

Abstract
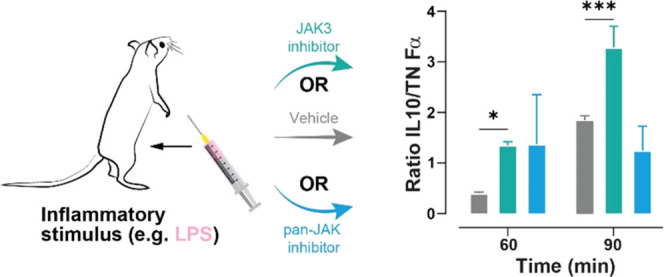
Janus kinase (JAK) inhibitors act at low doses (e.g., tofacitinib, 0.2–0.4 μmol/kg bid) in clinical use, suggesting an efficient underlying mode of action. We hypothesized that their effectiveness is due to their ability to raise the ratio of IL-10 to TNFα. Unlike other JAK isoforms, JAK3 is expressed mainly in hematopoietic cells and is essential for immune function. We used JAK3 selective inhibitors with preferential distribution to immune cells. Inhibition of JAK3 in human leukocytes reduced TNFα and IL-6 but maintained levels of IL-10, while pan-JAK inhibitors increased TNFα, IL-6, and IL-10. JAK1 is required for IL-10 receptor signaling, which suggests that, at exposure above the IC50 (55 nM for tofacitinib on JAK1), there is less feedback control of TNFα levels. This leads to self-limiting effects of JAK1 inhibitors and could place an upper limit on appropriate doses. In vivo, treating mice with JAK3 inhibitors before LPS administration decreased plasma TNFα and increased IL-10 above vehicle levels, suggesting that JAK3 inhibition may limit TNFα release by increasing IL-10 while leaving the IL-10 receptor functional. This mechanism should have general utility in controlling autoimmune diseases and can be conveniently observed by measuring the ratio of IL-10 to TNFα. In summary, our targeted, “leukotropic” inhibitors more effectively increased IL-10/TNFα ratios than unselective control compounds and could, therefore, be ideal for autoimmune therapy.
Keywords: inflammation, Janus kinase 3, cytokine profiling, IL-10, LPS challenge
The Janus kinase family consists of four isoforms: JAK1, JAK2, JAK3, and TYK2. They catalyze the transfer of γ-phosphate from ATP to a substrate tyrosine residue but differ in their associated type I/II cytokine receptors and their substrate peptides. After activation of transmembrane receptors by extracellular stimuli, JAKs bind, leading to transphosphorylation and formation of homodimers (only JAK2) or heterodimers. Signal transducer and activator of transcription (STAT) proteins associate with the JAK–receptor complex and are then phosphorylated, causing their dimerization, translocation to the nucleus, and transcription of target genes.1−3
Among the JAK isoforms, JAK3 is unusual in that it is found almost exclusively in leukocytes, forming heterodimers with JAK1 upon activation.4 It is constitutively expressed at low levels but can be upregulated by certain stimuli, e.g., Toll-like receptor (TLR) agonists or interferons.5,6 Dysfunctional, increased JAK signaling can lead to autoimmune disease and leukemia.7,8 On the other hand, loss-of-function mutations in the JAK3 gene are a cause of severe combined immunodeficiency disorder (SCID), leading to a phenotype of total depletion of T- and NK cells and impaired B cell function.9
The localization in immune cells makes JAK3 a potential drug target for treating autoimmune disorders and other inflammatory disorders. As JAK3 is mostly restricted to the hematopoietic system, the expectation is that specific inhibition will cause fewer adverse effects compared to targeting one or several of the other isoforms. Considering that JAK3 is co-localized with JAK1, some propose that inhibition of both isoforms may be necessary for more potent immunosuppression, which was the rationale for JAK1/3 inhibitor development.10,11 However, the observation that JAK1 is required for anti-inflammatory IL-10 signaling leads us to question the utility of this approach.
JAK3 binds to its associated receptors after their activation by common γ-chain cytokines (IL-2, IL-4, IL-7, IL-9, IL-15, IL-21, Figure 1).12 It is not involved in IL-10 signaling, so inhibition of JAK3 is not expected to interfere with IL-10 function. IL-10 levels are implicated in gut immune tolerance, where deficiencies in IL-10 are associated with colitis.13 Additionally, IL-10 is upregulated following excessive production of TNFα as a means to attenuate immune stimulation. Thus, IL-10 is considered to be an anti-inflammatory cytokine and as such, not interfering with IL-10 signaling would be essential in an anti-inflammatory therapy.
Figure 1.

JAKs associate as dimers or trimers with different cytokine or hormone receptors, depending on the isoform and the cell type they are expressed in. This leads to a variety of downstream effects, many of which are related to the immune system.4,14 JAK3 is exclusively found associating with common γ chain cytokine (IL-2, IL-4, IL-7, IL-9, IL-15, IL-21) receptors (indicated in cyan, right).12 Created with BioRender.com.
Among the JAK3 inhibitors employed in this study, several are covalently linked to carrier molecules based on the macrolide azithromycin (Figure 2). While mostly known for their use as antibiotics, macrolide drugs also possess immunomodulating properties that help support the management, attenuation, and resolution of inflammation.15,16 The basic, amphiphilic large scaffold also conveys a tendency to accumulate in acidic compartments and greatly increases uptake into tissues and immune cells.17,18 We used these scaffolds for the targeted delivery of JAK3 inhibitors to immune cells as a means to limit off-target effects.19−22 In combination with a target that is mostly located in immune cells, this represents a potentially useful mechanism to increase the potency of pharmacophores.
Figure 2.

Structure of the macrolide antibiotic azithromycin.
FM-381 (8, Figure 5) is a potent, selective inhibitor of JAK3 (JAK3 IC50 = 12 ± 1 nM).23,24 We recently reported the synthesis and characterization of a series of JAK3 inhibitors based on its scaffold (Figures 3 and 4).251–4 are structurally related analogues of 8. Their inhibitory potencies for JAK3 are within the same order of magnitude as 8 or slightly lower (see Supporting Information, Table S1), while possessing different pharmacokinetic profiles and in vivo stabilities.25
Figure 5.
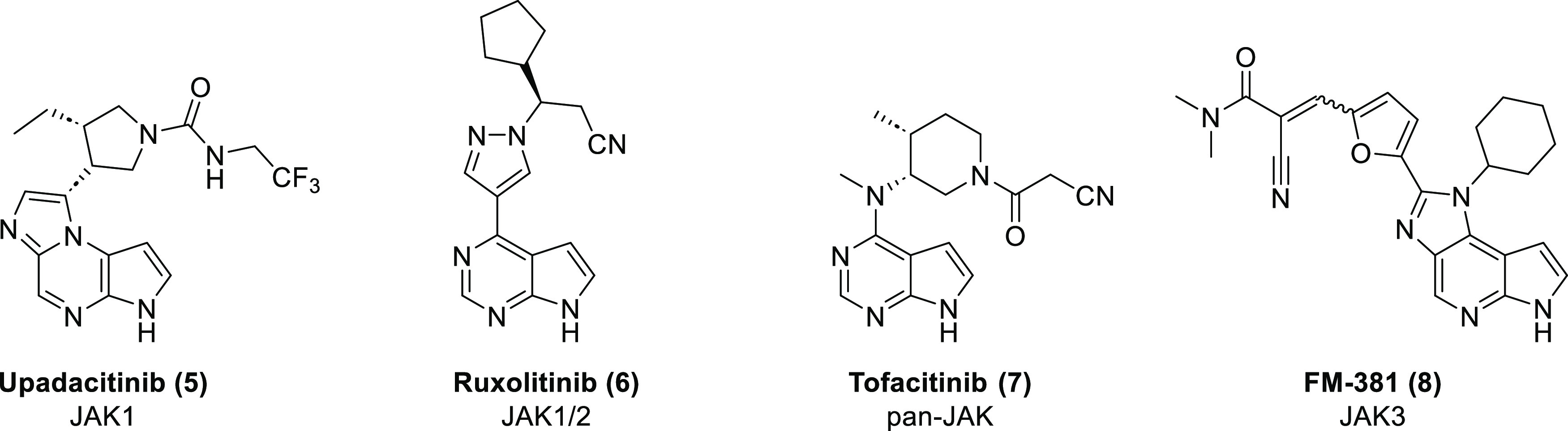
Structures of the control compounds used in this study and their selectivities within the JAK family.
Figure 3.

Structures and JAK3 inhibitory activity of the unconjugated JAK3 inhibitors 1 and 2.25 JAK family selectivity data have been obtained (Supporting Information, Table S2).
Figure 4.
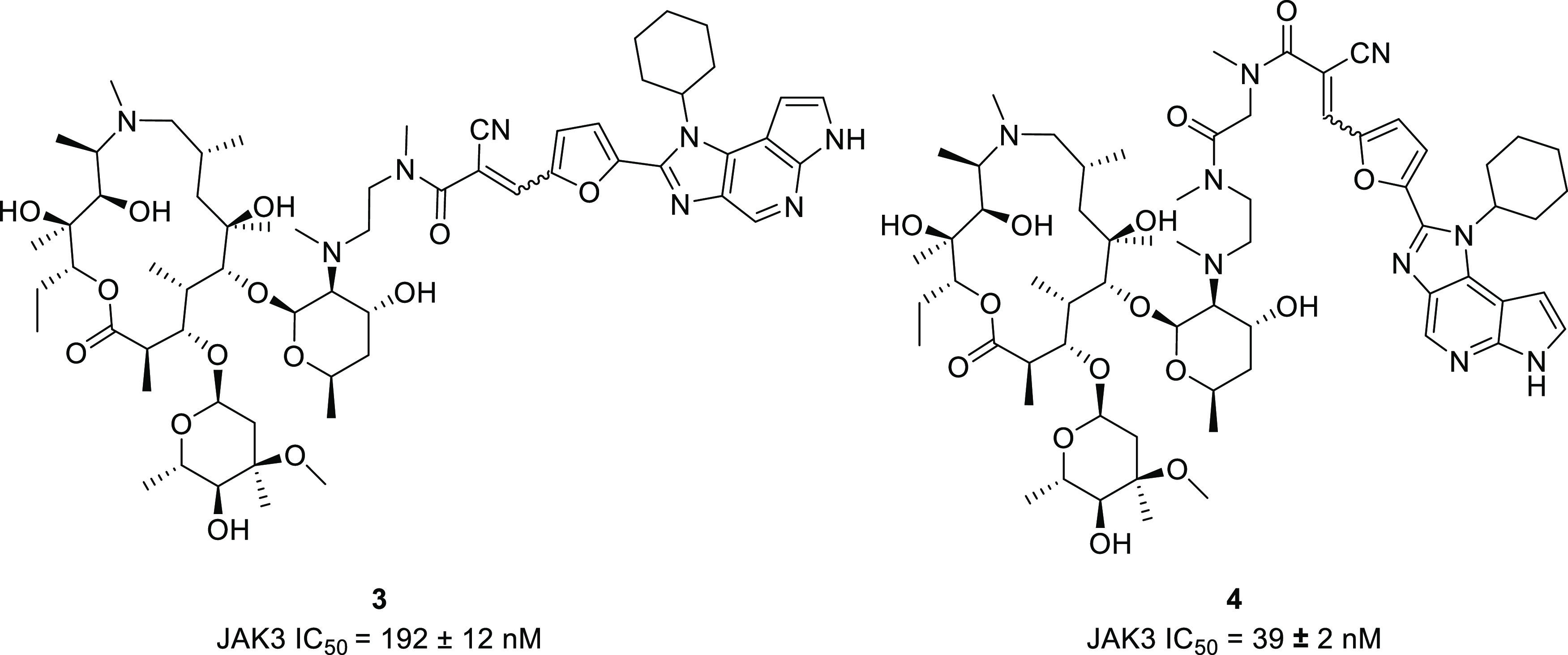
Structures of the azithromycin-derived JAK3 inhibitors 3 and 4.
3 and 4 (Figure 4) differ in that they are covalently linked to a macrocyclic carrier based on an azithromycin scaffold.
Upadacitinib (5) is a selective inhibitor of JAK1. It demonstrated efficacy in the treatment of rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, and atopic dermatitis.26
Ruxolitinib (6) is a selective inhibitor of JAK1 and JAK2. It was the first JAK inhibitor to receive FDA approval and is used for the treatment of proliferative disorders like polycythemia vera and myelofibrosis.27−29
Tofacitinib (7) is an orally available small-molecule JAK inhibitor. While initially considered JAK3 selective, later publications describe 6 to be a “pan-JAK” inhibitor, with preference for JAK1 and JAK3 and moderate, but pharmacologically relevant inhibition of JAK2 and TYK2.30,31 It is used in the treatment of ulcerative colitis, rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis.32,33
In this study, we first evaluated the anti-inflammatory effects of the aforementioned inhibitors of JAK3 in vitro in human peripheral blood leukocytes. To this end, cells were stimulated with bacterial lipopolysaccharides (LPS), which causes an inflammatory reaction based on toll-like receptor 4 (TLR4) signaling and the release of both pro- and anti-inflammatory cytokines like TNFα and IL-10, respectively.34,35 Innate immune cells and TLR signaling play a substantial role in several chronic autoimmune diseases like rheumatoid arthritis.36−39 Their significance is supported by the clinical success of anti-TNFα agents.40 Three FDA-approved JAK inhibitors (5–7) were included to compare the effects of blocking signaling via the other JAK isoforms. We then used an LPS-based in vivo peritonitis model with the same readout (levels of TNFα, IL-6, and IL-10) to select the most potent compounds for use in longer-term disease models.40 LPS peritonitis is a widely used reference model for general anti-inflammatory effects.
The most effective JAK3 inhibitors in the LPS challenge study were then assessed for their effects with i.v. treatment to compare their in vivo potency independently of oral availability. Again, 6–8 were included for comparison. Lastly, JAK3 inhibitor 4 was compared to 7 in a dose-response study to elucidate the effects of low-dosed JAK inhibitors in vivo, demonstrating efficacy even at low-dose ranges.
In this report, we demonstrate the in vitro and in vivo effects of a selection of JAK3 and mixed JAK inhibitors on inflammatory processes. These data demonstrate considerable differences in cytokine profiles between the inhibitor types in both cellular and a more complex murine in vivo setting.
Results
JAK3 Inhibitors Differentially Affect the Cytokine Response of Human Peripheral Leukocytes In Vitro Compared to JAK1 or Mixed JAK Inhibitors
First, we set out to assess the general “cytokine profiles” resulting from the inhibition of different JAKs in an inflammatory context. To that end, human peripheral leukocytes harvested from buffy coat were incubated with either 5 (selective for JAK1), 6 (JAK1/2), 7 (pan-JAK), or 8 (JAK3) and subjected to an LPS stimulus. Generally, the patterns for JAK1 or pan-JAK inhibitors were similar, suggesting that the effects are mainly a result of JAK1 inhibition. For cells treated with 5–7, a concentration-dependent increase of TNFα release was observed (up to 9-fold the levels of control cells). Dose response was bell-shaped for some compounds, e.g., 6. We suspect this to be a consequence of either off-target inhibition (given the high concentration) or by fully inhibiting JAK1 signaling via the IL-10 receptor, preventing feedback control of TNFα.41 In contrast to the JAK1 inhibitors, the JAK3 selective inhibitor 8 caused a potent dose-dependent reduction of TNFα release, with 2.5 μM causing 85% reduction.
As with TNFα, IL-6 was also produced and/or released at higher levels when cells were treated with 5–7 (up to 257 ± 13% of control at 5 μM 5). Effects were similar for the different JAK1 inhibitors, and a notable increase in IL-6 levels was already observed at a concentration of 0.1 μM. In contrast, JAK3 inhibition had little effect on the release of IL-6 with minor and variable nonsignificant reduction at most concentrations and significant reduction (p < 0.05) at 0.1 μM.
The effects on the release of the anti-inflammatory cytokine IL-10 were also similar for the JAK inhibitors already used in the clinical setting. Again, bell-shaped dose response was apparent (cJAKi = 0.01–5 μM). At most concentrations, IL-10 levels were elevated vs control in parallel with the TNFα levels. One possible reason for this effect is that JAK1 is required for IL-10 signaling. Excess TNFα release induces IL-10 release as a regulatory feedback signal. However, if IL-10 signaling is blocked, it is possible that there is no feedback control on either TNFα or IL-10 levels and IL-10 release overshoots. The bell-shaped drop in IL-10 at higher concentrations may reflect the increasing influence of off-target inhibition (e.g., JAK2, for which these substances have lower affinity than JAK1). Treatment of cells with 8 had no effect on IL-10 levels up to a concentration of 1 μM. Only at 2,5 and 5 μM was IL-10 elevated over control, albeit not significantly and to a lesser extent than for 5 (127 ± 15% at 5 μM 8). This phenomenon was observed with many compounds from this series, namely, that JAK3 inhibition is associated with increased IL-10 release and an increased ratio of IL-10 to TNFα.
In summary, cells treated with compounds 5–7, all of which share inhibition of JAK1, demonstrated similar cytokine responses after LPS stimulus. The JAK3 inhibitor 8 caused a distinctly different response with a potent decrease of TNFα levels and distinct patterns of IL-6 and IL-10 release.
Given these effects of JAK3 inhibition, we examined analogues of 8 in the same experimental conditions (Figure 7). 6–8 were included as controls.
Figure 7.
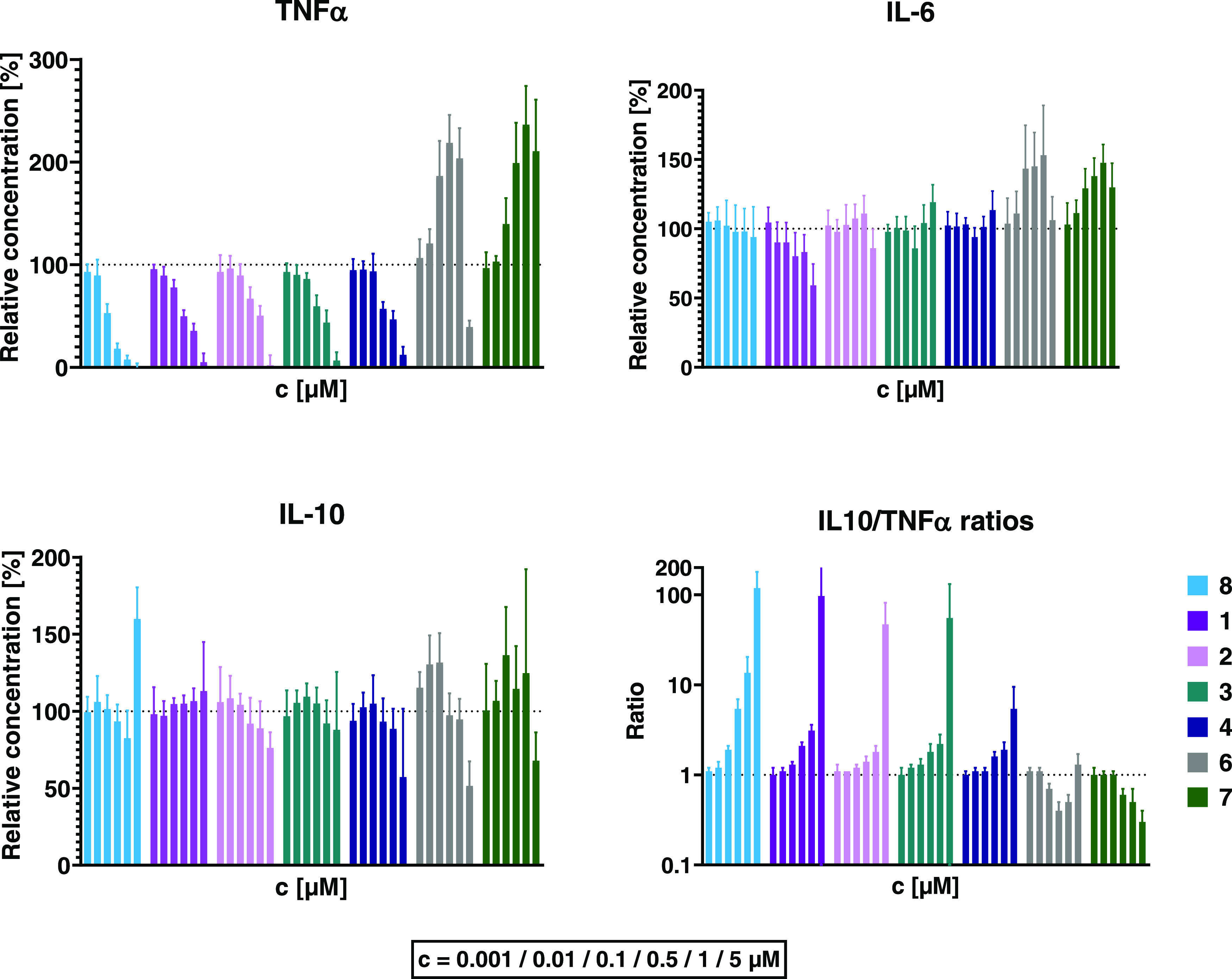
Effects of JAK inhibitors on LPS-induced production of TNFα, IL-6, and IL-10 in vitro. Peripheral blood leukocytes isolated from human donors were incubated with rising concentrations (from left to right: 0.001, 0.01, 0.1, 0.5, 1, and 5 μM) of JAK inhibitors before addition of 50 ng/mL LPS. In vitro data (n = 6) are displayed as mean ± 1 standard deviation, unless noted otherwise (2 donors, each n = 3 assays). IL-10/TNFα ratios are displayed in half-logarithmic scale.
Among the JAK3 inhibitors, 8 was the most effective at decreasing TNFα, (down to 8 ± 4% at 1 μM and 0 ± 5% at 5 μM relative to DMSO control). The other JAK3 inhibitors 1–4 exhibited similar effects but were less potent. This mirrors their respective in vitro JAK3 IC50 values (Supporting Information, Table S1), which are higher than 8.25
The pan-JAK inhibitor 7, on the other hand, increased TNFα levels at concentrations > 10 nM, up to 236 ± 38% compared to untreated cells at 1 μM. Similarly, TNFα production under treatment with 6 peaked at 0.5 μM, reaching 219 ± 27% vs control. As observed in previous studies, 6 dose response was bell-shaped and 5 μM caused TNFα to drop again, to 39 ± 6%.
These data show that JAK3 inhibitors inhibit TNFα release and differ from clinically used JAK inhibitors differing in selectivity.
JAK3 inhibition had little effect on IL-10 levels at most concentrations. However, at 5 μM, several compounds showed an effect that is, to the best of our knowledge, unique for this compound class: Pretreatment with the macrolide-linked compound 4 led to a drop of IL-10 levels (to 57 ± 44% relative to vehicle treatment) while 8 significantly elevated IL-10 release to 160 ± 21% (p < 0.0001). An increase was also observed at 5 μM 8 in the previous experiment (Figure 6), however to a lesser (nonsignificant) degree. Treatment with 7 reduced IL-10 levels at 5 μM (down to 68 ± 18% relative to vehicle) but caused increases of up to 36% over control between 0.001 and 1 μM. Low concentrations of 6 led to elevation of IL-10 production (up to 32 ± 19% at 0.1 μM vs vehicle); conversely, 5 μM had the opposite effect (decrease by 52 ± 16%). In general, all JAK3 inhibitors (1–4, 8) caused a concomitant inhibition of TNFα without interfering with IL-10 production. This led to a concentration-dependent increase of the IL-10/TNFα ratio (Figure 7). In contrast, the ratio decreased at higher concentrations of 7, while treatment with 6 caused an inverse bell-shaped response.
Figure 6.
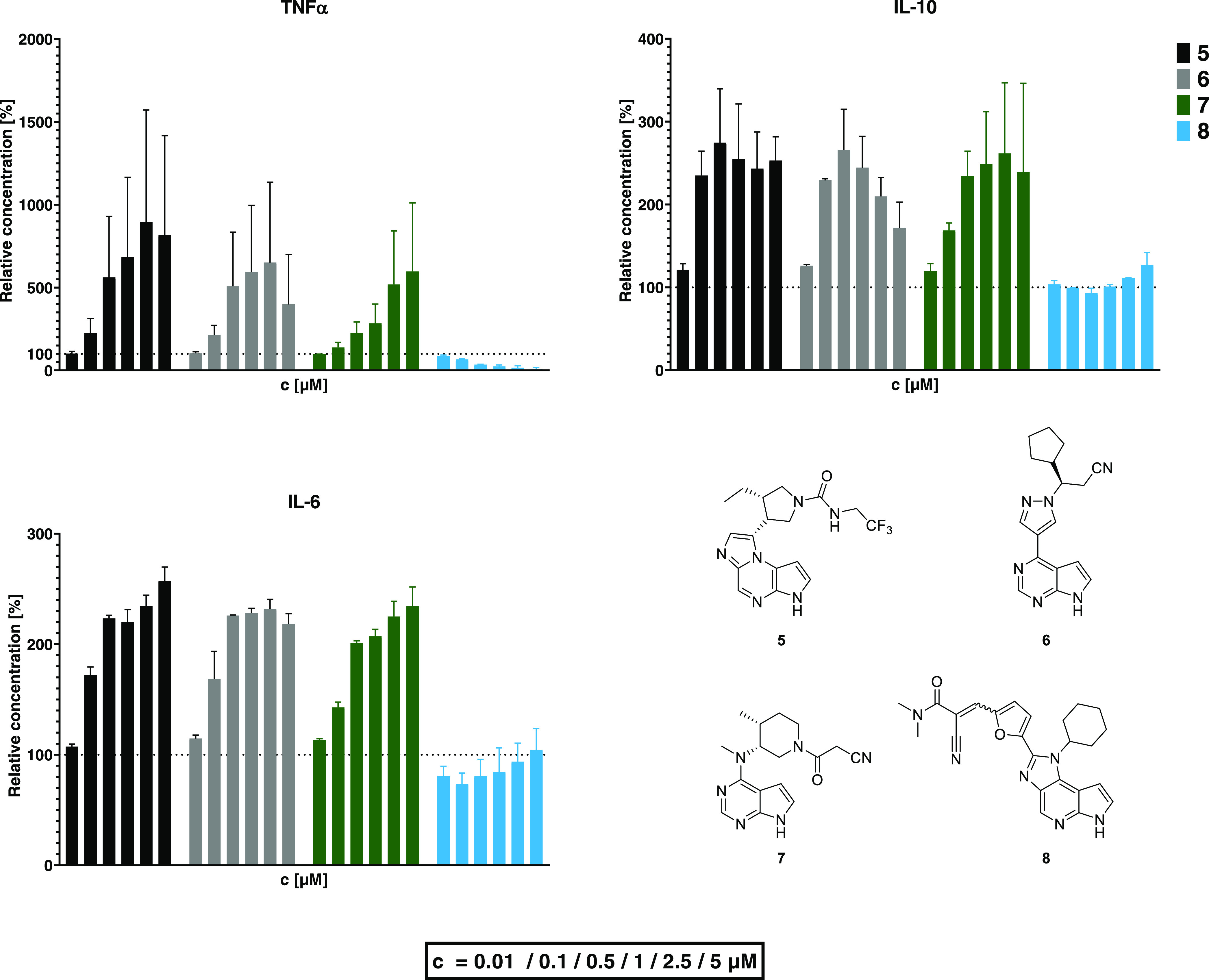
Effects of JAK inhibitors with different selectivity profiles on the cytokine release by human peripheral leukocytes. Cells were incubated with various concentrations of inhibitors before being subjected to 50 ng/mL LPS. In vitro data are the mean ± 1 standard deviation (2 biological replicates, each n = 3).
1 lowered IL-6-release in a dose-dependent way, all other JAK3 compounds had no effect on IL-6 levels. IL-6 release was inhibited to 59 ± 15% vs untreated control cells at 5 μM concentration of 1. While 8 displayed a different effect on IL-6 compared to the previous experiment (Figure 6), the effects were not significant compared to control cells. As with IL-10, we attribute the deviation from the previous experiments to minor, donor-specific variations of leukocyte populations.
Compared to JAK3 inhibitors, unselective inhibition of JAK isoforms by pan-JAK inhibitor 7 led to an elevation of IL-6 levels by 47 ± 13% at 1 μM and 30 ± 17% at 5 μM, while lower doses also caused a trend toward higher IL-6 levels. IL-6 concentrations in the samples treated with the JAK1/2 selective inhibitor 6 were analogous to those of TNFα: At concentrations between 0.001 and 1 μM, a concentration-dependent increase (up to 53 ± 36% at 1 μM) was observed, but at 5 μM the levels were close to those of untreated samples.
Treatment with JAK3 Inhibitors Attenuates the Inflammatory Response of BALB/c Mice to LPS Injection
In a first study to establish the dose range, the JAK3 inhibitor 3 was tested vs vehicle treatment. 3 is an example of the macrolide conjugate class with promising in vitro efficacy coupled with appropriate in vivo pharmacokinetics (oral availability and extended tissue half-life).25 Oral application of 3 decreased TNFα– and IL-6-release, while IL-10 levels remained constant or showed a trend toward increased IL-10 release.
In subsequent studies, 3 was replaced by 4, a similar conjugate compound with improved in vitro potency and pharmacokinetics (see Supporting Information, Figure S5 and Table S1). The other test compounds, 1 and 2, were selected based on pharmacokinetic properties: 1 has adequate JAK3 potency and distributes well into tissues including the CNS. It is intended as a potential candidate for CNS inflammation studies to the question whether partition to the CNS improves anti-inflammatory effects vs similar compounds without CNS exposure. 2 is more stable in vivo compared to the lead 8, but less potent vs JAK3, so it is intended to answer the question whether loss of (in vitro) potency could be outweighed by the increased in vivo stability.25
The JAK1/2 selective inhibitor 6, the pan-JAK inhibitor 7, and the JAK3 selective inhibitor 8 were included as control compounds.
Compound 3 Significantly Reduces TNFα Plasma Concentrations after LPS Injection While Sparing IL-10
In a small-scale study with p.o. treatment, the JAK3 inhibitor 3 was compared to vehicle treatment (Figure 8). The highest TNFα and IL-10 plasma concentrations were observed 90 min after LPS injection, while IL-6 peaked at 180 min. At a dose of 10 mg/kg (∼8.5 μmol/kg), 3 had no significant effect on IL-6 and IL-10 tail plasma concentrations, while reducing TNFα levels at 60 and 90 min (p < 0.01). As a parameter of anti-inflammatory potency in vivo, we calculated the ratio of IL-10 over TNFα at 60 and 90 min, and treatment with the JAK3 inhibitor 3 caused a significant (p < 0.05) shift toward an anti-inflammatory profile at both time points by suppressing TNFα release while maintaining IL-10 levels.
Figure 8.

Release of TNFα, IL-6, and IL-10 in mice after LPS stimulus. Balb/c female mice (n = 9) were treated p.o. with either vehicle or the JAK3 inhibitor 3 (10 mg/kg, ∼8.5 μmol/kg). After 15 min, 10 mg/kg LPS (S. enterica typhimurium) was administered i.p. and tail plasma was taken at 60, 90, and 180 min after LPS administration. Plasma samples were analyzed by ELISA. Data are shown as *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Data are the mean ± 1 SD.
Oral Pretreatment of Mice with Macrolide Conjugate 4 Significantly Increases the IL-10/TNFα Plasma Ratio Post LPS Injection
Treatment with the JAK3 inhibitors (1–4, 8) or 6/7 reduced LPS-induced release of TNFα 60 and 90 min post LPS injection (Figure 9). In contrast to in vitro experiments in human peripheral blood leukocytes, none of the compounds increased TNFα production over vehicle control. Greater reduction of TNFα at 90 min was observed for 1, 2, and 4 (4: 41% of vehicle at 60 and 21% at 90 min). Treatments with either 1 or 2 generated similar results, reducing TNFα at 90 min to 45 and 41%, respectively.
Figure 9.
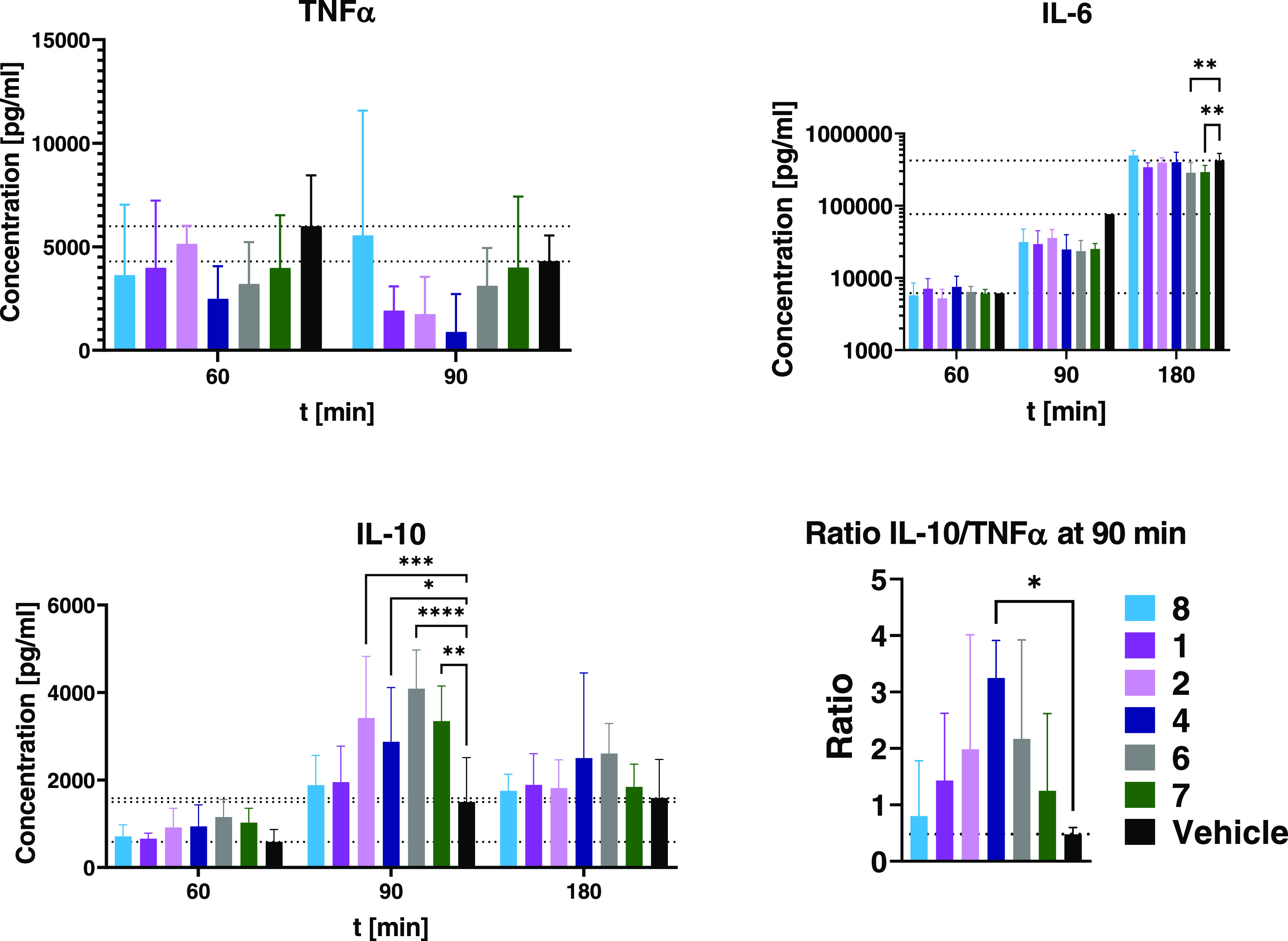
Tail plasma TNFα, IL-6 and IL-10 concentrations vs untreated control (n = 8). Animals were treated with 15 μmol/kg p.o. of JAK inhibitors or vehicle 30 min prior to intraperitoneal injection of 10 mg/kg LPS. Data are the mean ± 1 standard deviation. Concentrations of IL-6 are depicted in half-logarithmic scale.
All JAK inhibitors reduced plasma IL-6 concentrations 90 min after LPS administration, from 41 (8) to 28% (7) of vehicle. The control compounds 6 and 7 were the most effective treatments at the 180 min time point, lowering IL-6 levels down to 68 ± 28 and 69 ± 17% of vehicle, respectively (p < 0.01 each).
LPS-dependent release of IL-10 was enhanced by a selection of compounds from either class (Figure 10): Among the JAK3 inhibitors, both 2 and 4 led to a significant increase in plasma IL-10 at 90 min (173% with 2, p < 0.001), with 4 also effective at 180 min. Treatment with 1 and 8 did not affect IL-10. The control compounds 6 and 7 both elevated plasma levels of IL-10, each peaking at 90 min (207 ± 45 and 170 ± 41%, respectively, p < 0.01 and p < 0.0001).
Figure 10.

Cytokine levels of tail plasma taken 90 min (for TNFα) or 180 min (for IL-10) after intraperitoneal injection of 10 μg/kg LPS and 500 mg/kg Galactosamine. Animals were treated with 9.6 μmol/kg of each substance (equimolar to 3 mg/kg of 7) or vehicle i.v. 30 min before LPS injection. Data are shown as *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Data are the mean ± 1 standard deviation. Values for unstimulated mice are too low to be visible on these scales.
The ratio of tail plasma IL-10 to TNFα integrates effects on both cytokines (Figure 9). The macrolide conjugate 4 caused a significant increase (ratio of 3.2 ± 0.7, p < 0.05) at 90 min post LPS injection. The other compounds also increased the ratio compared to vehicle, but to a lesser degree. The differences were only apparent 90 min post treatment, whereas ratios were similar between treatments at 60 min (not depicted).
These data suggest that JAK3 inhibition, especially in immune cells, is associated with a shift in IL-10 to TNFα ratios toward an anti-inflammatory cytokine pattern.
Intravenous Pretreatment of Mice with JAK Inhibitors Significantly Lowers Plasma TNFα Levels Post LPS Injection
Intravenous application of JAK inhibitors 30 min prior to LPS exposure led to a significant reduction of tail plasma concentrations of TNFα compared to untreated animals (Figure 10). The JAK3 selective 8 has a JAK3 IC50 in a similar range to that reported for 7 (9 and 4 nM, respectively).237 is, in contrast to 8, not isoform selective. 8 was, however, more effective at reducing TNFα than 6 or 7. In contrast to the p.o. study, intravenously applied 8 caused the highest reduction of TNFα levels among all treatments (p < 0.001 vs vehicle). This is in line with the known pharmacokinetic limitations but high potency of 8. 1 and 2 also reduced plasma TNFα concentrations (p < 0.01 and < 0.001, respectively).
Increases in plasma IL-10 concentrations were observed in all treatment groups except for the group treated with 6, though none were statistically significant vs vehicle. These data are consistent with the general trend for JAK3 inhibitors to reduce TNFα and increase the IL-10/TNFα ratio, which was also observed in the p.o. studies.
JAK3 Inhibitor 4 Has a Complex Dose Response in the LPS Challenge Model in BALB/c Mice
Given that raising the dose of 7 increased TNFα levels in vitro and it was less potent than equally dosed JAK3 inhibitors in vivo, we reasoned that the dose optimum of the JAK1 inhibitors in respect of TNFα reduction may be lower in mice. We expanded the range of doses and included molar doses similar to human dosing regimens, which are typically 5–10 mg (or 0.23–0.46 μmol/kg) twice daily.42 Given that the uptake and metabolism of an oral dose may lead to significantly lower overall exposure in time (trough plasma concentrations in patients range from 2–10 ng/mL, equaling 6–32 nM on average depending on dose),43 we further extended the dose range to 0.03 μmol/kg i.v. As mice eliminate substances in the range of 8- to 12-fold faster than humans (approximate allometric scaling),44 this corresponds to ca. 0.003 μmol/kg in human subjects—below known trough levels. The JAK3 inhibitor 4 was selected as the comparator. By administering both substances i.v., differences in oral availability were avoided.
Treatment with 4 led to a bell-shaped dose response on TNFα levels (Figure 11). The two highest (10 and 3 μmol/kg) and lowest (0.1 and 0.03 μmol/kg) doses significantly reduced plasma concentrations of TNFα (p < 0.01 for 10 μmol/kg, p < 0.01 for 3/0.1/0.03 μmol/kg). In contrast, 7 was only associated with reductions in TNFα release at the 3 lower doses, with a trend for increased TNFα with dose: Plasma TNFα concentrations were always higher than for each corresponding dose of 4. The TNFα minimum in the groups treated with 7 was at 0.1 μmol/kg. These data suggest that the in vivo dose response to 7 in this model is complex, reflecting multiple signaling pathways and interactions. However, an optimum dose in the range of 0.1 μmol/kg is consistent with known doses in human use.
Figure 11.
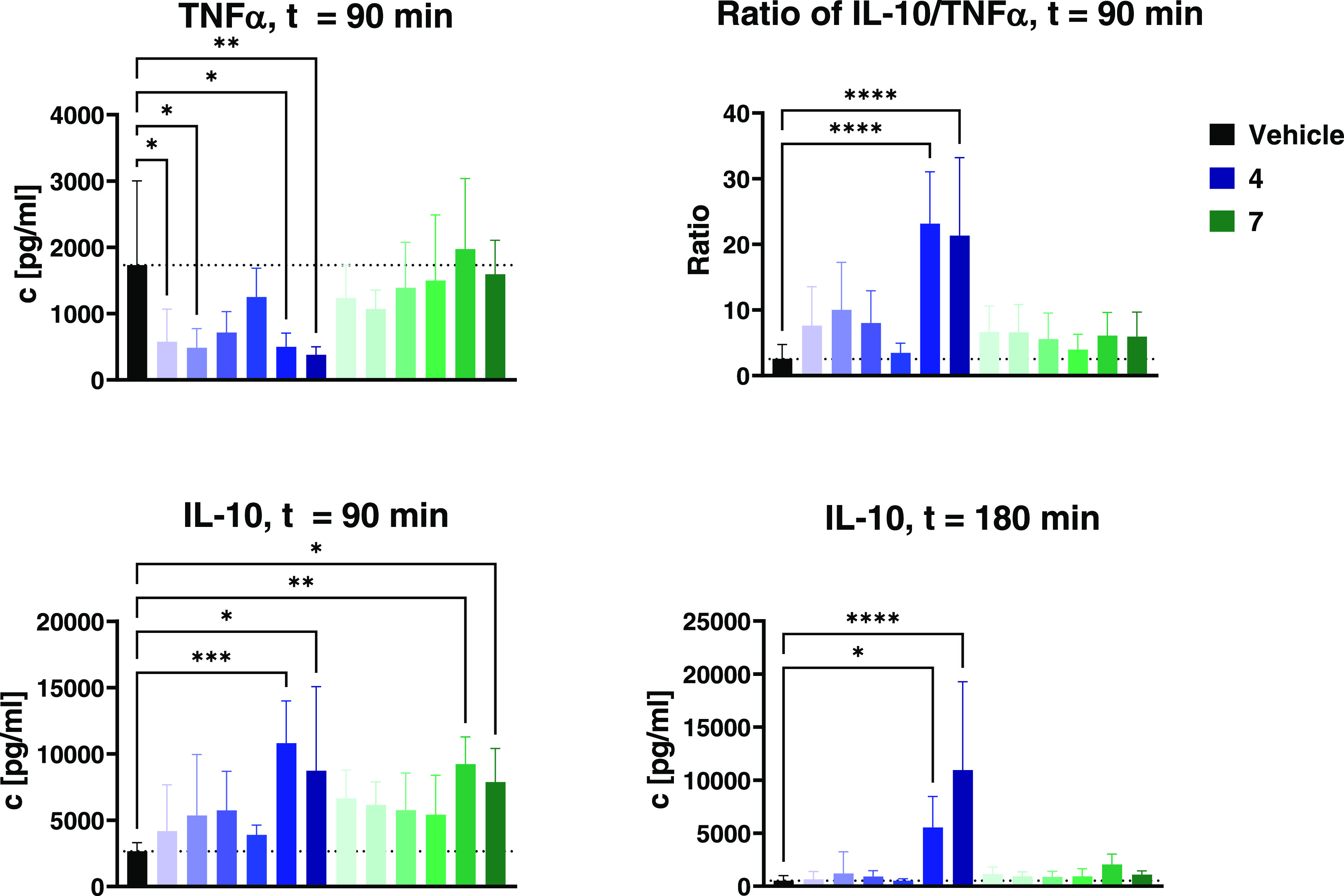
Cytokine levels of tail plasma taken 90 or 180 min after intraperitoneal injection of 10 μg/kg LPS and 500 mg/kg Galactosamine. Animals were treated with vehicle or various doses (from left to right: 0.03, 0.1, 0.3, 1, 3, 10 μmol/kg) of either 7 (shades of green) or 4 (shades of blue) 30 min before LPS injection. Data are shown as *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Data are displayed as mean ± 1 standard deviation.
At 90 min, both 4 and 7 elevated plasma levels of IL-10 at all doses. Treatment with either compound led to significant increases at 10 or 3 μmol/kg (p < 0.05 to p < 0.001); however, the dose response was again complex. From 1 to 0.03 μmol/kg, decreasing doses of 7 were associated with rising IL-10 levels, suggesting that another peak dose of 7 for the stimulus of IL-10 production is below 0.03 μmol/kg. Similarly, 4 exhibited two dose-response zones: A high-range response with strongly increasing IL-10 with dose and a low-range, bell-shaped response with 0.3 μmol/kg associated with the second IL-10 peak.
At 180 min post LPS, IL-10 levels were generally reduced, but still elevated vs vehicle in most treatment groups. At 10 μmol/kg 4, IL-10 was unchanged at ca. 10,000 pg/mL, suggesting that the substance in these amounts can maintain stimulus for a long period. At 3 μmol/kg 4ca. 5500 pg/mL IL-10 was still present, about half the 90 min level. The macrolide in 4 confers a long half-life (see Supporting Information, Figure S5) and is expected to maintain levels in host immune cells for a prolonged time in mice.25 For macrolide compounds, a typical pattern is rapid distribution into peripheral tissues after injection, followed by a longer phase of slow redistribution into the bloodstream.45−47 Additionally, inflammatory conditions like endotoxin shock are known to affect the pharmacokinetics of many drugs including macrolides, typically increasing AUC, plasma half-life, and cmax while lowering clearance.48−51
Quantification of terminal plasma levels of 4 by HPLC-MS indeed showed high concentrations of the compound, in contrast to 7, which was below the quantification limits in all groups (Figure S6).
Given that both TNFα and IL-10 levels are relevant to the resulting degree of inflammation, we also calculated the ratio of these cytokines. Given the rapid clearance of plasma TNFα, the ratio can only be accurately calculated at 90 min where the dose response suggests again that the optimal level of 7 is below 0.03 μmol/kg. For 4, beneficial effects are from 0.03 μmol/kg, with a potential optimum at 0.1 μmol/kg and between 3 and 10 μmol/kg. However, the trends for both substances suggest that the effect will be apparent well below 0.03 μmol/kg. This apparent potency is consistent with the doses and exposures known in the clinical use of 7.
In summary, the JAK3-specific inhibitor 4 reduced TNFα to lower levels than the unselective 7; however, both compounds reduced TNFα in the low-dose groups. The biphasic dose responses seen for both compounds and cytokines suggest that while high doses provide potent effects, the clinically relevant low doses can maintain favorable IL-10/TNFα ratios. These data seem consistent enough with the known clinical effects of 7 to suggest that JAK3 modulation at the lower doses could be the primary source of benefit of the compound.
The advantage of the JAK3-specific inhibitor 4 is that it retains beneficial ratios throughout its dose response and that there is less risk of unfavorable effects on inflammation (rising TNFα) should a subject have low clearance. Examining the high-dose (3 and 10 μmol/kg) effects shows that at these levels, the effects become even more favorable in terms of IL-10/TNFα ratio. This inherent therapeutic certainty over a long dose range in the mode of action would be reassuring for clinicians titrating doses in the elderly.
Discussion
Our efforts to investigate the mode of action of JAK3 inhibitors were primarily directed to the question “is selective JAK3 inhibition a more appropriate mode of action for the treatment of autoimmune diseases than JAK1, mixed JAK, or pan-JAK inhibitors?”. The rationale is found in the knockout phenotype of JAK3, namely, immune deficiency,52,53 and the leukocyte-specific expression of JAK3. A secondary rationale for more specific JAK3 inhibitors is in the limitation of adverse effects associated with existing JAK inhibitors which include cardiac effects reported for some pan- and JAK1 inhibitors and effects on spermatogenesis for filgotinib.54,55 The mechanism of these effects is not known. Adverse effects associated with infection or cancer progression may be due to immune suppression.
At first glance, adverse effects related to immune suppression are unlikely to be managed by a selective JAK3 inhibitor unless subtler dose ranges can be identified that do not impact anti-tumor surveillance. Indeed, one potential benefit of low-dose JAK3 inhibition in cancer has been reported: JAK3 mediates IL-2 signaling and it appears that moderate and sustained inhibition of the IL-2 receptor system prevents T-cell exhaustion in tumors.56 Prevention of exhaustion leads to a more active anti-tumor phenotype in T-cells and more effective activation by checkpoint antibodies. These data suggest that if JAK3-specific inhibitors can allow more nuanced dose ranges, there is the potential for risks in oncology to be manageable, or indeed turned to advantage at lower doses.
In Vitro
The in vitro observations reported here are both consistent with, and in contrast to, various literature reports of similar substances and challenge models. The effects of JAK1 and JAK3 inhibition in vitro are highly dependent on the experimental conditions: Both pro- and anti-inflammatory effects have been observed, depending on factors like stimulus, polarization of cells, and the concentration of inhibitors.41,57 This complicates the selection of an appropriate screening system. We used human primary peripheral blood leukocytes as a screen for attenuation of innate leukocyte-dependent inflammatory reaction.
Perhaps the most important aspect of our study was the potential for interference in IL-10 signaling. Pattinson et al. reported that interference with JAK1 signaling could change IL-10 reception and/or production.41 JAK isoform involvement in signaling is highly cell-dependent and in most cases, the same receptors associate with more than one JAK isoform.14,58 In particular, JAK3 is paired with JAK1 for receptors of γc cytokines.
The JAK3/STAT3 pathway can be activated in M2 macrophages, among other cells, by LPS stimulus.59 Quero et al. found that selective inhibition of JAK3 by 1 μM 8 in M2 macrophages isolated from human donors led to increased production of IL-6 and IL-8 and a decrease of IL-10 compared to control, whereas pan-JAK inhibition by 1 μM 7 caused even higher increases of IL-6 and IL-8. However, it did not affect IL-10 production at this concentration, doing so only at 5 μM. In M1 macrophages, both 7 and 8 decreased IL-6 output with similar efficiency.59
Our findings show that in a more complex environment of mixed immune cell types, the effects of both compounds on cytokine production can be quite different: In peripheral blood leukocytes, the JAK3 selective inhibitor 8 did not affect IL-6 concentrations vs control (Figures 6 and 7), in contrast to the data of Quero et al. in isolated M1 or M2 macrophage populations.59 While the mechanistic basis for this difference is not clear, it is reasonable to expect that mixed primary cells without a culture phase may react differently than differentiated cells. Similarly, macrophages have distinct signaling behavior and are not normally found in peripheral blood. The reported effects of high concentrations of 7 in stimulating pro-inflammatory cytokines in M2 cells do not provide any explanation of its in vivo potency and may not be relevant in terms of both T-cell effects or concentration ranges in vivo. Finally, 1 μM is almost certainly higher than the average concentration experienced in vivo given that the actual dose is at most 0.46 μmol/kg.60
In another report by Pattison et al., murine bone marrow-derived macrophages were incubated with 5 μM 7 for 1 h and stimulated with LPS for various times leading to a decrease in IL-10 production.41 IL-10 can be induced in macrophages by interferon/STAT1 signaling, which itself is inducible by LPS stimulus.61 As IL-10 induces both transcription of its own mRNA62 (via STAT3)63 and its negative regulator SOCS1,64−66 the overall effects on IL-10 levels are complex and depend on factors like time and cell type. Nonetheless, these data show that isolated cell types exposed to high concentrations of substance in vitro exhibit responses that differ from the in vivo setting.
The JAK3 inhibitors studied here exhibited the same ability to mildly decrease IL-10 levels at medium to high doses as seen in macrophages, although 8, at 5 μM, strongly increased IL-10. These observations are consistent with differential interaction with cytokine receptor feedback loops. However, at higher concentrations, off-target effects resulting from inhibition of other kinases potentially complicate interpretation.
The potential for JAK3 blockade to increase IL-10 is also apparent in JAK3–/– bone marrow-derived dendritic cells which have elevated IL-10 output vs wild type, both at baseline and as a reaction to inflammatory stimuli.5 On the other hand, signaling of the JAK1/JAK3-dependent IL-4 (e.g., post TLR4 stimulus) can either augment the production of IL-1067 or cause a decrease,62 once again emphasizing the complex mechanisms involved in the immune reaction.
JAK1 is responsible for the signal transduction by the IL-10 receptor68 and blockade of IL-10 signaling would counteract the IL-10-dependent attenuation of IL-6 production (by SOCS3, which is inducible by the IL-10/STAT3 pathway).64,69 In our assays, IL-10 production itself was amplified by medium concentrations (1–5 μM) of 7 (Figures 6 and 7).
Our findings on the effects of 7 are in line with a publication by Wang et al., where various concentrations of 7 or 10 μM WHI-P154, which is described as a JAK3 inhibitor, enhanced production of pro-inflammatory cytokines IL-6, IL-12, and TNFα by monocytes after LPS stimulus while decreasing production of IL-10.70 It should, however, be noted that WHI-P154 is not specific for JAK3, also being a potent inhibitor of at least EGFR, with lower activity against JAK3 (JAK3 IC50 = 1.8 μM).71 The effects were still reported to be JAK3-related, as similar results were achieved in models using siRNA or JAK3-knockout cell lines.70
There was a contrast in the JAK inhibitors’ effects on TNFα production depending on their selectivity profiles: While the JAK3 selective inhibitors all decreased TNFα in a concentration-dependent manner, the unselective 7 instead increased TNFα (Figure 6) as did 5 and 6. IL-10 depends on JAK1 for signaling, especially to inhibit the production of TNFα and IL-6 after LPS stimulus. Additionally, IL-10 induces the expression of SOCS3 genes via STAT3 phosphorylation (following application of LPS to monocytes), which in turn suppresses STAT1 activity, thus disrupting interferon signaling.72 In cells treated with 5–7, IL-10 signaling is hindered, resulting in increased levels of pro-inflammatory cytokines compared to untreated cells.41,68 Unlike 5–7, selective inhibition of JAK3 spared IL-10 function, leading to a decrease of TNFα, while IL-6 levels were largely unaffected (Figures 6 and 7). A precise mechanism linking JAK3 inhibition and reduced release of TNFα in the context of LPS-based inflammation has, to the best of our knowledge, not been clarified yet. Apart from the aforementioned effects of IL-10, the blockade of IL-2 signaling (which is dependent on JAK1/JAK3) may lead to decreased levels of TNFα, as IL-2 has been shown to induce TNFα production in various cell types.73−75 IL-2 itself is rapidly induced after TLR4 stimulus,76 which may explain the potent effects of the JAK3 inhibitors in our models. Furthermore, silencing of JAK3 in rats was shown to decrease their expression of TNFα and IL-6.77
While the isoform selectivity of the JAK3 inhibitors 3 and 4 has yet to be experimentally determined, they exhibited similar behavior in the assays compared to 8, which has been tested for its selectivity over the human kinome.783 and 4 are closely related structures and likely share the covalent-reversible binding mode. In addition, JAK selectivity has been assessed for 1 and 2 (Supporting Information, Table S2) in a radiometric assay, where both demonstrated selectivity for JAK3 over the other JAK family enzymes.
In Vivo
To investigate the relationship between in vitro potency and in vivo efficacy and the effects of stability and distribution, we compared a range of JAK3 inhibitors from the series in vivo using the LPS challenge model. The JAK3 inhibitors retained their anti-inflammatory potency in vivo, decreasing LPS-dependent production of TNFα and IL-6. However, there were changes in relative apparent potency due to pharmacokinetics. 8—which was the most potent JAK3 inhibitor in vitro—was less effective in vivo compared to 2 and 4 in terms of IL-10 and TNFα levels when given orally. Both 1 and 8 have short plasma half-lives and low peak concentrations after oral administration, while the more stable 2 and the macrolide conjugate 4 have higher tissue distribution and longer terminal half-lives.25 Among the compounds used in the p.o. study, the ratio of IL-10 to TNFα plasma concentrations was highest for 4 (Figure 9) at the relevant 90 min time point. This appears to be a promising anti-inflammatory cytokine profile and thus is the most suitable for the treatment of TNFα-mediated inflammation.
Our in vivo cytokine profile for 7 is in line with findings by Ghoreschi et al., where the substance was applied in the same dose of 5 mg/kg p.o. before LPS injection and increased plasma IL-10 concentrations while simultaneously lowering levels of TNFα and IL-6.796 gave similar results, suggesting that the effects are mostly JAK1-dependent. The efficacy of the selective JAK3 inhibitors in the in vivo model can be explained by the colocalization of JAK1 and JAK3, which form heterodimers during the signaling process.
The blockade of IL-10 signaling by the unselective inhibitors 6 and 7 had notably different effects in human cells compared to the murine models, especially on the production of TNFα. IL-10 induces the transcription of miR-187, an miRNA which suppresses the production of TNFα and IL-6.80,81 Its expression in human monocytes post TLR4 stimulus is potentiated by IL-10. However, this has not been observed in murine leukocytes, which appear to be unable to upregulate miR-187 transcription this way.80 Thus, blockade of IL-10 signaling may have more potent consequences in human cells than in murine cells or live mice, which would contribute to the increases in TNFα levels by 5–7 we observed in our in vitro studies.
Due to the subtler effects of 1 and 8 in the p.o. in vivo study compared to closely related compounds 2 and 4, we conducted another study where treatments were given intravenously (Figure 10). Unlike in the p.o. study, 8 had the strongest effect on TNFα secretion. As mentioned before, we attribute the different outcomes between the studies to the limited exposure to 8via the oral route. Oral administration of 15 μmol/kg of 8 did not lead to sufficient plasma concentrations over the course of that experiment. Meanwhile, the more stable analogue 2, while less potent in vitro, has an improved plasma half-life coupled with higher peak concentrations after oral treatment. The enhancement in exposure was sufficient for 2 to also be effective when administered orally. In the i.v. study, the higher JAK3 inhibitory potency of 8 compared to 2 led to more pronounced attenuation of TNFα production. Curiously 1, while more effective than 8 at reducing TNFα in the p.o. study, still had no effect on IL-10 production, unlike 2, 4, and the unselective control compounds.
A follow-up study, again with i.v. treatment, was carried out to assess the effects of JAK3 inhibitors at a lower dose range resembling trough exposures in human subjects. The strongest decrease of TNFα and concomitant elevation of IL-10 plasma levels were found in the high-dose groups (10 and 3 μmol/kg). 4, at low doses (e.g., 0.1 μmol/kg or 0.03 μmol/kg), also reduced TNFα plasma levels with only a minor increase of plasma IL-10 in these animals, suggesting that while the IL-10/TNFα ratio remains positive, another interaction is responsible for the very IL-10 high levels at doses of 3 μmol/kg and above. Selecting a more nuanced therapeutic dose is facilitated by the use of biomarkers. To this end, we have focused on IL-10 and TNFα as the prime indicators of immune homeostasis and JAK3 inhibitor effect. Clinical studies based on these biomarkers should then enable accurate dosing and treatment personalization. The ideal substance class would then have a linear dose response for effects on IL-10 and TNFα in the pharmacological range. As we have shown, dose response to pan-JAK or JAK1 inhibitors is not linear. We have attributed this to the potential for abrogation (or interruption) of IL-10 receptor (JAK1) mediated feedback control of TNFα production.66,69
The transition from in vitro to in vivo experiments revealed a change in trends for the control compounds 6 and 7 which was not observed in the JAK3 selective compounds: In the in vitro experiments with human peripheral leukocytes, 6 and 7 strongly increased production of TNFα IL-6 and IL-10 at most concentrations. In murine in vivo experiments the compounds instead caused a decrease of TNFα and IL-6 levels vs vehicle, while IL-10 was often increased (Figures 9 and11). While the in vitro environments were already complex (human mixed peripheral leukocytes) to better emulate clinical conditions, the change to in vivo models further increased complexity. An increased number of cell types and other factors (e.g., drug metabolism) can influence the results. Another factor is the switch from human cells to mice: There are potentially relevant differences in the effects of IL-10 on gene transcription depending on species. For example, as described above, IL-10 induces the transcription of an anti-inflammatory microRNA, miR-187, in human myeloid cells, but not in murine cells.80,81
In contrast, the effects of the JAK3 selective inhibitors translated more consistently between in vitro and in vivo experiments. In both settings, TNFα production was reduced by the compounds, while IL-10 levels remained similar to control or were increased. Comparing these observations to those from the mixed JAK inhibitors (or the JAK1 selective 5) gives rise to the question whether the inhibition of JAK1 has different effects in an in vitro setting compared to in vivo (unlike inhibition of JAK3).
Given the intrinsic risk that a JAK1 inhibitor can inhibit IL-10 signaling, the first question that we addressed was whether JAK3-specific inhibitors had the potential to preserve IL-10 feedback. We focused on the LPS model because, in our experience, TNFα and IL-10 responses are very consistent with LPS stimulus in naïve mice. In general, LPS challenge is considered largely a myeloid model;82,83 however, in vivo, all immune cell subsets are involved. In the dose-response study (Figure 11), JAK3 inhibitors provided greater regulation of TNFα levels and the stimulus and maintenance of IL-10 production, leading to a higher IL-10/TNFα ratio at all doses. The two are linked via downstream transcription factors and feedback loops and this is observed in wide-range dose-response studies.34,80,84
The second question we investigated was whether the efficacy of nanomolar doses of clinical JAK inhibitors could be due to effects on IL-10 upregulation. While many reported rodent studies of inflammation use high doses (10–30 mg/kg) of JAK inhibitors,85−87 we used the LPS stimulus model to investigate low-dose effects on the IL-10/TNFα ratio. These data show that the subtler effects of JAK1/2/3/TYK2 inhibition are apparent at nanomolar in vivo doses in mice. Human doses for this class are in the range of 5–10 mg (of 7) bd, which corresponds to 0.07–0.14 mg/kg or 0.23–0.46 μmol/kg bd.42 Using an allometric scaling factor of 8,44 this would imply a dose in mice in the range of 0.57–1.14 mg/kg or 1.76–3.52 μmol/kg twice daily. In this paper, we report on in vivo studies of this type ranging from 15 to 0.03 μmol/kg. We investigated effects observed at or below 1 μmol/kg, reasoning that this would approximate the human range. From Figure 11, it is apparent that 1 μmol/kg represents an inflection point for both the reference pan-JAK inhibitor 7 and the novel JAK3 compound 4 with dose-responsive effects down to 0.03 μmol/kg.
Conclusion
JAK1 inhibitors and JAK3 selective inhibitors differ in the apparent effects on cytokine release both in vitro and in vivo. A key difference is in the regulation of TNFα and IL-10. The most important difference seems to be in the stimulation of TNFα production at high concentrations in vitro. While there are clear differences between human primary cells and a murine system, the same trend of increased IL-10/TNFα ratios and differential regulation of IL-10 are maintained in the LPS challenge model in mice. Cytokine responses in mice reflected both pharmacokinetics and apparent target affinity. However, aspects of pharmacokinetics dominated results from oral application of JAK3 inhibitors. These data demonstrate that JAK3 inhibitors maintain their cytokine selectivity profiles in vivo which is consistent with a general anti-inflammatory mode of action.
Material and Methods
Test Compounds
Structures, synthesis, JAK3 IC50 data, and PK properties of 1–3 and 8 have been reported;25 the corresponding data for 4 are depicted in the Supporting Information. The compounds were tested for toxicity in an MTT assay. No toxicity was observed at the concentrations used for our in vitro experiments.
Tofacitinib (7) citrate and Ruxolitinib (6) phosphate were kindly provided by the University of Tübingen’s Department of Pharmaceutical Chemistry. 5 was obtained from Chempur.
Cytokine Determinations
Human peripheral blood leukocytes from human whole blood were kindly provided by ZKT Tübingen GmbH (Otfried-Müller-Straße 4/1, 72076 Tübingen). ELISAs were performed using Sarstedt 96-well flat-bottom plates. Assay diluent, coating buffer, and TMB substrate were purchased from Biolegend. Materials for ELISA were obtained from Biolegend (ELISA Max Standard Kit, mTNFα, hIL-6, hIL-10, mIL-6, mIL-10) and R&D Systems (Human TNF-α DuoSet ELISA, hTNFα)
In Vitro Stimulation of Cells
Human peripheral blood leukocytes were seeded into 96-well flat-bottom plates (Sarstedt), 2.5 × 105 cells/well, and stimulated with 50 ng/mL LPS (E. coli; Sigma). Then, cells were incubated with JAK inhibitors or DMSO at the indicated concentrations for 24 h at 37 °C and 5% CO2. Supernatant was taken after centrifugation at 400 rpm and immediately used for the determination of cytokine concentrations.
IL-10, IL-6, and TNFα levels were determined in the supernatants of the in vitro stimulation and in vivo tail plasma samples. ELISA was performed according to manufacturer’s instructions and OD was measured using a VersaMax Tunable Microplate Reader (Molecular Devices).
Experimental Animals
All animal experiments were carried out in accordance with German law and standard ethical provisions. Seven- to eight-week-old BALB/c female mice were purchased from Janvier Labs and maintained in our dedicated specific-pathogen-free (SPF) animal facility. Before each study, an acclimatization phase of at least one week was used. Animals had access to food and water ad libitum. Body weights were approx. 20 g per mouse, with each mouse being weighed right before treatment and doses adjusted accordingly to ensure equal doses for all animals.
LPS Challenge
Galactosamine and bacterial LPS from Salmonella enterica were obtained from Sigma-Aldrich.
Pre-Study
Balb/c female mice (n = 9) were treated with either vehicle or 10 mg/kg p.o. of 3 (in 0.5% citric acid, 10 ml/kg). After 15 min, a solution of 1 mg/mL (for 10 mg/kg) LPS (S. enterica typhimurium) in phosphate-buffered saline (10 mL/kg) was applied intraperitoneally. Blood was taken from the tail vein at 60, 90, and 180 min after LPS injection. After 180 min, the mice were sacrificed by CO2 inhalation.
LPS Challenge, p.o. Treatment
Balb/c female mice (n = 8) were treated with 15 μmol/kg p.o. of each of the JAK inhibitors or vehicle (0.5% citric acid in water, 10 mL/kg). After 30 min, 10 mg/kg LPS (S. enterica typhimurium) in phosphate-buffered saline was administered i.p. (10 mL/kg). Blood was taken from the tail vein at 60, 90, and 180 min after LPS injection. After 180 min, the mice were sacrificed by CO2 inhalation.
LPS Challenges, i.v. Treatment
Balb/c female mice (n = 6) were injected i.v. with either vehicle or 9.6 μmol/kg of JAK inhibitors (10% DMSO in murine serum, 2.5 mL/kg). After 30 min, a solution of 1 μg/mL LPS (for 10 μg/kg LPS, S. enterica typhimurium) and 500 mg/kg Galactosamine in 0.9% saline (10 ml/kg) was applied i.p. Blood was taken from the tail vein at 90 and 180 min after LPS injection. The mice were sacrificed by CO2 inhalation after 180 min.
Dose-Response i.v. Study
Balb/c female mice (n = 6) were injected i.v. with either 9.6, 3.2, or 1.06 μmol/kg of JAK inhibitors (10% DMSO in murine serum, 2.5 mL/kg). After 30 min, a solution of 1 μg/mL (for 10 μg/kg LPS, S. enterica typhimurium) and 500 mg/kg Galactosamine in 0.9% saline (10 mL/kg) was applied intraperitoneally. Blood was taken from the tail vein at 90 and 180 min after LPS injection. The mice were sacrificed by CO2 inhalation after 180 min.
In Vivo Samples
Blood taken from mice during in vivo studies was collected into heparinized tubes and centrifuged at 8000 rpm and 4 °C for 8 min. The supernatant plasma was then immediately used for the determination of cytokine concentrations.
Statistics
Statistical analysis of results was performed using GraphPad Prism. Unless otherwise noted, comparisons were carried out by t-test (in vivo pre-study) or one-way analysis of variance (ANOVA). Normality of data was tested with the Shapiro-Wilk test, heteroscedasticity was tested using the Brown–Forsythe test.
Acknowledgments
The authors thank Katharina Bauer for carrying out the JAK3 ELISA experiments, Gerd Helms for the recording of NMR spectra, Agne Klein-Vaičeliu̅naitė for the design of the Table of Contents graph, Anna Schwamborn for HPLC-MS measurements, and the members of the animal facility team at Synovo for performing the animal studies. Diagrams using BioRender were provided by Simon Strass. S.L. and iFIT are funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy–EXC 2180–390900677. TüCAD2 is funded by the Federal Ministry of Education and Research (BMBF) and the Baden-Württemberg Ministry of Science as part of the Excellence Strategy of the German Federal and State Governments.
Glossary
Abbreviations
- ELISA
enzyme-linked immunosorbent assay
- IL
interleukin
- i.p.
intraperitoneal
- i.v.
intravenous
- JAK
Janus kinase
- LPS
lipopolysaccharides
- PIAS
protein inhibitor of activated STAT
- PK
pharmacokinetics
- PTP
protein tyrosine phosphatase
- SOCS
suppressors of cytokine signaling
- SUMO
small ubiquitin-like modifier
- TNF
tumor necrosis factor
- TYK2
tyrosine kinase 2
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.3c00043.
Author Contributions
J.L. was responsible for the design, synthesis, and characterization of the JAK3 inhibitor test compounds, planning and coordination of in vitro and in vivo studies, and for writing. M.M. contributed to the planning and coordination of in vivo studies. N.S. contributed to the planning and coordination of in vitro studies and was responsible for the assessment of cytokine profiles in in vitro and in vivo studies. M.B. was responsible for general conceptualization of the therapeutic approach. F.M. and S.L. were responsible for general guidance and supervision of the project.
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
The authors declare the following competing financial interest(s): J.L., M.M, N.S., F.M., and M.B. are employees of Synovo GmbH, a pharmaceutical company that has an interest in the development of this class of compounds.
Supplementary Material
References
- Rawlings J. S.; Kristin M.; Harrison D. A. The JAK / STAT Signaling Pathway. J. Cell Sci. 2004, 117, 1281–1283. 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- Villarino A. V.; Kanno Y.; Shea J. J. O. Mechanisms and Consequences of Jak – STAT Signaling in the Immune System. Nat. Immunol. 2017, 18, 374–384. 10.1038/ni.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jatiani S. S.; Baker S. J.; Silverman L. R.; Reddy E. P. JAK / STAT Pathways in Cytokine Signaling and Myeloproliferative Disorders: Approaches for Targeted Therapies. Genes Cancer 2010, 1, 979–993. 10.1177/1947601910397187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard W. J.; O’Shea J. J. JAKs and STATs: Biological Implications. Annu. Rev. Immunol. 1998, 16, 293–322. 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- Yamaoka K.; Min B.; Zhou Y.-J.; Paul W. E.; O’Shea J. J. Jak3 Negatively Regulates Dendritic-Cell Cytokine Production and Survival. Blood 2005, 106, 3227–3233. 10.1182/blood-2005-02-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso T.; Johnston J. A.; Linnekin D.; Varesio L.; Rowe T. K.; O’Shea J. J.; McVicar D. W. Regulation of JAK3 Expression in Human Monocytes: Phosphorylation in Response to Interleukins 2, 4 and 7. J. Exp. Med. 1995, 181, 1425–1431. 10.1084/jem.181.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters D. K.; Mercher T.; Gu T.; O’Hare T.; Tyner J. W.; Loriaux M.; Goss V. L.; Lee K. A.; Eide C. A.; Wong M. J.; Stoffregen E. P.; Mcgreevey L.; Nardone J.; Moore S. A.; Crispino J.; Boggon T. J.; Heinrich M. C.; Deininger M. W.; Polakiewicz R. D.; Gilliland D. G.; Druker B. J. Activating Alleles of JAK3 in Acute Megakaryoblastic Leukemia. Cancer Cell 2006, 10, 65–75. 10.1016/j.ccr.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Virtanen A. T.; Haikarainen T.; Raivola J.; Silvennoinen O. Selective JAKinibs: Prospects in Inflammatory and Autoimmune Diseases. BioDrugs 2019, 33, 15–32. 10.1007/s40259-019-00333-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candotti F.; Oakes S. A.; Johnston J. A.; Giliani S.; Schumacher R. F.; Mella P.; Fiorini M.; Ugazio A. G.; Badolato R.; Notarangelo L. D.; Bozzi F.; Macchi P.; Strina D.; Vezzoni P.; Blaese R. M.; O’Shea J. J.; Villa A. Structural and Functional Basis for JAK3-Deficient Severe Combined Immunodeficiency. Blood 1997, 90, 3996–4003. 10.1182/blood.V90.10.3996. [DOI] [PubMed] [Google Scholar]
- Ghoreschi K.; Laurence A.; O’Shea J. J. Selectivity and Therapeutic Inhibition of Kinases: To Be or Not to Be?. Nat. Immunol. 2009, 10, 356–360. 10.1038/ni.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T.; Kawahara A.; Fujii H.; Nakagawa Y.; Minami Y.; Liu Z.; Oishi I.; Silvennoinen O.; Witthuhn B. A.; Ihle J. N.; Taniguchi T. Functional Activation of Jak1 and Jak3 by Selective Association with IL-2 Receptor Subunits. Science 1994, 266, 1045–1047. 10.1126/science.7973659. [DOI] [PubMed] [Google Scholar]
- Rochman Y.; Spolski R.; Leonard W. J. New Insights into the Regulation of T Cells by Γc Family Cytokines. Nat. Rev. Immunol. 2009, 9, 480–490. 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glocker E.-O.; Kotlarz D.; Klein C.; Shah N.; Grimbacher B. IL-10 and IL-10 Receptor Defects in Humans. Ann. N. Y. Acad. Sci. 2011, 1246, 102–107. 10.1111/j.1749-6632.2011.06339.x. [DOI] [PubMed] [Google Scholar]
- Choy E. H. Clinical Significance of Janus Kinase Inhibitor Selectivity. Rheumatology 2019, 58, 953–962. 10.1093/rheumatology/key339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnham M. J.; Erakovic V.; Giamarellos-bourboulis E. J.; Perletti G.; Verleden G. M.; Vos R. Azithromycin: Mechanisms of Action and Their Relevance for Clinical Applications. Pharmacol. Ther. 2014, 143, 225–245. 10.1016/j.pharmthera.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Kanoh S.; Rubin B. K. Mechanisms of Action and Clinical Application of Macrolides as Immunomodulatory Medications. Clin. Microbiol. Rev. 2010, 23, 590–615. 10.1128/CMR.00078-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier M.; Garcia-Luque I.; Montenez J.; Tulkens P.; Piret J. Accumulation, Release and Subcellular Localization of Azithromycin in Phagocytic and Non-Phagocytic Cells in Culture. Int. J. Tissue React. 1994, 16, 211–220. [PubMed] [Google Scholar]
- Retsema J. A.; Bergeron J. M.; Girard D.; Milisen W. B.; Girard A. E. Preferential Concentration of Azithromycin in an Infected Mouse Thigh Model. J. Antimicrob. Chemother. 1993, 31, 5–16. 10.1093/jac/31.suppl_E.5. [DOI] [PubMed] [Google Scholar]
- Gutke H.-J.; Burnet M.; Guse J.-H.. Macrocyclic Compounds and Methods of Use Thereof. U.S. Patent, US8461120, 2010.
- Burnet M.; Guse J.; Bauerlein C.; Hahn U.. Kinase Modulators for the Treatment of Cancer. U.S. Patent, US20130045938A1, 2013.
- Burnet M.; Guse J.-H.; Gutke H.-J.; Beck A.; Tsotsou G.; Droste-Borel I.; Reichert J.; Luyten K.; Busch M.; Wolff M.; Khobzaoui M.; Margutti S.; Meindl T.; Kim G.; Barker L.. Conjugates of Biologically Active Compounds, Methods for Their Preparation and Use, Formulation and Pharmaceutical Applications Thereof. U.S. Patent, US7579324B2, 2009.
- Burnet M.; Guse J.-H.; Gutke H.-J.; Guillot L.; Laufer S.; Hahn U.; Seed M.; Vallejo E.; Eggers M.; McKenzie D.; Albrecht W.; Parnham M.. Anti-Inflammatory Macrolides to Manage Chronic Neutrophilic Inflammation. In Macrocycles in Drug Discovery; Royal Society of Chemistry, 2014; pp 206–234. [Google Scholar]
- Forster M.; Chaikuad A.; Dimitrov T.; Do E.; Holstein J.; Berger B.; Gehringer M.; Ghoreschi K.; Mu S.; Knapp S.; Laufer S. A. Development, Optimization, and Structure–Activity Relationships of Covalent-Reversible JAK3 Inhibitors Based on a Tricyclic Imidazo[5,4-d]pyrrolo[2,3-b]pyridine Scaffold. J. Med. Chem. 2018, 61, 5350–5366. 10.1021/acs.jmedchem.8b00571. [DOI] [PubMed] [Google Scholar]
- Forster M.; Chaikuad A.; Bauer S. M.; Holstein J.; Robers M. B.; Corona C. R.; Gehringer M.; Pfaffenrot E.; Ghoreschi K.; Knapp S.; Laufer S. A. Selective JAK3 Inhibitors with a Covalent Reversible Binding Mode Targeting a New Induced Fit Binding Pocket. Cell Chem. Biol. 2016, 23, 1335–1340. 10.1016/j.chembiol.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laux J.; Forster M.; Riexinger L.; Schwamborn A.; Guezguez J.; Pokoj C.; Kudolo M.; Berger L. M.; Knapp S.; Schollmeyer D.; Guse J.; Burnet M.; Laufer S. A. Pharmacokinetic Optimization of Small Molecule Janus Kinase 3 Inhibitors to Target Immune Cells. ACS Pharmacol. Transl. Sci. 2022, 5, 573–602. 10.1021/acsptsci.2c00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y. A Review of Upadacitinib in Rheumatoid Arthritis. Mod. Rheumatol. 2020, 30, 779–787. 10.1080/14397595.2020.1782049. [DOI] [PubMed] [Google Scholar]
- Quintás-Cardama A.; Vaddi K.; Liu P.; Manshouri T.; Li J.; Scherle P. A.; Caulder E.; Wen X.; Li Y.; Waeltz P.; Rupar M.; Burn T.; Lo Y.; Kelley J.; Covington M.; Shepard S.; Rodgers J. D.; Haley P.; Kantarjian H.; Fridman J. S.; Verstovsek S. Preclinical Characterization of the Selective JAK1/2 Inhibitor INCB018424: Therapeutic Implications for the Treatment of Myeloproliferative Neoplasms. Blood 2010, 115, 3109–3117. 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe L. Ruxolitinib in Myelofibrosis and Polycythemia Vera. J. Adv. Pract. Oncol. 2016, 7, 436–444. 10.6004/jadpro.2016.7.4.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascarenhas J.; Hoffman R. Ruxolitinib: The First FDA Approved Therapy for the Treatment of Myelofibrosis. Clin. Cancer Res. 2012, 18, 3008–3014. 10.1158/1078-0432.CCR-11-3145. [DOI] [PubMed] [Google Scholar]
- Clark J. D.; Flanagan M. E.; Telliez J.-B. Discovery and Development of Janus Kinase (JAK) Inhibitors for Inflammatory Diseases. J. Med. Chem. 2014, 57, 5023–5038. 10.1021/jm401490p. [DOI] [PubMed] [Google Scholar]
- McInnes I. B.; Byers N. L.; Higgs R. E.; Lee J.; Macias W. L.; Na S.; Ortmann R. A.; Rocha G.; Rooney T. P.; Wehrman T.; Zhang X.; Zuckerman S. H.; Taylor P. C. Comparison of Baricitinib, Upadacitinib, and Tofacitinib Mediated Regulation of Cytokine Signaling in Human Leukocyte Subpopulations. Arthritis Res. Ther. 2019, 21, 183 10.1186/s13075-019-1964-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante M.; Sabino J. Efficacy of JAK Inhibitors in Ulcerative Colitis. J. Crohn’s Colitis 2020, 14, S737–S745. 10.1093/ecco-jcc/jjz202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelini J.; Talotta R.; Roncato R.; Fornasier G.; Barbiero G.; Dal Cin L.; Brancati S.; Scaglione F. JAK-Inhibitors for the Treatment of Rheumatoid Arthritis: A Focus on the Present and an Outlook on the Future. Biomolecules 2020, 10, 1002 10.3390/biom10071002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh L.; Kusnadi A.; Park S. H.; Murata K.; Park-Min K.-H.; Ivashkiv L. B. Opposing Regulation of the Late Phase TNF Response by MTORC1-IL-10 Signaling and Hypoxia in Human Macrophages. Sci. Rep. 2016, 6, 31959 10.1038/srep31959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanidworanun C.; Strober W. Predominant Role of Tumor Necrosis Factor-Alpha in Human Monocyte IL-10 Synthesis. J. Immunol. 1993, 151, 6853–6861. 10.4049/jimmunol.151.12.6853. [DOI] [PubMed] [Google Scholar]
- Drexler S. K.; Kong P. L.; Wales J.; Foxwell B. M. Cell Signalling in Macrophages, the Principal Innate Immune Effector Cells of Rheumatoid Arthritis. Arthritis Res. Ther. 2008, 10, 216 10.1186/ar2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler S. K.; Sacre S. M.; Foxwell B. M. Toll-like Receptors: A New Target in Rheumatoid Arthritis?. Expert Rev. Clin. Immunol. 2006, 2, 585–599. 10.1586/1744666X.2.4.585. [DOI] [PubMed] [Google Scholar]
- Dalbeth N.; Callan M. F. C. A Subset of Natural Killer Cells Is Greatly Expanded within Inflamed Joints. Arthritis Rheum. 2002, 46, 1763–1772. 10.1002/art.10410. [DOI] [PubMed] [Google Scholar]
- Edwards S. W.; Hallett M. B. Seeing the Wood for the Trees: The Forgotten Role of Neutrophils in Rheumatoid Arthritis. Immunol. Today 1997, 18, 320–324. 10.1016/S0167-5699(97)01087-6. [DOI] [PubMed] [Google Scholar]
- Feldmann M.; Brennan F. M.; Foxwell B. M. J.; Taylor P. C.; Williams R. O.; Maini R. N. Anti-TNF Therapy: Where Have We Got to in 2005?. J. Autoimmun. 2005, 25, 26–28. 10.1016/j.jaut.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Pattison M. J.; MacKenzie K. F.; Arthur J. S. C. Inhibition of JAKs in Macrophages Increases Lipopolysaccharide-Induced Cytokine Production by Blocking IL-10–Mediated Feedback. J. Immunol. 2012, 189, 2784–2792. 10.4049/jimmunol.1200310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon S. Tofacitinib: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1987–2001. 10.1007/s40265-017-0835-9. [DOI] [PubMed] [Google Scholar]
- Mukherjee A.; Hazra A.; Smith M. K.; Martin S. W.; Mould D. R.; Su C.; Niezychowski W. Exposure–Response Characterization of Tofacitinib Efficacy in Moderate to Severe Ulcerative Colitis: Results from a Dose-ranging Phase 2 Trial. Br. J. Clin. Pharmacol. 2018, 84, 1136–1145. 10.1111/bcp.13523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair A. B.; Jacob S. A Simple Practice Guide for Dose Conversion between Animals and Human. J. Basic Clin. Pharm. 2016, 7, 27 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulds G.; Shepard R. M.; Johnson R. B. The Pharmacokinetics of Azithromycin in Human Serum and Tissues. J. Antimicrob. Chemother. 1990, 25, 73–82. 10.1093/jac/25.suppl_A.73. [DOI] [PubMed] [Google Scholar]
- Cárceles C. M.; Fernández-Varón E.; Marín P.; Escudero E. Tissue Disposition of Azithromycin after Intravenous and Intramuscular Administration to Rabbits. Vet. J. 2007, 174, 154–159. 10.1016/j.tvjl.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Van Bambeke F.; Tulkens P. M. Macrolides: Pharmacokinetics and Pharmacodynamics. Int. J. Antimicrob. Agents 2001, 18, 17–23. 10.1016/S0924-8579(01)00406-X. [DOI] [PubMed] [Google Scholar]
- Elmas M.; Yazar E.; Uney K.; Karabacak A. Pharmacokinetics of Flunixin after Intravenous Administration in Healthy and Endotoxaemic Rabbits. Vet. Res. Commun. 2006, 30, 73–81. 10.1007/s11259-005-3227-7. [DOI] [PubMed] [Google Scholar]
- Yang K. H.; Lee M. G. Effects of Endotoxin Derived from Escherichia Coli Lipopolysaccharide on the Pharmacokinetics of Drugs. Arch. Pharmacal Res. 2008, 31, 1073–1086. 10.1007/s12272-001-1272-8. [DOI] [PubMed] [Google Scholar]
- Jaisue S.; Gerber J. P.; Davey A. K. Pharmacokinetics of Fexofenadine Following LPS Administration to Rats. Xenobiotica 2010, 40, 743–750. 10.3109/00498254.2010.506929. [DOI] [PubMed] [Google Scholar]
- Hao K.; Qi Q.; Hao H.; Wang G.; Chen Y.; Liang Y.; Xie L. The Pharmacokinetic-Pharmacodynamic Model of Azithromycin for Lipopolysaccharide-Induced Depressive-Like Behavior in Mice. PLoS One 2013, 8, e54981 10.1371/journal.pone.0054981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz P.; Toth S.; Falus A. Biological and Clinical Significance of the JAK-STAT Pathway; Lessons from Knockout Mice. Inflammation Res. 2001, 50, 435–441. 10.1007/PL00000267. [DOI] [PubMed] [Google Scholar]
- Cao X.; Shores E. W.; Hu-Li J.; Anver M. R.; Kelsail B. L.; Russell S. M.; Drago J.; Noguchi M.; Grinberg A.; Bloom E. T.; Paul W. E.; Katz S. I.; Love P. E.; Leonard W. J. Defective Lymphoid Development in Mice Lacking Expression of the Common Cytokine Receptor γ Chain. Immunity 1995, 2, 223–238. 10.1016/1074-7613(95)90047-0. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency. Increased Risk of Blood Clots in Lungs and Death with Higher Dose of Xeljanz (Tofacitinib) for Rheumatoid Arthritis, 2019.
- Safety Trial Finds Risk of Blood Clots in the Lungs and Death with Higher Dose of Tofacitinib (Xeljanz, Xeljanz XR) in Rheumatoid Arthritis Patients; FDA to Investigate, 2019. https//www.fda.gov/media/12048. Administration, U. S. F. and D.
- Dammeijer F.; van Gulijk M.; Klaase L.; van Nimwegen M.; Bouzid R.; Hoogenboom R.; Joosse M. E.; Hendriks R. W.; van Hall T.; Aerts J. G. Low-Dose JAK3 Inhibition Improves Antitumor T-Cell Immunity and Immunotherapy Efficacy. Mol. Cancer Ther. 2022, 21, 1393–1405. 10.1158/1535-7163.MCT-21-0943. [DOI] [PubMed] [Google Scholar]
- De Vries L. C. S.; Duarte J. M.; De Krijger M.; Welting O.; Van Hamersveld P. H. P.; Van Leeuwen-Hilbers F. W. M.; Moerland P. D.; Jongejan A.; D’Haens G. R.; De Jonge W. J.; Wildenberg M. E. A JAK1 Selective Kinase Inhibitor and Tofacitinib Affect Macrophage Activation and Function. Inflammatory Bowel Dis. 2019, 25, 647–660. 10.1093/ibd/izy364. [DOI] [PubMed] [Google Scholar]
- Leonard W. J.; O’Shea J. J. Jaks and STATs: Biological Implications. Annu. Rev. Immunol. 1998, 16, 293–322. 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- Quero L.; Tiaden A. N.; Hanser E.; Roux J.; Laski A.; Hall J.; Kyburz D. MiR-221-3p Drives the Shift of M2-Macrophages to a Pro-Inflammatory Function by Suppressing JAK3/STAT3 Activation. Front. Immunol. 2020, 10, 3087 10.3389/fimmu.2019.03087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamba M.; Wang R.; Fletcher T.; Alvey C.; Kushner J. IV; Stock T. C. Extended-Release Once-Daily Formulation of Tofacitinib: Evaluation of Pharmacokinetics Compared With Immediate-Release Tofacitinib and Impact of Food. J. Clin. Pharmacol. 2016, 56, 1362–1371. 10.1002/jcph.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W.; O’Garra A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. 10.1016/j.immuni.2019.03.020. [DOI] [PubMed] [Google Scholar]
- Staples K. J.; Smallie T.; Williams L. M.; Foey A.; Burke B.; Foxwell B. M. J.; Ziegler-Heitbrock L. IL-10 Induces IL-10 in Primary Human Monocyte-Derived Macrophages via the Transcription Factor Stat3. J. Immunol. 2007, 178, 4779–4785. 10.4049/jimmunol.178.8.4779. [DOI] [PubMed] [Google Scholar]
- Michée-Cospolite M.; Boudigou M.; Grasseau A.; Simon Q.; Mignen O.; Pers J.-O.; Cornec D.; Le Pottier L.; Hillion S. Molecular Mechanisms Driving IL-10-Producing B Cells Functions: STAT3 and c-MAF as Underestimated Central Key Regulators?. Front. Immunol. 2022, 13, 818814 10.3389/fimmu.2022.818814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey A. J.; Tan C. K.; Ulett G. C. Infection-Induced IL-10 and JAK-STAT: A Review of the Molecular Circuitry Controlling Immune Hyperactivity in Response to Pathogenic Microbes. JAK-STAT 2012, 1, 159–167. 10.4161/jkst.19918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y. P.; Brown J. R.; Sag D.; Zhang L.; Suttles J. Adenosine 5′-Monophosphate–Activated Protein Kinase Regulates IL-10–Mediated Anti-Inflammatory Signaling Pathways in Macrophages. J. Immunol. 2015, 194, 584–594. 10.4049/jimmunol.1401024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemand C.; Nimmesgern A.; Haan S.; Fischer P.; Schaper F.; Rossaint R.; Heinrich P. C.; Müller-Newen G. Activation of STAT3 by IL-6 and IL-10 in Primary Human Macrophages Is Differentially Modulated by Suppressor of Cytokine Signaling 3. J. Immunol. 2003, 170, 3263–3272. 10.4049/jimmunol.170.6.3263. [DOI] [PubMed] [Google Scholar]
- Ma X.; Yan W.; Zheng H.; Du Q.; Zhang L.; Ban Y.; Li N.; Wei F. Regulation of IL-10 and IL-12 Production and Function in Macrophages and Dendritic Cells. F1000Research 2015, 4, 1465 10.12688/f1000research.7010.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telliez J.-B.; Dowty M. E.; Wang L.; Jussif J.; Lin T.; Li L.; Moy E.; Balbo P.; Li W.; Zhao Y.; Crouse K.; Dickinson C.; Symanowicz P.; Hegen M.; Banker M. E.; Vincent F.; Unwalla R.; Liang S.; Gilbert A. M.; Brown M. F.; Hayward M.; Montgomery J.; Yang X.; Bauman J.; Trujillo J. I.; Casimiro-Garcia A.; Vajdos F. F.; Leung L.; Geoghegan K. F.; Quazi A.; Xuan D.; Jones L.; Hett E.; Wright K.; Clark J. D.; Thorarensen A. Discovery of a JAK3-Selective Inhibitor: Functional Differentiation of JAK3-Selective Inhibition over Pan-JAK or JAK1-Selective Inhibition. ACS Chem. Biol. 2016, 11, 3442–3451. 10.1021/acschembio.6b00677. [DOI] [PubMed] [Google Scholar]
- Hutchins A. P.; Diez D.; Miranda-Saavedra D. The IL-10/STAT3-Mediated Anti-Inflammatory Response: Recent Developments and Future Challenges. Briefings Funct. Genomics 2013, 12, 489–498. 10.1093/bfgp/elt028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Brown J.; Gao S.; Liang S.; Jotwani R.; Zhou H.; Suttles J.; Scott D. A.; Lamont R. J. The Role of JAK-3 in Regulating TLR-Mediated Inflammatory Cytokine Production in Innate Immune Cells. J. Immunol. 2013, 191, 1164–1174. 10.4049/jimmunol.1203084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changelian P. S.; Moshinsky D.; Kuhn C. F.; Flanagan M. E.; Munchhof M. J.; Harris T. M.; Whipple D. A.; Doty J. L.; Sun J.; Kent C. R.; Magnuson K. S.; Perregaux D. G.; Sawyer P. S.; Kudlacz E. M. The Specificity of JAK3 Kinase Inhibitors. Blood 2008, 111, 2155–2157. 10.1182/blood-2007-09-115030. [DOI] [PubMed] [Google Scholar]
- Ito S.; Ansari P.; Sakatsume M.; Dickensheets H.; Vazquez N.; Donnelly R. P.; Larner A. C.; Finbloom D. S. Interleukin-10 Inhibits Expression of Both Interferon α– and Interferon γ– Induced Genes by Suppressing Tyrosine Phosphorylation of STAT1. Blood 1999, 93, 1456–1463. 10.1182/blood.V93.5.1456. [DOI] [PubMed] [Google Scholar]
- O’Shea J. J.; Ma A.; Lipsky P. Cytokines and Autoimmunity. Nat. Rev. Immunol. 2002, 2, 37–45. 10.1038/nri702. [DOI] [PubMed] [Google Scholar]
- Hsu D.-H.; Moore K. W.; Spits H. Differential Effects of IL-4 and IL-10 on IL-2-Induced IFN-γ Synthesis and Lymphokine-Activated Killer Activity. Int. Immunol. 1992, 4, 563–569. 10.1093/intimm/4.5.563. [DOI] [PubMed] [Google Scholar]
- Reddy J.; Chastagner P.; Fiette L.; Liu X.; Thèze J. IL-2-Induced Tumor Necrosis Factor (TNF)-β Expression: Further Analysis in the IL-2 Knockout Model, and Comparison with TNF-α, Lymphotoxin-β, TNFR1 and TNFR2 Modulation. Int. Immunol. 2001, 13, 135–147. 10.1093/intimm/13.2.135. [DOI] [PubMed] [Google Scholar]
- Granucci F.; Feau S.; Angeli V.; Trottein F.; Ricciardi-Castagnoli P. Early IL-2 Production by Mouse Dendritic Cells Is the Result of Microbial-Induced Priming. J. Immunol. 2003, 170, 5075–5081. 10.4049/jimmunol.170.10.5075. [DOI] [PubMed] [Google Scholar]
- Hou Y.; Li H.; Huo W. MicroRNA-495 Alleviates Ulcerative Interstitial Cystitis via Inactivating the JAK–STAT Signaling Pathway by Inhibiting JAK3. Int. Urogynecol. J. 2021, 32, 1253–1263. 10.1007/s00192-020-04593-x. [DOI] [PubMed] [Google Scholar]
- Forster M.; Chaikuad A.; Dimitrov T.; Do E.; Holstein J.; Berger B.; Gehringer M.; Ghoreschi K.; Mu S.; Knapp S.; Laufer S. A.. Development, Optimization, and Structure–Activity Relationships of Covalent-Reversible JAK3 Inhibitors Based on a Tricyclic Imidazo[5,4-d]pyrrolo[2,3-b]pyridine Scaffold. 2018, 61, 5350–5366.. 10.1021/acs.jmedchem.8b00571. [DOI] [PubMed] [Google Scholar]
- Ghoreschi K.; Jesson M. I.; Li X.; Lee J. L.; Ghosh S.; Alsup J. W.; Warner J. D.; Tanaka M.; Steward-Tharp S. M.; Gadina M.; Thomas C. J.; Minnerly J. C.; Storer C. E.; LaBranche T. P.; Radi Z. A.; Dowty M. E.; Head R. D.; Meyer D. M.; Kishore N.; O’Shea J. J. Modulation of Innate and Adaptive Immune Responses by Tofacitinib (CP-690,550. J. Immunol. 2011, 186, 4234–4243. 10.4049/jimmunol.1003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossato M.; Curtale G.; Tamassia N.; Castellucci M.; Mori L.; Gasperini S.; Mariotti B.; De Luca M.; Mirolo M.; Cassatella M. A.; Locati M.; Bazzoni F. IL-10–Induced MicroRNA-187 Negatively Regulates TNF-α, IL-6, and IL-12p40 Production in TLR4-Stimulated Monocytes. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, E3101–E3110. 10.1073/pnas.1209100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comi M.; Amodio G.; Gregori S. Interleukin-10-Producing DC-10 Is a Unique Tool to Promote Tolerance via Antigen-Specific T Regulatory Type 1 Cells. Front. Immunol. 2018, 9, 682 10.3389/fimmu.2018.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.-P.; Yang K.; Xu G.-N.; Zhu L.; Hou L.-N.; Zhang W.-H.; Chen H.-Z.; Cui Y.-Y. Role of M3 MAChR in in Vivo and in Vitro Models of LPS-Induced Inflammatory Response. Int. Immunopharmacol. 2012, 14, 320–327. 10.1016/j.intimp.2012.07.020. [DOI] [PubMed] [Google Scholar]
- Guha M.; Mackman N. LPS Induction of Gene Expression in Human Monocytes. Cell. Signalling 2001, 13, 85–94. 10.1016/S0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- Chang B. Y.; Zhao F.; He X.; Ren H.; Braselmann S.; Taylor V.; Wicks J.; Payan D. G.; Grossbard E. B.; Pine P. R.; Bullard D. C. JAK3 Inhibition Significantly Attenuates Psoriasiform Skin Inflammation in CD18 Mutant PL/J Mice. J. Immunol. 2009, 183, 2183–2192. 10.4049/jimmunol.0804063. [DOI] [PubMed] [Google Scholar]
- Wagh A. D.; Sharma M.; Mahapatra J.; Chatterjee A.; Jain M.; Addepalli V. Investigation into the Role of PI3K and JAK3 Kinase Inhibitors in Murine Models of Asthma. Front. Pharmacol. 2017, 8, 82 10.3389/fphar.2017.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T. H.; Hegen M.; Quadros E.; Nickerson-Nutter C. L.; Appell K. C.; Cole A. G.; Shao Y.; Tam S.; Ohlmeyer M.; Wang B.; et al. Selective Functional Inhibition of JAK-3 Is Sufficient for Efficacy in Collagen-induced Arthritis in Mice. Arthritis Rheum. 2010, 62, 2283–2293. 10.1002/art.27536. [DOI] [PubMed] [Google Scholar]
- He L.; Pei H.; Lan T.; Tang M.; Zhang C.; Chen L. Design and Synthesis of a Highly Selective JAK3 Inhibitor for the Treatment of Rheumatoid Arthritis. Arch. Pharm. 2017, 350, 1700194 10.1002/ardp.201700194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.