


Abstract
Mammalian proteins expressed in Escherichia coli are used in a variety of applications. A major drawback in producing eukaryotic proteins in E.coli is that the bacteria lack most eukaryotic post-translational modification systems, including serine/threonine protein kinase(s). Here we show that a eukaryotic protein can be phosphorylated in E.coli by simultaneous expression of a mammalian protein kinase and its substrate. We show that in bacteria expressing SRPK1, ASF/SF2 becomes phosphorylated to a degree resembling native ASF/SF2 present in interphase HeLa cell nuclei. The E.coli phosphorylated ASF/SF2 is functional in splicing and, contrary to the unphosphorylated protein, soluble under native conditions.
INTRODUCTION
Methodological improvements have made it possible to study many cellular processes in vitro in cell-free extracts. This in turn has allowed dissection of the processes and the identification of individual components of the machinery, e.g. polypeptides and oligonucleotides. In many cases in vitro studies rely on access to relatively large amounts of recombinant proteins. Escherichia coli expression systems have been useful for this purpose and as a source of recombinant proteins they offer many advantages compared to eukaryotic expression systems (for a review see 1). It is, for instance, relatively easy and fast to set up an E.coli expression system; it is affordable, simple to scale up and the risk of contamination by other eukaryotic proteins is eliminated. The disadvantages with E.coli expression systems, compared to eukaryotic cells, are perhaps fewer, but nevertheless in some cases significant. For instance, it is not uncommon that mammalian proteins are insoluble when overexpressed in bacteria and E.coli lack several post-translational modification systems, such as serine/threonine phosphorylation.
Here we report functional coexpression of the mammalian SR protein kinase (SRPK1) and its substrate (ASF/SF2) in E.coli. ASF/SF2 is a pre-mRNA splicing factor that belongs to the SR family of proteins, which shares a highly phosphorylated region composed of repeats of arginine/serine residues, a so called RS-region (2). Phosphorylated SR-proteins are recognised by the monoclonal antibody mAb 104 (2), which is reactive against phosphopeptides in the RS-region. Studies have shown that the level of phosphorylation of the RS-region influences the subnuclear distribution of the SR-proteins and their function in splicing (3–6). Several protein kinases have been demonstrated to phosphorylate serine residues in the RS-region of ASF/SF2, including SRPK1 (7 and references therein). We show that ASF/SF2 isolated from SRPK1 expressing bacteria is phosphorylated in the same region and to a similar extent as the native protein isolated from HeLa cells. Moreover, the fraction of recombinant ASF/SF2 recovered in soluble form is greatly increased by coexpression of SRPK1. Our results are consistent with published observations that phosphorylated ASF/SF2 is more active than unphosphorylated ASF/SF2 in restoring splicing activity to cytoplasmic S100 extracts.
MATERIALS AND METHODS
Plasmids
A cDNA encoding full-length ASF/SF2 (8) was inserted into BamHI and HindIII sites in pQE-60 (Qiagen), resulting in construct pQE-ASF (lacks a his-tag) and into the same sites in vector pRSET A (Invitrogen), resulting in construct pRSET A-ASF, containing an N-terminal his-tag. For construction of pLS-SRPK1, an AflIII fragment from pRSET B-SRPK1 containing the complete SRPK1 coding sequence was cloned into one of the BspHI sites in pLys S (Novagen). pRSET B-SRPK1 was constructed by isolation of the entire SRPK1 coding sequence from pSP73-SRPK1 (a generous gift from X.-D. Fu), using complete EcoRI and partial NcoI digestion. The SRPK1 fragment was inserted into the same sites in pRSET B (Invitrogen).
Expression of rASF and SRPK1
Escherichia coli strain BL21 (DE3) was transfected with pLS-SRPK1 and either pQE-ASF, pLS-SRPK1 or pRSET A-ASF. Double transformants were selected on LB-plates containing 100 µg/ml ampicillin and 34 µg/ml chloramphenicol. Single colonies were picked and grown at 37°C, overnight, in LB-media containing the antibiotics. The overnight cultures were diluted 100-fold with LB-media containing antibiotics and incubated at 37°C with vigorous shaking for ~4 h. When OD600 reached 0.5, ASF/SF2 and SRPK1 expression were induced by addition of IPTG to a final concentration of 2.5 mM. The cells were incubated at 37°C for a further 4 h before they were collected by centrifugation.
ASF/SF2 purification
For expression of untagged ASF/SF, bacteria containing pLS-SRPK1 and pQE-ASF were grown in 2 l of cultured media. At the end of the incubation, after IPTG induction, the bacterial suspension was centrifuged and the bacteria pellet was dissolved in 28 ml lysis buffer (50 mM HEPES pH 7.9, 10 mM β-glycerophosphate, 0.3 M KCl, 0.5 mM β-mercaptoethanol, 5 µg/ml lysozyme, 5 µg/ml RNase A, 0.05% Triton X100). The suspension was kept on ice, sonicated and centrifuged at 8000 g for 20 min. The supernatant was collected and ammonium sulphate was added to 65% saturation. The mixture was stirred for 2 h at 4°C before it was centrifuged at 8000 g for 20 min. Once again, the supernatant was collected and ammonium sulphate added to 90% saturation. The solution was stirred and centrifuged as for the first ammonium sulphate cut. The pellet formed upon centrifugation of the 90% fraction contained the majority of soluble ASF/SF2 (data not shown) and this was washed with a lysis buffer with a 90% ammonia sulphate saturation. The ASF/SF2 pellet was then dissolved in 10 ml dialysis buffer (10 mM HEPES pH 7.6, 65 mM KCl, 15 mM NaCl, 1 mM EDTA, 2 mM DTT, 5 mM KF, 5 mM β-glycerophosphate, 0.2 mM PMSF) and dialysed for 4 h in total before MgCl2 was added to a final concentration of 20 mM, which causes aggregation of ASF/SF2. The mixture was incubated on ice for 1 h and ASF/SF2 was collected by centrifugation at 13 000 g for 15 min. The pellet was dissolved in 50 µl of 5% buffer D (20 mM HEPES pH 7.9, 5% glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM DTT) and stored in aliquots at –80°C.
Phosphorylated his-tagged ASF/SF2 was purified from 1 litre of media. ASF/SF2 and SRPK1 expression was induced with a final concentration of 2 mM IPTG and the bacteria were incubated for 2 h at 37°C. The pellet formed upon centrifugation was dissolved in 28 ml of lysis buffer (50 mM HEPES pH 7.9, 10 mM β-glycerophosphate, 0.3 M KCl, 0.05% Triton X100). The suspension was sonicated and centrifuged as for the untagged protein. The supernatant was incubated with 2 ml of nickel agarose at 4°C for 30 min and thereafter transferred into a column. The nickel agarose beads were first washed with 20 ml of lysis buffer containing 5 mM imidazole, thereafter with lysis buffer containing increasing concentrations of imidazole. The majority of ASF/SF2 elutes in the range 150–600 mM imidazole. Without making any attempts to optimise ASF/SF2 production we routinely observed that >10% of total ASF/SF2 is soluble in SRPK1 expressing bacteria and up to 33 mg of >95% pure ASF/SF2 was obtained per litre bacterial suspension. After dialysis to 20 mM HEPES pH 7.9, 0.1 M KCl, 20% glycerol, 1 mM DTT, ASF/SF2 was stored in aliquots at –20°C.
Unphosphorylated his-tagged ASF/SF2 was purified from E.coli transformed with only pRSET A-ASF, using a previously published protocol (9).
SDS–PAGE and western blotting
Protein samples were separated by SDS–PAGE and transferred to nitrocellulose membrane using a semi-dry transfer apparatus. The filter was blocked using TBS buffer (20 mM Tris pH 7.5, 150 mM NaCl, 10 mM β-glycerophosphate and 5% dry milk). His-tagged ASF/SF2 was detected using monoclonal antibody mAb 104, which recognises phosphorylated SR-proteins, and monoclonal anti-his antibody (Clontech). Untagged ASF/SF2 was detected using polyclonal antibodies developed against peptides from the extreme C-terminus of the protein (10). The his and mAb 104 antibodies were detected using HRP-conjugated anti-mouse antibodies (Amersham), ASF/SF2 antibodies were detected using HRP-conjugated anti-rabbit antibodies and visualised by ECL (Amersham).
In vitro splicing
HeLa nuclear extract was prepared as previously described (11) and the cytoplasmic fraction was used to prepare S100 extract as described (12). The total splicing reaction volume was 25 µl and contained 32P-labelled 52,55K-U1 RNA (13) (50 000 c.p.m./reaction), 2.5 mM MgCl2, 40 mM KCl, 2.6% polyvinylic alcohol, 20 mM creatinphosphate, 2 mM ATP, 15 U RNasin, 12 mM HEPES pH 7.9 and 40% (v/v) nuclear extract or S100 extract. SRPK1 phosphorylated and unphosphorylated ASF was either pre-incubated for 10 min on ice with pre-mRNA (Fig. 3, lanes 4 and 6), or for 10 min at room temperature with S100 extract (Fig. 3, lanes 5 and 7). Splicing was initiated at the end of the pre-incubation by addition of the remainder of the reaction mixtures. The splicing reactions were incubated at 30°C for 2 h before the RNA was isolated and analysed on 8% polyacrylamide gel, followed by autoradiography.
Figure 3.
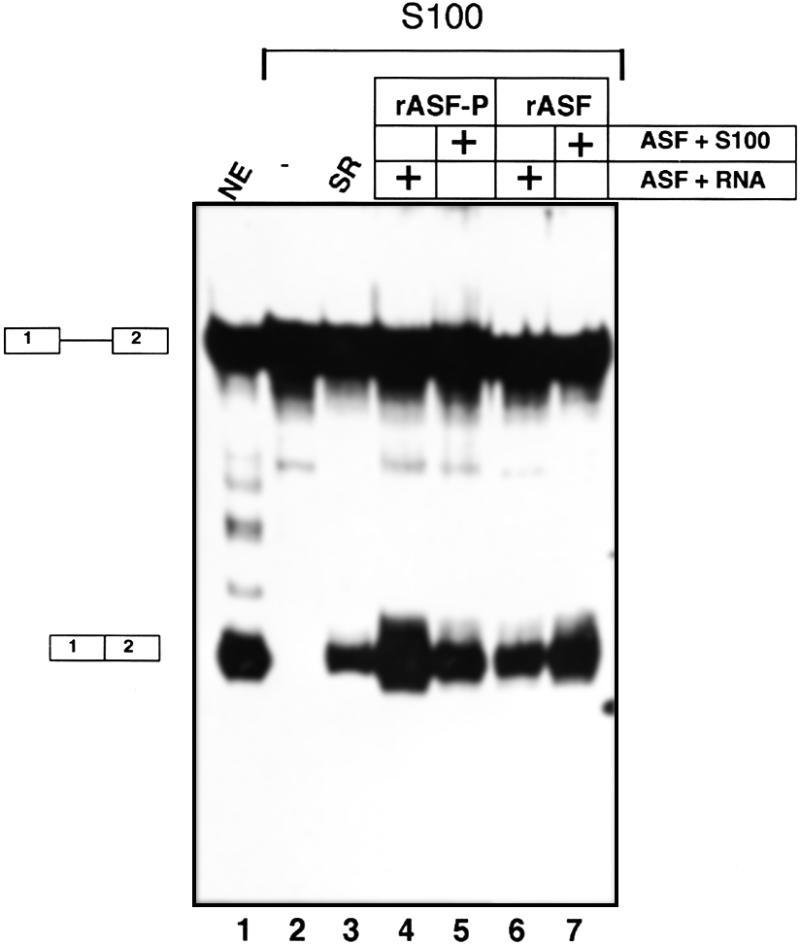
The effect of rASF-P on pre-mRNA splicing in vitro. 32P-labelled 52,55K-U1 pre-mRNA was incubated in HeLa cell nuclear extracts (lane 1) or cytoplasmic HeLa cell S100 extracts (lanes 2–7) under splicing conditions. S100 extracts were either supplemented with SR-proteins purified from HeLa cell nuclear extracts (lane 3), SRPK1 phosphorylated ASF/SF2 (lanes 4 and 5) or unphosphorylated ASF/SF2 (lanes 6 and 7). pre-mRNA and ASF/SF2 (lanes 4 and 6) or S100 extract and ASF/SF2 (lanes 5 and 7) were mixed first and incubated on ice, before initiation of splicing. Splicing products (depicted on the left) were analysed on an 8% denaturing polyacrylamide gel and revealed by autoradiography.
RESULTS
Phosphorylation of ASF/SF2 by SRPK1 in E.coli
ASF/SF2 has been widely studied in vitro and the protein is readily produced in E.coli, but unfortunately it is insoluble and therefore has to be purified under denaturing conditions (8,9). Another drawback is the lack of serine protein phosphorylation in the bacterial expression system. This is important because the activity of ASF/SF2 is affected by serine phosphorylation (3,6).
In an attempt to produce phosphorylated ASF/SF2, we decided to express ASF/SF2 and SRPK1 in the same bacterial cell. From previous studies it was known that recombinant SRPK1 expressed in E.coli has a substrate specificity that resembles that of the native kinase (14 and J.-P.Kreivi, unpublished data). In order to express the two proteins in the same bacteria, we subcloned ASF/SF2 and SRPK1 into two separate expression vectors with compatible origins of replication. In a first set of experiments we expressed an untagged ASF/SF2 together with SRPK1. Escherichia coli cells containing the two plasmids were grown in standard LB-media and, importantly, expression of both recombinant proteins were induced with IPTG.
We found that a substantial fraction of ASF/SF2 produced in bacteria expressing SRPK1 is soluble (data not shown) and the protein was therefore purified using a protocol that is used for purification of native SR-proteins from mammalian cells (2). As shown in Figure 1, ASF/SF2 antibodies detect two ASF/SF2 species after the final purification step: a slower migrating major form (rASF-P) and a faster migrating minor form (rASF) of ASF/SF2 (Fig. 1, lane 1). In order to determine whether the apparent difference in size between the two ASF/SF2 species reflected a difference in protein phosphorylation level, the protein fractions were treated with calf intestinal phosphatase (CIP). Indeed, CIP treatment of ASF/SF2 results in reduction of rASF-P and an increase in the amount of rASF (Fig. 1, lanes 1–4). The same shift in SDS–PAGE mobility upon CIP treatment was also observed for ASF/SF2 isolated from HeLa cell nuclei (Fig. 1, compare lanes 5 and 6). We also noticed that only rASF-P is reactive to a monoclonal antibody mAb 104 (data not shown, see also Fig. 2B), which specifically recognises phosphoepitope(s) present in SR-proteins (2).
Figure 1.
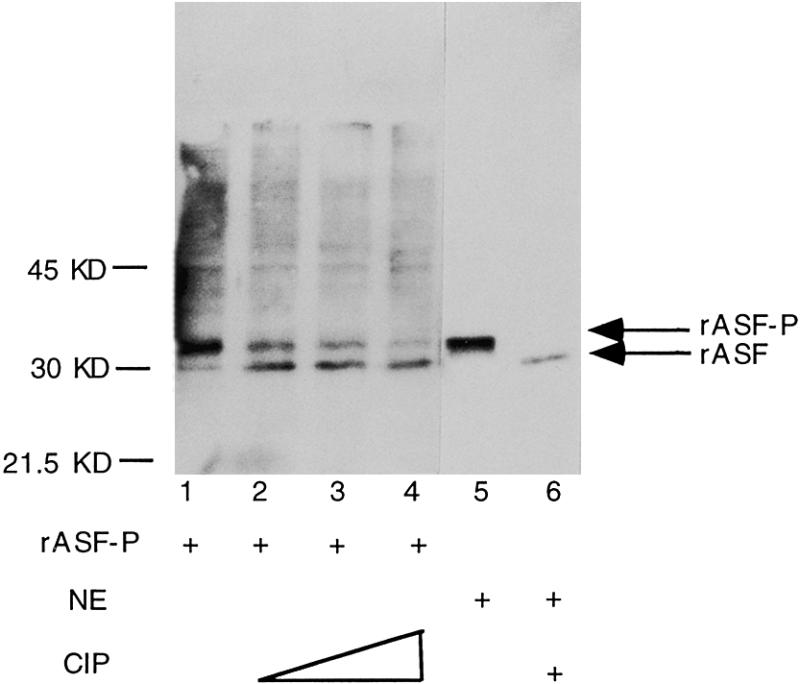
ASF/SF2 phosphorylation by SRPK1 in E.coli. Partially purified ASF/SF2 (lanes 1–4) and crude nuclear extracts from HeLa cells (lanes 5 and 6) were separated by SDS–PAGE, transferred to nitrocellulose membranes and probed with anti-ASF/SF2 antibodies. Prior to electrophoresis, protein samples in lanes 2–4 and 6 were treated with CIP. The arrows on the right side indicate the position of phosphorylated ASF/SF2 (rASF-P) and unphosphorylated ASF/SF2 (rASF), respectively.
Figure 2.
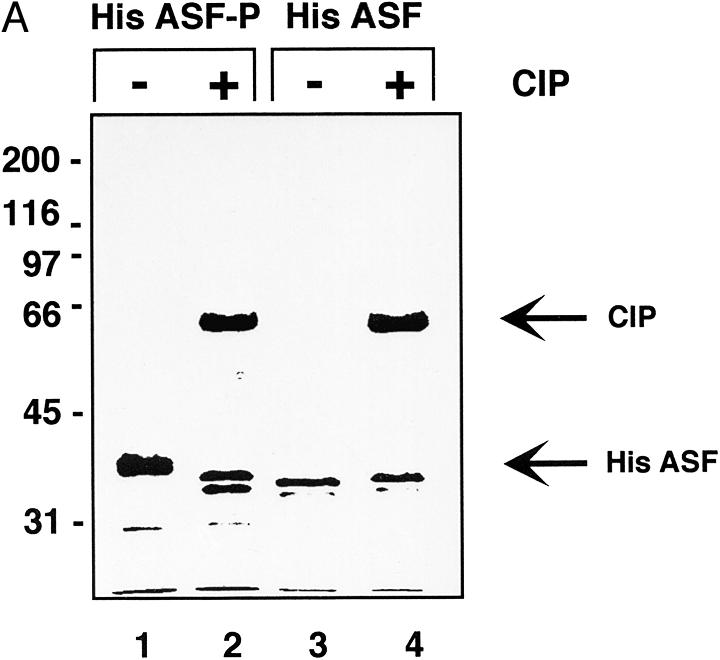
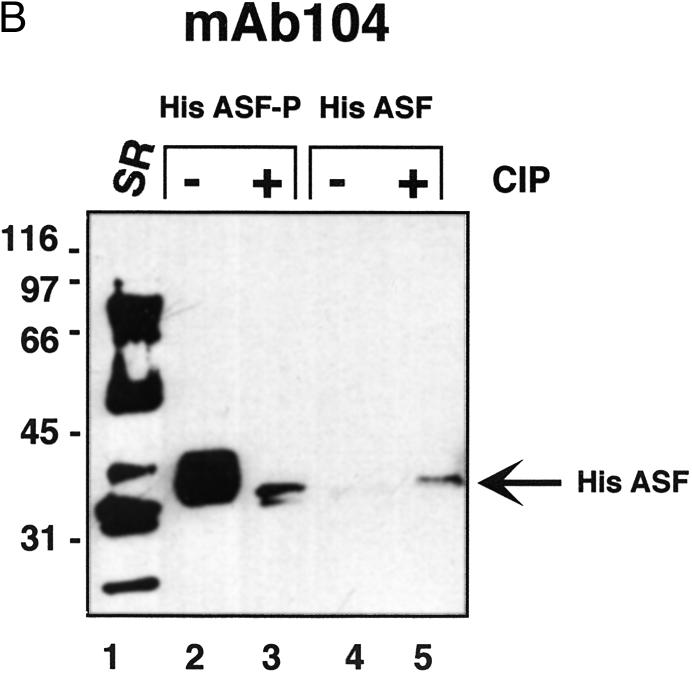
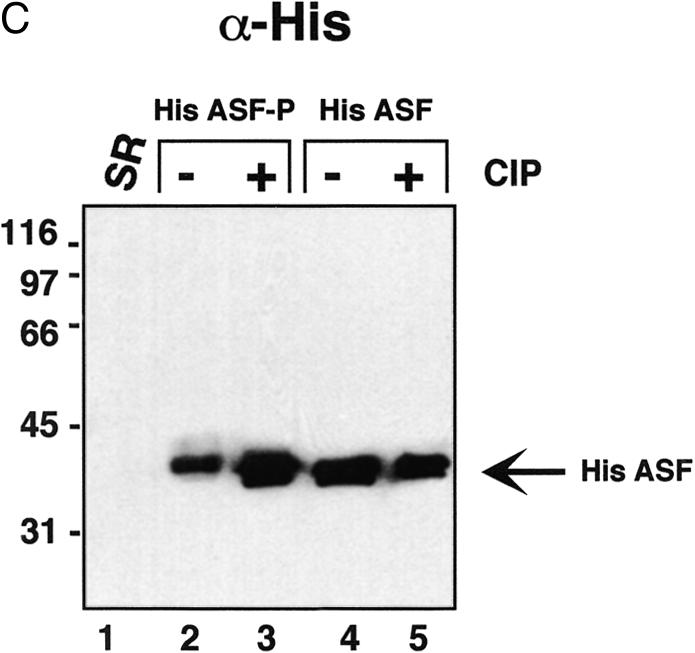
Characterisation of ASF/SF2 phosphorylated by SRPK1 in E.coli. (A) Coomassie Brilliant Blue staining. His-tagged ASF/SF2 from SRPK1 expressing E.coli (lanes 1 and 2) or from non-SRPK1 containing E.coli (lanes 3 and 4) were purified using nickel agarose chromatography and analysed by SDS–PAGE. ASF/SF2 was either untreated (lanes 1 and 3) or treated with 1 U of CIP for 30 min at 30°C. (B) SR-proteins purified from HeLa cell nuclear extracts (lane 1) or ASF/SF2 from SRPK1 containing E.coli (lanes 2 and 3) or from non-SRPK1 containing E.coli (lanes 4 and 5) were separated by SDS–PAGE, transferred to a nitrocellulose membrane and probed with mAb 104 antibody. Protein samples were either untreated (lanes 1, 2 and 4) or phosphatase treated with CIP (lanes 3 and 5). (C) The same filter as in (B) was stripped using SDS–buffer and thereafter probed with monoclonal anti-his antibody.
Taken together, these results show that ASF/SF2 is produced as a phosphoprotein in E.coli when coexpressed with SRPK1. Also, the fact that rASF-P is reactive to mAb 104 suggests that the native phosphoepitope in the RS-region is restored. The mobility of rASF-P in one-dimensional SDS–PAGE resembles that of ASF/SF2 present in HeLa cell nuclear extracts, suggesting that rASF-P is phosphorylated to similar extent as the native protein.
Although ASF/SF2 was phosphorylated by SRPK1 in E.coli, we encountered problems in reproducibly obtaining bacteria transformed by the two expression vectors used in the above-presented results. We believe the reason for this is due to small leakage of ASF/SF2 and SRPK1 expression, and that expression of either one or both of the proteins is toxic to the bacteria. In order to obtain a tighter control of ASF/SF2 expression, we subcloned the gene into a more suitable expression vector (pRSET from Invitrogen), where T7 RNA polymerase controls the ASF/SF2 expression. ASF/SF2 produced from this plasmid gives rise to a his-tagged protein which is slightly larger than the native protein (286 versus 248 amino acids), but it has the advantage that it is easy to purify efficiently to a high degree in a single purification step using nickel agarose chromatography.
The his-tagged ASF/SF2 was expressed in the presence or absence of SRPK1. Once again, when coexpressed with SRPK1, a large fraction of ASF/SF2 was recovered from the soluble bacterial cell lysate (>10% of total ASF/SF2, data not shown). ASF/SF2 synthesised in the absence of SRPK1, on the other hand, is insoluble (8,9) and >99% of the protein accumulates in inclusion bodies within the bacteria (data not shown). Moreover, ASF/SF2 remained soluble in a relatively concentrated solution (0.5 mg/ml) after purification and dialysis. As shown in Figure 2A, ASF/SF2 from SRPK1 expressing bacteria migrates slower than the same protein from non-SRPK1 expressing cells (Fig. 2A, compare lanes 1 and 3). As with untagged rASF-P, the his-tagged protein migrates faster in SDS–PAGE after phosphatase treatment, implying that ASF/SF2 coexpressed with SRPK1 indeed becomes phosphorylated (Fig. 2A, lanes 1 and 2). Unphosphorylated ASF/SF2 does not migrate faster upon incubation with phosphatase (Fig. 2A, lanes 3 and 4), thus demonstrating that CIP is not contaminated by proteases that may have been responsible for the mobility shift for ASF/SF2. In a duplicate experiment the proteins were blotted to nitrocellulose filter after SDS–PAGE and thereafter probed with monoclonal antibody mAb 104. As shown in Figure 2B, only ASF/SF2 from SRPK1 expressing E.coli is recognised by mAb 104 and we therefore conclude that also the his-tagged ASF/SF2 is phosphorylated in the RS-region (Fig. 2B, lane 2). As judged by the migration in SDS–PAGE and reactivity to mAb 104, phosphatase treatment of His ASF-P does not result in a complete dephosphorylation of the protein (Fig. 2B, lanes 2 and 3). This may imply that some phosphates in ASF/SF2 are less accessible to phosphatase treatment. The same phenomena has been observed for native ASF/SF2; treating nuclear extracts with high amounts of protein phosphatase results in loss of mAb 104 reactivity for all SR-proteins in nuclear extracts, except for the ASF/SF2 and SC35 fractions (15). After immunodetection with mAb 104 the filter was stripped and reprobed with a monoclonal anti-his antibody. As shown in Figure 2C, recombinant ASF/SF2 is present in equal amounts in all four lanes, demonstrating that the lack of mAb 104 recognition in lanes 3–5 is in fact due to the absence of a phosphorylated RS-region in those ASF species.
SRPK1 phosphorylated ASF/SF2 is active
We next performed a functional analysis of SRPK1 phosphorylated ASF/SF2. We used an assay that is based on cytoplasmic S100 extracts that contain all components required for splicing but have suboptimal concentrations of SR-proteins (16). S100 extracts have little or no splicing activity on their own (Fig. 3, lane 2), but are activated upon addition of a mixture of native SR-proteins purified from HeLa cells (Fig. 3, compare lanes 2 and 3). Importantly, the order of pre-incubation is important for the splicing activation capacity of ASF/SF2. Here we used an assay system that is routinely used in our laboratory, but differs slightly from the assay described by Xiao and Manley (3). As shown in Figure 3, SRPK1 phosphorylation of ASF/SF2 generates a protein that is more efficient than the unphosphorylated protein in activating splicing if the ASF/SF2 proteins are mixed with the pre-mRNA before initiation of splicing (Fig. 3, lanes 4 and 6). In contrast, pre-incubation of ASF/SF2 in S100 extract increases the activation capacity of the unphosphorylated ASF/SF2 (Fig. 3, lanes 6 and 7), and abolishes the difference between SRPK1 phosphorylated and unphosphorylated ASF/SF2. Thus, the order of addition is important to detect differences in biological activity. The increased activation potential of the unphosphorylated ASF/SF2 pre-incubated in S100 extract probably results from phosphorylation of the protein during the pre-incubation step (3). Conversely, the decrease in the activation capacity of phosphorylated ASF/SF2 potentially results from a partial dephosphorylation of ASF/SF2 and/or other modification of the protein that impairs its function. Collectively these results show that the SRPK1 phosphorylated ASF/SF2 is functional in its role in splicing.
DISCUSSION
In this study we demonstrate efficient production of a soluble, serine phosphorylated protein directly in E.coli. The method relies on the introduction and expression of a protein kinase (SRPK1) and its substrate (ASF/SF2) from two separate plasmid DNAs in the same E.coli cell. Our results show that SRPK1 is capable of phosphorylating ASF/SF2 in E.coli to a degree resembling the native protein present in interphase HeLa cells. Furthermore, SRPK1 phosphorylation of ASF/SF2 in bacteria restores mAb 104 reactivity to ASF/SF2, indicating that the same serine residues are phosphorylated in the RS-region of the recombinant and endogenous proteins. The phosphorylated form of ASF/SF2, in contrast to its unphosphorylated counterpart, is soluble in E.coli and it is thus possible to purify and study the protein under native conditions. We also show that SRPK1 phosphorylated ASF/SF2 restores splicing activity to cytoplasmic S100 extracts.
Several lines of evidence suggest that SRPK1 phosphorylates ASF/SF2 within the bacteria and not after lysis of the cells. First, the purification steps are performed at low temperatures (0–4°C), where SRPK1 activity is either low, or absent (data not shown). Secondly, unphosphorylated ASF/SF2 is insoluble in E.coli and it appears unlikely that ASF/SF2 present in inclusion bodies would become accessible for SRPK1 phosphorylation. Importantly, we have noticed that when SRPK1 and ASF/SF2 expression was induced at lower temperatures (30 instead of 37°C), fewer phosphates were added to ASF/SF2 as judged by SDS–PAGE mobility (data not shown).
Previous in vitro studies of ASF/SF2 (and other SR-proteins) have been complicated by the fact that the unphosphorylated form of the protein is insoluble under native conditions (8,9). With the above presented method it is now possible to obtain large amounts of soluble ASF/SF2 from E.coli that can be used in a number of biological assays. Also, by coexpressing ASF/SF2 with other SR-protein kinases (7 and references therein) it may be possible to establish the relevance of each kinase for the multiple activities associated with ASF/SF2.
We propose that the approach used here may perhaps be used for coexpression of other mammalian protein modifying enzymes, such as protein methylases and acetylaces and their substrates. Coexpression of such enzymes and their substrates could result in production of recombinant proteins that better resemble native proteins present in eukaryotic cells.
Acknowledgments
ACKNOWLEDGEMENTS
We want to thank Professor James Stévenin for generously providing us with polyclonal sera against ASF/SF2. We are also grateful to Dr C. Calvio for fruitful discussions at early stages of the project. J-P.K. and G.A. were supported by the Swedish Cancer Society and A.I.L and P.A were supported by the Wellcome Trust. A.I.L. is a Wellcome Trust Principle Research Fellow.
REFERENCES
- 1.Georgiou G. (1996) In Cleland,J.L. and Craik,C.S. (eds), Protein Engineering. Wiley-Liss Inc., New York, pp. 101–127.
- 2.Zahler A.M., Lane,W.S., Stolk,J.A. and Roth,M.B. (1992) Genes Dev., 6, 837–847. [DOI] [PubMed] [Google Scholar]
- 3.Xiao S.H. and Manley,J.L. (1997) Genes Dev., 11, 334–344. [DOI] [PubMed] [Google Scholar]
- 4.Kuroyanagi N., Onogi,H., Wakabayashi,T. and Hagiwara,M. (1998) Biochem. Biophys. Res. Commun., 242, 357–364. [DOI] [PubMed] [Google Scholar]
- 5.Duncan P.I., Stojdl,D.F., Marius,R.M., Scheit,K.H. and Bell,J.C. (1998) Exp. Cell. Res., 241, 300–308. [DOI] [PubMed] [Google Scholar]
- 6.Cao W., Jamison,S.F. and Garcia-Blanco,M.A. (1997) RNA, 3, 1456–1467. [PMC free article] [PubMed] [Google Scholar]
- 7.Okamoto Y., Onogi,H., Honda,R., Yasuda,H., Wakabayashi,T., Nimura,Y. and Hagiwara,M. (1998) Biochem. Biophys. Res. Commun., 249, 872–878. [DOI] [PubMed] [Google Scholar]
- 8.Ge H., Zuo,P. and Manley,J.L. (1991) Cell, 66, 373–382. [DOI] [PubMed] [Google Scholar]
- 9.Krainer A.R., Mayeda,A., Kozak,D. and Binns,G. (1991) Cell, 66, 383–394. [DOI] [PubMed] [Google Scholar]
- 10.Himmelspach M., Cavaloc,Y., Chebli,K., Stevenin,J. and Gattoni,R. (1995) RNA, 1, 794–806. [PMC free article] [PubMed] [Google Scholar]
- 11.Kreivi J.P., Zerivitz,K. and Akusjarvi,G. (1991) Nucleic Acids Res., 19, 2379–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krainer A.R., Conway,G.C. and Kozak,D. (1990) Cell, 62, 35–42. [DOI] [PubMed] [Google Scholar]
- 13.Kanopka A., Muhlemann,O. and Akusjarvi,G. (1996) Nature, 381, 535–538. [DOI] [PubMed] [Google Scholar]
- 14.Colwill K., Feng,L.L., Yeakley,J.M., Gish,G.D., Caceres,J.F., Pawson,T. and Fu,X.D. (1996) J. Biol. Chem., 271, 24569–24575. [DOI] [PubMed] [Google Scholar]
- 15.Mermoud J.E., Cohen,P.T. and Lamond,A.I. (1994) EMBO J., 13, 5679–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krainer A.R., Conway,G.C. and Kozak,D. (1990) Genes Dev., 4, 1158–1171. [DOI] [PubMed] [Google Scholar]