% SDS–PAGE separation and autoradiography (4 days at –70°C).
Transcriptional inhibition alters the cellular distribution and abundance of LmPAB1
The subcellular distribution of LmPAB1 was investigated by indirect immunofluorescence, using DAPI as a counter-stain for the nucleus and kinetoplast (extended mitochondrion) of the parasite cell. These experiments demonstrate that LmPAB1 is concentrated in a peri-nuclear region, a location consistent with the cytoplasmic distribution of mRNA and indicative of protein association with the endoplasmic reticulum (Fig. 5A). Concentration of PAB1 in the cytoplasm has also been demonstrated in mammalian cells using similar techniques (48–50) but functional evidence also supports a nuclear location for a proportion of PAB1 molecules (13,14,48). To confirm the presence or absence of PAB1 in Leishmania nuclei, cellular fractionation followed by immunoblotting with anti-LmPAB1 and anti-histone H1 antibodies (34) were performed (Fig. 5B). Low levels of LmPAB1 were detected in the nuclear fraction, correlating with the presence of histone H1, although minor cytoplasmic contamination of the nuclear fraction could not be discounted in this analysis.
Figure 5.
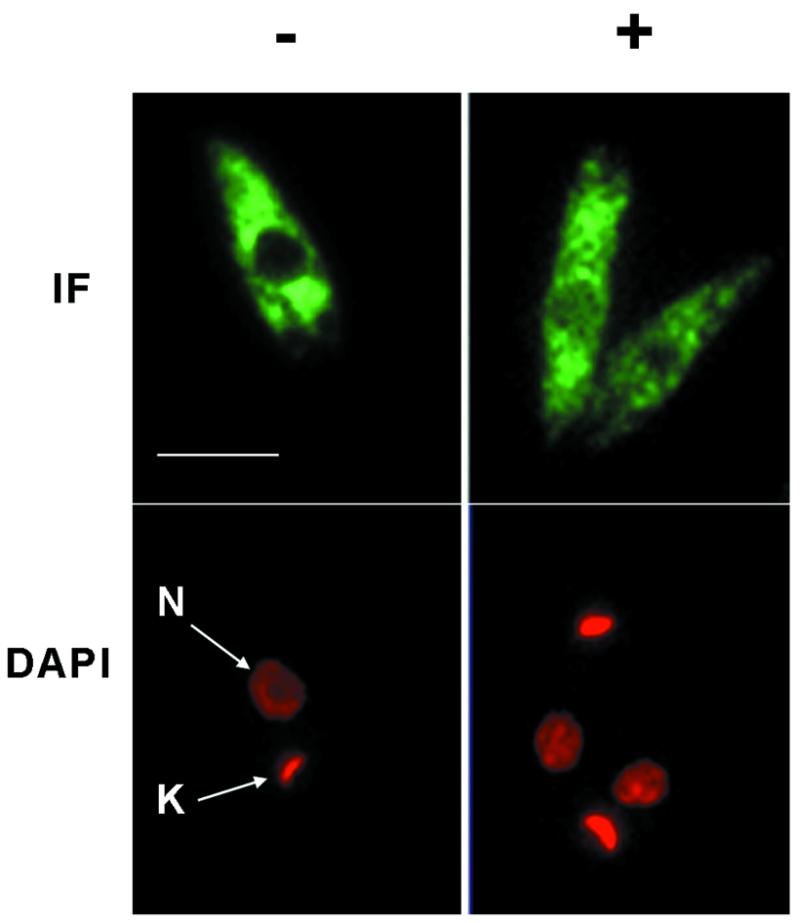
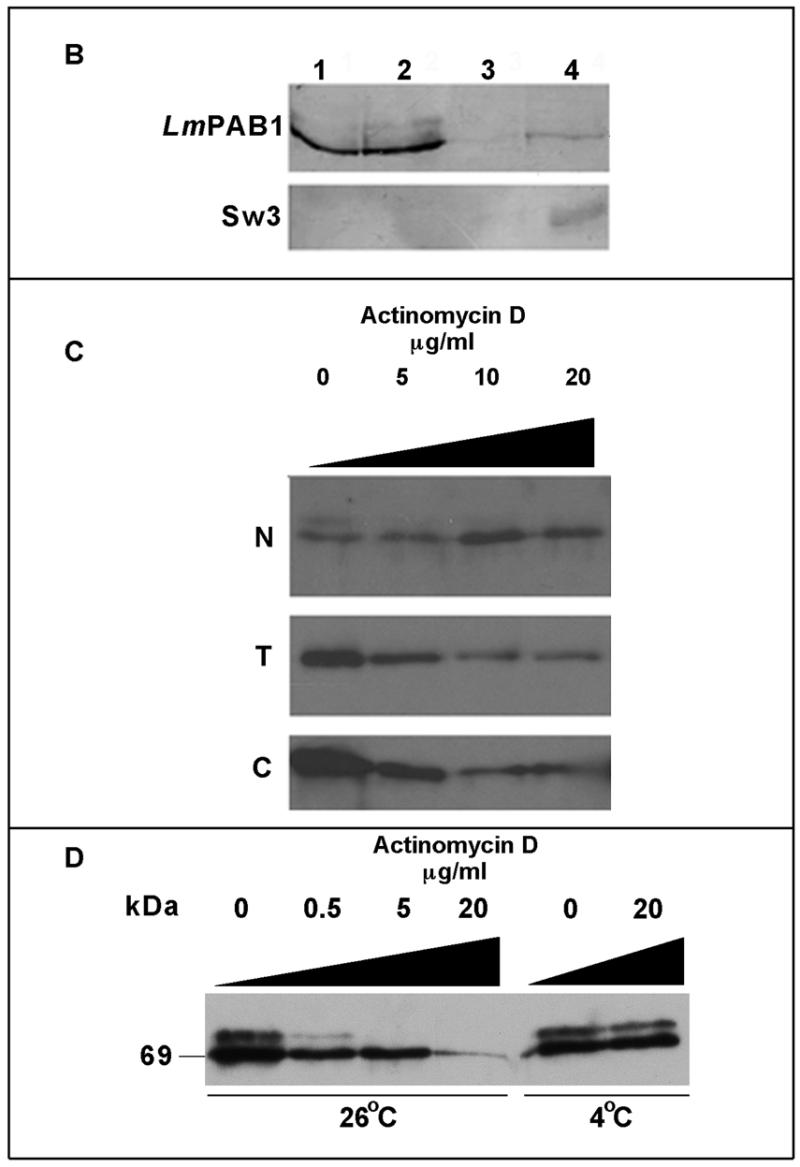
Transcriptional inhibition affects the cellular localisation of LmPAB1. (A) Immunofluorescent detection of LmPAB1 (with abSK375; IF) and DAPI staining of nuclear (N) and kinetoplast (K) DNA in late logarithmic phase parasites, untreated (–) or incubated with 10 µg/ml actinomycin D (+) for 10 h prior to fixing and permeabilisation. Scale bar = 5 µm. (B) Parasite fractions were separated (in the presence of protease inhibitors) and immunoblotted with anti-LmPAB1 (8% SDS–PAGE) or ab415 (Sw3, anti-histone H1; 10% SDS–PAGE). Lane 1, total lysate, 5 × 106 cells; lane 2, cytoplasmic fraction, 5 × 106 cells; lane 3, nuclear fraction, 5 × 106 cells; lane 4, nuclear fraction, 3 × 107 cells. (C) Parasites treated with 0–20 µg/ml of actinomycin D for 10 h were fractionated (in the absence of protease inhibitors) and analysed by 6% SDS–PAGE and immunoblotting with anti-LmPAB1. Chemiluminescent exposures were for 15 s [total (T) and cytoplasmic (C) fractions from 2.5 × 107 cells] or 1 min [nuclear fraction (N) from 4.5 × 107 cells]. (D) Cytoplasmic fractions from 2 × 106 parasites incubated with actinomycin D for 24 h at 26°C or 4°C as indicated. LmPAB1 was immunodetected by chemiluminescence, exposure time 8 min.
A recent study has shown that human PAB1 shuttles between the nucleus and the cytoplasm and only accumulates in the nucleus upon inhibition of transcription or during transient over-expression (48). A similar approach was adopted to demonstrate the accumulation of LmPAB1 in Leishmania nuclei. Late logarithmic parasites were incubated for 10 h under normal growth conditions in the presence of actinomycin D. Parasites were then lysed, fractionated and the fractions analysed by immunoblotting and indirect immunofluorescence (Fig. 5C).
The cellular content of LmPAB1 decreases with increasing inhibitor concentration, as shown by immunoblotting total cell lysate (T). Over the same time course, the level of total cellular protein was not depleted until the concentration of actinomycin D reached 20 µg/ml (data not shown). The drop in LmPAB1 abundance suggests that expression of the protein could be post-transcriptionally regulated in response to the abundance of mRNA within the cell. Analysis of fractionated, inhibitor-treated cells reveals an increase in the abundance of nuclear LmPAB1 with increasing concentration of actinomycin D and, at the same time, a decrease in total LmPAB1 is observed (Fig. 5C). These observations suggest that the cell fractionation methods used are robust and that cytoplasmic contamination of the nuclear fraction is minimal.
Accumulation of LmPAB1 is also apparent in the immunofluorescent analysis (Fig. 5A), in which partial occlusion of the nuclei is evident in actinomycin D-treated parasites. Under the experimental conditions used, no nuclear speckles (indicative of subnuclear localisation of LmPAB1) were observed, in contrast to the effects of transcriptional inhibition in HeLa cells, in which clear nuclear speckling is seen (48). It should be noted, however, that observation of speckles is dependent on the conditions used for indirect immunofluorescence microscopy and also on the transcriptional activity of the cell observed (reviewed in 51).
A feature of these transcriptional inhibitor studies is the disappearance of the 75 kDa isoform in the presence of actinomycin D. This is clearly seen in Figure 5D (an overexposed immunoblot of cytoplasmic proteins), in which it is also demonstrated that a drop in culture temperature to 4°C prevents depletion of this protein, indicative of an active process. This observation correlates with the demonstration that the 75 kDa protein is hyper-phosphorylated LmPAB1 (Fig. 3E). This molecule is also detectable in the nuclear fraction (Fig. 5B) but is undetectable following transcriptional inhibition, possibly indicative of processing within the nucleus. Cumulatively, these data support a nuclear role for LmPAB1 in the processing and/or transport of transcripts in Leishmania and suggest that modulation of LmPAB1 abundance and phosphorylation state occurs in response to RNA abundance.
LmPAB1 does not rescue the lethality of a pab1 deletion in S.cerevisiae
The pattern of conservation between the Leishmania and yeast PAB1 sequences suggested that heterologous functional complementation studies might elucidate essential functions of LmPAB1 and delineate relationships between the mechanisms of RNA metabolism in trypanosomatids and other eukaryotes. To this end, the lmpab1 open reading frame was cloned downstream of the galactose inducible promoter (GAL1) in the yeast expression vector p415 carrying the LEU2 selectable marker (28). The resulting plasmid, p415A, was transfected into the yeast strain YAS2031, a pab1 chromosomal null mutant containing a full length pab1 gene on a URA3 plasmid (pAS77). Immunoblotting of protein extracts from this and control transformants demonstrates induced expression of LmPAB1 from p415A in the presence of galactose and repression in the presence of glucose, whereas no protein expression is detected from the pAS77-encoded gene or in the presence of the vector p415 alone (Fig. 6A). The lack of cross-reactivity with wild-type yeast PAB1 confirms the results obtained in Figure 3E. A plasmid shuffle was then performed using 5′-fluoro-orotic acid to select against the presence of the URA3 selectable marker (36). Growth of a control strain, YDM146, which has the same genetic background as YAS2031 but carries the URA3 plasmid p416, acts as a positive control for the shuffle. Yeast strain YAS2031 pAS77 is unable to grow in the presence of 5′-fluoro-orotic acid and the presence of p415A does not rescue the lethality of the yeast knock-out (Fig. 6B), demonstrating that LmPAB1 is unable to complement the essential functions of PAB1 in S.cerevisiae.
Figure 6.
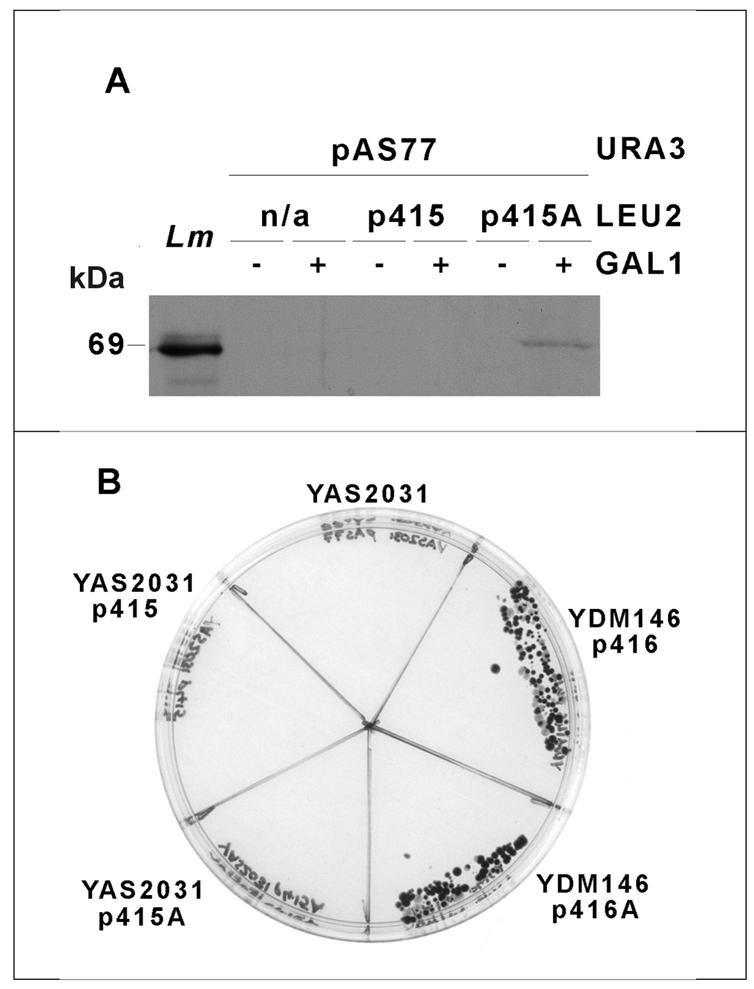
Functional complementation in S.cerevisiae. (A) Galactose inducible expression of LmPAB1 in yeast strain YAS2031. YAS2031 (pAS77) transformed with empty vector p415 or with vector carrying lmpab1 (p415A) and an untransformed control (n/a) were grown in the presence (+) or absence (–) of galactose. Expression of LmPAB1 was detected by immunoblotting and chemiluminescence. Lm, L.major total cell lysate (1 × 107 cells). (B) Yeast strain YAS2031 and control strain YDM146, transformed with empty vector (p416/p415) or vector plus lmpab1 (p416A/p415A) plated on to selective media including 5′-fluoro-orotic acid and galactose, as the sole carbon source.
DISCUSSION
The characterisation of LmPAB1 has identified several distinct features of the protein that may reflect the unusual mechanisms of gene expression utilised by trypanosomatids. Although LmPAB1 shares structural features with other eukaryotic PAB1s, the data presented here suggest that the function of this protein is divergent.
LmPAB1 is encoded by a single copy gene and there are no closely related proteins within the cell (Figs 2 and 3). In this respect, L.major contrasts with T.cruzi and T.brucei, which have two tandemly repeated genes encoding identical proteins (43,44). It is surprising that the sequence and genomic arrangement of the pab1 genes are not more highly conserved between trypanosomatid species: LmPAB1 is as closely related to its functional homologues in higher eukaryotes as it is to the other parasite proteins.
This divergence is unexpected, given the maintenance of identity both between the T.cruzi and T.brucei proteins and between the two copies of the gene within each organism, presumably due to strong selective pressure.
Antibodies raised against LmPAB1 recognise two proteins of 69 and 75 kDa apparent molecular mass. Their relative size and abundance, together with the disappearance of the larger band on transcriptional inhibition, suggest a role for modification by phosphorylation. This has been confirmed by in vivo radiolabelling, which has demonstrated hyperphosphorylation of the 75 kDa isoform of LmPAB1 and hypophosphorylation of the smaller protein (Fig. 3D), thus providing a possible explanation for the mobility difference observed. The presence of modified PAB1 isoforms has been noted in yeast, sea urchin and wheat (46,47) and yeast PAB1 interacts with protein kinase C (15). The Leishmania protein is the first PAB1 to be definitively demonstrated as a phosphorylated protein, however. Analysis of the amino acid sequence of LmPAB1 identifies seven potential sites for phosphorylation by protein kinase C (Fig. 1). Five of these are found within RBDs and three of the sites are highly conserved (although not with sites in the homologous proteins in T.brucei and T.cruzi). In addition, several casein kinase II sites are present in LmPAB1 but these are not well conserved. Phosphorylation of ribonucleoproteins has been implicated in RNA binding activity, cellular localisation and assembly of ribonucleoprotein complexes (52,53).
The level of LmPAB1 within the cell is responsive to the abundance of mRNA, as demonstrated by the depletion of total cellular LmPAB1 on inhibition of transcription and modest reduction of abundance in stationary phase cells. A small increase in the steady state level of lmpab1 transcripts in differentiating parasites does not correlate with an equivalent increase in abundance of the protein, suggesting that the regulation of expression of LmPAB1 is c-omplex. The 5′-UTR of the gene does not include the extensive A-rich tracts observed in other organisms, which are implicated in regulation of translation (39,40). The presence of A-rich tracts within the 3′-UTR of lmpab1 may be functionally significant, however.
The abundance of the hyperphosphorylated 75 kDa LmPAB1 isoform is depleted more rapidly than that of the 69 kDa protein upon transcriptional inhibition (Fig. 5C). However, the concentration of the 75 kDa isoform is maintained relative to the 69 kDa molecule in stationary phase Leishmania (Fig. 3B), in which constitutively transcribed housekeeping genes show decreased abundance of mature stable transcripts (J.K.Keen and D.F.Smith, unpublished). Clearly, the dynamics and function of LmPAB1 phosphorylation require further investigation but the data presented here suggest that this modification is responsive to mRNA abundance and transcription inhibition. In Aspergillus nidulans, over-expression of PAB1 results in inappropriate activation of differentiation (54). This effect has been attributed to disruption of the post-transcriptional regulation of development-specific genes. Consistent with this observation, tight modulation of LmPAB1 abundance and phosphorylation might play a role in maintaining the integrity of regulation in trypanosomatids.
The nuclear localisation of LmPAB1 is suggestive of a role in nuclear processing and/or transport of transcripts. However, LmPAB1 distribution within the nucleus is diffuse; there is no speckling indicative of association with a co-transcriptional processing complex. In contrast, distinct speckling of human PAB1 in the nucleus is observed in HeLa cells treated with actinomycin D and during transient over-expression of the protein (48). The observed nuclear speckling of T.brucei SR protein homologues (55,56) indicates that concentration of processing factors at sites of transcriptional activity does occur in trypanosomes. The diffuse distribution of LmPAB1 within the nucleus may reflect fundamental aspects of the dynamics and function of a putative trypanosomatid mRNA processing complex. The requirement of transcription for LmPAB1 nuclear export suggests that transcripts entering the cytoplasm are associated with LmPAB1.
The inability of LmPAB1 to rescue the lethality of the S.cerevisiae pab– mutant demonstrates that the Leishmania protein is unable to mediate the essential interactions of PAB1 with RNA and/or protein in yeast, an observation consistent with significant functional divergence between the two organisms. Current data suggest that there is no single essential interaction of eukaryotic PAB1 beyond the requirement that it be complexed with RNA (38). Rather, the contribution made to co-ordination of mRNA metabolism makes PAB1 essential for viability.
Whilst previous results have demonstrated that Arabidopsis and wheat PAB1s can partially complement yeast PAB1 (47,57), these studies were carried out in a yeast strain (YAS100) which differs from the one used in this work (YAS2031) in being more sensitive to mutation of PAB1 (38). Although the genetic basis for this difference has yet to be investigated, YAS2031 viability cannot be maintained by RBD4 alone (38,58), whereas this domain is sufficent for viability of YAS100 (59). Expression of human PAB1 from a 2 µ plasmid will maintain the viability of YAS2031, however, demonstrating that this protein can complement the essential functions of yeast when expressed in excess (60). Thus, it is probable that the inability of Lmpab1 to rescue the lethality of the yeast pab1 deletion is a consequence of gross differences in RNA metabolism between these organisms.
High specificity and affinity for poly(A) is a conserved feature of the Leishmania protein. However, specific high affinity binding of PAB1 to poly(A) is dispensable for viability in yeast (38). Sequence divergence within the RBDs of LmPAB1 may alter the RNA binding profile of the protein. Whereas poly(A) binding is a conserved and defining characteristic of LmPAB1, binding to other sequences and overall affinity for RNA may differ from the yeast protein. In this respect the divergence of RBD4 may be significant; this domain is responsible for non-specific binding to RNA (38).
Additionally it has been shown that binding of S.cerevisiae PAB1 to poly(A) is not sufficient to enhance poly(A)-dependent translation in vitro (58), while another study has uncoupled PAB1 poly(A)-binding activity from its effects on mRNA stability (59). In conclusion, our data support a model in which binding of PAB1 to RNA is necessary but not sufficient for the essential functions of this protein. Further analysis will elucidate the role of LmPAB1 in the assembly of RNA processing complexes in Leishmania.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Alan Sachs, Allan Jacobson and David Mangus for yeast vectors, strains and helpful advice, Nicholas Fasel for ab415, Al Ivens for cosmid library filters, Diane McGhie for parasite maintenance, SauFung Ma for macrophage culture, Helen Field and Tim Jeffries for T.brucei procyclics, and Jane Keen and Paul McKean for helpful discussion and critical reading of this manuscript. E.J.B. was the recipient of a Medical Research Council Postgraduate Studentship; E.K. holds a Department of Biochemistry Graduate Student Bursary. This work was supported by the Wellcome Trust (ref. no. 045493/Z/95/Z).
DDBJ/EMBL/GenBank accession no. AF093062
REFERENCES
- 1.Vanhamme L. and Pays,E. (1995) Microbiol. Rev. 59, 223–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ullu E., Tschudi,C. and Gunzl,A. (1996) In Smith,D.F. and Parsons,M. (eds), Molecular Biology of Parasitic Protozoa. IRL Press, Oxford, UK, pp. 115–133.
- 3.LeBowitz J.H., Smith,H.Q., Rusche,L. and Beverley,S.M. (1993) Genes Dev., 7, 996–1007. [DOI] [PubMed] [Google Scholar]
- 4.Matthews K.R., Tschudi,C. and Ullu,E. (1994) Genes Dev., 15, 491–501. [DOI] [PubMed] [Google Scholar]
- 5.Vassella E., Braun,R. and Roditi,I. (1994) Nucleic Acids Res., 22, 1359–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schurch N., Hehl,A., Vassella,E., Braun,R. and Roditi,I. (1994) Mol. Cell. Biol., 14, 3668–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hug M., Hotz,H.R., Hartmann,C. and Clayton,C. (1994) Mol. Cell. Biol., 14, 7428–7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapman A.B. and Agabian,N. (1994) J. Biol. Chem., 269, 4754–4760. [PubMed] [Google Scholar]
- 9.Neugebauer K.M. and Roth.M.B. (1997) Genes Dev., 11, 3279–3285. [DOI] [PubMed] [Google Scholar]
- 10.Hotz H., Hartmann,C., Huober,K., Hug,M. and Clayton,C. (1997) Nucleic Acids Res., 25, 3017–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weston D., La Flamme,A.C. and Van Voorhis,W.C. (1999) Mol. Biochem. Parasitol., 102, 53–66. [DOI] [PubMed] [Google Scholar]
- 12.McGwire B.S. and Chang,K. (1996) J. Biol. Chem., 271, 7903–7909. [DOI] [PubMed] [Google Scholar]
- 13.Minvielle-Sebastia L., Preker,P.J., Weiderkehr,T., Strahm,Y. and Keller,W. (1997) Proc. Natl Acad. Sci. USA, 94, 7897–7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amrani N., Minet,M., Le Gouar,M., Lacroute,F. and Wyers,F. (1997) Mol. Cell. Biol., 17, 3694–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mangus D., Aramani,N. and Jacobson,A. (1998) Mol. Cell. Biol., 18, 7383–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallie D.R. (1998) Gene, 216, 1–11. [DOI] [PubMed] [Google Scholar]
- 17.Hoshino S., Imai,M., Kobayiashi,T., Uchida,N. and Katada,T. (1999) J. Biol. Chem., 274, 16677–16680. [DOI] [PubMed] [Google Scholar]
- 18.Sachs A.B. and Davis,R.W. (1989) Cell, 58, 857–867. [DOI] [PubMed] [Google Scholar]
- 19.Decker C.J. and Parker,R. (1993) Genes Dev., 17, 1632–1643. [DOI] [PubMed] [Google Scholar]
- 20.Muhlrad D., Decker,C.J. and Parker,R. (1994) Genes Dev., 8, 855–866. [DOI] [PubMed] [Google Scholar]
- 21.Muhlrad D., Decker,C.J. and Parker,R. (1995) Mol. Cell. Biol., 15, 2145–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caponigro G. and Parker,R. (1995) Genes Dev., 9, 2421–2432. [DOI] [PubMed] [Google Scholar]
- 23.Couttet P., Fromont-Racine,M., Steel,D., Pictet,R. and Grange,T.L. (1997) Proc. Natl Acad. Sci. USA, 94, 5628–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molyneux D.H. and Killick-Kendrick,R. (1987) In Peters,W. and Killick-Kendrick,R. (eds), The Leishmaniases in Biology and Medicine. Academic Press, London, UK, Vol. 1, pp. 121–168.
- 25.Flinn H.M., Rangarajan,D. and Smith,D.F. (1994) Mol. Biochem. Parasitol., 65, 259–270. [DOI] [PubMed] [Google Scholar]
- 26.Langford C.K., Ullman,B. and Landfear,S.M. (1992) Exp. Parasitol., 74, 360–361. [DOI] [PubMed] [Google Scholar]
- 27.Ivens A.C., Lewis,S.M., Bagherzadeh,A., Zhang,L., Chan,H.M. and Smith,D.F. (1998) Genome Res., 8, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mumberg D., Muller,R. and Funk,M. (1994) Nucleic Acids Res., 22, 5767–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rangarajan D., Gokool,S., McCrossan,M.V. and Smith,D.F. (1995) J. Cell Sci., 108, 3359–3366. [DOI] [PubMed] [Google Scholar]
- 30.Chomczynski P. and Sacchi,N. (1987) Anal. Biochem., 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook J., Fritsh,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- 32.Goldstein L.S.B., Laymon,R.A. and MacIntosh,J.R. (1986) J. Cell Biol., 102, 2076–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKean P.G., Delahay,R., Pimenta,P.F. and Smith,D.F. (1997) Mol. Biochem. Parasitol., 85, 221–231. [DOI] [PubMed] [Google Scholar]
- 34.Noll T.M., Desponds,C., Belli,S.I., Glaser,T.A. and Fasel,N.J. (1997) Mol. Biochem. Parasitol., 84, 215–227. [DOI] [PubMed] [Google Scholar]
- 35.Rost B. and Sander,C. (1993) Proc. Natl Acad. Sci. USA, 90, 7558–7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaiser C., Michaelis,S. and Mitchell,A. (1994) Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- 37.Will C.L., Hastner,B. and Lührmann,R. (1994) In Higgins,S.J. and Hames,B.D. (eds), RNA Processing: A Practical Approach. Vol. II. IRL Press, Oxford, UK.
- 38.Deardorff J.A. and Sachs,A.B. (1997) J. Mol. Biol., 269, 67–81. [DOI] [PubMed] [Google Scholar]
- 39.De Melo Neto O.P., Standart,N. and de Sa,C.M. (1995) Nucleic Acids Res., 23, 2198–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bag J. and Wu,J. (1996) Eur. J. Biochem., 237, 143–152. [DOI] [PubMed] [Google Scholar]
- 41.Lefrere V., Vincent,A. and Amalric,F. (1990) Gene, 96, 219–225. [DOI] [PubMed] [Google Scholar]
- 42.Burd C.G., Mantunis,E.L. and Dreyfuss,G. (1991) Mol. Cell. Biol., 11, 3419–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hotchkiss T.L., Nerantzakis,G.E., Dills,S.C., Shang,L. and Read,L.K. (1998) Mol. Biochem. Parasitol., 98, 117–129. [DOI] [PubMed] [Google Scholar]
- 44.Batista J.A.N., Teixeira,S.M.R., Donelson,J.E., Kirchhoff,L.V. and Martins de Sa,C. (1994) Mol. Biochem. Parasitol., 67, 301–312. [DOI] [PubMed] [Google Scholar]
- 45.Coulson R.M., Connor,V., Chen,J.C. and Ajioka,J.W. (1996) Mol. Biochem. Parasitol., 82, 227–236. [DOI] [PubMed] [Google Scholar]
- 46.Drawbridge J., Grainger,J.L. and Winkler,M.M. (1990) Mol. Cell. Biol., 10, 3994–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Le H., Chang,S., Tanguay,R.L. and Gallie,D.R. (1997) Eur. J. Biochem., 243, 350–357. [DOI] [PubMed] [Google Scholar]
- 48.Afonina E., Stauber,R. and Pavlakis,G.N. (1998) J. Biol. Chem., 273, 13015–13021. [DOI] [PubMed] [Google Scholar]
- 49.Anderson J.T., Paddy,M.R. and Swanson,M.S. (1993) Mol. Cell. Biol., 13, 6102–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gorlach M., Burd,C.G. and Dreyfuss,G. (1994) Exp. Cell Res., 211, 400–407. [DOI] [PubMed] [Google Scholar]
- 51.Singer R.H. and Green,M.R., (1997) Cell, 91, 291–294. [DOI] [PubMed] [Google Scholar]
- 52.Walker J., Dale,M. and Standart,N. (1996) Dev. Biol., 173, 292–305. [DOI] [PubMed] [Google Scholar]
- 53.Hamilton B.J., Burns,C.M., Nichols,R.C. and Rigby,W.F.C. (1997) J. Biol. Chem., 272, 28732–28741. [DOI] [PubMed] [Google Scholar]
- 54.Marhoul J.F. and Adams,T.H. (1996) Genetics, 144, 1463–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manger I. and Boothroyd,J. (1998) Mol. Biochem. Parasitol., 97, 1–11. [DOI] [PubMed] [Google Scholar]
- 56.Ismaili N., Perez-Morga,D., Walsh,P., Mayeda,A., Pays,A., Tebabi,P., Krainer,A.R. and Pays,E. (1994) Mol. Biochem. Parasitol., 102, 103–115. [DOI] [PubMed] [Google Scholar]
- 57.Belotosky D.A. and Meagher,R.B. (1996) Plant Cell, 8, 1261–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kessler S.H. and Sachs,A.B. (1998) Mol. Cell. Biol., 18, 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sachs A.B., Davis,R.W. and Kornberg,R.D. (1987) Mol. Cell. Biol., 7, 3268–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Otero L.J., Ashe,M.P. and Sachs,A.B. (1999) EMBO J., 18, 3153–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith R.F., Wiese,B.A., Wojzynski,M.K., Davison,D.B. and Worley,K.C. (1996) Genome Res., 6, 454–462. [DOI] [PubMed] [Google Scholar]