


Abstract
We carried out a screen for mutants that arrest prior to premeiotic S phase. One of the strains we isolated contains a temperature-sensitive allele mutation in the fission yeast prp31+ gene. The prp31-E1 mutant is defective in vegetative cell growth and in meiotic progression. It is synthetically lethal with prp6 and displays a pre-mRNA splicing defect at the restrictive temperature. We cloned the wild-type gene by complementation of the temperature-sensitive mutant phenotype. Prp31p is closely related to human and budding yeast PRP31 homologs and is likely to function as a general splicing factor in both vegetative growth and sexual differentiation.
INTRODUCTION
The fission yeast Schizosaccharomyces pombe enters a sexual differentiation cycle in response to nutrient limitation (reviewed in 1,2). Cells of the opposite mating type conjugate with one another to form a transient diploid, which then proceeds directly to meiosis and sporulation. Meiotic progression in the fission yeast is regulated by a number of specific factors, most importantly the Mei2 protein. This RNA-binding protein is essential for the initiation of meiotic S phase and the subsequent meiotic divisions (3). Its activity is mediated by association with a small RNA molecule called meiRNA, encoded by the sme2+ gene (3). Mei2p is inhibited by the Pat1p protein kinase (4). During nitrogen starvation, expression of mei2+ is strongly induced (5). If the cells are diploid (determined by the presence of both mating pheromones), the mei3+ gene is also induced (6). Mei3p then inhibits Pat1p repression of Mei2p and the cells proceed into meiosis (2,7). However, if Pat1p is inactivated by a thermosensitive mutation in vegetative cells, cells enter meiosis inappropriately due to activation of Mei2p (7,8). This ectopic meiosis is independent of ploidy or nutrient conditions. Strikingly, the cells complete two abnormal meiotic divisions and form spores even from the haploid state, although the spores are inviable. pat1-induced meiosis has been used extensively to model the events of normal sexual development (9–12).
Meiotic progression also requires many vegetative cell cycle genes, including the p34cdc2 kinase (9,13). This is not unexpected, since many of the events of meiosis are similar to those in the mitotic cycle. Surprisingly, however, we have found that meiotic DNA replication occurs in the absence of several vegetative initiation proteins, including the MCM proteins and Cdc18p (14). We therefore carried out a screen for mutants defective in initiation of meiotic DNA replication. Our screen also isolated mutants required early in meiosis. In this paper we describe isolation of one such mutant and cloning of the cognate gene.
The fission yeast prp31-E1 mutant is temperature-sensitive for growth in vegetative cells, arresting with a cdc-like phenotype. A number of other prp (pre-mRNA processing) mutants identified in fission yeast also exhibit cell cycle defects (15–17). prp31+ encodes the fission yeast homolog of Saccharomyces cerevisiae PRP31, which is involved in assembly of the spliceosome by recruiting the U4/U6×U5 tri-snRNP to prespliceosome complexes (18). We show that Prp31p is essential for normal mRNA splicing. Although this mutant was isolated in a screen for meiotic factors, our data suggest that prp31+ is a general splicing factor required throughout the fission yeast life cycle.
MATERIALS AND METHODS
Strains and media
The S.pombe strains used in this study are listed in Table 1. Yeast were grown in YES medium or Edinburgh minimal medium (EMM) supplemented with adenine, leucine and uracil as required (19). Strains were constructed using standard genetic techniques (19).
Table 1. Strains used in this study.
| Strain | Genotype | Source |
|---|---|---|
| FY254 | h– can1-1 leu1-32 ade6-M210 ura4-D18 | (36) |
| FY255 | h+ can1-1 leu1-32 ade6-M210 ura4-D18 | (36) |
| FY261 | h+ can1-1 leu1-32 ade6-M216 ura4-D18 | (36) |
| FY527 | h– his3-D1 leu1-32 ade6-M216 ura4-D18 | (26) |
| FY528 | h+ his3-D1 leu1-32 ade6-M210 ura4-D18 | (26) |
| FY903 | h+ pat1-114 Δcdc19::[cdc19HA leu1+] ura4-D18 leu1-32 ade6-M210 | (14) |
| FY1138 | h– prp31-E1 ura4-D18 leu1-32 ade6-M210 | This study |
| FY1150 | h+ cdc5-120 | KGY162 |
| FY1162 | h+ pat1-114 cdc5-120 ura4-D18 ade6-M210 | This study |
| FY1182 | h+ prp31-E1 ura4-D18 leu1-32 ade6-M216 | This study |
| FY1183 | h– prp31-E1::[prp31+ura4+] leu1-32 ade6-M210 | This study |
| FY1184 | h– ura4-D18 leu1-32 ade6-M210 his3-D1 prp31+ | |
| h+ ura4-D18 leu1-32 ade6-M216 his3-D1 Δprp31::his3+ | This study | |
| FY1185 | h+ pat1-114 prp31-E1 ura4-D18 ade6-M210 | This study |
| KGY246 | h– leu1-32 ura4-D18 ade6-M210 | P. Nurse |
| KGY2457 | h– prp2-1 leu1-32 | (33) |
| KGY2457 | h– prp6-1 leu1-32 | (33) |
Mutagenesis and screen
The pat1-114 strain FY903 was plated on YES at a density of ~2000 cells/plate and mutagenized with 100 J/m2 UV in a Stratalinker to obtain a 50% survival rate. Plates were incubated in the dark at 25°C for 5 days to allow survivors to form colonies and replica-plated onto EMM plates. Plates were incubated overnight at 34°C to inactivate pat1-114 and induce a haploid meiosis. Cells were exposed to iodine vapor to determine sporulation efficiency, revealed by dark staining. We identified 114 mutant strains with reduced iodine staining out of ~20 000 colonies sampled. Cell morphology of the mutants was observed by microscopy and 46 strains which contained no visible spores were identified. These strains were tested for temperature sensitivity on YES at 34°C and reconfirmed for reduced iodine staining. A meiotic time course was performed (described below) and samples were collected for FACS analysis to analyze DNA content. Cells fixed for flow cytometry and rehydrated in 50 mM sodium citrate were stained with DAPI to assess nuclear divisions. Single gene mutations were isolated by backcrossing to the parent strain and performing a random spore analysis.
Physiological analysis (time courses)
For vegetative time courses, cultures were grown in EMM supplemented with uracil, leucine and adenine overnight at 25°C to mid-exponential phase (OD600 ~0.4). The cultures were split and incubated at 25 and 36°C and sampled every 2 h for FACS and DAPI staining. Samples were harvested by centrifugation and fixed for flow cytometry as described (20), except that we stained the cells with 1 µM Sytox Green (Molecular Probes). Data were collected on a Becton Dickinson FACScan and analyzed using Cell Quest software for the Macintosh.
For meiotic time courses, haploid strains containing the pat1-114 allele were grown to mid-log phase in EMM with uracil, leucine and adenine, then starved overnight in EMM lacking nitrogen to arrest in G1. Cultures were re-fed with an equal volume of preheated EMM-N supplemented with 1 g/l NH4Cl and 70 µg/ml required supplements at 36°C to release into meiosis. For meiotic time courses in diploids, appropriate strains were starved overnight in EMM without nitrogen and glucose to arrest in G2. We added glycerol to 1% and glucose to 0.1% to induce meiosis.
Cloning of prp31+
Yeast strain FY1138 containing the prp31-E1 mutation was transformed with a pUR19 genomic library (21). Cells were plated onto EMM supplemented with uracil, adenine and leucine at 25°C for 48 h to allow phenotypic expression, then shifted to 36°C to select for complementation. Plasmids were isolated from the ts+ candidates, transformed by electroporation into Escherichia coli and prepared for restriction analysis (22). All clones isolated contained overlapping restriction fragments representing two different plasmids. Approximately 700 nt at either end of the smallest plasmid (pDB8) were sequenced using plasmid primers (Salk Institute Sequencing Facility), which revealed two open reading frames (ORFs) (Fig. 3A). We subcloned each ORF. pDB2, containing ORF1 (the prp31+ gene), was generated by cleaving out a 2.2 kb SphI fragment from pDB8. pDB3, containing ORF2, was constructed by cleaving out a 2.3 kb SpeI–BamHI fragment. Each subclone was transformed into FY1138 to determine which ORF was responsible for complementing activity.
Figure 3.
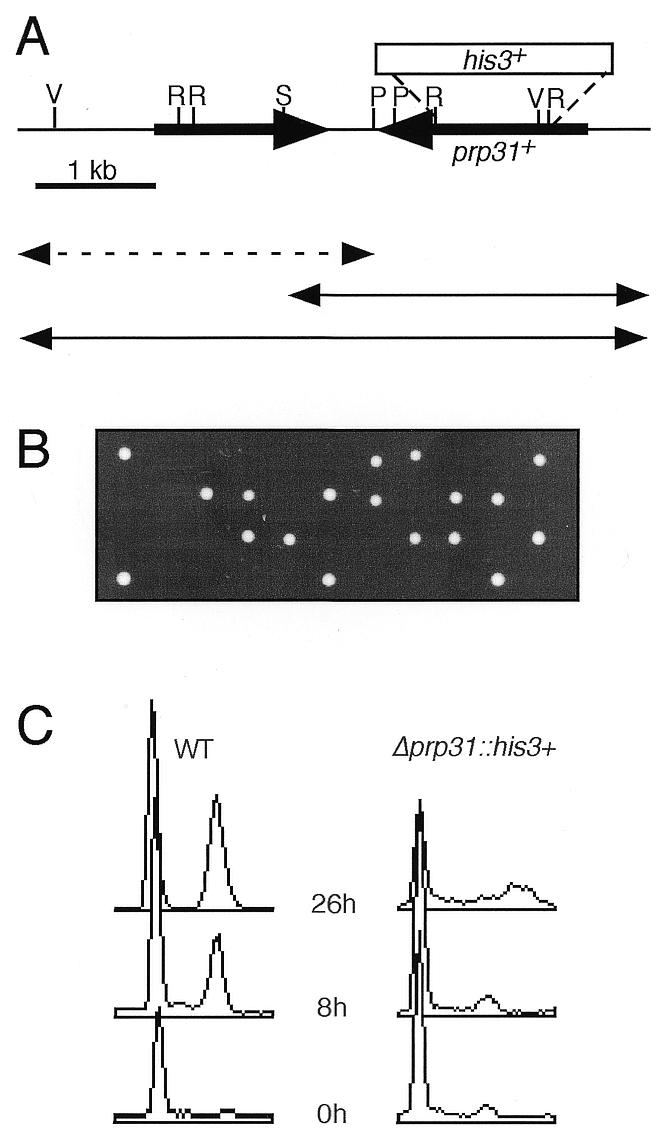
Analysis of the prp31+ locus and disruption phenotype. (A) Schematic of the genomic clone containing the prp31+ gene. Double-headed arrows represent subclones that were transformed into the prp31-E1 mutant strain FY1138 to test for complementation activity scored by the ability to form colonies at 36°C. Continuous lines, subclones that were able to complement at 36°C; broken line, subclone that failed to complement at 36°C. The box above the line indicates the location of the disruption/deletion with the his3+ marker. V, EcoRV; R, EcoRI; S, SphI; P, SpeI. (B) Tetrads dissected from the diploid prp31::his3+/prp31+ were grown on YES at 32°C for 2 days. All growing spore clones were His– (data not shown). (C) Flow cytometry of germinating wild-type (WT) and Δprp31::his3+. Samples were taken at 0, 8 and 26 h from cells growing in selective medium.
A 2.4 kb SspI–SacI fragment from pDB2 containing the prp31 gene was subcloned into the 2.9 kb EcoRV–SacI fragment of pBluescript II SK(+) to generate pDB4. A 2.4 SacI–KpnI fragment from this construct was subcloned into pJK210 (23) to generate prp31-E1::[prp31+ura4+]. This new construct (pDB7) was linearized with PstI and transformed into FY1138 to create FY1183. Integration at the prp31-E1 locus was verified by crossing against a wild-type strain and verifying that no temperature-sensitive segregants were recovered.
prp31 disruption
The Δprp31::his3+ disruption construct was generated by digesting pDB4 with EcoRI to release a 0.9 kb fragment within the ORF of prp31. A BglII linker was added following an end filling reaction and the his3+ cassette from pAF1 (24) was inserted, creating pDB6. The disruption cassette was excised with KpnI and SacI and transformed into a diploid strain (FY527/FY528) to create FY1184. Transformants were then sporulated and analyzed by tetrad dissection and random spore analysis.
Spore germination
We analyzed strain FY1184 (Δprp31-E1::his3+/prp31+ his3-D1/his3-D1) and a diploid heterozygous at the his3 locus (his3+/his3-D1) by spore germination (25,26). Spores were washed and partially purified by centrifugation through a 25% glycerol cushion. For the spore germination time course, spores were inoculated into EMM plus adenine, leucine and uracil to a final concentration of 1 × 108 spores/ml. Cultures were grown, with shaking, at 32°C. Samples were collected at 0, 8 and 26 h for FACS analysis and nuclear counts as previously described.
Splicing assays
Total RNA was isolated as described (19). Aliquots of 15 µg of total RNA were separated using formaldehyde–agarose gel electrophoresis (27), transferred to Duralon-UV™ membranes (Stratagene, La Jolla, CA) by capillary action and UV crosslinked by exposure to a Stratalinker™ UV source (Stratagene). Oligonucleotides were 32P-labeled by the StarFire method (Integrated DNA Technologies) according to the manufacturer’s directions and used to probe for precursor (TF2D intron, 5′-CTTGTTCCCTATCCAAAGAGGCACGACxxxxxx-3′) or mature (TF2D exon, 5′-GGAGTCATCCTCGGATTTGCCACCCxxxxxx-3′) tf2d+ RNA. The filter was prehybridized in 10 ml of Perfecthyb™ hybridization buffer (Sigma, St Louis, MO) and hybridized overnight at 60°C in 10 ml of Perfecthyb™ hybridization buffer with 107 c.p.m. of StarFire labeled probe (106 c.p.m./ml). The filter was washed at 25°C for 10 min twice in 2× SSC (0.3 M NaCl, 0.03 M Na citrate), 0.5% SDS and twice in 0.5× SSC, 0.5% SDS. To detect his3+ RNAs, a 484 bp fragment was excised from pKG594 (28) using the restriction endonucleases EcoRV and BamHI. This DNA fragment was labeled with 32P using the Prime It II kit (Stratagene) according to the manufacturer’s instructions. The filter was prehybridized in 10 ml of ExpressHyb™ hybridization buffer (Clontech) and hybridized for 1 h at 68°C in 10 ml of ExpressHyb™ hybridization buffer with 107 c.p.m. of probe (106 c.p.m./ml). Filters were washed twice at 50°C for 10 min in 2× SSC, 0.5% SDS. After exposure, the filters were visualized on a PhosphoImager™ with ImageQuant™ v.3.3 software (Molecular Dynamics, Sunnyvale, CA).
RESULTS
Isolation of mutants defective in meiotic S phase
Recently, we found that several vegetative DNA replication initiation proteins are not required for meiotic progression (14). Therefore, we designed a screen for mutants that block premeiotic S phase to identify any meiosis-specific factors. Wild-type cells that complete meiosis and sporulation are stained dark brown by iodine vapor. Colonies that do not complete meiosis or sporulation have reduced iodine staining. The Pat1p kinase represses meiotic progression; the temperature-sensitive pat1-114 mutation allows even haploid cells to enter meiosis at 34°C, where they complete sporulation and stain dark with iodine (8). Therefore, we employed a pat1-114 mutant to induce meiosis in response to a shift in temperature. We mutagenized pat1 mutant cells by UV treatment (see Materials and Methods) and isolated colonies that showed reduced iodine staining at the restrictive temperature. These iodine-minus mutants were further screened for those that maintained temperature sensitivity, to ensure that the pat1 lesion was not suppressed, and by FACS analysis, to determine whether they entered meiotic S phase. We isolated four mutants that were iodine minus, still temperature sensitive and blocked meiotic progression prior to bulk DNA synthesis. This paper will discuss the first of the four mutants, which we designate prp31-E1 for reasons that will be explained below.
We characterized the meiotic defect of prp31-E1 by carrying out a meiotic time course. We arrested pat1-114 prp31-E1 cells in the G1 phase of the cell cycle by nitrogen starvation, then released them directly to meiosis by re-feeding and shifting to the restrictive temperature. This method is specific for meiotic phenotypes and is unaffected by any vegetative phenotype of the gene under study. We harvested and fixed samples at each time point. For each time point we determined DNA content by FACS analysis and monitored progression through both meiotic divisions by counting the number of nuclei per cell. The parental pat1-114 strain usually completes DNA replication after 4 h at the restrictive temperature; it completes the second meiotic division with 60% efficiency at the end of 8 h, as determined by the fraction of cells with three or more nuclei (Fig. 1A–C). The pat1-114 prp31-E1 mutant did not undergo normal meiotic DNA replication and arrested with a single nucleus, indicating a block prior to meiotic S phase (Fig. 1A–C).
Figure 1.
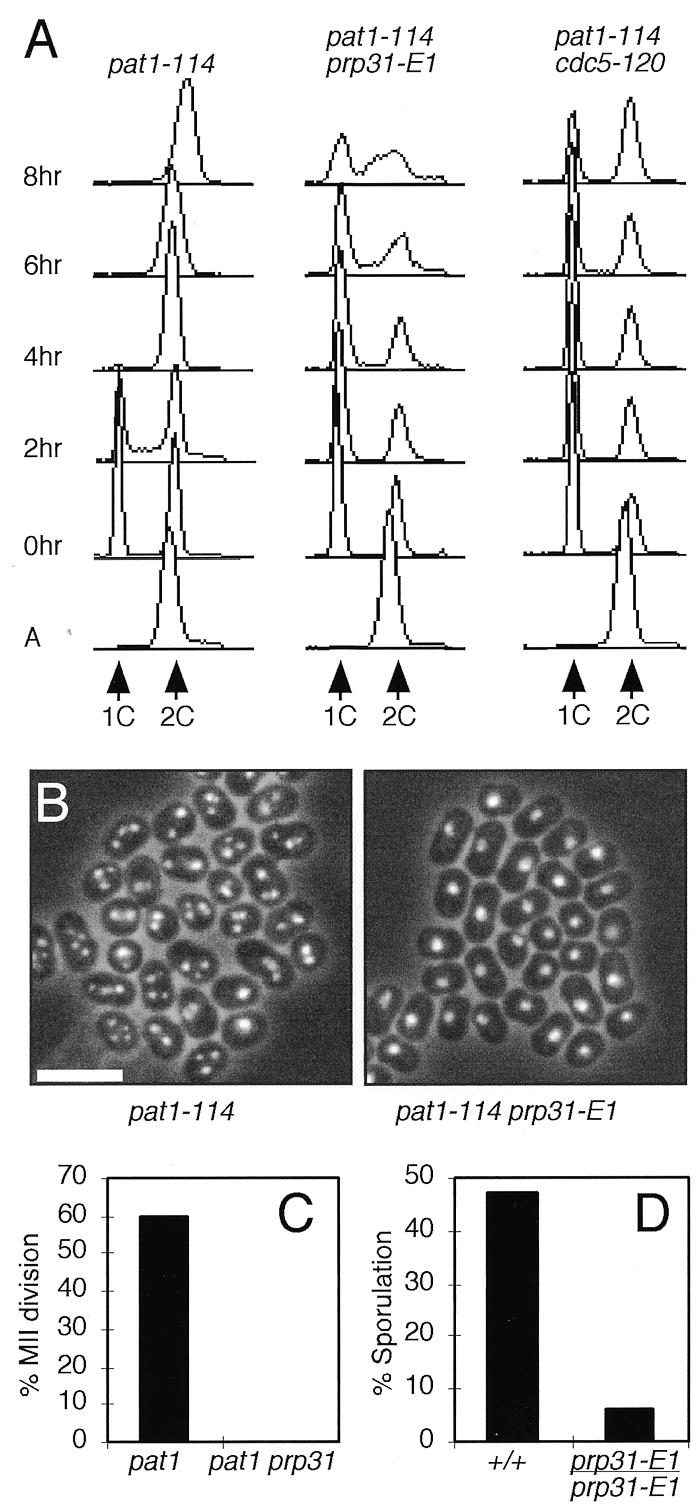
prp31-E1 is defective in meiotic S phase. (A) pat1-114, pat1-114 prp31-E1 and pat1-114 cdc5-120 strains were arrested in G1 and released to meiosis as described in Materials and Methods. Samples were harvested every 2 h for flow cytometry. Positions of 1C (unreplicated) and 2C (replicated) DNA content are indicated. A, asynchronous. (B) Nuclear morphology of cells in (A) after 8 h. Cells were stained with DAPI to visualize nuclei. Cells with two nuclei have completed the first meiotic division and cells with more than three nuclei have completed both meiotic divisions. Bar, 10 µm. (C) The percentage of cells in (B) that completed the second meiotic (MII) division after 8 h was determined by counting cells with more than three nuclei. (D) Percentage of diploid cells completing sporulation was determined by counting asci under phase contrast microscopy, following release of diploid strains to meiosis as described in Materials and Methods.
To verify that these results reflected a role for prp31 in meiosis and were not an artifact of inducing meiosis using the pat1 mutant, we constructed a homozygous prp31 mutant diploid using complementation of the ade6-M210 and ade6-M216 markers (19). The cells were arrested in G2 of the vegetative cell cycle by starvation for glucose and nitrogen, then released to meiosis at 34°C by addition of glucose and glycerol (29). Again, this protocol separates vegetative effects from meiotic requirements by inducing meiosis at the same time as the prp31 mutant protein is inactivated. Completion of meiosis was monitored by counting the percentage of asci formed after a day long incubation at restrictive versus permissive temperatures (Fig. 1D). Whereas the wild-type diploid was able to complete meiosis efficiently at both temperatures (ratio ≥1), the mutant diploid was severely defective at the restrictive temperature. We conclude that prp31 is required for normal meiotic progression.
prp31-E1 has a vegetative phenotype and encodes a putative splicing factor
To analyze the phenotype of prp31-E1 in vegetative cells, we crossed the pat1 prp31-E1 strain to wild-type and dissected tetrads to segregate it from the pat1 mutation. We found that prp31-E1 in a wild-type background was temperature sensitive for growth at 36°C (Fig. 2A). FACS analysis showed that prp31-E1 arrests with a 2C DNA content and somewhat heterogeneously elongated cells (Fig. 2B and C). This indicates that prp31 is essential for growth in vegetative cells and suggests that it may have a cell cycle defect. There was no evidence for a specific defect in S phase in these asynchronous cultures.
Figure 2.
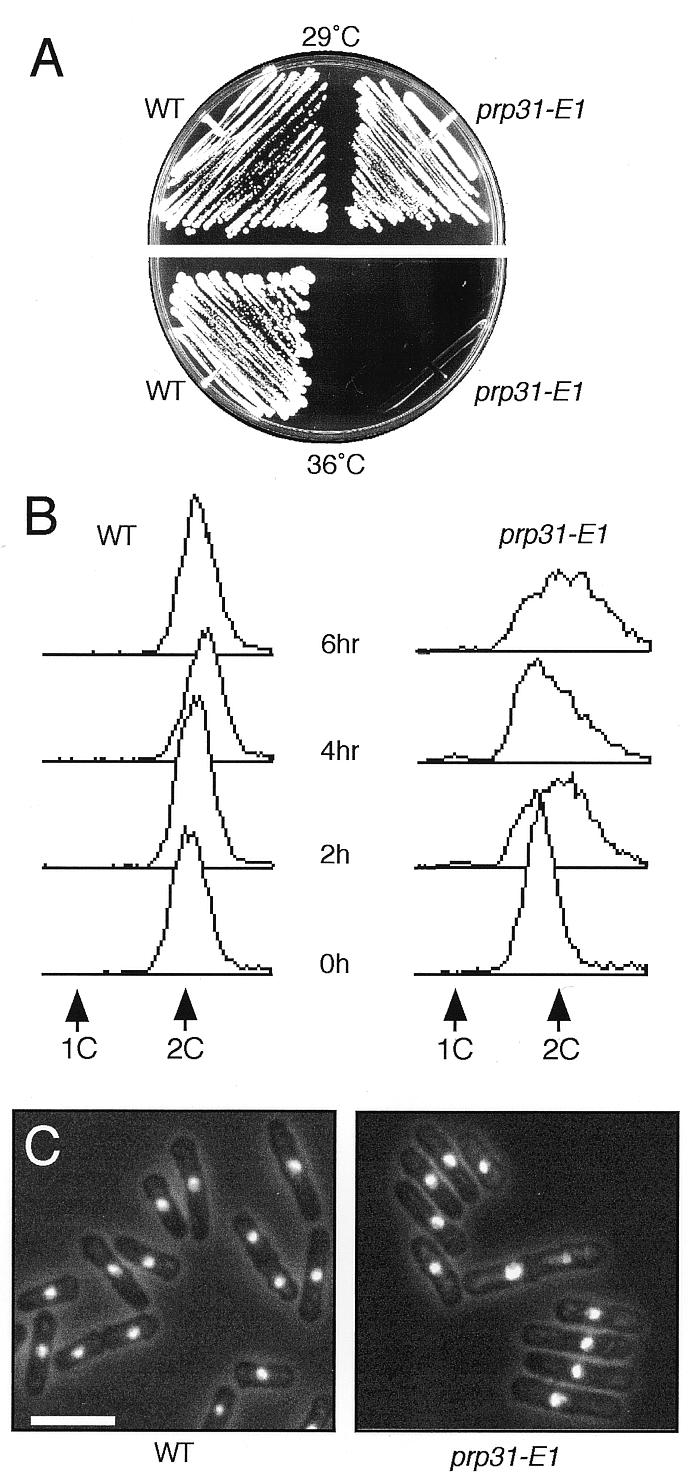
prp31-E1 has a vegetative phenotype. (A) Wild-type (FY261) and prp31-E1 strains were streaked onto YES plates and incubated for 3 days at 29 and 36°C. (B) Wild-type and prp31-E1 strains were grown overnight at the permissive temperature of 25°C and shifted to 36°C as described in Materials and Methods. Samples were harvested every 2 h for flow cytometry. Positions of 1C (unreplicated) and 2C (replicated) DNA content are indicated. (C) Nuclear morphology of cells in (B) after 6 h. Cells were stained with DAPI to visualize the nuclei. Bar, 10 µm.
We cloned the prp31+ gene by complementation of the temperature-sensitive phenotype. We transformed the mutant prp31-E1 strain with a genomic library and isolated clones that supported growth at 36°C (see Materials and Methods). Two independent plasmids with overlapping fragments were isolated. The smallest fragment was partially sequenced and compared to the fission yeast genome database BLAST server (http://www.sanger.ac.uk/Projects/S_pombe/ ). This revealed two ORFs on the insert (Fig. 3A). These were separately subcloned and each subclone was transformed back into the original prp31-E1 mutant and tested for complementation at 36°C. Only the subclone containing the prp31+ ORF complemented the mutant.
We verified that the sequence cloned corresponded to the mutant locus by targeting a plasmid containing prp31+ and ura4+ for integration at prp31-E1. The resulting strain prp31-E1::[prp31+ura4+] was no longer temperature sensitive. When crossed against the wild-type, no temperature-sensitive segregants were observed. When crossed against prp31-E1, 50% of the segregants were wild-type for growth and all these were ura4+. This indicates that the cloned sequence is tightly linked to the original mutant allele.
The sequence of this ORF was used for a GenBank BLAST search (http://www.ncbi.nlm.nih.gov/Genbank/ ) and found to be homologous to a number of sequences, including proteins from humans and Arabidopsis and the S.cerevisiae gene PRP31, encoding a pre-mRNA splicing factor (Fig. 4). PRP31 is required for assembly of the spliceosome, specifically in recruitment of the U4/U6×U5 tri-snRNP to prespliceosome complexes (18). Interestingly, fission yeast Prp31p showed significant similarity (34%) across the entire sequence to both the human and Arabidopsis genes, while sharing a smaller core of homology (28%) with the budding yeast homolog. Based on this comparison, we named our fission yeast gene prp31+.
Figure 4.

Alignment of prp31+-related protein sequences. Sequences were aligned using MEGALINE by DNASTAR. Identical residues are shown in white text with a black background. Sp, S.pombe (accession no. CAA17928); Hs, human (accession no. CAB43677); At, Arabidopsis thaliana (accession no. 3249066); Pf, Plasmodium falciparum (accession no. CAB62868); Sc, S.cerevisiae (accession no. S64386).
A number of prp mutants with potential cell cycle defects have been previously characterized (16,30). These mutants affect mRNA processing and often express cdc-like phenotypes. We tested for genetic interactions between prp31-E1 and alleles of previously identified prp genes, prp1-prp7 and prp11, as well as cdc5 (16,24). prp31-E1 showed a significant genetic interaction with only one mutant that was tested, prp6-1. In this case, the two mutations were synthetically lethal. Of the 12 tetrads examined, two were parental ditypes, one was a non-parental ditype and nine were tetratypes. Although the identity of prp6+ has not yet been determined, these data indicate that Prp6p and Prp31p may be involved in the same biochemical step of pre-mRNA splicing. prp6+ may also be involved in spliceosome assembly or may interact physically with prp31+. Importantly, these data also indicate that prp31+ is not allelic to other prp mutants already defined in fission yeast.
prp31+ is essential
We determined the phenotype of cells lacking prp31+ by constructing a deletion/disruption. We replaced the majority of the prp31+ ORF with a fragment containing his3+ (Fig. 3A; Materials and Methods). A linear fragment containing this construct was integrated into a diploid strain and His+ transformants were isolated. Upon sporulation, we found that no viable His+ haploids were recovered. Upon tetrad dissection, we isolated eight complete tetrads which showed 2:2 segregation of viable:inviable spores (Fig. 3B); all viable spores were His–. We transformed the parent diploid with a plasmid containing the cloned prp31+ and tested whether the plasmid could rescue the lethal phenotype. Random spore analysis of this strain showed that His+ segregants were recovered if they also contained the prp31+ura4+ plasmid. Microscopic analysis revealed that spores which did not form colonies underwent one division.
We analyzed the phenotype of the Δprp31 spores using a spore germination procedure. We prepared spores from the Δprp31/prp31+ diploid and inoculated them into medium lacking histidine. Under these conditions, only the spores containing the gene disruption are able to germinate. We sampled this preparation for DNA content, by FACS, and cell morphology (Fig. 3C). The Δprp31 cells have a mixed phenotype, with a large number of elongated cells. A fraction of the cells produce a 2C DNA content, although this appears to be far less than half.
Prp31p is involved in pre-mRNA splicing
To investigate the role of prp31+ in pre-mRNA splicing, we determined whether prp31-E1 mutant cells accumulate unspliced messages. We prepared total RNA from prp31-E1 grown at the permissive temperature or at the restrictive temperature for 2 and 4 h. Total RNA prepared from the known pre-mRNA splicing mutant prp2-1 and wild-type cells served as controls. We probed northern blots for the intron-containing transcripts tf2d+ and his3+. In both cases, we observed an accumulation of unspliced precursor RNA and a reduction in mature transcript in the prp31-E1 mutant cells at the restrictive temperature (Fig. 5). These data indicate that S.pombe prp31+ function is required for pre-mRNA splicing in vivo.
Figure 5.
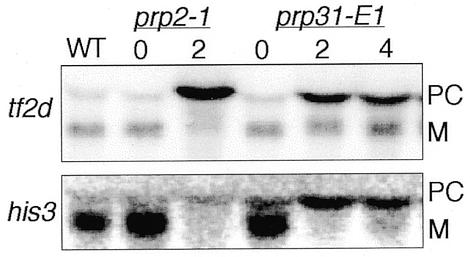
prp31+ is required for pre-mRNA splicing in vivo. RNA was prepared from wild-type (WT), prp2-1 cells at either the permissive temperature of 25°C (0) or after 2 h at the restrictive temperature of 36°C (2) and prp31-E1 cells at the permissive temperature of 25°C (0) or after 2 (2) and 4 (4) h at the restrictive temperature of 36°C. Northern blots were probed for the intron-containing RNAs, tf2d+ and his3+. Precursor RNAs (PC) and the mature messages (M) are indicated.
Other splicing factors are also required for meiosis
The phenotype of prp31-E1 suggests that there may be a general requirement for mRNA splicing factors in meiotic progression as well as in the vegetative cell cycle. We tested this by analyzing the requirement for cdc5+, which encodes a Myb-related splicing factor (24,31,32). cdc5ts mutants also show a strong cell cycle phenotype (24). We constructed a pat1-114 cdc5-120 double mutant strain and carried out the same analysis done previously, using FACS analysis to monitor meiotic S phase and visual inspection to determine meiotic divisions. Similarly to prp31-E1 in the pat1-114 background, the cdc5-120 mutation blocked meiotic DNA replication and meiotic divisions at a high temperature (Fig. 1A), suggesting that there is a general requirement for mRNA splicing in meiosis.
DISCUSSION
In this report, we describe a new mutant isolated in a screen for S.pombe mutants that block an early stage of meiotic progression. We cloned the corresponding gene prp31+, which encodes a protein homologous to proteins in S.cerevisiae (PRP31), human, Arabidopsis and Plasmodium. Budding yeast PRP31 has been shown to be involved in assembly of the spliceosome by recruiting the U4/U6×U5 tri-snRNP to prespliceosome complexes (18). Schizosaccharomyces pombe prp31-E1 cells are defective in normal vegetative growth and prp31-E1 is synthetically lethal in combination with prp6, another presumed splicing factor. The prp31-E1 mutant shows a splicing defect in vegetative fission yeast cells comparable to that of prp2-1 (33). Both prp2 and prp6 were isolated from a screen for temperature-sensitive mutants exhibiting pre-mRNA splicing defects (16,33).
prp31+ is essential for viability of vegetative cells and Δprp31::his3+ results in a cdc-like arrest with limited accumulation of DNA. In meiosis, prp31 blocks cells prior to completion of S phase. Similar effects on meiotic progression were observed for another splicing mutant, cdc5. We conclude that other general splicing factors are likely to be required early in meiosis. However, prp31-E1 does not show an S phase defect in vegetative cells when asynchronously growing cells are shifted to the restricted temperature. Therefore we conclude that its arrest prior to meiotic S phase and the S phase defects of the disruption merely reflect the assay used. We anticipate that prp31+ probably affects multiple targets throughout the cell cycle in both vegetative and meiotic cells.
Interestingly, there is evidence that fission yeast undergoes meiosis-specific splicing of the mes1+ gene, required for the MII division (34). However, because the arrest point of prp31-E1 is significantly earlier in meiosis than that of mes1 and because prp31+ is required for vegetative growth as well, it is probable that Prp31p is required for splicing of other meiotic substrates. Whether the general splicing apparatus is also required for mes1+ splicing remains to be determined. Meiosis-specific splicing has also been reported for budding yeast and does require general splicing genes (35). The interplay of general splicing factors with meiosis-specific elements may provide a general method of regulating differentiation.
What are the likely targets of the splicing apparatus in meiosis? A number of vegetative genes are required for premeiotic DNA synthesis (9,14); in addition, there are meiosis-specific factors (reviewed in 1). Recognizable introns have been identified in several genes required for meiotic S phase, including res2+ (transcription factor; accession no. D1776), cdc17+ (DNA ligase; accession no. X05107), pol1+ (catalytic subunit of DNA polymerase α; accession no. X69673), cdc22+ (large subunit of ribonucleotide reductase; accession no. X65116), rpa1+ (large subunit of RPA; accession no. U75446) and pol3+ (catalytic subunit of DNA polymerase δ; accession no. X59278). Mutations in res2 and cdc22 in particular block cells prior to meiotic S phase and bulk DNA synthesis (9,13). These introns may explain the requirement for general splicing factors early in the meiotic pathway. It is likely that genes required later in meiosis will also require the activity of the cellular splicing machinery, which is thus ubiquitously required in the life cycle of fission yeast.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Anna Feoktistova for expert technical assistance and Debbie Liang for advice on cloning. We thank Hilary Snaith, Debbie Liang and Eliana Gomez for critical reading of the manuscript. This work funded by NIH grants GM54797 (S.L.F.) and GM47728 (K.L.G.). W.H.M. was supported by NCI grant T32 CA09592. K.L.G. is an Associate Investigator of the Howard Hughes Medical Institute. S.L.F. is a scholar of the Leukemia Society of America.
REFERENCES
- 1.Yamamoto M. (1996) Trends Biochem. Sci., 21, 18–22. [PubMed]
- 2.Yamamoto M., Imai,Y. and Watanabe,Y. (1997) In Pringle,J.R., Broach,J.R. and Jones,E.W. (eds), The Molecular and Cellular Biology of the Yeast Saccharomyces. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, Vol. 3, pp. 1037–1106.
- 3.Watanabe Y. and Yamamoto,M. (1994) Cell, 78, 487–498. [DOI] [PubMed]
- 4.Watanabe Y., Shinozaki-Yabana,S., Chikashige,Y., Hiraoka,Y. and Yamamoto,M. (1997) Nature, 386, 187–190. [DOI] [PubMed]
- 5.Watanabe Y., Iino,Y., Furuhata,K., Shimoda,C. and Yamamoto,M. (1988) EMBO J., 7, 761–767. [DOI] [PMC free article] [PubMed]
- 6.Willer M., Hoffmann,L., Styrkarsdottir,U., Egel,R., Davey,J. and Nielsen,O. (1995) Mol. Cell. Biol., 15, 4964–4970. [DOI] [PMC free article] [PubMed]
- 7.McLeod M. and Beach,D. (1988) Nature, 332, 509–514. [DOI] [PubMed]
- 8.Iino Y. and Yamamoto,M. (1985) Proc. Natl Acad. Sci. USA, 82, 2447–2451. [DOI] [PMC free article] [PubMed]
- 9.Iino Y., Hiramine,Y. and Yamamoto,M. (1995) Genetics, 140, 1235–1245. [DOI] [PMC free article] [PubMed]
- 10.Li Y.F. and Smith,G.R. (1997) Genetics, 146, 146–157.
- 11.Lin Y. and Smith,G.R. (1994) Genetics, 136, 769–779. [DOI] [PMC free article] [PubMed]
- 12.Bähler J., Schuchert,P., Grimm,C. and Kohli,J. (1991) Curr. Genet., 19, 445–451. [DOI] [PubMed]
- 13.Grallert B. and Sipiczki,M. (1991) Curr. Genet., 20, 199–204. [DOI] [PubMed]
- 14.Forsburg S.L. and Hodson,J. (2000) Nature Genet., in press.
- 15.Lundgren K., Allan,S., Urushiyama,S., Tani,T., Ohshima,Y., Frendewey,D. and Beach,D. (1996) Mol. Biol. Cell, 7, 1083–1094. [DOI] [PMC free article] [PubMed]
- 16.Potashkin J., Kim,D., Fons,M., Humphrey,T. and Frendewey,D. (1998) Curr. Genet., 34, 153–163. [DOI] [PubMed]
- 17.Burns C.G. and Gould,K.L. (1999) In Chew,S.L. (ed.), Frontiers in Hormone Research, Post-Transcriptional Processing and the Endocrine System. Karger, Basel, Switzerland, Vol. 25, pp. 59–82.
- 18.Weidenhammer E.M., Ruiz-Noriega,M. and Woolford,J.L.,Jr (1997) Mol. Cell. Biol., 17, 3580–3588. [DOI] [PMC free article] [PubMed]
- 19.Moreno S., Klar,A. and Nurse,P. (1991) Methods Enzymol., 194, 795–823. [DOI] [PubMed]
- 20.Sazer S. and Sherwood,S.W. (1990) J. Cell Sci., 97, 509–516. [DOI] [PubMed]
- 21.Barbet N., Muriel,W.J. and Carr,A.M. (1992) Gene, 114, 59–66. [DOI] [PubMed]
- 22.Becker D.M. and Guarente,L. (1991) Methods Enzymol., 194, 182–187. [DOI] [PubMed]
- 23.Keeney J.B. and Boeke,J.D. (1994) Genetics, 136, 849–856. [DOI] [PMC free article] [PubMed]
- 24.Ohi R., McCollum,D., Hirani,B., Denhaese,G.J., Zhang,X., Burke,J.D., Turner,K. and Gould,K.L. (1994) EMBO J., 13, 471–483. [DOI] [PMC free article] [PubMed]
- 25.Kelly T.J., Martin,G.S., Forsburg,S.L., Stephen,R.J., Russo,A. and Nurse,P. (1993) Cell, 74, 371–382. [DOI] [PubMed]
- 26.Liang D.T., Hodson,J.A. and Forsburg,S.L. (1999) J. Cell Sci., 112, 559–567. [DOI] [PubMed]
- 27.Sambrook J. and Gething,M.J. (1989) Nature, 342, 224–225. [DOI] [PubMed]
- 28.Burke J.D. and Gould,K.L. (1994) Mol. Gen. Genet., 242, 169–176. [DOI] [PubMed]
- 29.Horie S., Watanabe,Y., Tanaka,K., Nishiwaki,S., Fujioka,H., Abe,H., Yamamoto,M. and Shimoda,C. (1998) Mol. Cell. Biol., 18, 2118–2129. [DOI] [PMC free article] [PubMed]
- 30.Urushiyama S., Tani,T. and Ohshima,Y. (1996) Mol. Gen. Genet., 253, 118–127. [DOI] [PubMed]
- 31.Burns C.G., Ohi,R., Krainer,A.R. and Gould,K.L. (1999) Proc. Natl Acad. Sci. USA, 96, 13789–13794. [DOI] [PMC free article] [PubMed]
- 32.McDonald W.H., Ohi,R., Smelkova,N., Frendewey,D. and Gould,K.L. (1999) Mol. Cell. Biol., 19, 5352–5362. [DOI] [PMC free article] [PubMed]
- 33.Potashkin J., Li,R. and Frendewey,D. (1989) EMBO J., 8, 551–559. [DOI] [PMC free article] [PubMed]
- 34.Kishida M., Nagai,T., Nakaseko,Y. and Shimoda,C. (1994) Curr. Genet., 25, 497–503. [DOI] [PubMed]
- 35.Engebrecht J.A., Voelkel-Meiman,K. and Roeder,G.S. (1991) Cell, 66, 1257–1268. [DOI] [PubMed]
- 36.Forsburg S.L. and Nurse,P. (1994) J. Cell Sci., 107, 2779–2788. [DOI] [PubMed]