

ABSTRACT
Acute pancreatitis (AP) is an abdominal inflammatory disease initiated by damaged pancreatic acinar cells and developed by systemic inflammation. SQSTM1 (sequestosome 1) has an intracellular function in mediating substrate degradation during macroautophagy/autophagy, and it can be released by macrophages and monocytes to trigger lethal inflammation during bacterial infection. Here, we report that extracellular SQSTM1 acts as a mediator of AP by enhancing the sensitivity to autophagy-dependent ferroptotic cell death. Serum SQSTM1 is elevated in AP patients as well as in mice that have cerulein-induced AP. The administration of SQSTM1-neutralizing antibodies protects against experimental AP in mice. Mechanistically, recombinant SQSTM1 protein (rSQSTM1) increases AGER (advanced glycosylation end-product specific receptor)-dependent ACSL4 (acyl-CoA synthetase long chain family member 4) expression, leading to polyunsaturated fatty acid production for autophagosome formation and subsequent ferroptosis. The rSQSTM1-elicited pathological responses during AP are attenuated in mice with the conditional deletion of Ager in the pancreas. These findings may provide not only new insights into the mechanism of autophagy-dependent cell death, but also suggest that targeting the extracellular SQSTM1 pathway is a potential strategy for the treatment of AP.
Abbreviations
5-HETE, 5-hydroxyeicosatetraenoic acid; ACSL4, acyl-CoA synthetase long chain family member 4; AP, acute pancreatitis; ATG, autophagy related; AGER, advanced glycosylation end-product specific receptor; DAMPs, danger/damage-associated molecular patterns; FTH1, ferritin heavy chain 1; GPX4, glutathione peroxidase 4; IL, interleukin; INSR, insulin receptor; MAP1LC3B, microtubule associated protein 1 light chain 3 beta; MDA, malondialdehyde; MPO, myeloperoxidase; PRRs, pattern recognition receptors; PUFA, polyunsaturated fatty acid; RNAi, RNA interference; SQSTM1, sequestosome 1; TNF, tumor necrosis factor; TLR, toll like receptor
KEYWORDS: Autophagy, ferroptosis, inflammation, lipid peroxidation, pancreatitis, SQSTM1
Introduction
Acute pancreatitis (AP) is an acute response to injury of the pancreas that can be fatal or lead to serious complications [1]. The global incidence of AP has continued to increase over the last several decades, with gallstone migration and alcohol abuse being the two major risk factors for AP in humans [2]. Although the pathogenesis of sterile inflammation in AP remains largely unclear, increasing evidence suggests that danger/damage-associated molecular pattern (DAMP) molecules play a critical role in linking local trypsin-induced acinar cell damage to subsequent systemic inflammation and multiorgan failure [3]. DAMPs are endogenous molecules that can be released by dead or dying cells to induce or amplify inflammatory responses through various pattern recognition receptors (PRRs) expressed by immune and nonimmune cells [4,5]. Certain DAMPs (e.g., HMGB1 [high mobility group box 1]) and DAMP receptors (e.g., AGER [advanced glycosylation end-product specific receptor] and TLRs [toll like receptors]) have been identified as therapeutic targets for AP [6–8].
In mammalian cells, SQSTM1/p62 (sequestosome 1) acts as a receptor protein involved in signal transduction, oxidative stress, and macroautophagy (hereafter referred to as autophagy) [9]. As a representative autophagy receptor, SQSTM1 sorts ubiquitinated substrates (including SQSTM1 itself) for lysosome-mediated degradation [10,11]. In addition to these intracellular functions, we recently demonstrated that SQSTM1 can be released by innate immune cells to mediate lethal inflammation during bacterial infection [12]. Specifically, we demonstrated that INSR (insulin receptor) is a PRR responsible for the cytokine-like activity of extracellular SQSTM1 in macrophages and monocytes, providing a potential therapeutic target for sepsis and septic shock [12]. However, it remains unknown whether extracellular SQSTM1 plays a similar role in AP caused by sterile inflammation.
Ferroptosis is a type of regulated cell death that was first described in cancer cells harboring RAS mutations [13,14]. Unlike apoptosis, which relies on caspase activation, ferroptosis typically requires the activation of oxidative stress to trigger lipid peroxidation and subsequent plasma membrane rupture [15,16]. Ferroptosis is often associated with the overactivation of autophagy, and the inhibition of autophagy reduces ferroptotic responses, including aberrant immune activation [17–19]. Targeting ferroptotic pathways may be a strategy to prevent inflammation-associated diseases or pathological conditions [20], including pancreatitis [21–23].
In this study, we provide the first evidence that extracellular SQSTM1 is involved in the development of AP. We found that serum SQSTM1 is elevated in human and mouse AP and that blocking extracellular SQSTM1 activity protects against experimental AP in mice. We further revealed that AGER, but not INSR, is essential for extracellular SQSTM1 activity in acinar cells to drive polyunsaturated fatty acid (PUFA) synthesis, resulting in autophagy-dependent ferroptotic damage.
Results
Serum SQSTM1 is elevated in AP
Cerulein, a cholecystokinin analog, is the most used experimental stimulant to analyze early pathological events in AP because of its rapidity, ease of induction, and reproducibility [24]. To determine whether serum SQSTM1 is a biomarker to predict disease severity in AP, we assayed the serum level of SQSTM1 in cerulein-induced experimental AP in mice. As expected, hematoxylin and eosin (H&E) stains showed that cerulein time-dependently caused pancreatic damage with the typical histopathological features of AP (e.g., edema, vacuolization, inflammatory cell infiltration, and necrosis) (Figure 1A,B). Serum AMY (amylase) and PNLIP (pancreatic lipase) remain more sensitive and specific biochemical markers for the diagnosis of AP [25]. In addition to serum AMY and PNLIP, serum SQSTM1 concentrations were time-dependently increased in mice by the administration of cerulein (Figure 1C-1E).
Figure 1.
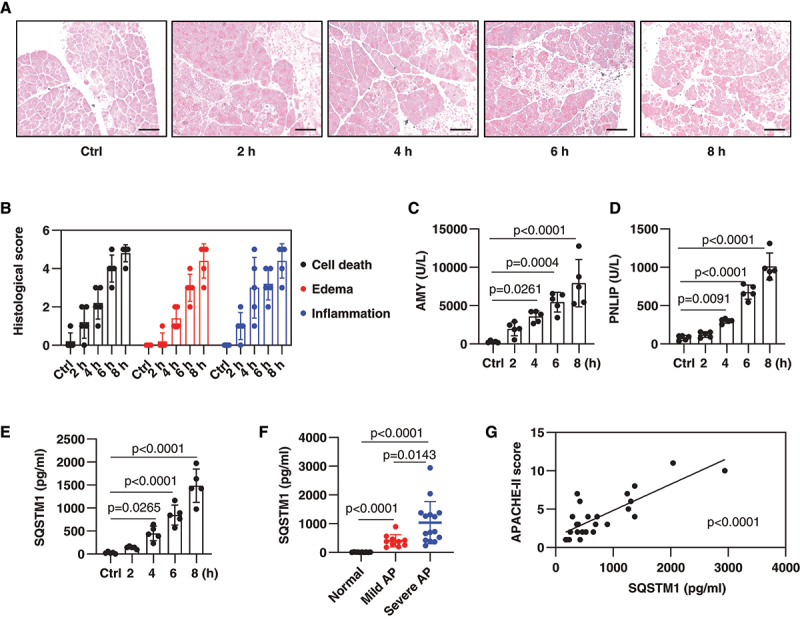
Serum SQSTM1 is elevated in AP. (A) Representative samples of pancreas H&E staining in mice after the administration of cerulein for 2–8 h. Scale bar: 100 μm. (B) Bar graph shows quantification of histological scores of cell death, edema, and inflammation (n = 5 mice/group; data are presented as means ± SD). (C-E) Analysis of serum concentration of AMY, PNLIP, or SQSTM1 in mice after the administration of cerulein for 2–8 h (n = 5 mice/group; one-way ANOVA with Tukey’s multiple comparisons test on all pairwise combinations; data are presented as means ± SD). (F) Analysis of serum SQSTM1 concentration in normal healthy people and AP patients (n = 10–15 cases/group; one-way ANOVA with Tukey’s multiple comparisons test on all pairwise combinations; data are presented as means ± SD). (G) Correlation analysis of serum SQSTM1 levels with APACHE-II scores in AP patients (linear regression t test; data are presented as scatter plots).
Furthermore, compared with normal healthy controls, serum SQSTM1 was elevated in AP patients, especially in severe AP patients (Figure 1F). The acute physiology and chronic health evaluation (APACHE-II) is a prognostic scoring system for predicting the severity of AP [25]. Statistical analysis showed that there was a positive correlation between SQSTM1 and APACHE-II scores (Figure 1G). Thus, elevated serum SQSTM1 correlates with AP disease severity.
Blocking SQSTM1 prevents experimental AP
To determine whether elevated serum SQSTM1 contributes to experimental AP, we used an SQSTM1-neutralizing antibody (termed αSQSTM1) previously shown to effectively block extracellular SQSTM1 activity in mice [12]. H&E staining revealed that αSQSTM1, in comparison to control IgG, dose-dependently prevented pancreatic tissue damage caused by cerulein (Figure 2A). Other pancreatitis experimental parameters, including serum AMY (Figure 2C), serum PNLIP (Figure 2D), pancreatic trypsin activity (Figure 2E), pancreatic neutrophil recruitment (as measured by pancreatic MPO [myeloperoxidase] activity) (Figure 2F), and pancreatic necrosis (as reflected by HMGB1) (Figure 2G) were ameliorated by the administration of αSQSTM1. TNF (tumor necrosis factor), IL1B (interleukin 1 beta), IL6, and IL17 are important cytokines in the development of inflammation in AP. Additionally, αSQSTM1 restricted the production of these pro-inflammatory cytokines in serum during cerulein-induced AP (Figure 2H).
Figure 2.
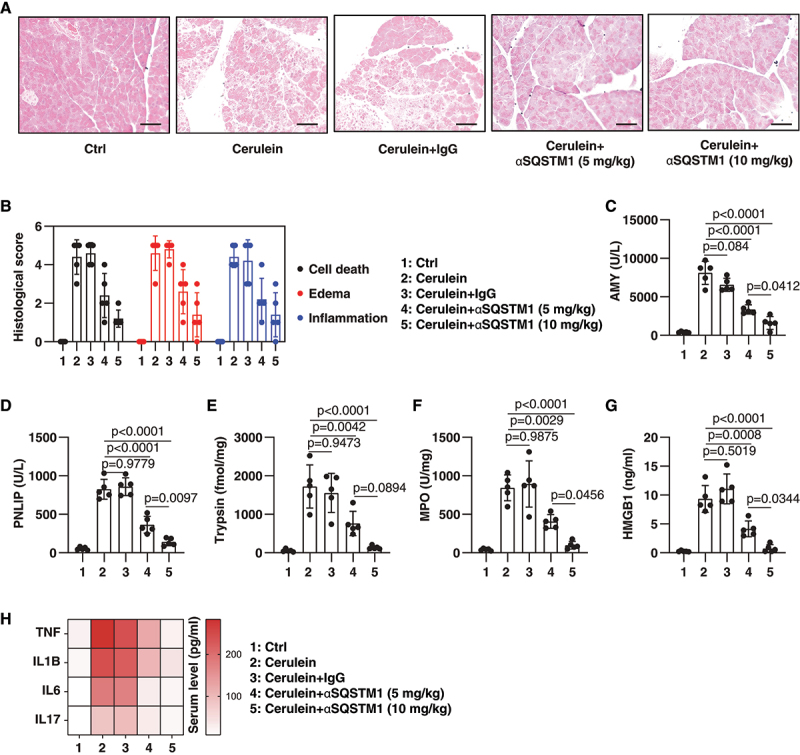
Blocking SQSTM1 prevents cerulein-induced AP. (A) Representative samples of pancreas H&E staining in mice after the administration of cerulein in the absence or presence of αSQSTM1 (5–10 mg/kg) for 8 h. Scale bar: 100 μm. (B) Bar graph shows quantification of histological scores of cell death, edema, and inflammation (n = 5 mice/group; data are presented as means ± SD). (C-H) Analysis of serum AMY, serum PNLIP, pancreatic MPO, serum HMGB1, and indicated serum cytokines in mice after the administration of cerulein in the absence or presence of αSQSTM1 (5–10 mg/kg) for 8 h (n = 5 mice/group; one-way ANOVA with Tukey’s multiple comparisons test on all pairwise combinations; data are presented as means ± SD in C-G; data are presented as a heat map of the mean in H).
To further understand the utility of αSQSTM1 in AP therapy, we investigated a different model of L-arginine-induced acute necrotizing pancreatitis. Previous studies by us and others have shown that peak pathological responses in L-arginine-induced AP occur at 72 h, whereas cerulein-induced AP occurs at 8 h [26–28]. The αSQSTM1 treatment also protected mice from L-arginine-induced AP, reducing pancreatic histological and experimental responses at 72 h (Fig. S1). Collectively, these neutralizing antibody experiments demonstrate that extracellular SQSTM1 is a mediator of experimental AP.
Autophagy-dependent ferroptosis is enhanced by rSQSTM1
One of the primary mechanisms of cerulein- or L-arginine-induced AP is the triggering of acinar cell death [3]. To determine the direct effects of extracellular SQSTM1 on acinar cells, we compared cerulein- or L-arginine-induced cell death in 266–6 cells (a mouse acinar pancreatic cell line) in the absence or presence of recombinant SQSTM1 protein (rSQSTM1). The rSQSTM1 alone had no significant effect on the induction of cell death in 266–6 cells (Figure 3A). However, cerulein- or L-arginine-induced cell death was enhanced by rSQSTM1 (Figure 3A). To determine the type of increased cell death sensitivity caused by rSQSTM1, we used classical cell death inhibitors. Our analysis showed that this death-sensitizing effect of rSQSTM1 on 266–6 cells could be reversed by the addition of the ferroptosis inhibitor liproxstatin-1, but not by the addition of the necroptosis inhibitor necrosulfonamide or the apoptosis inhibitor Z-VAD-FMK (Figure 3B).
Figure 3.
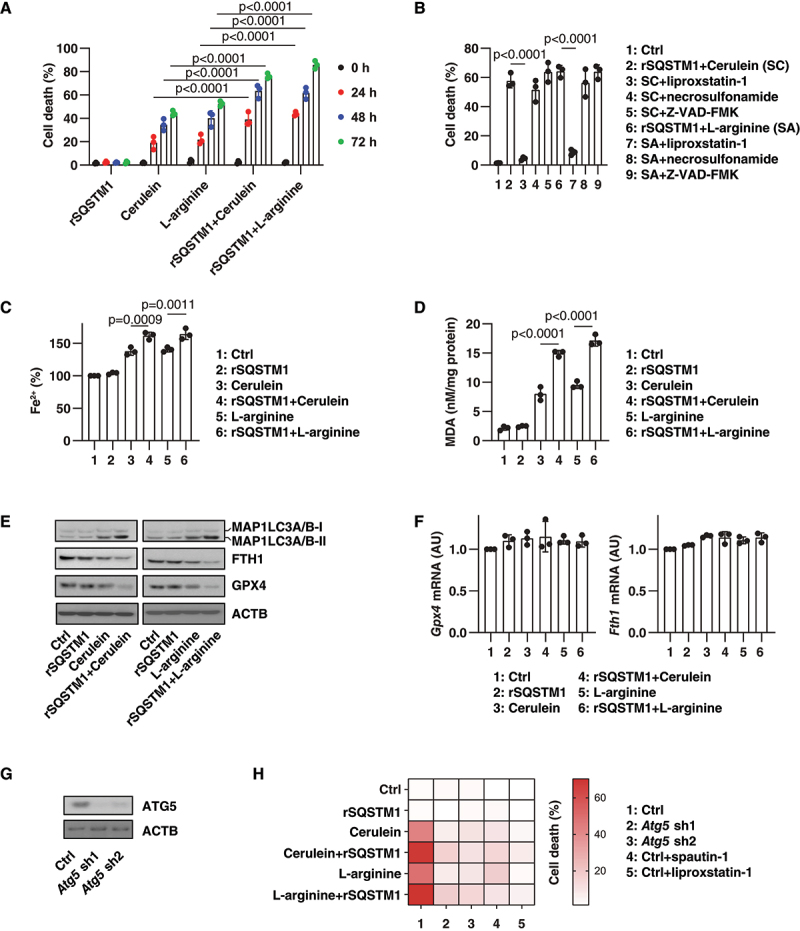
Autophagy-dependent ferroptosis is enhanced by rSQSTM1. (A) Analysis of the level of cell death in 266–6 acinar cell line after treatment with cerulein (100 nM) or L-arginine (5 mg/mL) in the absence or presence of rSQSTM1 (2 ng/ml) for 24–72 h (n = 3 biologically independent samples; two-way ANOVA with Tukey’s multiple comparisons test on all pairwise combinations; data are presented as means ± SD). (B) Analysis of the level of cell death in 266–6 acinar cell line after treatment with cerulein (100 nM) or L-arginine (5 mg/mL) in the absence or presence of rSQSTM1 (2 ng/ml), liproxstatin-1 (500 nM), Z-VAD-FMK (10 µM), or necrosulfonamide (1 µM) for 48 h (n = 3 biologically independent samples; one-way ANOVA with Tukey’s multiple comparisons test on all pairwise combinations; data are presented as means ± SD). (C, D) Analysis of the concentration of intracellular Fe2+ or MDA in 266–6 acinar cell line after treatment with cerulein (100 nM) or L-arginine (5 mg/mL) in the absence or presence of rSQSTM1 (2 ng/ml) for 48 h (n = 3 biologically independent samples; one-way ANOVA with Tukey’s multiple comparisons test on all pairwise combinations; data are presented as means ± SD). (E, F) Analysis of indicated protein or gene expression in 266–6 acinar cell line after treatment with cerulein (100 nM) or L-arginine (5 mg/mL) in the absence or presence of rSQSTM1 (2 ng/ml) for 24 h (data are presented as means ± SD in F). (G) Western blot analysis of ATG5 expression in Atg5-knockdown 266–6 cells. (H) Analysis of the effect of Atg5 knockdown, spautin-1 (5 µM), or liproxstatin-1 (500 nM) on the level of cell death in 266–6 acinar cell line after treatment with cerulein (100 nM) or L-arginine (5 mg/mL) in the absence or presence of rSQSTM1 (2 ng/ml) for 48 h (data are presented as a heat map of the mean of 3 biologically independent samples).
Because ferroptosis is a type of iron-dependent cell death caused by lipid peroxidation [15,29], we determined the effect of rSQSTM1 on iron accumulation and lipid peroxidation in the cells. Cerulein- or L-arginine-induced iron accumulation was increased by rSQSTM1 (Figure 3C). Quantitative determination of malondialdehyde (MDA, one of the end products of lipid peroxidation) showed that rSQSTM1 increased lipid peroxidation in response to cerulein or L-arginine treatment (Figure 3D). The rSQSTM1 alone failed to induce MDA production, suggesting that rSQSTM1 may not directly cause lipid peroxidation (Figure 3D).
Because ferroptosis has excessive autophagy responses [17], we also examined the levels of MAP1LC3B (microtubule-associated protein 1 light chain 3 beta) and the degradation substrates GPX4 (glutathione peroxidase 4) and ferritin, which are repressors and autophagic substrates of ferroptosis [30–32]. Western blot analysis showed that rSQSTM1 increased cerulein- or L-arginine-induced MAP1LC3B-II protein upregulation, GPX4 protein downregulation, and FTH1 (ferritin heavy polypeptide 1) protein downregulation (Figure 3E). In contrast, the mRNAs of Gpx4 and Fth1 were not remarkedly altered by cerulein or L-arginine in the absence or presence of rSQSTM1 (Figure 3F). As expected, the knockdown of Atg5 using two specific shRNAs and the administration of the autophagy inhibitor spautin-1 blocked cell death induced by cerulein or L-arginine in the presence of rSQSTM1 (Figure 3G,H). These findings indicate that rSQSTM1 enhances the sensitivity to autophagy-dependent ferroptosis.
ACSL4-mediated autophagy and ferroptosis are induced by rSQSTM1
Although there are many types and sources of lipids, the synthesis of PUFA is an important initial step to enhance subsequent lipid peroxidation during ferroptosis [33]. We and others previously demonstrated that ACSL4 (acyl-CoA synthetase long chain family member 4) is a driver of ferroptosis because of its involvement in the metabolism and formation of PUFA-containing phospholipids [34,35]. Western blot and qPCR analysis demonstrated that rSQSTM1 alone upregulated ACSL4 protein and mRNA (Figure 4A,B). Consequently, the levels of 5-hydroxyeicosatetraenoic acid (5-HETE), one of the products of arachidonic acid catalyzed by ACSL4, were increased (Figure 4C). In contrast, the expression of ALOX5 (arachidonate 5-lipoxygenase), a mediator of lipid peroxidation in the pancreas [36], was not affected by rSQSTM1 (Figure 4A).
Figure 4.
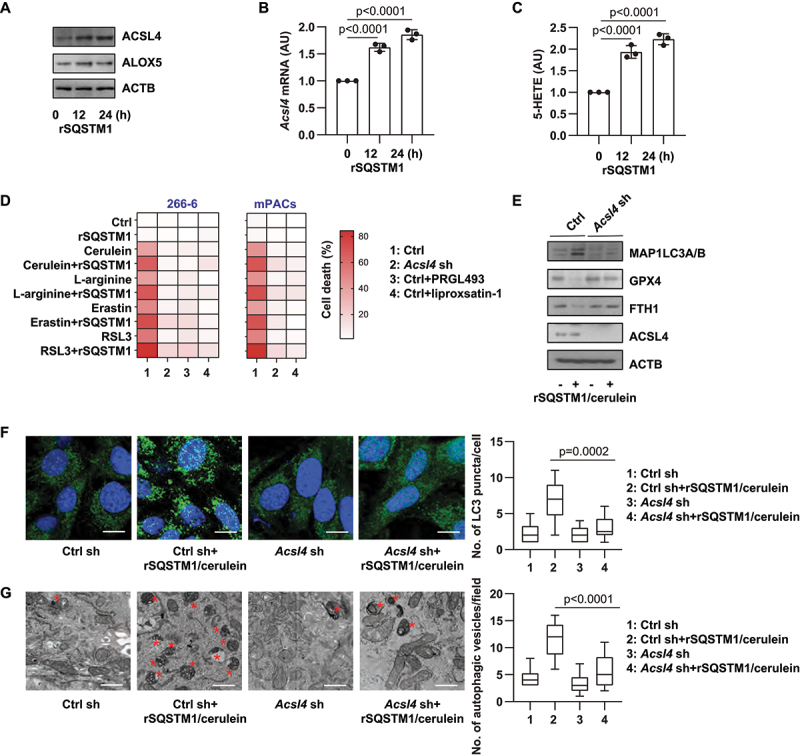
ACSL4-mediated autophagy and ferroptosis are induced by rSQSTM1. (A) Western blot analysis of ACSL4 and ALOX5 protein expression in 266–6 cells after treatment with rSQSTM1 (2 ng/ml) for 12–24 h. (B, C) Analysis of Acsl4 mRNA expression and intracellular 5-HETE levels in 266–6 cells after treatment with rSQSTM1 (2 ng/ml) for 12–24 h (n = 3 biologically independent samples; one-way ANOVA with Tukey’s multiple comparisons test on all pairwise combinations; data are presented as means ± SD). (D) Analysis of the effect of Acsl4 knockdown, PRGL493 (25 µM), or liproxstatin-1 (500 nM) on the level of cell death in 266–6 acinar cell line or primary mouse acinar cells (mPACs) after treatment with cerulein (100 nM), L-arginine (5 mg/mL), erastin (5 µM), or RSL3 (500 nM) in the absence or presence of rSQSTM1 (2 ng/ml) for 48 h (data are presented as a heat map of the mean of 3 biologically independent samples). (E) Western blot analysis of indicated protein expression in WT and Acsl4 knockdown 266–6 cells after treatment with cerulein (100 nM) plus rSQSTM1 (2 ng/ml) for 24 h. (F, G) Analysis of LC3 puncta formation and autophagic vacuoles in WT and Acsl4 knockdown 266–6 cells after treatment with cerulein (100 nM) plus rSQSTM1 (2 ng/ml) for 24 h (n = 10 fields/group; one-way ANOVA with Tukey’s multiple comparisons test on all pairwise combinations; data are presented as median value [black line], interquartile range [box], and minimum and maximum of all data [black line]; scale bar: 20 µm in F, 500 nm in G; red stars show autophagic vesicle).
In addition to cerulein or L-arginine, rSQSTM1 also increased the toxicity of classical ferroptosis inducers (erastin and RSL3) in 266–6 cells and primary acinar cells (mPACs) (Figure 4D). These processes were reversed by the ACSL4 inhibitor PRGL493 or by the genetic knockdown of Acsl4 (Figure 4D). Because PUFAs are also essential fatty acids that participate in autophagosome formation [37], we assayed the impact of the knockdown of Acsl4 on rSQSTM1 plus cerulein-induced MAP1LC3B-II expression and LC3 puncta formation. The knockdown of Acsl4 by shRNA inhibited rSQSTM1 plus cerulein-induced MAP1LC3B-II protein upregulation (Figure 4E), and it increased LC3 puncta formation (Figure 4F). Accordingly, Acsl4 silencing inhibited rSQSTM1 plus cerulein-induced degradation of GPX4 and FTH1 proteins (Figure 4E). Transmission electron microscopy analysis of autophagic vacuoles further confirmed the role of ACSL4 in mediating autophagic responses to rSQSTM1 plus cerulein (Figure 4G). Altogether, these findings suggest that rSQSTM1 selectively upregulates the expression of ACSL4, which contributes to the production of PUFAs for autophagosome formation and subsequent autophagy-dependent ferroptosis.
AGER mediates rSQSTM1 activity in acinar cells
We first investigated the effect of INSR in mediating rSQSTM1 activity in ferroptosis. Consistent with the previous notion that INSR is a receptor for rSQSTM1 in macrophages [12], rSQSTM1-induced Tnf (tumor necrosis factor) and Il6 (interleukin 6) mRNA expression was restricted in insr−/− macrophages (Figure 5A,B). However, rSQSTM1-induced Acsl4 mRNA expression and 5-HETE production was unaffected in insr−/− acinar cells (Figure 5C,D), suggesting that INSR is not essential for extracellular SQSTM1-mediated ACSL4 expression and activity.
Figure 5.
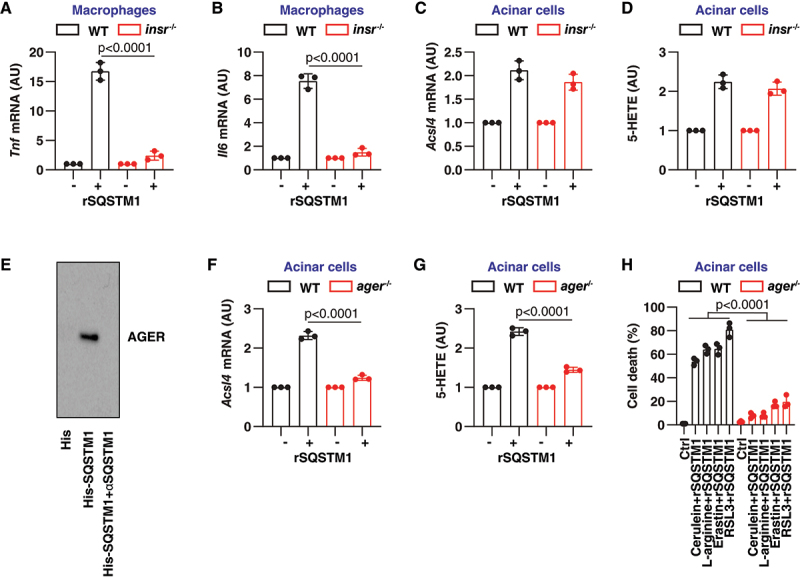
AGER mediates rSQSTM1 activity in acinar cells. (A, B) Analysis of Tnf and Il6 mRNA expression in WT and insr−/− peritoneal macrophages after treatment with rSQSTM1 (2 ng/ml) for 24 h (n = 3 biologically independent samples; two-way ANOVA with Tukey’s multiple comparisons test on all pairwise combinations; data are presented as means ± SD). (C, D) Analysis of Acsl4 mRNA expression and intracellular 5-HETE level in WT and insr−/− acinar cells after treatment with rSQSTM1 (2 ng/ml) for 24 h (n = 3 biologically independent samples; data are presented as means ± SD). (E) His-tag affinity-isolation analysis of the binding of SQSTM1 to AGER in the absence or presence of αSQSTM1. (F, G) Analysis of Acsl4 mRNA expression and intracellular 5-HETE levels in WT and ager−/− acinar cells after treatment with rSQSTM1 (2 ng/ml) for 24 h (n = 3 biologically independent samples; two-way ANOVA with Tukey’s multiple comparisons test on all pairwise combinations; data are presented as means ± SD). (H) Analysis of the level of cell death in WT and ager−/− acinar cells after treatment with cerulein (100 nM), L-arginine (5 mg/mL), erastin (5 µM), or RSL3 (500 nM) in the absence or presence of rSQSTM1 (2 ng/ml) for 48 h (n = 3 biologically independent samples; two-way ANOVA with Tukey’s multiple comparisons test on all pairwise combinations; data are presented as means ± SD).
AGER belongs to a class of PRRs that recognize a variety of ligands to drive pancreatitis and pancreatic cancer [38,39]. Next, we examined whether AGER is a receptor for SQSTM1 in acinar cells. Indeed, His tag affinity-isolation experiments demonstrated a direct interaction between AGER and SQSTM1 proteins, which was inhibited by αSQSTM1 (Figure 5E). Functionally, rSQSTM1-induced upregulation of Acsl4 mRNA was blocked in ager−/− acinar cells compared with wild-type acinar cells (Figure 5F). Quantitative analysis of 5-HETE also confirmed the role of AGER in mediating rSQSTM1 activity in arachidonic acid metabolism in acinar cells (Figure 5G). Moreover, the depletion of Ager in acinar cells reversed the rSQSTM1-increased toxicity of cerulein, L-arginine, erastin, and RSL3 (Figure 5H), indicating that rSQSTM1 amplifies ferroptosis sensitivity in acinar cells through the receptor AGER.
AGER depletion prevents rSQSTM1-mediated AP
To determine whether AGER is required for SQSTM1 activity in vivo, we treated wild-type and conditional pancreatic Ager-depleted mice (hereafter ager−/−) with rSQSTM1 in cerulein-induced AP. Histopathological analysis revealed that rSQSTM1 increased cerulein-induced inflammatory, edema, and necrotic responses in pancreatic tissue (Figure 6A,B). Accordingly, serum AMY (Figure 6C), serum PNLIP (Figure 6D), pancreatic trypsin activity (Figure 6E), pancreatic MPO activity (Figure 6F), and serum HMGB1 (Figure 6G) were upregulated by rSQSTM1 in cerulein-induced AP. The rSQSTM1-induced pathological responses of AP were reduced in ager−/− mice. As expected, rSQSTM1-induced pancreatic MDA production (Figure 6H), pancreatic ACSL4 expression (Figure 6I), and serum DCN (decorin; a marker of ferroptosis) (Figure 6J) were limited in ager−/− mice. These animal studies demonstrated the pathological role of AGER in driving rSQSTM1-mediated AP with an increase in ACSL4 expression and ferroptosis sensitivity.
Figure 6.
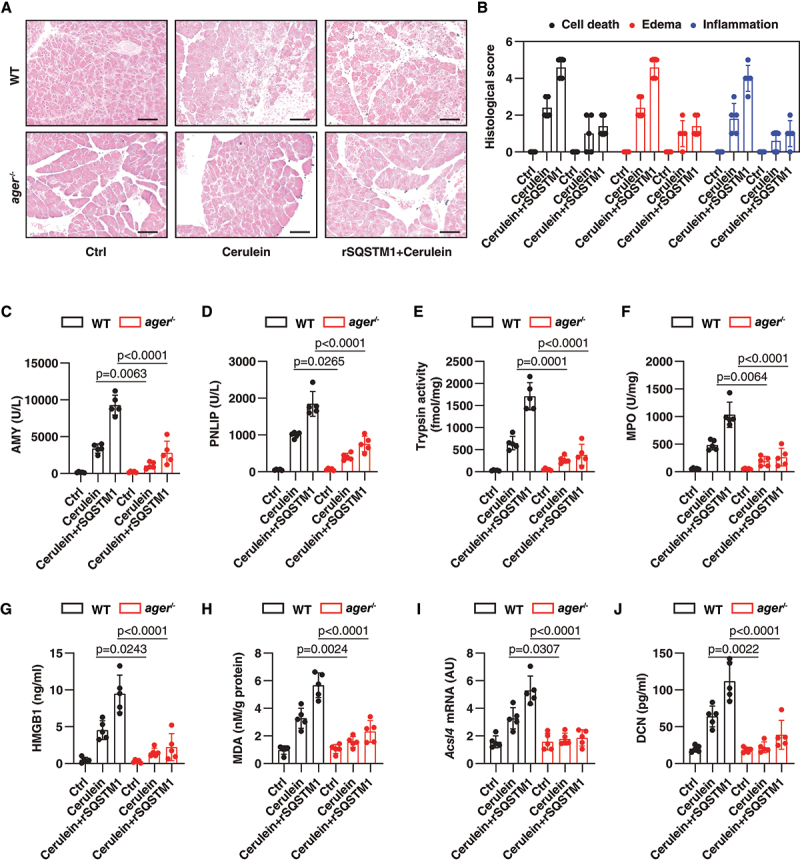
AGER depletion prevents rSQSTM1-mediated AP. (A) Representative samples of pancreas H&E staining in WT mice and in mice with conditional deletion of the Ager gene in the pancreas (ager−/−) after the administration of cerulein in the absence or presence of rSQSTM1 for 8 h. Scale bar: 100 μm. (B) Bar graph shows quantification of histological scores for cell death, edema, and inflammation (n = 5 mice/group; data are presented as means ± SD). (C-J) Analysis of serum AMY, serum PNLIP, pancreatic trypsin, pancreatic MPO, serum HMGB1, pancreatic MDA, pancreatic Acsl4 mRNA, and serum DCN in WT and ager−/− mice after the administration of cerulein in the absence or presence of rSQSTM1 for 8 h (n = 5 mice/group; two-way ANOVA with Tukey’s multiple comparisons test on all pairwise combinations; data are presented as means ± SD).
Discussion
Sterile inflammation occurs in the absence of microorganisms and is often associated with PRRs that recognize DAMPs released by damaged cells [4]. Unrestricted sterile inflammation is the fundamental pathological event leading to various human diseases [4]. In this study, we established an SQSTM1-AGER signaling pathway that drives experimental AP by activating ferroptosis-related sterile inflammation. Our findings also provide new insights into the immune consequences of ferroptotic death [40], which could be useful for the development of anti-inflammation therapeutic strategies.
Impaired autophagic flux in pancreatitis can lead to the accumulation of SQSTM1 [41]. We demonstrated that SQSTM1 can be released into the extracellular space and act as a mediator to restore autophagic flux and subsequent autophagy-dependent ferroptosis in acinar cells. Thus, there is a complex dynamic of autophagic activity that controls acinar cell injury during pancreatitis in a SQSTM1-dependent manner. Most DAMPs are endogenous proteins that have important functions during cellular stress, including cell death stimulation [42]. Once they are released into the extracellular space, they can individually or synergistically trigger an inflammatory response [4]. We found that serum SQSTM1 was significantly upregulated during AP in humans or mice compared with normal groups, although we cannot exclude that non-acinar cells also contribute to the release of SQSTM1 in vivo. Furthermore, serum SQSTM1 levels were time-dependently upregulated with the development of experimental AP. Whether serum SQSTM1 can be used to assist in AP staging needs to be further investigated in large clinical studies [43]. Nevertheless, a comparison of the sensitivity and specificity of SQSTM1 with other biomarkers (e.g., serum AMY and PNLIP) may be important for the early diagnosis and assessment of the course of AP [25].
Our study highlights the fact that lipid peroxidation-mediated ferroptotic damage accelerates AP by releasing SQSTM1. There are two mechanisms for releasing SQSTM1 into the extracellular environment: passive release and active release [44]. In response to bacterial lipopolysaccharide, SQSTM1 is actively secreted from activated macrophages and monocytes by secretory lysosome-mediated exocytosis [12]. This process requires the phosphorylation of SQSTM1 through the activation of the lipopolysaccharide-TLR4-STING1 (stimulator of interferon response cGAMP interactor 1) pathway [12]. In addition, pyroptosis caused by bacterial infection can lead to the passive release of SQSTM1 in immune cells [12]. Although the pathogenesis of pancreatitis involves the induction of multiple distinct cell death pathways, trypsin from pancreas extracts plays a positive role in susceptibility to ferroptosis, resulting in increased pancreatitis severity in mice. Our study further demonstrated that extracellular SQSTM1 also can activate autophagy-dependent ferroptosis. The inhibition of SQSTM1 activity by neutralizing antibodies prevents experimental forms of AP, including those associated with ferroptosis injury and cytokine production. These data also support previous clinical observations that lipid peroxidation is a common pathological event in patients with pancreatitis [45–47].
Further, we demonstrated that AGER is a new receptor for SQSTM1 in acinar cells. Increased cytokines may accelerate acinar cell damage, resulting in positive feedback through immune cell infiltration that leads to systemic inflammation [48]. Our current study shows that INSR is not required for SQSTM1-triggered ACSL4 expression in acinar cells, although INSR favors SQSTM1 activity in mediating cytokine production in macrophages [12]. INSR also protects acinar cells from AP-stimulated injury, including cerulein or fatty acid plus ethanol [49]. We showed that AGER mediates extracellular SQSTM1 activity in acinar cells during ACSL4 production. The lipid metabolizing enzyme ACSL4 is one of the most widely recognized mediators of ferroptosis, and the expression of ACSL4 by SQSTM1 increases autophagosome formation and the sensitivity to ferroptosis. The deletion of Ager and pharmacological interventions that interrupt the AGER-ligand interaction also reduce pyroptosis-mediated release of IL1B and HMGB1 during AP [6]. AGER is also required for DCN-mediated immune responses in AP during ferroptosis [50]. Therefore, AGER plays a central role in organizing multiple cell death-related inflammatory and metabolic pathways [38,39].
In summary, we revealed that activating the SQSTM1-AGER pathway in acinar cells contributes to ferroptosis and sterile inflammation in experimental AP. Targeting SQSTM1 release or activity may represent a potential therapeutic strategy for the treatment of AP. Future directions for study include research to determine how different DAMPs (including SQSTM1) shape the inflammatory response to different cell death modalities (including ferroptosis) to drive AP [51]. It is also important to understand how different regulatory necrosis, including ferroptosis and necroptosis [52], synergistically lead to acute pancreatitis. One possibility is that while ferroptosis and necroptosis differ in their entire signaling pathways [53], they may share some common regulators (e.g., receptor interacting serine/threonine kinase 3 [RIPK3] and GPX4) that cross-affect cell death types [54,55]. To determine whether extracellular SQSTM1 is the cause of AP, future clinical trials are needed to block SQSTM1 activity to see if AP progression in patients is improved. Achieving this goal requires the collaboration of various disciplines to develop specific SQSTM1 antagonistic antibodies or small-molecule drugs that can be used in clinical trials.
Materials and methods
Reagents
Erastin (S7242), RSL3 (S8155), liproxstatin-1 (S7699), Z-VAD-FMK (S7023), necrosulfonamide (S8251), and spautin-1 (S7888) were obtained from Selleck Chemicals. PRGL493 (32,748) was obtained from Cayman Chemical. Cerulein (C9026) and L-arginine (A5006) were obtained from Sigma-Aldrich. Dimethyl sulfoxide (DMSO; 472,301) was obtained from Sigma-Aldrich and was used to prepare the stock solution of most drugs. The final concentration of DMSO in the drug working solution in the cells was <0.01%. Additionally, 0.01% DMSO was used as a vehicle control in the corresponding cell culture assays.
The neutralizing antibodies to SQSTM1 (814,801) and mouse IgG1 control antibodies (400,101) were obtained from BioLegend. The antibodies to ACTB (3700) and MAP1LC3A/B (4108) were obtained from Cell Signaling Technology. The antibodies to GPX4 (ab125066) and FTH1 (ab183781) were obtained from Abcam. The antibodies to ACSL4 (PA5-27,137) and ALOX5 (PA5-78,762) were obtained from Thermo Fisher Scientific. The rSQSTM1 (TP506359) was obtained from OriGene.
Cell culture
The mouse 266–6 (CRL-2151) cell line was obtained from the American Type Culture Collection and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific, 11,995,073) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Thermo Fisher Scientific, A3840001) and 1% penicillin and streptomycin (Thermo Fisher Scientific, 15,070–063) at 37°C, 95% humidity, and 5% CO2. Primary mouse acinar cells were cultured as described previously [56]. Briefly, the pancreas from male C57BL/6 J mice (8–10 weeks) was removed and minced for 5 min in Hanks’ balanced salt solution (HBSS; Sigma-Aldrich, H8264) plus 0.1% bovine serum albumin (BSA; Sigma-Aldrich, 05470) and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; Sigma-Aldrich, PHG0001). After washing, the pancreatic segments were incubated in 10 mL collagenase IA solution (HBSS 1× containing 10 mM HEPES, 200 U/ml of collagenase IA [Sigma-Aldrich, C9891], and 0.25 mg/ml of trypsin inhibitor) for 20–30 min at 37°C. The solution containing collagenase was then removed and replaced with DMEM supplemented with 1% penicillin and streptomycin, 10% FBS, 0.25 mg/ml of trypsin inhibitor, and 25 ng/ml of recombinant mouse EGF (epidermal growth factor; Sigma-Aldrich, E5160) at 37°C, 95% humidity, and 5% CO2. Cell line identity was validated by short tandem repeat profiling, and routine mycoplasma testing was negative for contamination.
Animal models
Our protocol for animal use was reviewed and approved by our institutional animal care and use committees. Pancreatic-specific ager-knockout mice were produced and identified in our laboratory by crossing floxed Ager (produced by the Rui Kang laboratory [57]) and Pdx1-Cre (the Jackson Laboratory, 014647) transgenic mice (C57BL/6 J background). Mouse were kept under standard pathogen-free conditions with an artificial 12-h light/dark cycle (lights on: 08:00) and constant 50%-60% humidity. Mouse were allowed access to tap water and free (ad libitum) access to standard laboratory chow during the experimental period.
For cerulein-induced AP, male mice (8–10 weeks) received 7 hourly intraperitoneal (i.p.) injections of 50 μg/kg cerulein in sterile saline [58]. In some experiments, rSQSTM1 was administered by i.p. at a dose of 250 μg/mouse at 3 h after the first cerulein injection. For L-arginine-induced AP, a sterile solution of L-arginine (8%) was prepared in normal saline and the pH was adjusted to 7.0. Mice received 2 hourly i.p. injections of L-arginine (4 g/kg), while controls were administered saline i.p. as described previously [26]. For the treatment group, αSQSTM1 monoclonal antibodies (5 or 10 mg/kg) or control IgG1 (10 mg/kg) were repeatedly administered intraperitoneally to mice at 1 and 6 h after the first cerulein or L-arginine injection. The parameters of AP were assessed 8 or 72 h after the last cerulein or L-arginine treatment, respectively.
Animals were sacrificed at the indicated time by CO2 asphyxia, and a blood sample and tissue were collected. Serum was further collected immediately after centrifugation at 10,000 g for 5 min at 4°C. Tissue samples were collected, snap-frozen in liquid nitrogen, and stored at −80°C. Formalin-fixed pancreas samples were processed, and 5-μm-thick paraffin sections were stained with H&E for histological analysis. Pancreatitis was scored according to a revised scoring standard [59], with an independent pathologist evaluating histological scores for acinar cell death, leukocyte infiltration, or edema in a blind manner. Histological images were acquired using an EVOS FL Auto Cell Imaging System (Thermo Fisher Scientific).
Western blot analysis
Cells were lysed 3 times with 1× cell lysis buffer (Cell Signaling Technology, 9803) containing protease inhibitor cocktail (Sigma-Aldrich, P8340) on ice for 10 min. Protein was quantified using the bicinchoninic acid assay (Thermo Fisher Scientific, 23,225) and 20–30 μg of each sample was resolved on 4%-12% Criterion XT Bis-Tris gels (Bio-Rad, 3,450,124) in XT MES running buffer (Bio-Rad, 1,610,789) and transferred to polyvinylidene difluoride membranes (Bio-Rad, 1,620,233) using the Trans-Blot Turbo Transfer Pack and System (Bio-Rad, 1,704,150) [60]. Membranes were blocked with Tris-buffered saline with 0.1% Tween 20 detergent (Cell Signaling Technology, 9997; TBST) containing 5% nonfat milk (Cell Signaling Technology, 9999) for 1 h and incubated overnight at 4°C with various primary antibodies (1:500–1:1000). Following 3 washes in TBST, membranes were incubated with goat anti-rabbit or anti-mouse IgG horseradish peroxidase-conjugated secondary antibody (1:1000; Cell Signaling Technology, 7074 or 7076) at room temperature for 1 h. After being washed with TBST, the signals were visualized using enhanced chemiluminescence (Thermo Fisher Scientific, 32,106) and then visualized and analyzed with a ChemiDoc Touch Imaging System (Bio-Rad).
Cytotoxicity assays
A Countess II FL Automated Cell Counter (Thermo Fisher Scientific) was used to assay the percentages of dead cells after cell staining with 0.4% trypan blue solution (Thermo Fisher Scientific, T10282). In short, samples were prepared by adding 10 μl of cell suspension to 10 μl of 0.4% trypan blue stain.
The qPCR assay
Total RNA was extracted and purified from cultured cells using the RNeasy Plus Mini Kit (QIAGEN, 74,136). First-strand cDNA was synthesized from 1 µg of RNA using the iScript cDNA Synthesis Kit (Bio-Rad, 1,708,890). Briefly, 20-μl reactions were prepared by combining 4 μl of iScript Select reaction mix, 2 μl of gene-specific enhancer solution, 1 μl of reverse transcriptase, 1 μl of gene-specific assay pool (20×, 2 μM), and 12 μl of RNA diluted in RNase-free water. Then cDNA from various cell samples was amplified by real-time qPCR with specific primers using a CFX96 Touch Real-Time PCR Detection System (Bio-Rad) with CFX Manager software 2.0 (Bio-Rad). The data were normalized to Actb and the fold change was calculated via the 2−ΔΔCt method [61]. The relative concentrations of mRNA were expressed in arbitrary units based on the untreated group, which was assigned a value of 1. The primers were used as below. Gpx4: CCTCTGCTGCAAGAGCCTCCC and CTTATCCAGGCAGACCATGTGC; Fth1: GCCGAGAAACTGATGAAGCTGC and GCACACTCCATTGCATTCAGCC; Acsl4: CCTTTGGCTCATGTGCTGGAAC and GCCATAAGTGTGGGTTTCAGTAC; Tnf: GGTGCCTATGTCTCAGCCTCTT and GCCATAGAACTGATGAGAGGGAG; Il6: TACCACTTCACAAGTCGGAGGC and CTGCAAGTGCATCATCGTTGTTC; and Actb: CATTGCTGACAGGATGCAGAAGG and TGCTGGAAGGTGGACAGTGAGG.
RNAi
The pre-desigined Atg5 shRNA1 (TRCN0000375819), Atg5 shRNA2 (TRCN0000099432), and Acsl4 shRNA (TRCN0000011987) were obtained from Sigma-Aldrich. The shRNA was transfected into cells using Lipofectamine 3000 (Invitrogen, L3000-015) when cells were at approximately 60%-70% confluence. Puromycin (InvivoGen, ant-pr-1) was used to generate stable knockdown cell lines. The efficiency of RNAi was verified by western blotting.
ELISA analysis
The concentrations or activity of AMY (amylase; Abcam, ab102523), MPO (Thermo Fisher Scientific, EMMPO), trypsin (Abcam, ab102531), PNLIP (pancreatic lipase; CUSABIO, CSB-E16930m), HMGB1 (Sino-Test Corporation, 326,054,329), TNF (Thermo Fisher Scientific, A43658), IL1B (Thermo Fisher Scientific, BMS6002), IL6 (Thermo Fisher Scientific, A43656), IL17 (Thermo Fisher Scientific, BMS6001), MDA (Abcam, ab118970), iron (Sigma-Aldrich, MAK025), SQSTM1 (ADI-900-212, Enzo Life Sciences), DCN (R&D Systems, DY1060), and 5-HETE (Abbexa, abx251443) in the indicated samples were measured using enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s guidelines.
Serum SQSTM1 levels in patients or mice were measured by ELISA using 35–50 µl serum/sample. Briefly, samples and standards were added to wells coated with SQSTM1-specific monoclonal antibodies (supplied with the ELISA kit). The plate was then incubated and washed to remove excess antibody. A blue solution of horseradish peroxidase (HRP) conjugate was added to each well bound to the SQSTM1 antibody. The plate was incubated again, washed and 3,3’,5,5’ tetramethylbenzidine substrate solution was added. Finally, stop solution was added to stop the substrate reaction. The resulting yellow color was read at 450 nm, which was proportional to the level of SQSTM1 in the sample.
Immunofluorescence analysis
Cells were cultured on glass coverslips and fixed in 3% formaldehyde for 30 min at room temperature prior to detergent extraction with 0.1% Triton X-100 (Cell Signaling Technology, 39,487) for 10 min at 25°C. Coverslips were saturated with 2% BSA in phosphate-buffered saline (PBS; Cell Signaling Technology, 9872) for 1 h at room temperature and processed for immunofluorescence with primary antibodies, followed by Alexa Fluor 488-conjugated secondary antibodies (Thermo Fisher Scientific, A32731). Nuclear morphology was analyzed with the fluorescent dye Hoechst 33,342 (Thermo Fisher Scientific, 62,249). Images were taken with a ZEISS LSM 800 confocal microscope.
Transmission electron microscopy analysis
Transmission electron microscopy analysis of autophagic vacuoles was performed as previously described [62]. In brief, cells were fixed with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 mol/L phosphate buffer (pH 7.4), followed by postfixation for 6 h in 1% OsO4. After dehydration with graded alcohol solutions, each sample was embedded in epoxy resin (Sigma-Aldrich, 45,359). The cut-thin sample (70 nm) was mounted on a copper mesh (Sigma-Aldrich, TEM-74357) and post-stained with 2% uranyl acetate and 1% lead citrate, dried, and analyzed with a transmission electron microscope (JEOL).
His tag-based assays for protein-protein interactions
A Pierce His Protein Interaction Pull-Down Kit (Thermo Fisher Scientific, 21,277) was used to assay the interaction between SQSTM1 and AGER according to the manufacturer’s guidelines. Briefly, His-tag human SQSTM1 protein (OriGene, AR09432PU-L) was used as bait, while A549 (ATCC, CCL-185) cell lysate was used as the source of prey protein. The prey protein was captured through the Pierce Spin Column containing the immobilized polyhistidine-tagged bait protein. The Pierce Spin Column was then rinsed with wash and binding buffer to reduce nonspecific interactions. The resulting complex could then be recovered and analyzed by western blotting.
Patient samples
Serum from patients with AP were collected from the University of Pittsburgh. The collection of samples was approved by the University of Pittsburgh institutional review board. The severity of AP was defined according to the revised Atlanta classification [63].
Statistical analysis
Data are presented as mean ± SD except where otherwise indicated. GraphPad Prism 8.4.3 was used to collect and analyze data. A one-way or two-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test was used for comparison among the different groups. A p value of <0.05 was considered statistically significant. We did not exclude samples or animals.
Supplementary Material
Acknowledgments
We thank David C. Whitcomb (Division of Gastroenterology, Hepatology and Nutrition, University of Pittsburgh) for providing serum samples from AP patients and Dave Primm (Department of Surgery, University of Texas Southwestern Medical Center) for his critical reading of the manuscript.
Funding Statement
D.J.K. was supported by the National Institute of General Medical Sciences of USA (GM131919). J.L. was supported by grants from the National Natural Science Foundation of China (32200594).
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2152209
References
- [1].Petrov MS, Shanbhag S, Chakraborty M, et al. Organ failure and infection of pancreatic necrosis as determinants of mortality in patients with acute pancreatitis. Gastroenterology. 2010;139:813–820. [DOI] [PubMed] [Google Scholar]
- [2].Iannuzzi JP, King JA, Leong JH, et al. Global incidence of acute pancreatitis is increasing over time: a systematic review and meta-analysis. Gastroenterology. 2022;162:122–134. [DOI] [PubMed] [Google Scholar]
- [3].Kang R, Lotze MT, Zeh HJ, et al. Cell death and DAMPs in acute pancreatitis. Mol Med. 2014;20(1):466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gong T, Liu L, Jiang W, et al. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20:95–112. [DOI] [PubMed] [Google Scholar]
- [5].Liu L, Yang M, Kang R, et al. DAMP-mediated autophagy contributes to drug resistance. Autophagy. 2011;7:112–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kang R, Chen R, Xie M, et al. The receptor for advanced glycation end products activates the AIM2 inflammasome in acute pancreatitis. J Immunol. 2016;196(10):4331–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li G, Wu X, Yang L, et al. TLR4-mediated NF-kappaB signaling pathway mediates HMGB1-induced pancreatic injury in mice with severe acute pancreatitis. Int J Mol Med. 2016;37:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Regel I, Raulefs S, Benitz S, et al. Loss of TLR3 and its downstream signaling accelerates acinar cell damage in the acute phase of pancreatitis. Pancreatology. 2019;19:149–157. [DOI] [PubMed] [Google Scholar]
- [9].Sanchez-Martin P, Saito T, Komatsu M.. p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J. 2019;286:8–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20:233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kirkin V, Rogov VV. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol Cell. 2019;76:268–285. [DOI] [PubMed] [Google Scholar]
- [12].Zhou B, Liu J, Zeng L, et al. Extracellular SQSTM1 mediates bacterial septic death in mice through insulin receptor signalling. Nat Microbiol. 2020;5:1576–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tang D, Kroemer G. Ferroptosis. Curr Biol. 2020;30:R1–R6. [DOI] [PubMed] [Google Scholar]
- [14].Dai C, Chen X, Li J, et al. Transcription factors in ferroptotic cell death. Cancer Gene Ther. 2020;27:645–656. [DOI] [PubMed] [Google Scholar]
- [15].Chen X, Li J, Kang R, et al. Ferroptosis: machinery and regulation. Autophagy. 2021;17:2054–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kuang F, Liu J, Tang D, et al. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front Cell Dev Biol. 2020;8:586578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu J, Kuang F, Kroemer G, et al. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. 2020;27(4):420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kang R, Tang D. Autophagy and Ferroptosis—What Is the Connection? Curr Pathobiol Rep. 2017;5(2):153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kang R, Zhu S, Zeh HJ, et al. BECN1 is a new driver of ferroptosis. Autophagy. 2018;14:2173–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu Y, Wang Y, Liu J, et al. The circadian clock protects against ferroptosis-induced sterile inflammation. Biochem Biophys Res Commun. 2020;525(3):620–625. [DOI] [PubMed] [Google Scholar]
- [22].Liu J, Song X, Kuang F, et al. NUPR1 is a critical repressor of ferroptosis. Nat Commun. 2021;12(1):647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen X, Huang J, Yu C, et al. A noncanonical function of EIF4E limits ALDH1B1 activity and increases susceptibility to ferroptosis. Nat Commun. 2022;13(1):6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yang X, Yao L, Fu X, et al. Experimental acute pancreatitis models: history, current status, and role in translational research. Front Physiol. 2020;11:614591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mederos MA, Reber HA, Girgis MD. Acute Pancreatitis: a Review. JAMA. 2021;325(4):382–390. [DOI] [PubMed] [Google Scholar]
- [26].Kang R, Zhang Q, Hou W, et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology. 2014;146(4):1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kui B, Balla Z, Vasas B, et al. New insights into the methodology of L-arginine-induced acute pancreatitis. PLoS One. 2015;10(2):e0117588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Luthen R, Grendell JH, Haussinger D, et al. Beneficial effects of L-2-oxothiazolidine-4-carboxylate on cerulein pancreatitis in mice. Gastroenterology. 1997;112(5):1681–1691. [DOI] [PubMed] [Google Scholar]
- [29].Chen X, Comish P, Tang D, et al. Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol. 2021. DOI: 10.3389/fcell.2021.637162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gao M, Monian P, Pan Q, et al. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wu Z, Geng Y, Lu X, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A. 2019;116(8):2996–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lin Z, Liu J, Kang R, et al. Lipid metabolism in ferroptosis. Advanced Biology. 2021;5(8):e2100396. [DOI] [PubMed] [Google Scholar]
- [34].Yuan H, Li X, Zhang X, et al. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478(3):1338–1343. [DOI] [PubMed] [Google Scholar]
- [35].Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kuang F, Liu J, Xie Y, et al. MGST1 is a redox-sensitive repressor of ferroptosis in pancreatic cancer cells. Cell Chem Biol. 2021;28(6):765–75.e5. [DOI] [PubMed] [Google Scholar]
- [37].Yang B, Zhou Y, Wu M, et al. Omega-6 Polyunsaturated fatty acids (linoleic acid) activate both autophagy and antioxidation in a synergistic feedback loop via TOR-dependent and TOR-independent signaling pathways. Cell Death Dis. 2020;11:607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hudson BI, Lippman ME. Targeting RAGE signaling in inflammatory disease. Annu Rev Med. 2018;69:349–364. [DOI] [PubMed] [Google Scholar]
- [39].Kang R, Tang D, Lotze MT, et al. AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL6-pSTAT3 pathway. Autophagy. 2012;8:989–991. 3rd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chen X, Kang R, Kroemer G, et al. Ferroptosis in infection, inflammation, and immunity. J Exp Med. 2021;218(6):e20210518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gukovskaya AS, Gukovsky I. Autophagy and pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2012;303:G993–G1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang Q, Kang R, Zeh HJ 3rd, et al. DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy. 2013;9(4):451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Luo Y, Fan L, Huang L, et al. Expression of serum autophagy-related protein P62 in patients with severe pancreatitis and its correlation with prognosis. Am J Transl Res. 2022;14:1376–1383. [PMC free article] [PubMed] [Google Scholar]
- [44].Zou B, Liu J, Klionsky DJ, et al. Extracellular SQSTM1 as an inflammatory mediator. Autophagy. 2020;16:2313–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Schoenberg MH, Buchler M, Pietrzyk C, et al. Lipid peroxidation and glutathione metabolism in chronic pancreatitis. Pancreas. 1995;10:36–43. [DOI] [PubMed] [Google Scholar]
- [46].Grigor’eva IN, Romanova TI, Ragino Iu I. Lipid peroxidation in patients with acute and chronic pancreatitis. Eksp Klin Gastroenterol. 2011;7:24–27. [PubMed] [Google Scholar]
- [47].Tsai K, Wang SS, Chen TS, et al. Oxidative stress: an important phenomenon with pathogenetic significance in the progression of acute pancreatitis. Gut. 1998;42:850–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gukovsky I, Li N, Todoric J, et al. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144(6):1199–209 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bruce JIE, Sanchez-Alvarez R, Sans MD, et al. Insulin protects acinar cells during pancreatitis by preserving glycolytic ATP supply to calcium pumps. Nat Commun. 2021;12(1):4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Liu J, Zhu S, Zeng L, et al. DCN released from ferroptotic cells ignites AGER-dependent immune responses. Autophagy. 2022;18(9):2036–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chen X, Zeh HJ, Kang R, et al. Cell death in pancreatic cancer: from pathogenesis to therapy. Nat Rev Gastroenterol Hepatol. 2021;18(11):804–823. [DOI] [PubMed] [Google Scholar]
- [52].He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell. 2009;137(6):1100–1111. [DOI] [PubMed] [Google Scholar]
- [53].Tang D, Kang R, Berghe TV, et al. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Muller T, Dewitz C, Schmitz J, et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci. 2017;74(19):3631–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Canli Ö, Alankus YB, Grootjans S, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127(1):139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Liu K, Liu J, Zou B, et al. Trypsin-mediated sensitization to ferroptosis increases the severity of pancreatitis in mice. Cell Mol Gastroenterol Hepatol. 2022;13(2):483–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chen R, Zhu S, Zeng L, et al. AGER-mediated lipid peroxidation drives caspase-11 inflammasome activation in sepsis. Front Immunol. 2019;10:1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mareninova OA, Hermann K, French SW, et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest. 2009;119:3340–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Schmidt J, Rattner DW, Lewandrowski K, et al. A better model of acute pancreatitis for evaluating therapy. Ann Surg. 1992;215:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Deng W, Zhu S, Zeng L, et al. The circadian clock controls immune checkpoint pathway in sepsis. Cell Rep. 2018;24(2):366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhang H, Zeng L, Xie M, et al. TMEM173 drives lethal coagulation in sepsis. Cell Host Microbe. 2020;53:444–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Banks PA, Bollen TL, Dervenis C, et al. Classification of acute pancreatitis–2012: revision of the Atlanta classification and definitions by international consensus. Gut. 2013;62:102–111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.