

Abstract
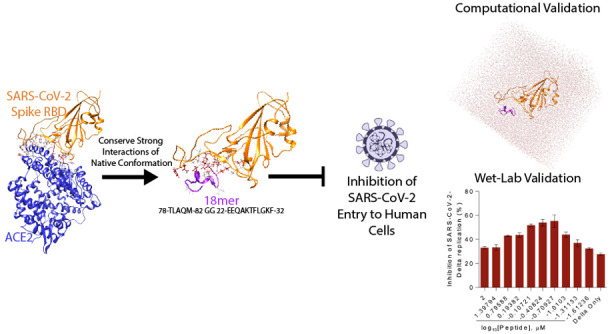
Entry inhibitors are an important resource in the response against emerging pathogens like the novel SARS-CoV-2, which enters human cells via interaction between the surface spike glycoprotein and the cellular membrane receptor angiotensin-converting enzyme 2 (ACE2). Using a combination of comparative structural analyses of the binding surface of the spike to ACE2, docking experiments, and molecular dynamics simulations, we identified a stable fragment of ACE2 that binds to the spike, is soluble, and is not predicted to bind to its physiological ligand angiotensin II. From this fragment we computationally designed and experimentally validated a smaller, stable peptide that disrupts ACE2-spike interaction at nanomolar concentrations, suggesting its potential use as a decoy that could interfere with viral binding by competition.
Keywords: entry inhibitors, ACE2, SARS-CoV-2, spike, decoy
The entry of the novel SARS-CoV-2 into the host cell is mediated by the interaction between the viral transmembrane spike glycoprotein and the cellular membrane receptor angiotensin-converting enzyme 2 (ACE2) .1 The spike is synthesized as a precursor of about 1300 amino acids that is then cleaved into an amino N-terminal S1 subunit of about 700 amino acids and a carboxyl C-terminal S2 subunit of about 600 amino acids. Three S1–S2 heterodimers assemble to form a trimer protruding from the viral surface. The S1 subunit contains a receptor-binding domain (RBD), while the S2 subunit contains a hydrophobic fusion peptide. Upon receptor binding, the S1 subunit is cleaved, and the fusion S2 subunit undergoes a conformational rearrangement to form a six-helix bundle that mediates viral and cellular membrane fusion.2
The angiotensin-converting enzyme (ACE)-related carboxypeptidase, ACE2, is a type I integral membrane protein receptor of about 805 amino acids that contains one HEXXH + E zinc-binding consensus sequence. ACE2 is a close homolog of the somatic angiotensin-converting enzyme (ACE; EC 3.4.15.1), a peptidyl dipeptidase that plays an important role in the renin–angiotensin system. ACE2 sequence includes an N-terminal signal sequence (amino acids 1 to 18), a potential transmembrane domain (amino acids 740 to 763), and a potential metalloprotease zinc-binding site (amino acids 374 to 378, HEMGH). The internal cavity hosts the angiotensin I substrate (consisting of amino acids 1 to 10 of the angiotensinogen precursor) that ACE2 converts into angiotensin II (amino acids 1 to 8) .3,4 ACE2 is expressed mainly in heart, kidney, testis, smooth muscle, and in coronary vessels and it seems to increase in lately differentiated epithelial tissues.5 The expression of ACE2 seems inversely regulated by the expression levels of ACE, a key regulator of blood pressure and the target of the pharmacological ACE inhibitors that control blood pressure.6
X-ray and cryo-electron microscopy (cryo-EM) structures of complexes between the viral spike from different Coronaviruses both alone and in complex with ACE2 have been solved.7−12 We studied the structural features of the binding of ACE2 to spike by analyzing the crystallographic and cryo-EM structures of the spikes from SARS-CoV and SARS-CoV-2, human ACE2, and their complexes.
In addition to small molecules inhibitors,13 several groups have proposed decoy inhibitors based on the first two α-helices of ACE2 using different techniques. Some of the publications rely on a linear component of the ACE2 sequence from either the first or second α-helix.14−17 Others also investigated mutations and modifications to the linear sequence of the α-helices.15,18−22 There are only two other publications we are aware of that combine two fragments of ACE2 with a linker but with different sequences.23,24
By performing structural analyses and molecular docking experiments, we identified the most important residues of ACE2 involved in the interaction with spike. These residues are mostly located along two α-helices, as other studies pointed out,23,25 and do not seem to interfere with ACE2 physiological ligand, angiotensin II. Using molecular dynamics (MD) simulations we confirmed that the ACE2-derived peptide that spans the two α-helices remains in complex with the spike and it appears to be stable in an aqueous environment. From this fragment, we engineered a smaller, stable peptide of 18 amino acids and experimentally confirmed that it specifically disrupts ACE2-spike interaction at nanomolar concentrations. We conclude by discussing scalability and cost related to the synthesis of this peptide, and we highlight how the molecule could be further studied for potential applications in a diagnostic and clinical context.
Results
Structural Determinants of ACE2 Binding to the SARS-CoV-2 and SARS-CoV Spike Viral Protein
Using the coordinates of the crystal structure of the ACE2 in complex with the spike (PDB: 6M0J), we determined the per-residue interaction energies with RosettaDock.26,27 The results are shown in Figure 1A and 1B, where we highlight the key residues involved in binding, according to their interaction energy values. Additional tables listing the interacting amino acids between the human receptor ACE2 and viral proteins from SARS-CoV-2 and SARS-CoV, along with the corresponding interaction energies, can be found in the Supporting Information.
Figure 1.

Structural analysis of viral spike binding to ACE2. Panel A highlights the residues in ACE2 (blue) that are involved in binding to the SARS-CoV-2 spike (orange backbone - black labels). The residues are colored by interaction energy as predicted by RosettaDock on a sliding scale: from −4.645 Rosetta Energy Units (REU) (dark red) to >0 REU (white). Panel B gives a surface representation of the structures in panel A. Panel C shows a superposition of SARS-CoV (gray backbone - red labels) and SARS-CoV-2 (orange backbone - black labels) spike in complex with ACE2 (blue). Panel D shows NL63-CoV spike (green backbone - black labels) in complex with ACE2 (blue).
In addition, we compared the same complex in another coronavirus, CoV-NL63 (PDB: 3KBH), which targets the ACE2 as well. The structure of CoV-NL63 spike shows limited structural similarity in binding to ACE2 as compared to the other two SARS viruses (Figure 1D). CoV-NL63 spike appears to bind to a narrower area of ACE2, centered around Lys353, which interacts with the conserved viral amino acid Tyr498.
The amino acids of the SARS-CoV-2 spike involved in binding to ACE2 span a poorly structured region from Leu455 to Tyr505 (including a disulfate bridge between Cys480 and Cys488), which corresponds to the Tyr442-Tyr491 region in SARS-CoV.
The amino acids of ACE2 involved in viral protein binding span two back-to-back helices, α-helices 1 and 2, from Ser19 to Tyr83. Based on the per-residue energy breakdown (Figure 2), this area contributes almost 80% of the total interaction energy calculated with RosettaDock. An additional, point-wise binding feature is represented by Lys353 located in the connecting loop of a downstream beta-hairpin. Lys353 is anchored to the N-terminal ACE2 helix by Tyr41 and Asp38, and it contributes 17% of the total interaction energy. An additional small downstream helix contributes negligibly to the total binding energy. In addition, Lys31 in ACE2 comes into contact with the spike loop encompassing amino acids (aa) 452–454, whose corresponding sequences in SARS-CoV have been shown to be important for binding by mutagenesis experiments.28
Figure 2.

Energy breakdown of crystal structure 6M0J interface. Energy values are calculated with Rosetta’s Residue Energy Breakdown application and curated into the heat map. Interaction energies are labeled, and negative interactions are colored according to the REU scale shown on the right. Values are calculated after the structure has undergone refinement with 100 rounds of Rosetta’s standard relax protocol.
The ACE2 Double Helix Appears Stable in Complex with the Viral Spike and in an Aqueous Environment
The comparative structural analyses and the interaction energy calculations suggest that most ACE2 key residues involved in spike binding are clustered in the back-to-back N-terminal part of α-helix 1 and C-terminal part of α-helix 2.
To determine whether the two helices do in fact remain stably in complex with the spike, we performed a 100 ns MD simulation of this complex. We visually inspected the trajectory and computed the root-mean-square deviation (RMSD) over time with respect to the energy-minimized initial conformer (Figure 3A, blue curve). In addition, we calculated the fraction of residues that remain in α-helix conformation over time (Figure 3B, blue curve). These results suggest that the double helix in complex with the spike is stable.
Figure 3.

Molecular dynamics simulations of the ACE2 single and double helix alone and in complex with spike. Panel A shows the root-mean-square deviation (RMSD) over time with respect to the initial minimized and equilibrated structures (as determined by GROMACS rms). Panel B shows the fraction of residues in α-helix (as determined by GROMACS do_dssp) for the two ACE2 fragments over time. All residues are extracted from PDB 6M0J. The lines correspond to double-helix residues in isolation (aa 19–83, green), double helix with spike with residues beyond aa 83 of ACE2 removed (blue), and single helix in isolation (aa 19–52, orange).
Further, we analyzed whether the double helix is stable in an aqueous environment in the absence of the spike, a feature that would be desirable for a viable inhibitor. We carried out a 100 ns MD simulation of the double helix alone, visually inspected the trajectory, and compared the RMSD with respect to the initial conformer (Figure 3A, green curve). We also computed the fraction of residues that remain in α-helix conformation over time (Figure 3B, green curve). The results of this analysis indicate that the double helix is fairly stable in an aqueous environment.
We also compared the stability of the double helix versus that of α-helix 1 alone. As shown in Figures 3A and 3B (orange curves), the single helix is substantially less stable than the double helix, with almost half of the residues losing the α-helical conformation over time. Although the helix might correctly refold upon substrate binding, this observation suggests that a double-helical conformation might enhance the peptide stability.
The ACE2 Double Helix Does Not Appear to Interfere with the Physiological Substrate Angiotensin II
To determine whether the double-helix fragment has the potential to bind to angiontensin II, possibly interfering with that physiological system, we docked the angiotensin II ligand extracted from the crystal structure of the complex with the ACE receptor (PDB: 4APH, chain P) and docked onto the crystal structure of ACE2 active site by RosettaDock (Figure 4). The angiotensin II binding site is located in an internal cavity of the receptor, quite far from the surface where the viral spike binds. Most of the residues of ACE2 involved in binding to angiotensin II are internal to the core of the protein. Only two amino acids (Asp51 and Ser47) in the C-terminal part of α-helix 1 have appreciable interactions with angiotensin II, and contribute about 15% of the total interaction energy (not shown).
Figure 4.

Docking of the physiological ligand angiotensin II onto ACE2. The molecule surfaced in pink is angiotensin II docked onto ACE2. The residues depicted in red and yellow in the ACE2 double helix make contact with angiotensin II and reside at the C-terminus of the α-helix 1. The residues highlighted in green represent the bulk of the ACE2 physiological binding site and do not encompass the double-helix fragment.
These results suggest that the double-helix fragment is highly unlikely to interfere with angiotensin II in a physiological context.
Engineering an 18 Amino Acid Peptide That Binds the Viral Spike at Nanomolar Concentration
Having identified ACE2 residues interacting with the spike as clustered in a double helix and assessed that the double-helical arrangement conferred structural stability to this ACE2 binding site, we engineered an 18 amino acid peptide (18-mer) out of the back-to-back ACE2 double-helical fragment. We fused the C-terminal part of the α-helix 2 (aa 78 to 82) to the N-terminal part of the αred-helix 1 (aa 22 to 32) using a two glycine linker, obtaining the following 18-mer sequence: 78-TLAQM-82 GG 22-EEQAKTFLDKF-32, with numbers delimiting the amino acids in the two contributing helices.
A tertiary structure of the 18-mer peptide predicted by PEPstrMOD was superimposed by Chimera MatchMaker to the crystal structure of ACE2 in complex with the spike and then docked by RosettaDock (Figure 5A). The subsequence EEQAKT shows good superposition with ACE2. The N-terminal part of the 18-mer (corresponding to the C-terminus of α-helix 2) appears to be unstructured, but correctly follows the orientation of the α-helix 2. In contrast, the C-terminal part of the 18-mer, FLDKF, is unstructured and does not encompass the corresponding sequence in α-helix 1, being somehow folded back and assuming a sort of “globular” fold, apparently guided by the hydrophobic interaction among Phe28-Thr78 and Leu79.
Figure 5.

18-mer predicted interaction with ACE2 and structural analysis Panel A shows the tertiary structure of the 18-mer (gray), predicted by PEPstrMOD, superimposed to the corresponding ACE2 crystal structure (blue, 60% transparency) in complex with spike (orange) by Chimera, (MatchMaker tool) with docked position (purple) as predicted by RosettaDock. Panel B shows the interactions between the 18-mer and spike. The residues are colored by interaction energy as predicted by RosettaDock, on a sliding scale from −2.858 REU (dark red) to >0 REU (white)). Panel C (front - aligned to panel B) and D (rear) show the surface representation of the hydrophobic interactions as calculated by Chimera (kdHydrophobicity tool). The peptide is shown with 70% transparency. Hydrophobic regions are displayed in magenta while hydrophilic ones are displayed in cyan.
Figure 6 displays the per-amino acid interaction energy between the docked peptide and spike.
Figure 6.

Interaction energy breakdown for 18-mer residues in complex with SARS-CoV-2 spike. Energy values are calculated with Rosetta’s Residue Energy Breakdown and curated into the heat map. Interaction energies are labeled and negative (favorable) interactions are colored according to the scale. Positive values are shown but not colored.
PBSA Analysis of Docked Peptide Position
To further characterize the interaction between the 18-mer and spike, we performed an MD simulation of the native crystal structure and predicted docked complex of the 18-mer with the spike RBD using GROMACS.29,30 Using this MD simulation, we then performed a Molecular Mechanics/Poisson–Boltzmann (Generalized-Born) surface area (MMPBSA) analysis to predict the binding free energy using the tool gmx_MMBPSA.31 The average free energy binding calculated for the native spike+ACE2 interaction (PDB: 6M0J) was −46.38 ± 10.44 kcal/mol. The average free energy binding calculated for the docked spike+18-mer interaction was −38.84 ± 8.15 kcal/mol, indicating a slight decrease in binding energy with respect to the full double helix, as expected. Full plots are available in Supporting Information Figure S1.
Experimental Validation
The 18-mer Disrupts Spike/ACE2 Binding at Low Concentrations
We performed immunoprecipitation experiments to assess the binding efficiency of the 18-mer. We used an in vitro system where the spike, overexpressed from HEK-293 cell lysate, immunoprecipitates ACE2 (Figure 7, upper panel). We assessed the stability of this binding by using nanomolar concentrations of three different peptides (Figure 7, lower panel): the physiological N-terminal part of the α-helix 1 (P1, aa 22 to 42), this same peptide with a single mutation as a control (P2, aa 22 to 42 with Aspartate 38 mutated to Glycine), and the engineered 18-mer (P3). Increasing concentrations of these peptides were incubated with the spike before immunoprecipitation with ACE2. The 18-mer efficiently inhibits ACE2-spike binding, better than the physiological N-terminal part of the α-helix 1. The Aspartate mutation in ACE2 physiological peptide dramatically reduces this binding.
Figure 7.

Interaction of ACE2 with viral spike is inhibited by the 18-mer in immunoprecipitation. Spike, overexpressed from HEK-293 cell lysate, immunoprecipitates ACE2 protein (upper panel). Physiological ACE2 peptide (P1), its singly mutated counterpart (P2), and the 18-mer peptide (P3) incubated at increasing, nanomolar concentration and in different combinations with the immuno-immobilized spike protein that was overexpressed from HEK-293 cell lysate, followed by incubation with HEK-293 cell lysate expressing ACE2. The interaction with ACE2, as detected by immunoprecipitation, is only weakly inhibited by P1, not affected by P2 and extensively disrupted by the 18-mer peptide. Controls with IgG not bound to spike was added.
The 18-mer Inhibits SARS-CoV-2 Replication in Calu-3 Cells at Low Concentrations
To understand whether the peptide has any antiviral effect on the SARS-CoV-2 life cycle, the human lung bronchial epithelial cell line Calu-3 was pretreated with increasing concentrations of the 18-mer peptide 2 h before inoculation with a Delta and Omicron variant of SARS-CoV-2 (lineage: B.1.617.2 and lineage: BA.1.529) at a multiplicity of infection (MOI) 1 and then followed for infection progression (Figure 8A). After 48 h, the cells were fixed and stained after the supernatant has been collected. Immunofluorescence assay (IFA) confirmed a productive infection (Figure 8B); the viral kinetics was measured by RT-qPCR.
Figure 8.

Effect of peptide on SARS-CoV-2 replication in Calu-3 cells. (A) Schematic representation of replication kinetics of SARS-CoV-2 (Delta variant; lineage: B.1.617.2 and Omicron variant; lineage: BA.1.529) measured in Calu-3 cells treated with the peptide. Representative immunofluorescence images of SARS-CoV-2-Delta variant infected (positive control), or uninfected (negative control) Calu-3 cells treated without or with different concentrations of peptide (1.56 to 0.48 μM). (B) Cells were probed with antibodies against SARS-CoV-2 S protein (green). Scale bar; 100 mm. Percentage inhibition SARS-CoV-2 infection Delta (C) and Omicron (D) were quantified from immunofluorescence results. SARS-CoV-2 viral load Delta (E) and Omicron (F) was quantified by RT-qPCR in Calu-3 cell culture supernatant treated without or with different concentrations of peptide and IC50 was determined. (G) Cytotoxicity of peptide was measured in Calu-3 cells for 48 h. Data represents mean SEM from three replicas.
Concerning the IFA, the treatment with the 18-mer led to a decrease in SARS-CoV-2 spike protein staining (green), indicating that the peptide treatment blocked the virus infection/replication to a certain extent (Figure 8B).
The 18-mer inhibited SARS-CoV-2 infection in Calu-3 cells by more than 40% in Delta variant and by more than 35% in Omicron variant at a concentration of 0.1 μM on IFA based assay (Figure 8C–D): at 0.1 μM peptide concentration Calu-3 cell viability was around 90% (Figure 8G). At 0.2 μM peptide concentration, inhibition was even higher (around 50%) and Calu-3 cell viability was still around 80%, with some variance. Concerning the viral kinetics, viral loads in the culture supernatant were measured by RT-qPCR method at different peptide concentrations. We found that the peptide interfered with the Delta and Omicron variant SARS-CoV-2 replication at a half inhibitory concentration (IC50) of 0.19 μM and 0.23 μM, respectively (Figure 8E,F), cross-checking and confirming the results of the IFA.
Experimental Section
Structural Data for Comparative Analyses and Sequences
For the structural analyses we used the following PDB structures:
6M0J: crystal structure of the spike in complex with ACE2 from SARS-CoV-212
6ACK: cryo-EM structure of the spike in complex with ACE2 from SARS-CoV (chain D and aa 23–512 of chain C, corresponding to CTD1 of spike; extracted from complex between spike-trimer and ACE2)9
3KBH: crystal structure of the spike in complex with ACE2 from CoV-NL63 (chains A and E)11
4APH: crystal structure of ACE1 in complex with angiotensin II32
The sequence of the two wild type α-helices (aa 19 to 82) joined by a short loop (aa 52 to 56):
19-STIEEQAKTFLDKFNHEAEDLFYQSSLASWNYN-52-TNIT-56-EENVQNMNNAGDKWSAFLKEQSTLAQMY-83
The sequences indicated as P1, P2, P3 are, respectively:
22-EEQAKTFLDKFNHEAEDLFYQ-42
22-EEQAKTFLDKFNHEAEGLFYQ-42
78-TLAQM-82 GG 22-EEQAKTFLGKF-32
The structures were analyzed and rendered in UCSF Chimera.34 The tertiary structure predictions of peptides 1, 2, and 3 were obtained with PepStrMod with standard parameters.35,36 The structure of the full α-helices 1 and 2 (aa 19 to 82) pulled from the PDB crystal structure 6M0J. Structure superpositions was performed by the MatchMaker module as implemented in UCSF Chimera.34
Docking Experiments
Structure preparation for docking was performed by superimposing the peptide being docked to the ACE2 receptor (PDB: 6M0J, chain A) and then removing chain A. The PDB file was then cleaned using the in-house program PDB_Tools_V3.py with the argument -renum. The cleaning step removes everything but the PDB record types of HEADER, ATOM, TER, and END and then renumbers the atoms and residue numbers in consecutive order as required by Rosetta. The numbering can be rematched to the crystal structure after docking.
Docking experiments were carried out using RosettaDock v4.0.26,27,37 RosettaDock v4.0 is an improvement over v3.2 with an updated scoring method that allows for lower computational time of ensemble docking. This allowed us to provide 100 alternative poses of the peptide being docked to the spike protein which has an individual relaxed pose. The 100 ensembles were generated with three different relax protocols: 40 produced by Normal Mode relax, 30 by Rosetta’s standard Relax protocol, and 30 Backrub ensembles. For the spike protein, 100 rounds of Rosetta’s standard Relax protocol were performed and the structure with the lowest total score was selected. After generating the relaxed structures, prepacking was performed prior to performing 5,000 docking permutations. The permutations with the lowest overall interface score (I_sc) then underwent 100 rounds of refinement using the Rosetta’s standard refinement protocol. All of the steps listed for RosettaDock v4.0 were automated by an in-house script (flexauto_rosetta.py).
The selected docked structure can then have the original crystal structure numbering retained by using the in-house script PostCoupler.py.
Energy Breakdown
Pairwise energy breakdown of the interfaces of the crystal structures and docked peptides were calculated by the Rosetta program Residue Energy Breakdown. Output from Residue Energy Breakdown was displayed as a heat map and table (Figure 2, Figure 6, Supporting Information Tables S1 and S2). These steps were automated by the in-house script Rosetta_Breakdown.py. For structures that did not undergo docking, Rosetta’s standard Relax protocol was performed and the best scoring structure of 100 was utilized.
Molecular Dynamics Simulations
Molecular dynamics simulations were performed using GROMACS29,30 with the all-atom OPLS force field. Nonprotein atoms were removed from the PDB files. The protein structures were placed in a cubic box with 10 Å from the box edge and solvated with spc216.gro, an equilibrated 3-point solvent model.
Next, we added ions to the system with the genion module and performed 50,000 steps of energy minimization. We then carried out equilibration in two steps: NVT (canonical) ensemble followed by NPT (isothermic–isobaric). For the NVT equilibration phase, we set the target temperature to 300 K and inspected the temperature graph over time (100 ps) to make sure the system was stabilized around the target temperature value. For the NPT equilibration phase, we used the Parrinello–Rahman barostat, and inspected the density values over time (100 ps) to check the stability of the system.
After the equilibration steps, we ran a 100 ns production run on a GPU-accelerated machine. All MD simulations were performed with the same protocol and a time step of 2 fs. The trajectories were visually inspected in UCSF Chimera,34 and the RMSD with respect to the initial energy-minimized conformer was obtained with the rms module in the GROMACS suite. Fraction of residues in α-helix was computed with the GROMACS module do_dssp. Data was then processed and plotted with the in-house script helix_plot.py.
PBSA Calculations
PBSA calculations were performed with gmx_MMPBSA.31 This tool was selected due to its support of current versions of GROMACS and its included analysis program gmx_MMPBSA_ana. Single Trajectory Protocol (STP) was performed for the 100 ns MD run of the docked peptide bound to spike and compared to the spike+ACE2 complex.31 Several files were collected from the MD simulations of the spike+ACE2 (PDB: 6M0J) and spike+18-mer docked position such as .tpr, .ndx, .xtc, and .top files. These files were then submitted to gmx_MMPBSA for Single Trajectory Protocol (STP) free energy binding decomposition calculations. All calculations were performed with Generalized Born (GB) solvation and the default “oldff/leaprc.ff99SB,leaprc.gaff” force field. gmx_MMPBSA was run with MPI and example command-line input can be found in the public GitHub repository that accompanies this study, along with Ubuntu Linux setup instructions.
Immunoprecipitation Experiments
The HEK293 cells (2× 106) cultured in DMEM media supplemented with 10% fetal bovine serum, penicillin, and streptomycin were transiently transfected with the spike protein expressing plasmid (Addgene, Cat. No. 145032) using TurboFect (Cat. No. R0533, ThermoFisher, Waltham, MA). For coimmunoprecipitation, anti-spike monoclonal antibodies (Cat. No. 7G12, ThermoFisher) or correspondent control IgG antibodies were immune-immobilized on 50 μL magnetic beads (Dynabeads, Cat. No. 10003D, ThermoFisher) at 40 °C for 4h. The spike protein collected from transfected HEK293 cells was concentrated and incubated with the antibody-conjugated magnetic beads for 4h at 40 °C. The spike protein bound magnetic beads were washed three times with the buffer [20 mM Tris-HCl pH8, 137 mM NaCl, 2 mM EDTA, 1% Nonidet P-40 (NP-40), 1 mM NaF, 1 mM sodium orthovanadate, 1 mM PMSF, 5 mg/mL protease inhibitor cocktail], and incubated with untransfected HEK293 cell lysate expressing ACE2 overnight at 40 °C on a rotator. The next day, the immunoprecipitated protein bound magnetic beads were washed with the lysis buffer, followed by wash buffers with low (150 mM) and high (300 mM) NaCl concentrations and a final wash with phosphate-buffered saline. The pulled down products were eluted with 2× Laemmli sample buffer with β-mercaptoethanol at 95 °C for 10 min. The pulled down proteins were resolved using 10% SDS-polyacrylamide gel electrophoresis, transferred onto the PVDF membrane, and probed with ACE2 primary antibody (Cat. No. SKU A00756, Boster, Pleasanton, CA, USA). For competition assay, spike protein bound magnetic beads were first incubated with peptides (P1, P2, and P3) at the indicated concentration for 2h and then incubated with the HEK293 cell lysate overnight at 40 °C.
Cell Culture and SARS-CoV-2 Infection
Calu-3 cells, a human bronchial epithelial line (ATCC, Manassas, VA, USA), were maintained in a complete medium [Eagle’s Minimum Essential Medium (EMEM) ATCC] supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin (P/S).
Treatment with Peptide and SARS-CoV-2 Infection
Calu-3 cells were plated in a 96-well transparent bottom black color plate (Greiner) at the rate of 20,000 cells/well and cultured for 48h before infection. On the day of infection, the cells were treated with different concentrations of peptide at 0.024 μM to 100 μM 2 h before infection. The cells were infected with SARS-Related Coronavirus 2, Isolate hCoV-19/USA/PHC658/2021 (Lineage B.1.617.2; Delta Variant; no. NR-55611) and SARS-Related Coronavirus 2, Isolate hCoV-19/USA/GA-EHC-2811C/2021 (Lineage B.1.1.529; Omicron Variant; no. NR-56481) obtained through BEI Resources with 1 MOI of viral titer or left uninfected (negative control). At 24 h postinfection virus inoculum was removed from the cells, washed 3 times with 1× PBS, and replenished with fresh media. The reaction was terminated 24h later. The culture supernatant was collected to measure the viral replication kinetics and the cells were washed and fixed with 50 μL of 4% PFA for immunofluorescence analysis.
Immunofluorescence Assay
The 4% PFA fixed Calu-3 cells were washed three times with 1× PBS and permeabilized by adding 50 μL of 0.1% Triton X-100. The cells were blocked with 3% bovine serum albumin-phosphate buffered saline for 2h. Then 50 μL of the primary antibody against the SARS-CoV-2 spike (Sino biological, Cat. No. MA14AP0204) was added at a 1:1,000 dilution and cells were incubated overnight at 4 °C on a shaker. Next day cells were washed three times with 1× PBS and 50 μL of secondary antibody Alexa Fluor 488 Goat anti-rabbit (Thermo Fisher, Cat. No. A-11034) was added per well at a 1:2,000 dilution. The reactions were incubated on a shaker for 1h in the dark at room temperature. The cells were then washed once with 1X PBS, stained with Hoechst 33258 (Invitrogen, Cat. No. H3570) and Cell Mask (Invitrogen, Cat. No. C10046) to visualize the nucleus and cytoplasm. Images were captured using a high throughput at 20× air on an Operetta CLS. Percentage SARS-CoV-2 cells infection were analyzed with the Harmony analysis software.
Real-Time PCR (RT-qPCR)
The viral load in the supernatant was determined to understand the virus kinetics. Briefly, RT-qPCR was performed on a set of primers targeting the E gene of SARS-CoV-2 using PrimeDirect Probe RT-qPCR Mix (TaKaRa Bio USA, Inc.) and Applied Biosystems QuantStudio3 real-time PCR system (Applied Biosystems, Waltham, MA, USA). Primers and probes used for SARS-CoV-2 RNA quantification were as follows:
E_Sarbeco_F1: 5′-ACAGGTACGTTAATAGTTAATAGCGT-3′ (400 nM)
E_Sarbeco_R2: 5′-ATATTGCAGCAGTACGCACACA-3′ (400 nM)
E_Sarbeco_P1: FAM-ACACTAGCCATCCTTACTGCGCTTCG-BHQ1-3′ (200 nM)
as recommended by the WHO. The SARS-CoV-2 genome equivalent copies were calculated using quantitative PCR control RNA from heat-inactivated SARS-CoV-2, isolating USA-WA1/2020 (BEI, Cat. No. NR-52347). The percent inhibition of SARS-CoV-2 replication by peptide treatment was measured based on viral concentration in positive control wells (considered 0% inhibition) and negative control wells (uninfected cells). IC50 values were calculated using four-parameter variable slope sigmoidal dose–response models using GraphPad Prism 9.0, San Diego, California, USA, www.graphpad.com.
Discussion and Conclusions
In the context of a pandemic, when the scientific community and the pharmaceutical industry are hard pressed to discover and develop life-saving treatments in a short time,38 this work suggest an easy-to-test and potentially viable strategy to interfere with the first step of SARS-CoV-2 and SARS-CoV entry into human cells.
By performing comparative structural analyses, molecular docking and molecular dynamics simulations we identified the key residues that are involved in the binding between the viral spike and the human ACE2 receptor. These key residues are clustered into the two N-terminal back-to-back helices 1 and 2, spanning about 60 amino acids and contributing most of the predicted interaction energy between ACE2 and the SARS-CoV-2 and SARS-CoV spikes.
An ideal fragment would also include the downstream beta-hairpin carrying the conserved Lys353. However, this residue is too far in the primary structure from the double helices. A cut and seal between these two binding regions is unlikely to work due to the unpredictable folding of the resulting chimeric structure.
Compared to a shorter fragment comprising only helix 1, the ACE2 double-helix fragment remains stable in an aqueous environment. Further, except for two residues in its apical loop, the double helix does not appear to interfere with the physiological substrate of ACE2 (angiotensin II), which is part of the renin-angiotensin system involved in the regulation of blood pressure.
Therefore, we searched for a candidate inhibitor peptide inside the double helix, maintaining the stabilizing, double-helical arrangement. We engineered an 18-mer peptide encompassing both ACE2 helices of the identified double-helical fragment (78-TLAQM-82 GG 22-EEQAKTFLGKF-32). This 18-mer shows efficient inhibition on ACE2-spike interaction, as detected in immunoprecipitation experiments. Tertiary structure prediction of this 18-mer shows a fold that is globally similar to the physiological counterpart. When superimposed to the ACE2 in complex with the spike, the 18-mer shows a global fold correspondence, except for a marked deviation in its C-terminal part, where a newly formed hydrophobic cleft can accommodate the spike.
Given the diffusion of other coronaviruses among the human population, included the almost innocuous CoV-NL63, that target the same human ACE2 receptor, one might expect a strong immunological memory against this family of viruses. However, recent data seems to suggest that the immunological memory triggered by vaccines might not be long lasting.39 CoV-Nl63 narrower binding area (not encompassed by our construct) seems to suggest that coronaviruses diverged by extending or narrowing the binding surface with ACE2, with consequences for their binding affinity and infectivity. In agreement with this observation, the viral spike protein is poorly structured in its binding surface, probably allowing high flexibility and quick “adaptability” in target binding.
To understand whether this newly engineered 18-mer peptide showed inhibition of ACE-2 and spike interaction, we confirmed this interaction for the SARS-CoV-2 life cycle and showed a decrease in viral replication. Our data suggest that the peptide may have the capacity to interfere with ACE-2 and spike interaction during virus replication. Compared to other peptidic inhibitors,15,17,18,21 our 18-mer has IC50 values in the submicromolar range, and is also considerably shorter than other published inhibitors.18,21
We expect our 18-mer to be relatively easy to either synthesize or express and purify at a large-scale level. Possible clinical applications would require, of course, further analysis regarding stability. In addition, the fact that CC50 and IC50 are of the same order of magnitude (Figure 8) suggests a narrow therapeutic index, and indicates that further modifications to the peptide are required to lower toxicity. Our future studies will address such modifications to improve the therapeutic index. Since the peptide is engineered from a protein that is normally expressed in human tissues, except for the poly glycine linker, we expect low immunogenicity. The fragment could potentially be administered directly by aereosol, avoiding the systemic route and possible degradation by proteases, adding to the growing arsenal of inhibitory strategies40 against the infection.
A possible extension to this work is the application of a similar discovery pipeline to design entry interaction inhibitors for other viruses. To this end, we applied the computational pipeline developed for this investigation to the epidemic virus Middle East respiratory syndrome coronavirus (MERS-CoV). The preliminary results, shown in Supporting Information Figure S2, suggest that candidate inhibitors for this other virus could be derived following the same principles outlined in this study.
Acknowledgments
This work was supported in part by a COVID-19 Rapid Response grant to S.K. and D.G. and by a UNO GRACA grant to A.S. Docking and molecular dynamics simulations were performed on a GPU-accelerated high-performance computing machine partly funded by a Nebraska EPSCoR grant to D.G. Thanks to Dr. Antonello De Santis for informatics support to F.R. S.N.B. is supported by NIH grants AI129745 and DA052845 and by the Nebraska Research Initiative (NRI) of the University of Nebraska System. We acknowledge the UNMC BSL-3 core facility at DRC-1, where live SARS-CoV-2 experiments were performed; further, the UNMC BSL-3 core facility is administered by the Office of the Vice-Chancellor for Research and supported by the Nebraska Research Initiative (NRI). We thank the St Patrick Reid laboratory for allowing us to use the Operetta CLS instrument.
Data Availability Statement
All in-house scripts mentioned in the text are available in a GitHub repository at https://github.com/Aseamann/ACEdecoy. Additional data such as docking run information and MD starting poses are also provided.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.2c00180.
Interaction energy breakdown table of ACE2 interactions with SARS-CoV and SARS-CoV-2 spike; interaction energy breakdown table of 18-mer and SARS-CoV-2 spike from docked position; binding free energy calculation plots from 18-mer/spike MD; analysis of MERS-CoV interaction with DPP4 (PDF)
Author Contributions
‡ F.R. and A.S. contributed equally. F.R.: idea, research design, structural analyses, manuscript. A.S.: engineered peptide design, docking, MD simulations, manuscript. K.G.: bench experiments. K.P.: viral bench experiments. S.B.: viral bench experiments. S.K.: bench experiments. D.G.: docking, MD simulations, engineered peptide design, manuscript.
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Pharmacology & Translational Science virtual special issue “New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science”.
Supplementary Material
References
- Belouzard S.; Millet J. K.; Licitra B. N.; Whittaker G. R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. 10.3390/v4061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorici M. A.; Veesler D.. Structural insights into coronavirus entry. Complementary Strategies to Understand Virus Structure and Function; Advances in Virus Research 105; Elsevier, 2019; pp 93–116. 10.1016/bs.aivir.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue M.; Hsieh F.; Baronas E.; Godbout K.; Gosselin M.; Stagliano N.; Donovan M.; Woolf B.; Robison K.; Jeyaseelan R.; Breitbart R. E.; Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000, 87, E1–9. 10.1161/01.RES.87.5.e1. [DOI] [PubMed] [Google Scholar]
- Towler P.; Staker B.; Prasad S. G.; Menon S.; Tang J.; Parsons T.; Ryan D.; Fisher M.; Williams D.; Dales N. A.; Patane M. A.; Pantoliano M. W. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H. P.; Look D. C.; Shi L.; Hickey M.; Pewe L.; Netland J.; Farzan M.; Wohlford-Lenane C.; Perlman S.; McCray P. B. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005, 79, 14614–14621. 10.1128/JVI.79.23.14614-14621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koka V.; Huang X. R.; Chung A. C. K.; Wang W.; Truong L. D.; Lan H. Y. Angiotensin II up-regulates angiotensin I-converting enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway. Am. J. Pathol. 2008, 172, 1174–1183. 10.2353/ajpath.2008.070762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A. C.; Park Y.-J.; Tortorici M. A.; Wall A.; McGuire A. T.; Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui M.; Song W.; Zhou H.; Xu J.; Chen S.; Xiang Y.; Wang X. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017, 27, 119–129. 10.1038/cr.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.; Gui M.; Wang X.; Xiang Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018, 14, e1007236 10.1371/journal.ppat.1007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J.; Ye G.; Shi K.; Wan Y.; Luo C.; Aihara H.; Geng Q.; Auerbach A.; Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K.; Li W.; Peng G.; Li F. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 19970–19974. 10.1073/pnas.0908837106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J.; Ge J.; Yu J.; Shan S.; Zhou H.; Fan S.; Zhang Q.; Shi X.; Wang Q.; Zhang L.; Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Acharya A.; Pandey K.; Thurman M.; Klug E.; Trivedi J.; Sharma K.; Lorson C. L.; Singh K.; Byrareddy S. N. Discovery and Evaluation of Entry Inhibitors for SARS-CoV-2 and Its Emerging Variants. J. Virol. 2021, 95, e0143721 10.1128/JVI.01437-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odolczyk N.; Marzec E.; Winiewska-Szajewska M.; Poznański J.; Zielenkiewicz P. Native Structure-Based Peptides as Potential Protein-Protein Interaction Inhibitors of SARS-CoV-2 Spike Protein and Human ACE2 Receptor. Molecules 2021, 26, 2157. 10.3390/molecules26082157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitsike L.; Krstenansky J.; Duerksen-Hughes P. J. ACE2:S1 RBD Interaction-Targeted Peptides and Small Molecules as Potential COVID-19 Therapeutics. Adv. Pharmacol. Pharmaceutical Sci. 2021, 2021, 1828792. 10.1155/2021/1828792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S. K.; Sen Gupta P. S.; Biswal S.; Ray A. K.; Rana M. K. ACE-2-Derived Biomimetic Peptides for the Inhibition of Spike Protein of SARS-CoV-2. J. Proteome Res. 2021, 20, 1296–1303. 10.1021/acs.jproteome.0c00686. [DOI] [PubMed] [Google Scholar]
- Larue R. C.; Xing E.; Kenney A. D.; Zhang Y.; Tuazon J. A.; Li J.; Yount J. S.; Li P.-K.; Sharma A. Rationally Designed ACE2-Derived Peptides Inhibit SARS-CoV-2. Bioconjugate Chem. 2021, 32, 215–223. 10.1021/acs.bioconjchem.0c00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curreli F.; Victor S. M. B.; Ahmed S.; Drelich A.; Tong X.; Tseng C.-T. K.; Hillyer C. D.; Debnath A. K. Stapled Peptides Based on Human Angiotensin-Converting Enzyme 2 (ACE2) Potently Inhibit SARS-CoV-2 Infection In Vitro. mBio 2020, 11, e02451–20. 10.1128/mBio.02451-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpoot S.; Ohishi T.; Kumar A.; Pan Q.; Banerjee S.; Zhang K. Y. J.; Baig M. S. A Novel Therapeutic Peptide Blocks SARS-CoV-2 Spike Protein Binding with Host Cell ACE2 Receptor. Drugs R&D 2021, 21, 273–283. 10.1007/s40268-021-00357-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal G.; Yaduvanshi S.; Kumar V. A potential peptide inhibitor of SARS-CoV-2S and human ACE2 complex. J. Biomol. Struct. Dyn. 2022, 40, 6671–6681. 10.1080/07391102.2021.1889665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoyan P.; Vieillard V.; Gómez-Morales L.; Odile E.; Guihot A.; Luyt C.-E.; Denis A.; Grondin P.; Lequin O. Human ACE2 peptide-mimics block SARS-CoV-2 pulmonary cells infection. Commun. Biol. 2021, 4, 197. 10.1038/s42003-021-01736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee P.; Ponnapati M.; Kramme C.; Plesa A. M.; Church G. M.; Jacobson J. M. Targeted intracellular degradation of SARS-CoV-2 via computationally optimized peptide fusions. Commun. Biol. 2020, 3, 715. 10.1038/s42003-020-01470-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Pearce R.; Zhang Y. De novo design of protein peptides to block association of the SARS-CoV-2 spike protein with human ACE2. Aging 2020, 12, 11263–11276. 10.18632/aging.103416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basit A.; Karim A. M.; Asif M.; Ali T.; Lee J. H.; Jeon J. H.; Rehman S. U.; Lee S. H. Designing Short Peptides to Block the Interaction of SARS-CoV-2 and Human ACE2 for COVID-19 Therapeutics. Front. Pharmacol. 2021, 12, 731828. 10.3389/fphar.2021.731828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R.; Boorla V. S.; Maranas C. D. Computational biophysical characterization of the SARS-CoV-2 spike protein binding with the ACE2 receptor and implications for infectivity. Comput. Struct. Biotechnol. J. 2020, 18, 2573–2582. 10.1016/j.csbj.2020.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J. J.; Moughon S.; Wang C.; Schueler-Furman O.; Kuhlman B.; Rohl C. A.; Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003, 331, 281–299. 10.1016/S0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- Chaudhury S.; Berrondo M.; Weitzner B. D.; Muthu P.; Bergman H.; Gray J. J. Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS One 2011, 6, e22477 10.1371/journal.pone.0022477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. K.; Li W.; Moore M. J.; Choe H.; Farzan M. A. 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 2004, 279, 3197–3201. 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C.; van der Spoel D.; van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- Pronk S.; Páll S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; Hess B.; Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdés-Tresanco M. S.; Valdés-Tresanco M. E.; Valiente P. A.; Moreno E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. 10.1021/acs.jctc.1c00645. [DOI] [PubMed] [Google Scholar]
- Masuyer G.; Schwager S. L. U.; Sturrock E. D.; Isaac R. E.; Acharya K. R. Molecular recognition and regulation of human angiotensin-I converting enzyme (ACE) activity by natural inhibitory peptides. Sci Rep. 2012, 2, 717. 10.1038/srep00717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Li W.; Farzan M.; Harrison S. C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Singh S.; Singh H.; Tuknait A.; Chaudhary K.; Singh B.; Kumaran S.; Raghava G. P. S. PEPstrMOD: structure prediction of peptides containing natural non-natural and modified residues. Biol. Direct 2015, 10, 73. 10.1186/s13062-015-0103-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur H.; Garg A.; Raghava G. P. S. PEPstr: a de novo method for tertiary structure prediction of small bioactive peptides. PPL 2007, 14, 626–631. 10.2174/092986607781483859. [DOI] [PubMed] [Google Scholar]
- Marze N. A.; Roy Burman S. S.; Sheffler W.; Gray J. J. Efficient flexible backbone protein-protein docking for challenging targets. Bioinformatics 2018, 34, 3461–3469. 10.1093/bioinformatics/bty355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothan H. A.; Byrareddy S. N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmunity 2020, 109, 102433. 10.1016/j.jaut.2020.102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinands J. M. Waning 2-Dose and 3-Dose Effectiveness of mRNA Vaccines Against COVID-19-Associated Emergency Department and Urgent Care Encounters and Hospitalizations Among Adults During Periods of Delta and Omicron Variant Predominance - VISION Network. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 255–263. 10.15585/mmwr.mm7107e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vann K. R.; Acharya A.; Jang S. M.; Lachance C.; Zandian M.; Holt T. A.; Smith A. L.; Pandey K.; Durden D. L.; El-Gamal D.; Côté J.; Byrareddy S. N.; Kutateladze T. G. Binding of the SARS-CoV-2 envelope E protein to human BRD4 is essential for infection. Structure 2022, 30, 1224–1232.e5. 10.1016/j.str.2022.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All in-house scripts mentioned in the text are available in a GitHub repository at https://github.com/Aseamann/ACEdecoy. Additional data such as docking run information and MD starting poses are also provided.