

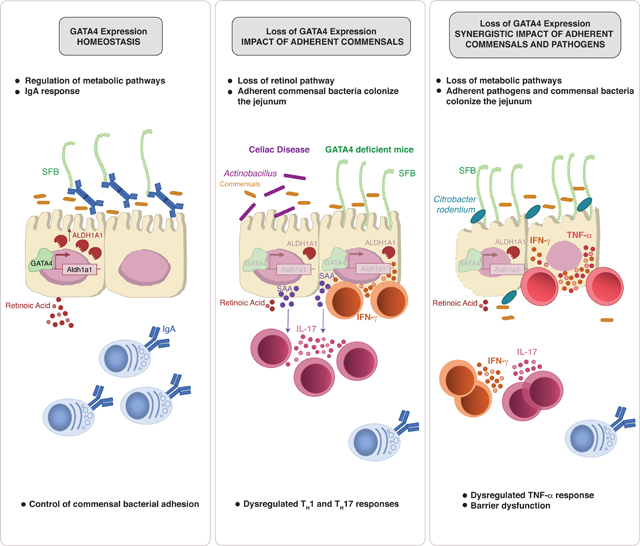
Summary:
There is growing recognition that regionalization of bacterial colonization and immunity along the intestinal tract has an important role in health and disease. Yet, the mechanisms underlying intestinal regionalization and its dysregulation in disease are not well understood. This study found that regional epithelial expression of the transcription factor GATA4 controls bacterial colonization and inflammatory tissue immunity in the proximal small intestine by regulating retinol metabolism and luminal IgA. Furthermore, in mice without jejunal GATA4 expression, the commensal segmented filamentous bacteria promoted pathogenic inflammatory immune responses that disrupted barrier function and increased mortality upon Citrobacter rodentium infection. In celiac disease patients, low GATA4 expression was associated with metabolic alterations, mucosal Actinobacillus, and increased IL-17 immunity. Taken together, these results reveal broad impacts of GATA4-regulated intestinal regionalization on bacterial colonization and tissue immunity, highlighting an elaborate interdependence of intestinal metabolism, immunity, and microbiota in homeostasis and disease.
Keywords: Intestinal regionalization, intestinal epithelial cells, GATA4, bacterial colonization, segmented filamentous bacteria, retinoic acid, IgA, infection, immunopathology, celiac disease
In brief:
Regulators of intestinal regionalization are vital, yet poorly understood. Here, Earley et al. demonstrate how the transcription factor GATA4 regulates metabolic pathways and luminal IgA to control adherent bacteria colonization. Proper gut regionalization and commensal colonization is critical in preventing dysregulated TH17 responses and immunopathology in humans and mice.
Graphical Abstract
Introduction:
Each region of the gastrointestinal tract performs distinct physiological functions, with the proximal small intestine optimized to digest and absorb critical nutrients, the distal small intestine to reabsorb bile acids and vitamin B12, and the colon to absorb water and electrolytes1. There is growing recognition that bacterial colonization2 and immune phenotypes3 are also spatially distributed along the gastrointestinal tract. Yet, little is known about the pathophysiological implications of this regionalization, or the molecular mechanisms regulating it. A key challenge in addressing these questions has been a lack of in vivo models that allow changes to the tissue environment in one specific intestinal compartment. Previous studies have shown that in the gut, expression of the transcription factor GATA4, is restricted to duodenal and jejunal intestinal epithelial cells (IECs), and that, in its absence, jejunal IECs acquire an ileum-like transcriptional program4,5. However, these studies did not address key questions motivating our study, namely, whether and how a jejunal shift to ileal identity impacts bacterial colonization, tissue immunity, or host susceptibility to pathology.
Results:
GATA4 controls regionalization of intestinal metabolism and immunity
To assess region-specific immune regulation, we analyzed cytokine production in T cells from the intestinal track of specific-pathogen-free (SPF) and germ-free (GF) mice (Figure 1A). The data revealed that the ileum is uniquely permissive for microbiota-dependent development of inflammatory T cell responses (Figure 1A). To investigate whether GATA4 plays a role in the regionalization of inflammatory immune responses, we performed total jejunal and ileal tissue RNA-sequencing (RNA-seq) in GATA4ΔIEC (Vil1cre+Gata4fl/fl) and littermate control wild-type (WT, Gata4fl/fl) mice. We first confirmed that GATA4 was expressed in duodenal and jejunal, but not ileal or colonic, IECs (Figure S1A). In addition, as previously shown5, in the absence of GATA4, jejunal IECs acquired an ileum-like transcriptional program (Figure S1B; Table S1). In particular, GATA4 strongly repressed ileal genes (Fabp6, Slc10a2) involved in the enterohepatic circulation of bile acids and induced jejunal expression of lipid metabolic genes involved in retinol metabolism (Adh1), fat digestion and absorption (Cd36, Fabp1, Dgat2, Apoa4), and uptake of vitamins and folate (Slc46a1, Pdxk) (Figure S1C; Table S1). By comparing the transcriptional profiles of WT jejunum with both WT ileum and GATA4ΔIEC jejunum, we identified 2,964 GATA4-regulated region-specific genes.
Figure 1. Epithelial GATA4 controls regionalization of tissue immunity in the proximal small intestine.

(A) Percentage of IFNγ+, IL-17a+, or IL-10+ cells among CD4+ or CD8αβ+ T cells from intraepithelial lymphocytes (IEL) or the lamina propria (LP) of each intestinal segment in specific-pathogen-free (SPF) or germ-free (GF) mice. ****, P<0.0001, effect due to region; oooo P<0.0001, ooo P<0.001, oo P< 0.01, effect due to microbiota; two-way ANOVA of microbiota and region impact on cytokine levels. N= 5–7 mice/group.
(B) Tissue samples plotted by the top two principal components (PCs) of the expression of the 500 most variable immune genes as measured by RNA-seq. N= 8 mice/group.
(C) Heatmap of the z-scored expression of region-specific, GATA4-regulated immune genes (rows) in jejunum and ileum tissue samples (columns) of wild-type (WT) and GATA4ΔIEC mice. Of 625 genes, 145 are uniquely in the IFNγ module, 54 are uniquely in the IL-17 module, 39 are in both modules, and 387 are in neither (annotation column). N= 8 mice/group.
(D) Representative (left) and summary (right) plots of the frequencies of IFN-γ+ cells among CD8αβ+ T cells in the IELs. N= 6 mice/group.
(E) Representative (left) and summary (right) plots of the frequencies of IL-17a+ cells among CD4+ T cells in the LP. N= 6 mice/group.
(F) Heatmap of the z-scored expression of 50 selected microbiota-dependent and -independent (right annotation column), region-specific, GATA4-regulated immune genes in jejunum tissue samples from SPF and GF WT and GATA4ΔIEC mice. Gene modules (left annotation column) as in C.
(G, H) Frequency (y axis) of IFNγ+ cells among CD8αβ+ T cells from the IEL (G) or of IL-17a+ cells among CD4+ T cells from the LP (H) in the jejunum of SPF and GF WT and GATA4ΔIEC mice. N= 6 mice/group.
All data in this figure are pooled from at least two independent experiments. **** P<0.0001 , *** P<0.001, ** P<0.01, * P<0.05, paired t-test (D and E), ANOVA with Tukey multiple comparison test (G and H).
To hone in on immune impacts, we focused on the 21% (625) of GATA4-regulated region-specific genes that were among 4,279 immune genes we curated from public databases6,7 (Table S1). The results revealed distinct immune signatures of WT jejunum and ileum, a regionalization of tissue immunity that was lost in GATA4ΔIEC mice (Figure 1B). Among the immune genes thus identified, more than a third (238) were potential targets of IFNγ or IL-17 regulation (Figure 1C; Table S2). Consistent with GATA4-regulated regionalization of IFNγ and IL-17 immune pathways, in the absence of epithelial GATA4, the frequency of intraepithelial IFNγ+ CD8αβ+ T cells in the jejunum increased to the levels observed in the ileum (Figure 1D). Furthermore, GATA4 deficiency led to a heightened TH17 response in the jejunum, with frequencies of IL-17+ CD4+ T cells surpassing those in the ileum (Figure 1E). The high levels of IL-17 may be related to additional changes imparted by GATA4 downregulation, such as an increase in serum amyloid A expression, which is known to amplify TH17 responses in the gut (Figure 1F)8. To determine whether GATA4 was sufficient to induce the jejunal immune signature, we compared previously obtained transcriptional data of ileal epithelial scrapings from either WT or Rosa26LsL Gata4 Vil1cre (GATA4TG) mice, which selectively overexpress GATA4 in IECs9. We observed that the ileum of GATA4TG mice expressed characteristic jejunal immune genes (Il15ra, B2m) and repressed ileal immune genes (Saa1/2, Nlrc5, Cxcr5) (Figure S1D), indicating that GATA4 is both necessary and sufficient for controlling compartmentalization of immune responses in the small intestine.
We next asked whether the increased IFNγ and IL-17 T cell responses in the jejunum of GATA4ΔIEC mice were microbiota dependent. Analyzing the expression of region-specific GATA4-regulated immune genes in the jejunum of GF GATA4ΔIEC mice revealed that the microbiota were required to drive the elevated IFNγ- and IL-17-associated genes and T cell responses seen in SPF mice (Figure 1F–H; Figure S1E and F; Table S1). While a few immune genes, such as Csf2 (GM-CSF), anti-viral response genes Ifnar2 and Mavs, and the tissue alarmins Il33 and Il15, were GATA4 regulated in a microbiota-independent manner (Figure 1G), most of the microbiota-independent genes were involved in lipid and cholesterol metabolism (Figure S1G and S1H). Furthermore, microbiota-independent genes were more enriched, compared to the microbiota-dependent subset, in direct targets of GATA4 (Figure S1H, Table S1), as indicated by GATA4 binding of promoter regions in published ChIP-seq data (hypergeometric test; P <10-7) (Figure S1H) 10.
Taken together, these results suggest that GATA4 is necessary and sufficient for regulating regional tissue immunity between the proximal and distal small intestine, both by directly controlling the transcription of immune genes in IECs, and by blocking the development of microbiota-dependent inflammatory T cell responses in the jejunum.
GATA4 prevents adherent bacteria from colonizing the jejunum
To investigate which microbiota trigger inflammatory immune responses in the absence of GATA4 in the proximal small intestine, we performed 16S ribosomal RNA sequencing of luminal- and mucosal-associated bacterial communities in the jejunum and ileum. This analysis revealed a striking expansion of segmented filamentous bacteria (SFB, Candidatus arthromitus) to WT ileum levels in the GATA4ΔIEC jejunum (Figure 2A and 2B; Figure S2A), where SFB adhered to IECs (Figure 2C). In WT mice, SFB colonize only the ileum, where they adhere to epithelial cells and induce an antigen-specific TH17 response11,12. Consistent with the lack of GATA4 expression in the WT ileum, GATA4ΔIEC mice demonstrated no changes in ileal bacterial composition (Figure S2A). To assess other commensal bacteria, we transplanted GF mice with altered Schaedler flora (ASF), a defined eight-member bacterial community, which resulted in an expansion of mucus-associated Mucispirillum schaedleri13 in the jejunum of GATA4ΔIEC, but not littermate control WT, mice (Figure S2B). These data suggest that epithelial GATA4 expression limits colonization by mucus-resident or adherent bacteria.
Figure 2. GATA4 prevents commensal and pathogenic bacteria from colonizing the jejunum.

(A) SFB load, as measured by qPCR, relative to the amount of host DNA in mucosal scrapings of jejunum and ileum from WT and GATA4ΔIEC mice. N= 18–19 mice/group.
(B) FISH staining using universal 16s rRNA probes (Alexa 546, red-orange), SFB 16s probe (Alexa 488), and counterstained with DAPI (blue). The overlay of the 16s probes (yellow-orange) represents SFB. Figure is a representative image from 4 independent WT and GATA4ΔIEC mice.
(C) Transmission electron microscopy of SFB adhering to jejunal IECs of GATA4ΔIEC mice. Figure is a representative image from 3 separate mice and a minimum of 5 different areas of view.
(D) C. rodentium load, measured by qPCR relative to host DNA, in distinct intestinal in WT and GATA4ΔIEC mice. N= 13 mice/group.
(E) Bacterial loads of wild-type C. rodentium and the ΔEAE mutant, measured by qPCR relative to host DNA, in distinct intestinal segments of GF WT or GATA4ΔIEC mice. N= 7–9 mice/group. All data in this figure are pooled from at least two-independent experiments and represented as mean ± SEM. **** P<0.0001, *** P<0.001, ** P<0.01, * P<0.05, Kruskal-Wallis with Dunn multiple comparison test (A, E), Mann-Whitney test (D).
To assess whether GATA4 indeed plays such a role, we analyzed the colonization pattern of rat SFB, which cannot adhere to mouse IECs but can stably colonize the lumen of GF mice14. In contrast to mouse SFB, rat SFB showed no difference in its capacity to colonize the lumen of WT and GATA4ΔIEC mice (Figure S2C). The capacity of SFB to adhere may be a critical feature driving its regionalization in a GATA4-dependent manner; however, this cannot be formally demonstrated because rat SFB is only 86% identical to mouse SFB, and other biochemical activities may be involved15. We therefore extended our analysis to Citrobacter rodentium, an adherent pathogen that preferentially colonizes the colon16,17 and can be genetically modified. In the absence of GATA4, the niche for C. rodentium was altered such that, by seven days after infection, the pathogen colonized the small intestine at levels approaching those in the colon of WT mice (Figure 2D). Using the mutant ΔEAE C. rodentium, which lacks the gene intimin required for adherence to IECs, we confirmed that C. rodentium’s capacity to colonize the small intestine of GATA4ΔIEC mice depended on its ability to adhere to IECs (Figure 2E).
Taken together, these results suggest that a key role of GATA4 is to prevent adherent bacteria from interacting with IECs of the proximal small intestine, whose primary function is to ensure the absorption of nutrients. They also suggest that the tissue microenvironment in the jejunum, created through the expression of GATA4, actively prevents colonization of the small intestine by colonic bacteria.
Changes in SFB colonization enhance inflammatory T cell immunity to C. rodentium
We next sought to determine whether the presence of SFB adhering to jejunal IECs was required and sufficient for the observed increase in inflammatory host immunity in the jejunum of GATA4ΔIEC mice (Figures 1B–E). To evaluate necessity, microbial communities lacking mouse SFB were transplanted into GF GATA4ΔIEC mice. Specifically, GF GATA4ΔIEC mice were transplanted with: (i) ASF, (ii) jejunal microbiota from a WT donor within our colony in which SFB was undetectable, and (iii) fecal microbiota from SFB-free C57BL/6J mice from Jackson laboratory (JAX). In all instances, these microbes failed to induce IFNγ+ CD8αβ+ T cells and TH17 cells in the jejunum of GATA4ΔIEC mice (Figures 3A and 3B; Figures S3A and S3B). To determine the sufficiency of SFB, we supplemented JAX microbiota or monoassociated GF GATA4ΔIEC mice with SFB and observed a significant induction of IFNγ+ CD8αβ+ T cells and TH17 cells in the jejunum of GATA4ΔIEC mice to SPF levels (Figures 3A and 3B). Furthermore, non-adherent rat SFB was unable to induce appreciable T cell responses in the jejunum of GATA4ΔIEC mice (Figures 3C and 3D). Finally, to exclude the possibility that another microbe intrinsic to the GATA4ΔIEC microbiota was driving inflammation, we transplanted GATA4ΔIEC and littermate control WT microbiota into GF WT and GF GATA4ΔIEC hosts, respectively. We found that the host genotype determined the immune outcome, irrespective of the input microbial community (Figures S3A and S3B). These data conclusively demonstrate that jejunal colonization of SFB drives the loss of compartmentalization of inflammatory T cell immunity seen in GATA4ΔIEC mice.
Figure 3. SFB colonization of the proximal small intestine drives excessive inflammatory T cell responses to C. rodentium infection.

(A, B) Frequency of IFNγ+ cells among CD8αβ+ T cells (A) or IL-17a+ cells among CD4+ T cells (B) in the jejunum from SPF, GF, Jackson (JAX) microbiota transfer, and Jackson microbiota + SFB transfer into GF WT and GATA4ΔIEC mice. N= 4–6 mice/group.
(C, D) Frequency of IFNγ+ cells among CD8αβ+ T cells (I) and of IL-17+ cells among CD4+ T cells (J) in jejunum of GATA4ΔIEC mice monocolonized with rat or mouse SFB. N= 5 mice/group.
(E) C. rodentium load, measured by qPCR, in distinct intestinal segments in SFB free (open circles) or SFB colonized (filled circles) GATA4ΔIEC mice. N= 5–6 mice/group.
(F) Representative (left) plots and summarized (right) of IFNγ+ and TNF+ CD8αβ+ IEL T cells from the jejunum of GATA4ΔIEC mice that are colonized with JAX (open circle) or JAX + SFB (solid circle) and either uninfected (− C.r) or infected (+ C.r) with C. rodentium. Red box indicates double IFNγ+ TNF+ CD8αβ+ T cells which are summarized (right). Mice were analyzed 5 days after infection. N= 4–5 mice/group.
All data in this figure are pooled from at least two-independent experiments and represented as mean or mean± SEM. **** P<0.0001, *** P<0.001, ** P<0.01, * P<0.05, ns P> 0.05, t-test (A-D), Mann-Whitney test (E), ANOVA with Tukey multiple comparison test (F).
Since SFB colonization induces an antigen-specific TH17-cell response against the 3340 epitope of SFB 12, we asked if the increased TH17 cell response observed in the jejunum was a consequence of altered T cell priming. Congenically marked CD45.1+ 7B8+ CD4+ T cells, specific to the 3340 epitope of SFB, differentiated into RORγt+ Foxp3− CD4+ T cells selectively in the draining ileal mesenteric lymph nodes (MLNs) of WT mice, as previously reported3. In contrast, in GATA4ΔIEC mice, RORγt+ Foxp3− CD4+ T cells were expanded in the jejunal MLNs, indicating a change in regional T cell priming against SFB (Figure S3C). Furthermore, nine days after transfer, 7B8+ T cells expanded (Figure S3D) and decreased expression of their TCR in the jejunum of GATA4ΔIEC versus WT mice (Figure S3E). These data indicate that the jejunal bacterial colonization resulting from GATA4 deficiency causes priming of SFB-specific T cells in the jejunal draining lymph nodes, as well as their expansion and activation in the proximal portion of the small intestine.
We next investigated whether altered regionalization of SFB led to dysregulated host immune responses to a competing pathogen in the proximal small intestine. In line with previous studies11, the presence of SFB decreased the load of C. rodentium in the colon of WT mice (Figure S3F). However, in GATA4ΔIEC mice, SFB did not decrease the load of C. rodentium in either the small intestine or the colon (Figure 3E). Furthermore, the presence of SFB promoted excessive inflammatory immune responses to C. rodentium in the jejunum of GATA4ΔIEC mice (Figure 3F; Figure S3G). Specifically, there was a marked increase in TNF+ IFNγ+ CD8αβ+ IEL T cells (Figure 3F) and IFNγ+ CD4+ T cells from the lamina propria (LP) (Figure S3G) as early as 5 days post infection, a time at which C. rodentium did not induce an adaptive immune response in the jejunum in SFBnegative GATA4ΔIEC mice (Figure 3F; Figure S3G). In contrast, SFB-dependent homeostatic TH17 and CD8αβ+ IFNγ+ responses were not synergistically elevated by C. rodentium (Figures S3H and S3I).
Altogether, these results suggest that changes in SFB colonization in the jejunum alter the intestinal immune response to C. rodentium, in particular, by driving the expansion of inflammatory TNF+ IFNγ+ CD8αβ+ T cell immune responses.
Dysregulated immune responses to SFB drive TNF-induced immunopathology after infection
We asked whether the heightened and altered inflammatory immune response to C. rodentium infection observed in SFB-colonized GATA4-deficient mice led to increased pathology. GATA4ΔIEC mice developed severe colitis and villous atrophy in the ileum ten days after infection, symptoms not associated with the infection in WT littermate control mice (Figure 4A; Figure S4A). Furthermore, the intestinal barrier was compromised in GATA4ΔIEC mice with C. rodentium translocating to systemic sites, including the MLNs, liver, and spleen (Figure 4B). By day 12 post-infection, 87.5% of GATA4ΔIEC mice had died (Figure 4C), punctuating the critical role of GATA4-dependent intestinal regionalization in controlling host disease susceptibility to an enteric pathogen. Consistent with SFB driving dysregulated inflammatory immune responses to C. rodentium, albeit without altering C. rodentium colonization, the increased mortality observed in GATA4ΔIEC mice was dependent on the presence of SFB (Figure 4D).
Figure 4. Dysregulated SFB colonization of the proximal intestine promotes loss of barrier function and TNF induced immunopathology upon C. rodentium infection.

(A) Representative H&E staining of each intestinal region 10 days after C. rodentium infection.
(B) CFUs of C. rodentium translocation to MLN, liver, and spleen. N= 8–10 mice/group.
(C) Percent survival of WT and GATA4ΔIEC mice 0–15 days post C. rodentium infection. N= 8–9 mice/ group.
(D) Percent survival of JAX colonized WT (blue) and GATA4ΔIEC (red) in SFB associated (solid lines) or SFB free mice (dashed lines) 0–20 days post C. rodentium infection. N= 6 WT mice/group, N= 9 GATA4ΔIEC – SFB mice/group, N= 10 GATA4ΔIEC + SFB mice/group.
(E) Relative expression as measured by qPCR of tight junction proteins to GAPDH in the jejunum of SFB free (open circles) or SFB colonized (filled circles) GATA4ΔIEC mice 5 days after infection. N= 5–6 mice/group.
(F) CFUs of C. rodentium translocation to MLN of SFB free or SFB colonized GATA4ΔIEC mice 5 days after infection. N= 5–6 mice/group.
(G) CFUs of C. rodentium translocation to MLN of SFB positive WT isotype, GATA4ΔIEC isotype, or GATA4ΔIEC αTNF treated mice 5 days after infection. N= 4 mice/group.
(H) Percent survival of WT isotype treated, and GATA4ΔIEC isotype treated, or αTNFα treated mice 0–15 days post C. rodentium infection. N= 7–9 mice/ group.
All data in this figure are pooled from at least two-independent experiments and represented as mean or mean± SEM. **** P<0.0001 , *** P<0.001, ** P<0.01, * P<0.05, Mann-Whitney test (B), Mantel-Cox test (C, D, H), t-test (E), Mann-Whitney test (F), ANOVA with Tukey multiple comparison test (G).
We next pursued how SFB increased the mortality of GATA4ΔIEC mice infected by C. rodentium. TNF and IFN-γ can disrupt intestinal epithelial barrier function18–20. We hypothesized that the synergistic effect of SFB and C. rodentium on inflammatory TNF and/or IFN-γ immune responses (Figure 3F) disrupted epithelial barrier function and thereby caused bacterial translocation. In accordance with that hypothesis, SFB-colonized GATA4ΔIEC mice infected with C. rodentium had decreased expression of tight junction and barrier proteins (Figure 4E), and increased translocation of C. rodentium to the MLNs (Figure 4F), compared to SFB-free, infected GATA4ΔIEC mice. In line with other studies21,22, treatments that neutralize IFN-γ and IL-17a increased mouse mortality (Figure S4B). In contrast, anti-TNF treatment reduced C. rodentium translocation (Figure 4G), restored expression of Tjp2 (Figure S4C), and increased survival (Figure 4H), consistent with a previous finding that TNF-neutralizing antibodies restore barrier function in Crohn’s disease patients20.
Taken together, these data highlight how, in the context of GATA4-deficiency, SFB promotes C. rodentium-induced immunopathology by increasing dysregulated TNF-producing T cell responses. More generally, this observation reveals a previously unknown role of commensal bacteria regionalization in promoting pathogenic versus protective immune responses to pathogens.
GATA4 regulates retinol metabolism and luminal IgA levels to control colonization of SFB
Based on our finding that SFB colonization of the proximal small intestine in GATA4ΔIEC mice was responsible for the severe immunopathology observed upon C. rodentium-infection, we sought to understand how GATA4 restricts SFB colonization of the WT proximal small intestine. A previous report revealed that B-cell deficient mice display lipid metabolic defects in the jejunum, as well as a gene expression signature associated with GATA4ΔIEC mice23. We therefore asked whether B-cell deficient JH mice, which lack the JH gene segments necessary for BCR recombination24, recapitulate the bacterial colonization defect observed in GATA4 deficient mice (Figure 2). Monocolonization of GF JH deficient mice with C. rodentium or SFB led to their expansion in the jejunum (Figures S5A–S5C), phenocopying GATA4ΔIEC mice. Since a substantial proportion of the microbiota, and in particular, SFB, is coated by IgA25–27, we asked whether the change in bacterial colonization in B-cell deficient mice was mediated through IgA. In agreement with previous studies28, SFB expanded in the ileum of IgA-deficient (Igha−/−) mice (Figure S5D). More importantly, monocolonization of GF Igha−/− mice with SFB led to an expansion in the jejunum to levels equivalent to those found in the ileum of WT mice (Figures 5A and 5B). In contrast, C. rodentium was not altered in IgA deficient mice (Figure S5E). This finding is consistent with C. rodentium being coated in the intestinal lumen by IgG and not IgA29, and suggests that GATA4, through yet unknown mechanisms that may involve changes in the metabolic milieu, prevents C. rodentium colonization of the small intestine.
Figure 5. GATA4 regulates regionalization of retinol metabolism and IgA to limit SFB colonization in the proximal intestine.

(A) FISH staining of SFB (Cy5) in monocolonized IgA deficient (Igha−/−) and littermate control (Igha+/−) mice and counterstained with DAPI.
(B) SFB load, as measured by qPCR, in mucosal scrapings from the jejunum and ileum of control (Igha+/−) mice and the jejunum of IgA deficient (Igha−/−) mice. N= 7–8 mice/group.
(C) Number of IgA+ B220− plasma cells, in the jejunum and ileum tissue of WT and GATA4ΔIEC mice. N= 5 mice/group.
(D) Amount of sIgA, as determined by enzyme-linked immunoassay (ELISA), in contents of the jejunum.
(E) Frequency of IgA coated bacteria after staining of Rag1−/− feces with supernatant from WT and GATA4ΔIEC jejunal contents. N= 4–5 mice/group.
(F) SFB loads, in jejunal mucosal scrapings of PBS-treated WT or GATA4ΔIEC mice, and IgAsupplemented GATA4ΔIEC mice. N= 5–7 mice/group.
(G) Heatmap of z-scored expression of region-specific GATA4-regulated genes in the KEGG retinol metabolism pathway, from RNA-seq on epithelial cells. Compared to other genes in the pathway expressed in epithelial samples, these genes are significantly enriched in GATA4-bound promoters, as determined by ChIP-seq (black squares in the annotation column) (Table S1; odds ratio 2.6, P < 0.005; Fisher’s exact test).
(H) Top, representative histogram of ALDH activity by ALDEFLUOR staining in jejunal epithelial cells. WT epithelial cells treated with ALDH inhibitor are shown as negative control for background fluorescence. Bottom, summary plots show the normalized geometric mean fluorescence intensity (gMFI) of ALDEFLUOR staining in epithelial cells from the jejunum and ileum of WT and GATA4ΔIEC mice. N= 6 mice/group.
(I) Total IgA in the jejunal contents of WT, GATA4ΔIEC vehicle-treated, and GATA4ΔIEC RA-treated mice after 14 days. N= 4 mice/group.
(J) SFB loads, in jejunal mucosal scrapings of GF WT mice fed a control or vitamin A deficient diet and subsequently colonized with SFB. N= 5 mice/group.
(K) Pigr expression as measured by qPCR relative to Gapdh, in the jejunum of WT and GATA4ΔIEC mice. N= 7 mice/group.
All data in this figure are pooled from at least two-independent experiments and represented as mean± SEM. **** P<0.0001 , *** P<0.001, ** P<0.01, * P<0.05, Kruskal-Wallis with Dunn multiple comparison test (B), ANOVA with Tukey multiple comparison test (C, F, H, I), t-test (D, E, K), Mann Whitney test (J).
Given these observations, we asked whether GATA4 may control SFB colonization by regulating the regional distribution of IgA+ plasma cells in the small intestine. We found that the jejunum contained approximately three times as many IgA+ B220− plasma cells as the ileum (Figure 5C), and that the higher numbers of IgA-producing plasma cells were associated with a greater capacity to produce IgA in tissue explants (Figure S5F). This regionalization of IgA response was GATA4-dependent, as evidenced by significantly reduced numbers of IgA+ B220− plasma cells (Figure 5C) and IgA production (Figure S5F) in the jejunum of GATA4ΔIEC versus WT mice. The overall result was a substantial decrease in luminal secretory IgA in the jejunum of GATA4ΔIEC mice (Figure 5D). Reduced luminal IgA was also observed in the jejunum of GF GATA4ΔIEC mice, indicating that the reduction is independent of microbiota (Figure S5G). Moreover, free-IgA in the jejunal luminal content from GATA4ΔIEC mice had less capacity than that of littermate-control WT mice to coat an IgA-unbound microbiota taken from the feces of Rag1−/− mice (Figure 5E; Figure S5H). We next tested whether exogenous luminal IgA was sufficient to rescue the luminal IgA defect and prevent colonization of the jejunum by SFB in GATA4ΔIEC mice. Polyclonal luminal sIgA, capable of strongly coating microbes from Rag1−/− feces, was isolated from the intestinal contents of WT mice with protein L magnetic beads (Figure S5I). This luminal polyclonal IgA or phosphate-buffered saline (PBS) was gavaged to GF WT or GATA4ΔIEC mice prior to and after colonization with SFB (Figure S5J). This IgA was indeed sufficient to prevent SFB from colonizing the jejunum of GATA4ΔIEC mice (Figure 5F). These results indicate that GATA4 dependent regulation of luminal IgA in the proximal small intestine prevents SFB colonization.
Since the IgA defect was microbiota independent, we hypothesized that epithelial GATA4 mediated IgA levels by controlling region-specific metabolic processes. To identify potential candidates, we performed gene set enrichment analysis of the differentially expressed, i.e., GATA4-dependent region-specific, genes in our epithelial RNA-seq data and in published GATA4TG data (Figures S1B and S1D), which revealed retinol metabolism as a top enriched KEGG pathway (Figures S5K and S5L). Genes in the retinol metabolic pathway were elevated in the jejunum of WT mice, relative to ileum-like tissues (Figure 5G), supporting previous reports that the proximal intestine facilitates greater vitamin A uptake and metabolism30,31. Conversely, GATA4TG mice induced retinol metabolic genes in the ileum, indicating that GATA4 was both necessary and sufficient to control its regionalization (Figure S5L).
In fact, direct transcriptional regulation of retinol metabolism by GATA4 in IECs was suggested by our analysis of published ChIP-seq data9,10, which found an enrichment of GATA4-binding promoters among the differentially expressed, versus not differentially expressed, epithelial genes in the retinol metabolism pathway (Figure 5G; Table S1; odds ratio 2.6, P < 0.005; Fisher’s exact test). The rate limiting enzyme in RA production, Aldh1a1 (RALDH1), was directly bound by GATA4 in a ChIP-seq study10 (Figure 5G). Concordantly, in the absence of GATA4, jejunal epithelial cells exhibited impaired aldehyde dehydrogenase (ALDH) activity, indicating a decreased capacity to produce retinoic acid (RA) (Figure 5H). RA regulates intestinal B cell responses, including intestinal homing receptors on B cells (CCR9, α4β7) and IgA class switching32. In addition, epithelial RARα/β regulates the number of IgA-producing B cells33,34. To determine whether the defect in luminal IgA in the small intestine of GATA4ΔIEC mice could be rescued with exogenous RA, we injected GATA4ΔIEC mice intraperitoneally with RA for two weeks. RA augmented luminal IgA levels in the jejunum of GATA4ΔIEC but not WT mice (Figure 5I; Figure S5M). Conversely, in mice fed a vitamin-A deficient diet, SFB colonized the jejunum (Figure 5J). Exogenous RA did not fully restore WT levels of IgA and failed to reduce jejunal SFB in GATA4ΔIEC mice (Figure S5N). This result suggests that GATA4 regulates luminal IgA levels through additional mechanisms. In line with this hypothesis, we observed that GATA4 induces jejunal expression of the polymeric immunoglobulin receptor (PIGR) (Figure 5K), which regulates IgA transcytosis35.
Taken together, these data indicate that, by controlling regional retinol metabolism, expression of PIGR, and potentially other aspects of IECs, GATA4 regulates luminal IgA, which in turn restricts SFB colonization of the proximal small intestine.
Loss of GATA4 expression in celiac disease is associated with regional tissue defects and increased IL-17 immunity.
Celiac disease (CeD) is an auto-immune–like TH1-mediated enteropathy of the duodenum caused by dietary gluten in genetically susceptible individuals36. A long-standing conundrum has been the increase of gluten-dependent, but not gluten-specific, duodenal TH17 responses in CeD patients37–39. During active CeD (ACeD), patients exhibit alterations in IECs36,40, as well as lipid41 and vitamin deficiencies42. We therefore hypothesized that ACeD patients have decreased expression of GATA4 in IECs, and that this decrease could be associated with reported changes in their microbiota43,44 and increased IL-17 immunity.
To investigate this possibility, we compared the transcriptional profiles of duodenal biopsies from ACeD patients, CeD patients on a gluten free diet (GFD), and control patients with non-inflammatory intestinal symptoms that required upper endoscopies45. The analysis revealed that a subset of active celiac patients had lower GATA4 expression, compared to controls, which was restored by GFD (Figure 6A). Immunohistochemistry staining confirmed at the protein level that there are ACeD patients with low and high GATA4 expression (Figure 6B). Furthermore, in ACeD, loss of GATA4 protein production was specifically seen in apical epithelial cells, whereas intestinal crypts cells retained GATA4 production (Figure 6B). Overall GATA4 expression was inversely correlated with severe tissue damage in ACeD, as measured by the APOA4/KI67 ratio46 (Figure S6A). However, we also observed that GATA4 expression could be preserved in ACeD with severe villous atrophy (Figure 6B). This observation suggests that other factors in addition to GATA4 influence the degree of tissue damage, and that IECs lining the damaged mucosa may exhibit distinct transcriptional programs, independently of the severity of villous atrophy.
Figure 6. Loss of GATA4 expression is associated with lipid metabolic dysfunction and increased IL-17 signaling in celiac disease.

(A) Normalized GATA4 expression in duodenal biopsies from healthy controls, active celiac disease patients (ACeD), and gluten free diet celiac patients (GFD).
(B) Representative IHC staining for GATA4 in healthy control, GATA4-hi and GATA4-lo active celiac disease patients, and gluten free diet celiac patients.
(C) Bar plot shows the percentages of human-mouse homologous genes specific to GATA4-hi or GATA4-lo individuals, which are also either GATA4-regulated and specific to the jejunum (purple) or to the ileum-like tissues (yellow), or not (gray). *** P <10−51, ** P <10−6, NS not significant.
(D) Bar plot shows the most significantly enriched gene ontologies and their significance (x axis, negative log FDR-adjusted P-values) in the intersection of genes specific to GATA-hi individuals and WT mouse jejunum.
(E) Single-sample gene set enrichment analysis (ssGSEA) scores for the retinol metabolism (left) and IL-17 downstream signaling (right) pathways in GATA4-hi and GATA4-lo individuals from all patient groups.
(F) Left, heatmap displays the scaled effect size of the absence or presence of five relevant bacteria (Fig. S4I) on GATA4 expression and on the ssGSEA scores of metabolic and immune pathways. Right, bar plot shows the numbers of detectable bacteria in ACeD samples.
(G) Box plots show GATA4 expression and ssGSEA scores for the retinol metabolism, IL17 downstream signaling, and IFNγ pathways in ACeD patients, grouped by the absence or presence of Actinobacillus.
**** P<0.0001 , *** P<0.001, ** P<0.01, * P<0.05, · P <0.1, Wilcoxon rank test (A, E, G), Fisher’s exact test (C), t-test (F).
To gain insight into the impact of low GATA4 expression in ACeD, we identified genes that were differentially expressed between 42 “GATA4-hi” individuals (18 control, 6 ACeD, and 18 GFD), defined by GATA4 expression above the 70th percentile of the entire cohort, and 15 “GATA4-lo” ACeD patients, defined by GATA4 expression below the 30th percentile of ACeD patients (Figure S6B; Table S3). Genes expressed in GATA4-lo patients were enriched, relative to GATA4-hi individuals, in the human orthologs of genes specifically expressed in mouse ileum-like tissues (WT ileum and GATA4ΔIEC jejunum), versus the jejunum (Figure 6C; Figure S6C). Similarly, GATA4-hi specific genes were enriched in human orthologs of genes specific to the mouse jejunum, relative to ileum-like tissues (Figures 6C; Figure S6C). Thus, during ACeD, intestinal regionalization may be lost as the duodenum decreases expression of GATA4-dependent jejunum-specific genes and increases expression of ileum-specific genes.
Many of the jejunal genes increased in GATA4-hi patients are direct targets of GATA4 and involved in lipid metabolic processes, such as cholesterol absorption and retinol metabolism (Figure 6D). Compared to GATA4-hi individuals, GATA4-lo ACeD patients demonstrated reduction in retinol metabolic genes and increased IL-17 signaling genes (Figure 6E; Figure S6D–S6H). In fact, expression of both pathways was correlated (or anti-correlated, in the case of IL-17 signaling genes) with GATA4 expression across all ACeD patients (Figures S6F and S6G). Together, these data reveal that loss of GATA4 expression in ACeD may play a role in regional defects, such as low retinol metabolism, low plasma cholesterol47, and increased IL-17 immunity. Several reports indicate that only a subset of ACeD patients have metabolic defects, and that they are not directly correlated with the degree of villous atrophy42,48. Our results suggest that heterogeneity in levels of GATA4 expression may contribute to the heterogeneity in metabolic defects observed in ACeD patients.
Similarly, while the IL-17 signaling pathway was overall significantly more highly expressed in GATA4-lo versus GATA4-hi individuals, heterogeneity in its expression across GATA4-lo patients (Fig. 6E) suggests that factors beyond loss of GATA4 expression contributed to TH17 cell responses in ACeD. In mice, the presence of TH17 cells in the ileum is dependent on the microbiota11 and bacteria can drive distinct context-dependent immune responses49. To determine whether particular microbes were implicated in promoting IL-17 responses in ACeD patients, we leveraged a quantitative framework to detect absolute abundances of individual bacterial taxa in duodenal biopsies50 along with concomitant host transcriptional analysis. Digital PCR anchoring of 16S rRNA amplicon sequencing revealed no change in overall bacterial load in ACeD patients versus controls (Figure S6I) and only a trend of stratification between ACeD patients and control in the second and third principal components (Figure S6J). We observed an expansion in ACeD patients of Neisseria, the only microbe with significant changes of abundance in the disease state (Figures S6K and S6L; Table S4), confirming a previous report51. Increased abundance of Neisseria was not, however, associated with decreased GATA4 expression, metabolic defects, or IL-17 signaling in ACeD patients (Figure 6F; Table S5).
In contrast, we found that Actinobacillus was associated with lower GATA4 expression, lower retinol metabolism, and higher IL-17 signaling in ACeD patients, but not controls (Figures 6F and 6G; Figure S6M; Table S5). We did not observe a significant association with the prototypical gluten-specific TH1 IFNγ pathway in ACeD patients, suggesting that Actinobacillus may play a specific role in IL-17 immunity (Figure 6F and 6G). Intriguingly, Actinobacillus was also associated with other metabolic defects in ACeD patients, such as in xenobiotic, bile acid, and heme metabolism, whereas other microbes, such as Streptococcus, had inverse patterns and positive associations with bile and heme metabolism (Figure 6F). These data also agree with the increased enrichment in IL-17 signaling genes in GATA4-lo ACeD patients but not controls.
The discovery of mucosal-associated bacteria, such as Actinobacillus, associated with the loss of GATA4, metabolic dysfunction, and IL-17 immunity in ACeD patients, highlights the potential importance of GATA4 dependent intestinal regionalization in the regulation of host microbial interactions and the pathogenesis of celiac disease.
Discussion
In this report, we identified mechanisms controlling regionalization of the proximal and distal small intestine and revealed the importance of this segregation in homeostasis and disease. We found that by regulating retinol metabolism and local IgA responses, the epithelial transcription factor GATA4 limited the colonization of the proximal small intestine by the commensal SFB, which in turn restricted inflammatory immune responses. Both GATA4-deficient mice and GATA4-lo ACeD patients showed signs of microbiota-dependent dysregulated inflammatory T cell responses after infection or gluten ingestion, respectively. In particular, failure to restrict SFB colonization of the jejunum in GATA4ΔIEC mice altered the immune response to the pathogen C. rodentium and promoted severe TNF-induced pathology and mortality. Furthermore, the intriguing association between loss of GATA4 expression in IECs, metabolic defects, and microbial-associated dysregulated IL-17 immune responses in ACeD patients poses the question of the role of GATA4 and IL-17 in regulating the severity of tissue damage and driving at least some of the clinical heterogeneity observed in ACeD patients. Beyond CeD, it raises the general question of whether a decrease in GATA4 in IECs of the proximal intestine, by altering host-microbial interactions and triggering dysregulated immune responses to bacteria, may play a role in other complex immune disorders. In contrast, the ileum, lacking GATA4 expression and producing less IgA, permits adherent microbes, such as SFB, to induce inflammatory IL-17+ T cell responses required for controlling pathogenic infections11,52. Therefore, there is a change in the tradeoff between the protective host immune responses (IgA) and inflammatory immune responses (IFNγ and IL-17) in the proximal versus distal small intestine. Our work suggests that proper compartmentalization of epithelial programs and commensal bacteria colonization in the small intestine is critical for establishing immune homeostasis and preventing disease. Previous reports identified decreased IgA coating of fecal bacteria in celiac patients, and an increased prevalence of celiac disease in IgA-deficient patients53,54. Whether GATA4 deficiency drives this IgA defect in celiac patients remains to be investigated.
Another reason for bacterial and immune regionalization could be that the proximal small intestine evolved to maximize nutrient digestion and absorption to increase host fitness. Preventing adherent bacterial colonization and the development of inflammatory immune responses in the proximal small intestine may support these vital digestive functions. Together, our findings emphasize the need to integrate signals of regional changes in host cells and in commensal bacteria colonization to decipher the mechanisms underlying the development of complex immune disorders.
Limitations of the Study
Here we find that GATA4 is an essential regulator of metabolic, immune, and microbial regionalization between the proximal and distal small intestine. To maintain this regionalization, GATA4 directly controls many biological pathways. While IgA was necessary and sufficient to control SFB colonization, retinol metabolism was not. Furthermore, supplementing retinoic acid to GATA4 deficient mice was not sufficient to fully restore luminal IgA to WT levels. Finally, we have evidence that GATA4 prevents C. rodentium from colonizing the proximal small intestine independently from IgA through yet unknown mechanisms. Together these data suggest that other GATA4-dependent mechanisms are involved in regulating adherent bacteria colonization and luminal IgA, the identification of which will require future studies.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bana Jabri (bjabri@bsd.uchicago.edu).
Materials availability
All reagents generated or used in this study are available on request from the lead contact with a completed Materials Transfer Agreement. Information on reagents used in this study is available in the key resources table.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD45 Pac Blue (30-F11) | Biolegend | Cat#103126 |
| TCRgd FITC (eBioGL3) | Thermo Fisher | Cat#11–5711-82 |
| CD4 BV785 (GK1.5) | Biolegend | Cat#100453 |
| CD4 BV605 (GK1.5) | Biolegend | Cat#100451 |
| CD8β BUV395 (H35–17.2) | BD | Cat#740278 |
| CD8α PerCp/Cy5.5 (53–6.7) | BD | Cat#551162 |
| NK1.1 PE-CF594 (PK136) | BD | Cat#562864 |
| TCRβ BUV737 (H57–597) | BD | Cat#612821 |
| TCRβ BV711 (H57–597) | BD | Cat#563135 |
| CD3ε BUV737 (145–2C11) | BD | Cat#612771 |
| IFNγ APC (XMG1.2) | BD | Cat#554413 |
| TNF BB700 (MP6-XT22) | BD | Cat#566510 |
| IL10 PEcy7 (JES5–16E3) | Biolegend | Cat#505026 |
| IL17a PE (ebio17B7) | Thermo Fisher | Cat#12–7177-81 |
| CD45.1 Pac Blue (A20) | Biolegend | Cat#110722 |
| CD45.2 BUV395 (104) | BD | Cat#553772 |
| vβ14 TCR FITC (14–2) | BD | Cat#553258 |
| RORγt BV786 (Q31–37) | BD | Cat#564723 |
| FOXP3 eflour450 (FJK-16s) | Thermo Fisher | Cat#48–5773-82 |
| FOXP3 FITC (FJK-16s) | Thermo Fisher | Cat#11–5773-82 |
| FOXP3 PE-cy7 (FJK-16s) | Thermo Fisher | Cat#25–5773-82 |
| Tbet PE (4B10) | Biolegend | Cat#644810 |
| CD44 PE-CY7 (IM7) | Biolegend | Cat#103030 |
| CD62L PE (MEL-14) | Biolegend | Cat#104408 |
| Epcam PerCp/Cy5.5 (G8.8) | Biolegend | Cat#118220 |
| CD19 FITC (1D3/CD19) | Biolegend | Cat#152404 |
| NK1.1 BV605 (PK136) | Biolegend | Cat#108753 |
| CD11C BV605 (N418) | Biolegend | Cat#117334 |
| TER119 BV605 (TER-119) | Biolegend | Cat#116239 |
| F4/80 BV605 (BM8) | Biolegend | Cat#123133 |
| CD3ε BV605 (145–2C11) | Biolegend | Cat#100351 |
| Ly6g BV605 (1A8) | Biolegend | Cat#127639 |
| B220 PE-cy7 (RA3–6B2) | Biolegend | Cat#103222 |
| IgA PE (mA-6E1) | Thermo Fisher | Cat#12–4204-81 |
| IgA AF647 goat polyclonal | Southern Biotech | Cat#1040–31 |
| Anti-mouse IFNγ (XMG1.2) | BioXCell | Cat#BE0055 |
| Rat IgG1 isotype anti-HRP (HRPN) | BioXCell | Cat#BE0088 |
| Anti-mouse IL-17a (17F3) | BioXCell | Cat#BE0173 |
| Mouse IgG1 isotype (MOPC-21) | BioXCell | Cat#BE0083 |
| Anti-mouse TNFa (TN3–19.12) | BioXCell | Cat#BE0244 |
| Polyclonal hamster IgG | BioXCell | Cat#BE0091 |
| Anti-GATA4 (G4) | SantaCruz | Cat#sc-25310 |
| Bacterial and virus strains | ||
| Citrobacter rodentium DBS120 pler-lux | Alexander Chervonsky | 55 |
| Citrobacter rodentium DBS100 | Gabriel Nuñez | 14 |
| Citrobacter rodentium DBS100 ΔEAE | Gabriel Nuñez | 14 |
| Mouse segmented filamentous bacteria | Kenya Honda | 14 |
| Rat segmented filamentous bacteria | Kenya Honda | 14 |
| Biological samples | ||
| Fetal Bovine Serum | Biowest | Cat#S01520; Lot#A11504E |
| Normal Goat Serum | JacksonImmunoResearch | Cat#005–000-121 |
| Chemicals, peptides, and recombinant proteins | ||
| EDTA, 0.5M, pH8.0 | Corning | Cat#46–034-CI |
| 1M MgCl2 | Thermo Fisher | Cat#AM9530G |
| Cytiva Percoll™ Centrifugation Media | GE Healthcare | Cat#45–001-747 |
| RNAprotect Tissue Reagent | Qiagen | Cat#76106 |
| 2-Mercaptoethanol (BME) | Sigma-Aldrich | Cat#M7154 |
| Phorbol Myristate Acetate | Sigma-Aldrich | Cat#P1585 |
| Ionomycin Calcium Salt from Streptomyces conglobatus | Sigma-Aldrich | Cat#10634 |
| BD GolgiStop Protein Transport Inhibitor | BD | Cat#554724 |
| Ethanol 200 Proof | Decon Labs Inc | Cat#DSP-MD 43 |
| Inhibitex Buffer | Qiagen | Cat#19593 |
| Nuclease-free Water | Ambion | Cat#AM9932 |
| Carnoy Solution | Ricca Chemical | Cat#R18510004C |
| 10% Formalin Solution | Thermo Fisher | Cat#SF98–4 |
| RPMI 1640 with L-Glutamine | Corning | Cat#MT-10043CV |
| Collagenase from Clostridium histolyticum | Sigma-Aldrich | Cat#C2139–500MG |
| 1M TRIS-HCL pH 7.5 | Thermo Fisher | Cat#15567027 |
| 10% SDS solution | Thermo Fisher | Cat#15553027 |
| Sodium chloride | Sigma-Aldrich | Cat#S9888 |
| All-trans-Retinoic acid | Sigma-Aldrich | Cat#R2625 |
| Critical commercial assays | ||
| SytoBC | Thermo Fisher | Cat#S34855 |
| LIVE/DEAD® Fixable Aqua Dead Cell Stain Kit | Thermo Fisher | Cat#L34966 |
| Zombie NIR™ Fixable Viability Kit | Biolegend | Cat#423106 |
| BD Cytofix/Cytoperm Plus Fixation/Permeabilization Solution Kit | BD | Cat#554714 |
| eBioscience™ Foxp3 / Transcription Factor Staining Buffer Set | Thermo Fisher | Cat#00–5523-00 |
| QIAamp Fast DNA Stool Mini Kit | Qiagen | Cat#51604 |
| RNeasy Plus Mini Kit | Qiagen | Cat#74136 |
| RNeasy Micro Kit | Qiagen | Cat#74004 |
| GoScript Reverse Transcriptase Kit | Promega | Cat#A5001 |
| SYBR Advantage qPCR Premix | Clontech | Cat#639676 |
| ProLong™ Diamond Antifade Mountant with DAPI | Thermo Fisher | Cat#P36962 |
| Pierce™ Protein L Magnetic Beads | Thermo Fisher | Cat#88850 |
| Pierce™ IgG Elution Buffer, pH 2.0 | Thermo Fisher | Cat#21028 |
| 1.0 M Tris HCl pH 8.5 | VWR | Cat#76236–402 |
| ALDEFLUOR™ Kit | Stemcell technologies | Cat#01700 |
| IgA mouse ELISA | Thermo Fisher | Cat#88–50450-86 |
| QX200 droplet digital PCR system | BioRad | Cat#1864001 |
| QX200 ddPCR EvaGreen Supermix | BioRad | Cat#1864034 |
| 5Prime Hotstart Mastermix | QuantaBio | Cat#2200410 |
| Evagreen | Biotium | Cat#31000 |
| KAPA library quantification kit | Roche | Cat#07960140001 |
| AmpureXP beads | Beckman Coulter | Cat#A63880 |
| Deposited data | ||
| RNA sequencing data | This paper | GEO: GSE205743 |
| 16S rRNA sequencing data | This paper | BioProject: PRJNA797871 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Mouse: GATA4fl/fl villin-cre | Polly Matzinger laboratory | 23 |
| Mouse: germ-free GATA4fl/fl villin-cre | This study | N/A |
| Mouse: B6 GATA4fl/fl villin-cre | This study | N/A |
| Mouse: C57BL/6J | Jackson laboratory | JAX: 000664 |
| Mouse: B6-Tg(Tcra, Tcrb)2Litt/J | Jackson laboratory | JAX: 027230 |
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ | Jackson laboratory | JAX: 002014 |
| Mouse: B.6129S7-Rag1tm1mom/J | Jackson laboratory | JAX: 002216 |
| Mouse: CD-1 IGS | Charles River | Crl: CD1 |
| Mouse: B6 Jh-/- | Albert Bendelac laboratory | N/A |
| Mouse: B6 IgA-/- | Albert Bendelac laboratory | N/A |
| Oligonucleotides | ||
| GAPDH Forward 5′-AGGTCGGTGTGAACGGATTTG-3′ | 45 | N/A |
| GAPDH Reverse 5′-TGTAGACCATGTAGTTGAGGTCA-3′ | 45 | N/A |
| IFNγ Forward 5′-GGATGCATTCATGAGTATTGC-3′ | 45 | N/A |
| IFNγ Reverse 5′-CCTTTTCCGCTTCCTGAGG -3′ | 45 | N/A |
| IL-17a Forward 5’-TTTAACTCCCTTGGCGCAAAA-3’ | This study | N/A |
| IL-17a Reverse 5’-CTTTCCCTCCGCATTGACAC-3' | This study | N/A |
| TNFa Forward 5’-TGGGAGTAGACAAGGTACAACCC-3’ | This study | N/A |
| TNFa Reverse 5’-CATCTTCTCAAAATTCGAGTGACAA-3’ | This study | N/A |
| ASL Forward 5’-TCTTCGTTAGCTGGCAACTCACCT-3’ | 56 | N/A |
| ASL Reverse 5’-ATGACCCAGCAGCTAAGCAGATCA-3’ | 56 | N/A |
| Uni 16S 340F 5’-ACTCCTACGGGAGGCAGCAGT-3’ | 57 | N/A |
| Uni 16S 514R 5’-ATTACCGCGGCTGCTGGC-3’ | 57 | N/A |
| SFB 736F 5’-GACGCTGAGGCATGAGAGCAT-3’ | 57 | N/A |
| SFB 844R 5’-GACGGCACGGATTGTTATTCA-3’ | 57 | N/A |
| ASF457F 5’ -TGCAAGAATGAAACTCAAAGGAAT-3’ | 58 | N/A |
| ASF457R 5’- TAAGGTTCTTCGGTTAGCATCGA-3’ | 58 | N/A |
| C. rodentium espBF 5’- ATGCCGCAGATGAGACAGTTG-3’ | 59 | N/A |
| C. rodentium espBR 5’-CGTCAGCAGCCTTTTCAGCTA-3’ | 59 | N/A |
| EUB338 5'-Alexa546-GCTGCCTCCCGTAGGAGT-3' | 60 | N/A |
| EUB338 5'-Cy3-GCTGCCTCCCGTAGGAGT-3' | 60 | N/A |
| SFB1008 5'-Alexa488-GCGAGCTTCCCTCATTACAAGG-3' | 57 | N/A |
| SFB1008 5'-Cy5-GCGAGCTTCCCTCATTACAAGG-3' | 57 | N/A |
| Uni 16S 519F 5'-CAGCMGCCGCGGTAA-3’ | 50 | N/A |
| Uni 16S 806R 5'-GGACTACHVGGGTWTCTAAT-3’ | 50 | N/A |
| Tjp1F 5'-AGGACACCAAAGCATGTGAG-3’ | 61 | N/A |
| Tjp1R 5'-GGCATTCCTGCTGGTTACA-3’ | 61 | N/A |
| Tjp2F 5'-ATGGGAGCAGTACACCGTGA-3’ | 61 | N/A |
| Tjp2R 5'-TGACCACCCTGTCATTTTCTTG-3’ | 61 | N/A |
| Tjp3F 5'-TCGGCATAGCTGTCTCTGGA-3’ | 61 | N/A |
| Tjp3R 5'-GTTGGCTGTTTTGGTGCAGG-3’ | 61 | N/A |
| OclnF 5'-GCTGTGATGTGTGTGAGCTG-3’ | 61 | N/A |
| OclnR 5'-GACGGTCTACCTGGAGGAAC-3’ | 61 | N/A |
| Cldn2F 5'-GGCTGTTAGGCACATCCAT-3’ | 61 | N/A |
| Cldn2R 5'-TGGCACCAACATAGGAACTC-3’ | 61 | N/A |
| Jam1F 5'-ACCCTCCCTCCTTTCCTTAC-3’ | 61 | N/A |
| Jam1R 5'-CTAGGACTCTTGCCCAATCC-3’ | 61 | N/A |
| Cdh1F 5'-TCCTTGTTCGGCTATGTGTC-3’ | 61 | N/A |
| Cdh1R 5'-GGCATGCACCTAAGAATCAG-3’ | 61 | N/A |
| Pigr F 5’-GTAACCGAGGCCTGTCCTTC-3’ | This study | N/A |
| Pigr R 5’-GTAGACGTGGGTGTCACTCG-3’ | This study | N/A |
| Recombinant DNA | ||
| Software and algorithms | ||
| QIIME2: 2020.2.0 | 62 | |
| Silva 132 SSURef NR99 | 63 | |
| Python 3.7.6 | https://www.python.org/ | |
| Scipy 1.4.1 | https://www.scipy.org/ | |
| Statsmodels 0.10.1 | https://www.statsmodels.org/stable/index.html | |
| Numpy 1.18.1 | https://numpy.org/ | |
| Pandas 1.0.3 | https://pandas.pydata.org/ | |
| GraphPad Prism 8 | https://www.graphpad.com | |
| FlowJo 10 | https://www.flowjo.com | |
| Other | ||
| Bead ruptor elite bead mill homogenizer | Omni International | Cat#19–040E |
| Tissue-Tearor | Biospec Products | Cat#985370-XL |
| Glass Beads | Biospec Products | Cat#11079101 |
| LightCycler® 480 System | Roche | N/A |
| LSRFortessa™ X-20 Flow Cytometer | BD | N/A |
| Cytek® Aurora | Cytek | N/A |
| CRi Pannoramic SCAN 40x Whole Slide Scanner | 3DHistech | N/A |
| EasyEights™ EasySep™ Magnet | Stemcell technologies | Cat#18103 |
| CFX96 Touch RT-PCR detection system | BioRad | Cat#1855196 |
| Mouse RNAseq analysis | This paper | http://doi.org/10.5281/zenodo.7255834. |
| Human RNAseq analysis | This paper | https://doi.org/10.5281/zenodo.7272314 |
Quantification and statistical analysis
Data were first analyzed for normal distribution using D’Agostino and Pearson omnibus normality tests. Normally distributed data were analyzed with unpaired two-tailed Student’s t-test when comparing two groups, one-way ANOVA followed by Tukey’s post-hoc test for multiple comparisons, or two-way ANOVA for comparing two groups against multiple variables. Not normally distributed data were analyzed using two-tailed Mann-Whitney test when comparing two groups, or Kruskal-Wallis with Dunn’s multiple comparison test for multiple comparisons. Data in all figures displayed are pooled from a minimum of two-independent experiments and represented as mean ± SEM when possible. Number of samples are reported in each figure legend. The statistical test used and P values are indicated in each figure legend and performed with GraphPad Prism 8. P values <0.05 were considered statistically significant. ****P<0.0001 , *** P<0.001, ** P<0.01, * P<0.05., or oooo P<0.0001 , ooo P<0.001, oo P<0.01, o P<0.05.
Data and code availability
All the data supporting the findings of the article are available within the main text or supplementary information. The published article includes datasets generated during this study. Original RNA-seq data has been deposited in GEO: GSE205743. Original 16S rRNA sequencing datasets analyzed in this study are available at the NCBI BioProject: PRJNA797871. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. Original code for analyzing these datasets have been deposited in Zenodo and is publicly available. DOIs are listed in the key resources table.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
7–12 week old mice were used for experiments, co-housed in specific pathogen-free conditions, and kept Helicobacter hepaticus, murine norovirus free at the University of Chicago. Some mice were also housed in gnotobiotic isolators and routinely checked for sterility by culture and 16S PCR or kept SFB monocolonized at the University of Chicago Gnotobiotic Research Animal Facility. GATA4fl/fl villin-cre SPF mice were previously generated in the CD1 background and obtained from the Matzinger laboratory 23.This line was rederived GF for this study and backcrossed for 10 generations to C57BL/6J background for T cell transfers. C57BL/6J, B6-Tg(Tcra, Tcrb)2Litt/J SFB TCRtg, B6.SJL-Ptprca Pepcb/BoyJ, B.6129S7-Rag1tm1mom/J were obtained from the Jackson Laboratory. CD-1 IGS mice were obtained from Charles River Laboratories. B cell deficient mice deficient for IgH J segment locus (JH), recreating the previously described model 24, were generated at University of Chicago and obtained from Dr. Bendelac at the University of Chicago on a C57BL/6 background using Cas9 with the protospacers GCTACTGGTACTTCGATGTC and GCCATTCTTACCTGAGGAGA. IgA deficient mice where the Sα (IgA switch region) and C1α (first exon) were deleted, as previously described 64, were generated at University of Chicago and obtained from Dr. Bendelac on a C57BL/6 background using Cas9 with the protospacers AAGCGGCCACAACGTGGAGG and TCAAGTGACCCAGTGATAAT. Jh and IgA deficient mice were rederived GF at Taconic Biosciences. Littermate controls of GATA4, Jh, and IgA were used for all experiments in this study. Mice were fed a standard chow diet, vitamin A control diet (Harlan TD.91280), or vitamin A deficient diet (Harlan TD. 86143). Animal husbandry and experimental procedures were performed in accordance with Public Health Service policy and approved by the University of Chicago Institutional Animal Care and Use Committees.
Patients
A duodenal biopsy was obtained from 166 individuals undergoing upper gastrointestinal endoscopy at the University of Chicago and at Mayo Clinic as previously reported 45. There were 64 control patients, 56 untreated patients with active celiac disease, and 46 patients treated with a gluten free diet. All control patients underwent endoscopies for issues unrelated to celiac disease and had normal intestinal histology, no family history of celiac disease, and no significant levels of anti-TG2 antibodies in the serum. Patients with active celiac disease contained positive anti-TG2 antibodies and small intestinal enteropathies with increased IEL infiltration, crypt hyperplasia, and villous atrophy according to the accepted diagnostic guidelines 65. The subjects signed an informed consent as provided by the Institutional Review Board of each institution (IRB-12623B for the University of Chicago, and IRB-1491–03 for the Mayo Clinic). DNA and RNA were isolated from each biopsy as described previously 45 using the AllPrep DNA/RNA mini kit (Qiagen).
Citrobacter rodentium infections
C. rodentium strains DBS100, DBS120 pler-lux, or DBS100 ΔEAE were grown at 37 °C in Luria broth under agitation 14,55. The cultures were diluted 100X and grew to log phase until the OD600nm reached 0.75. For gavage, 200 μl of bacteria were used, which gave a dose of 2.5X109 CFU/mouse. Mice were separated into cages based on genotype for infections, and male mice were used for survival studies. DBS100 or DBS100 ΔEAE strains were given to GF mice and DBS120 pler-lux was given to SPF mice. The DBS120 strain has a genomic kanamycin resistance cassette inserted through Tn5. To determine CFUs of DBS120, 2 fecal pellets/mouse were resuspended in 1 ml of PBS and plated on MacConkey agar containing 50 μg/ml of kanamycin. The CFU/mg feces concentration was determined as: (#CFU counted*Dilution factor/(vol plated in ml))/mg feces. To determine the amount of bacterial translocation, the MLN, liver, and spleen were aseptically dissected, weighed, and homogenized with the Tissue-Tearor rotor (BioSpec) in 500 μl of PBS. Then 200 μl of homogenate was plated on MacConkey agar containing 50μg/ml of kanamycin. For infections with SFB free mice, JAX colonized WT and GATA4ΔIEC mice were colonized with SFB as described below at approximately 6 weeks of age and infected with C. rodentium two weeks later. For cytokine neutralizations, WT and GATA4ΔIEC mice were treated i.p with 250μg of either isotype control or αTNF, αIL-17a, or αIFNγ neutralizing antibodies on days 3, 5, 7, and 9 after infection.
Microbial transfers
To colonize mice with SFB 55 or rat SFB 14, 3–4 fresh fecal pellets from SFB monocolonized mice were homogenized in 1 ml of PBS, vortexed for 3 min, and spun at 300 g to remove large debris. Then 200 μl of the homogenate were gavaged to recipient mice. When possible, SFB donor pellets were taken from monocolonized Jh or IgA deficient mice, which harbor 10-fold higher levels of SFB. To colonize mice with SFB-free microbiota, C57BL6 mice from Jackson Labs, which lack SFB in the microbiota, were used as donor mice. Small intestinal and cecal contents were pooled for one donor mouse homogenized in PBS, and gavaged to recipient mice with or without SFB supplemented. For WT and GATA4ΔIEC microbiota transfer to GF WT and GATA4ΔIEC hosts, jejunal content was pooled from two donor SPF GATA4ΔIEC mice or littermate WT mice. Colonization of ASF strains (Taconic) was performed as described previously 66 and gavaged to recipient WT and GATA4ΔIEC mice. For all microbial transfers, mice were colonized at 4 weeks of age and analyzed at 8 weeks.
Vitamin A deficient diet
GF C57BL6 mice were placed on control (Harlan TD.91280) or vitamin A deficient (Harlan TD. 86143) diets from 4 to 8 weeks of age. At 8 weeks, mice were monocolonized with SFB for one week as described above, and the amount of SFB in jejunal mucosal scrapings was quantified by qPCR.
SFB TCRtg adoptive transfer
Naïve SFB TCRtg Vβ8 CD4+ T cells were isolated from LNs and spleen of congenically marked CD45.1 Vβ8+/− female mice using the naïve CD4 T cell isolation kit (Miltenyi), and 2x105 cells/100μl mouse were injected retroorbitally into CD45.2 WT and GATA4ΔIEC. Three days after transfer, the mice were euthanized to assess T cell priming and activation in the jejunal and ileal draining MLN as described previously (Esterházy et al., 2019). To assess T cell expansion in the LP of the jejunum nine days after transfer, 50,000 cells were injected/mouse.
METHOD DETAILS
Isolation of intestinal epithelial cells (IEC), intraepithelial lymphocytes (IEL), and lamina propria (LP) cells
The segments of the intestine were excised as follows to isolate cells for flow cytometry: duodenum was taken 12 cm from the stomach, jejunum 12 cm from the middle, and ileum 12 cm from the cecum. Any leftover segments were discarded. The entire colon was taken after the cecum to the rectum. To isolate IEL and IECs, Peyer’s patches were first removed from the small intestine, and then the segments were opened longitudinally and washed briefly in phosphate-buffered saline (PBS). Epithelial cells, including IELs and LP cells, were isolated as previously described 67 using ethylenediaminetetraacetic acid (EDTA) containing calcium-free media and collagenase VIII (Sigma-Aldrich, C2139), respectively. The IEL and LP compartments were then subjected to a 40% percoll density gradient centrifugation step to remove dead cells and debris as previously described 45. The IEL and LP cells were then counted on a hemocytometer.
Cytokine stimulation
Up to 2x106 cells were collected and resuspended in RPMI 1640 media with 10% fetal bovine serum (FBS) and cultured in 48-well plates in the presence of 750 ng/ml of ionomycin, 50 ng/ml of Phorbol 12-myristate 13-acetate (Sigma-Aldrich), and golgi-stop (BD). The cells were incubated for 2 hours at 37C with 5% CO2. After stimulation, the reaction was quenched with ice cold FACS buffer, and the cells were subsequently stained with antibodies for flow cytometry.
Flow cytometry
The cells were first stained with FC block (CD16/32) to block nonspecific binding and then were stained with dead dye to exclude dead cells (Aqua, ThermoFisher or Zombie NIR, Biolegend) for 15 min at 4 °C, followed by staining with cell surface markers for 20 min at 4 °C. For intracellular cytokine staining, the BD cytofix/cytoperm kit was used, and cells were incubated with the antibodies for 40 min at 4 °C. For intracellular transcription factors, the Foxp3 eBioscience kit was used according to the manufacturer’s instructions. The antibodies used are indicated in Key resources table. For ALDH staining of IECs, the ALDEFLUOR kit was used (StemCell Technologies), following the manufacturer’s protocol. All cells were gated FSC, SSC, singlets, and live cells. IECs were gated CD45− EpCAM+. 100,000 IECs from the jejunum or ileum were sorted with Aria Fusion (BD Biosciences) into RLT buffer (Qiagen) with β-mercaptoethanol for downstream sequencing analysis. IgA plasma cells were gated as described previously 26, i.e. EpCAM−, CD45+/dim, lineage negative (Ter119, F4/80, CD3, Ly6G, NK1.1, CD19), IgA+, B220−. CD8αβ IELs were gated TCRβ+, CD4−, CD8α+, CD8β+. CD4+ LP T cells were gated TCRβ+, CD4+, CD8α. The following antibodies and clones were purchased from Biolegend: CD45 Pacific Blue (30-F11), CD4 BV785 (GK1.5), CD4 BV605 (GK1.5), IL10 PE-Cy7 (JES5–16E3), CD45.1 Pacific Blue (A20), Tbet PE (4B10), CD44 PE-Cy7 (IM7), CD62L PE (MEL-14), Epcam PerCP-Cy5.5 (G8.8), CD19 FITC (1D3/CD19), NK1.1 BV605 (PK136), CD11C BV605 (N418), TER119 BV605 (TER-119), F4/80 BV605 (BM8), CD3ε BV605 (145–2C11), Ly6G BV605 (1A8), B220 PE-Cy7 (RA3–6B2). The following antibodies and clones were purchased from BD: CD8β BUV395 (H35–17.2), CD8α PerCP-Cy5.5 (53–6.7), NK1.1 PE-CF594 (PK136), TCRβ BUV737 (H57–597), TCRβ BV711 (H57–597), CD3ε BUV737 (145–2C11), IFN-g APC (XMG1.2), TNF BB700 (MP6-XT22), CD45.2 BUV395 (104), vβ14 TCR FITC (14–2), RORγt BV786 (Q31–37). The following antibodies and clones were purchased from Thermo Fisher: TCRgd FITC (eBioGL3), IL17a PE (ebio17B7), FOXP3 eFluor450 (FJK-16s), FOXP3 FITC (FJK-16s), FOXP3 PE-Cy7 (FJK-16s), IgA PE (mA-6E1). The cells were run on the LSRFortessa X-20 Flow Cytometer (BD Biosciences) or the Cytek Aurora and data were analyzed using FlowJo software (Treestar).
DNA isolation
For mucosal scrapings for DNA isolation, 5 cm of tissue proximal to the middle of the intestine was taken for the jejunum, and 5 cm from the ileocecal valve was taken for the ileum. The entire colon was used for mucosal scrapings. The tissue was excised, opened longitudinally, scraped with a glass slide, transferred to 2 ml screw cap tube containing 0.1 mm glass beads (Bio-spec), and snap frozen on dry ice. For luminal content, 50–100 mg of content was taken from as close to the middle of the jejunum as possible and from the last 5–7 cm of the ileum. Homogenization was performed after adding 1 ml of inhibitex buffer (Qiagen) using the Bead Ruptor Elite bead mill homogenizer (Omni, 19040E) on speed 6 for 3 min. DNA was then extracted using the QIAmp Fast DNA stool mini kit (Qiagen) following the manufacturer’s protocol with the optional high temp (95 °C) lysis step. DNA concentration was determined using the nanodrop UV spectrophotometer (ThermoFisher).
RNA isolation
For eventual RNA purification, 1 cm of tissue was excised from the beginning of the duodenum, the middle of the jejunum, the end of the ileum, and the center of the colon and preserved in RNAprotect (Qiagen) overnight at 4 °C and then transferred to −80 °C for long-term storage. The tissue was transferred to 600 μl of RLT buffer containing β-mercaptoethanol (Qiagen) and homogenized for 30 sec with a hand held rotor (Tissue-Tearor, BioSpec). RNA was purified using RNAeasy plus mini kit (Qiagen) following the manufacturer’s protocol with the optional on column DNase digest (Qiagen).
qPCR
RNA was first reverse-transcribed to cDNA using GoScript Reverse Transcriptase kit (Promega) following the manufacturers protocol. For qPCR, 10 ng of cDNA or 20 ng of DNA from mucosal scrapings and content was used. TB green Advantage qPCR Premix (Takara) was used, and the target gene was quantified and normalized to the housekeeping gene as described previously 45 using 1000*2-(Ct target- Ct housekeeping) formula. For host gene expression, the target gene was normalized to GAPDH. For bacterial load, the target gene was normalized to either host DNA as described previously 57 with primers specific for host argininosuccinate lyase (ASL) gene or universal 16S primers. The qPCR was performed on the LightCycler 480 System (Roche). The primer pairs and DNA sequences are included in Key resources table.
Histology
The tissue was collected in the same manner as for RNA, placed in cassettes and fixed in 10% formalin for H&E staining or Carnoy solution (ThermoFisher) for fluorescent in situ hybridization (FISH) staining overnight at room temperature. Cassettes were transferred to 70% ethanol for formalin or 100% ethanol after Carnoy fixation to wash out the fixative. The tissue was embedded in paraffin, and slides were cut at 5 μm thickness. The H&E staining was performed by the Human Tissue Resource Center at the University of Chicago. For FISH staining, the paraffin was first removed by running the slides through four 3-min incubations in xylene and four 3-min incubations in 100% ethanol. The slides were then moved to a polypropylene slide container and filled with hybridization solution containing the diluted 16S probe (0.9M NaCl, 20mM Tris-HCL pH 7.5, 0.1% SDS with 0.2ng of probe specific for SFB 16S or universal 16S) 68. The 16S probes used are included in Key resources table. The slides were incubated overnight at 50 °C in the dark. The slides were washed three times with the hybridization buffer, briefly rinsed in H20, and then mounted with Prolong diamond antifade with DAPI (ThermoFisher). The slides were scanned with the CRi Pannoramic SCAN 40x Whole Slide Scanner at the University of Chicago Integrated Light Microscopy core.
Transmission Electron Microscopy
2cm of jejunum tissue was open longitudinally and fixed in 2% glutaraldehyde, 4% paraformaldehyde, in 0.1M sodium cacodylate buffer for 2 hours. The fixative was then replaced with 1% osmium tetroxide in 0.1M sodium cacodylate buffer for 60min. The tissue was subsequently washed 2X for 5 min with sodium cacodylate buffer and finally with maleate buffer (pH 5.1). 1% uranyl acetate in maleate buffer was added for 60 min, and the tissue was washed again with maleate buffer 3X for 5min. The tissue was next dehydrated by running through 25%, 50%, 70%, 95% ethanol for 2X 5min each and ending on 100% ethanol for 3X 15min. Finally 100% propylene oxide was added for 3X 15min. 2:1 propylene oxide spurr resin was added 2X 30 min, and 1:1 propylene oxide spurr resin was added 2X 30 min and overnight, next day 100% spur resin was added 6X 60min. The polymerized spur with embedded tissue was put into a 60C oven for 1–2 days. 90nm sections were cut by Leica EM UC6, stained with uranyl acetate and lead citrate. Images were examined under 300KV at FEI Tecnai F30 Gatan CCD digital micrograph.
ELISA
To quantify luminal IgA levels, content was collected from the jejunum and ileum and weighed in 2-ml bead beating tubes containing 0.1mm glass beads. After adding 1 ml of 1X cell lysis buffer with protease inhibitors (Cell Signaling Technologies), the content was homogenized on a vortex for 5 min. The debris were pelleted at 13000 rpm for 10 min, and the supernatant was collected for ELISA. For tissue explants, 1 cm of tissue was excised and opened longitudinally, washed in PBS, and placed in complete RPMI at 37 °C for 24 h. The culture supernatant was collected and used for ELISA. The supernatant was diluted in 1X assay diluent A (ThermoFisher), and the dilution in the middle of the standard curve was used to quantify IgA levels. IgA mouse uncoated ELISA kit (ThermoFisher) was used following the manufacturer’s protocol, and absorbance was read at 450 nm. The amounts of IgA were back calculated to the original sample and normalized relative to the weight of the content or to ml of culture supernatant.
Luminal IgA isolation and in vivo treatment
To isolate luminal polyclonal sIgA from the intestine, luminal content was pooled from the small intestine, large intestine, and cecum from SFB+ 8–12 week old WT CD1 mice (Charles river). Content was transferred to falcons containing 1X Tris-Buffered Saline, 0.1% Tween 20 (TBST) buffer with proteinase inhibitor (Roche). Falcons were then vortexed for 5 min on max speed and centrifuged for 10 min at 5000 rpm. The supernatant was collected and spun again two times to further remove bacteria and debris. Pierce Protein L Magnetic Beads (Thermo Scientific) were added and incubated for 1 h at room temperature while shaking. After 1 h, beads selectively bound to IgA through kappa light chain were separated from the supernatant with EasySep magnetic stand (StemCell technologies). Supernatant was discarded and beads washed 3 times. IgA was separated from the beads with Pierce IgG Elution Buffer pH 2.0 (ThermoFisher). Elution buffer was incubated with the beads for 10 min at room temperature on a shaker. Tris-HCl 1M pH 8.5 was added to neutralize the solution. IgA protein concentration was measured using NanoDrop. The isolated IgA preparation was then filtered with 0.22 μm sterile syringe filter unit. The IgA preparation was kept up to one week at 4 °C. When IgA were administered to the mice by gavage, the isolated IgA preparation was further concentrated with Amicon Ultra-4 Centrifugal Filter Units (MilliporeSigma) until 250–350μg/0.1ml final concentration was achieved. To optimize the treatment protocol to restore luminal IgA levels in vivo to GATA4ΔIEC mice, we first treated RAG−/− mice with 250 μg of the IgA preparation. After 1 h, we assessed the frequency of IgA+ bacteria in small intestinal contents by flow cytometry as described below, and noted 10–20% of bacteria were IgA+ after gavage. The bacterial coating was transient due to intestinal flow and undetectable after two hours. Therefore, continuous IgA gavages were necessary to sustain luminal IgA and bacterial coating in the small intestine. To administer the IgA preparation and analyze SFB colonization, GF, WT and GATA4ΔIEC mice were gavaged with 100 μl of IgA or PBS. After 1 h, the mice were colonized with SFB and gavaged again with IgA or PBS. Three more gavages were performed at 2-h intervals. The mice were euthanized 24 h after the gavage of SFB, and the regionalization of SFB load was assessed in jejunal and ileal mucosal scrapings by qPCR.
In-vivo retinoic acid treatment
All-trans-retinoic acid (Sigma) was resuspended in DMSO at a concentration of 20mg/ml, diluted in corn oil, and administered to mice at a dose of 300μg i.p every other day for 14 days.
Bacterial staining with luminal IgA
Luminal content was taken from WT, GATA4ΔIEC, or RAG−/− mice and resuspended in 1X PBS with protease inhibitors at a concentration of 0.1mg/μl, vortexed for 5 min, and spun at 8000 rpm for 5 min. Three fecal pellets from RAG−/− mice were homogenized and pelleted. The bacterial pellet was resuspended in 50μl PBS and combined with 50μl of luminal supernatant containing IgA. The IgA was incubated with the bacteria for 1 h at 4 °C. The bacteria were then washed, pelleted, and stained with SYTO BC (ThermoFisher) diluted 1:5000 and anti-IgA APC (Southern Biotech) diluted 1:200 for 30 min. Bacteria were gated on FSC, SSC, SYTOBC+, and IgA+.
Microbial 16S sequencing: Library generation and initial data processing
Extracted DNA was amplified, barcoded and sequenced as described previously 50,69,70. Briefly, amplification of the variable 4 (V4: 519F-806R) region of the 16S rRNA gene was performed with total DNA input (determined by NanoDrop) limited to 400ng to prevent inefficient amplification. Amplification was stopped in late exponential phase to minimize chimera formation. Amplified libraries were combined at equimolar concentrations and sequenced on an Illumina MiSeq (2x300bp). Fastq files were processed with QIIME 2 2020.2 62 using dada2 for amplicon sequence variant (ASV) determination and the Silva 132 99% OTUs reference database for taxonomy assignment. Rarefaction to the lowest read depth present in all samples (48,305 reads) was performed to decrease biases from varying sequencing depth between samples 71.
Microbial 16S sequencing: Absolute abundances
The total microbial load (bacteria and archaea) of each sample and the absolute abundance of each taxon in individual samples was determined as described previously 50,69. Briefly, the Bio-Rad QX200 droplet dPCR system (Bio-Rad Laboratories) with primers targeting the V4 (519F-806R) region was utilized to measure the number of 16S rRNA gene copies per sample. The final concentration of 16S rRNA gene copies in each sample was normalized to the extracted sample total DNA measurement from NanoDrop. Total DNA levels provide a good proxy for tissue mass in biopsy samples. The input-DNA-normalized total microbial load was multiplied by each ASV’s relative abundance to determine the absolute abundance of each ASV.
Microbial 16S sequencing data analysis: Poisson quality filtering
Poisson quality filtering of low abundance taxa was performed as previously described 72. Briefly, the relative abundance limit of detection (LOD) was determined for each sample. Relative abundance LOD is a function of two Poisson sampling steps: one based on the number of 16S molecules input into the library amplification reaction, and the other by the number of sequencing reads generated from the amplicon library. In each case, the relative abundance LOD was set at the point where 95% confidence of detection was observed and then the minimum of the two described LODs was used. For each sample, the relative abundance of each ASV detected below the LOD was set to zero.
Microbial 16S sequencing data analysis: Statistical analysis
Group comparisons were analyzed using the non-parametric Kruskal-Wallis rank sums tests with Benjamini–Hochberg multiple hypothesis testing correction using SciPy.stats Kruskal function and statsmodels.stats.multitest multipletests function with the fdr_bh option.
RNA-seq of purified IECs from mice
To perform RNA-seq on IECs, 100,000 EPCAM+ CD45- cells were cell sorted from the jejunum and ileum of WT and GATA4ΔIEC mice. Three independent cell sorting experiments were performed and the libraries and sequencing were done on the same batch with 8 mice per group. The SMART-Seq v4 Ultra Low Input RNA Kit (TaKaRa) was used to generated amplified cDNA, using either 7500pg of RNA input. The cDNA was generated and purified according to the manufacturer’s specifications. cDNA was amplified 12 cycles based on empiric testing. The Nextera XT DNA Library Preparation Kit (Illumina) was used to generate the RNA-seq libraries, with an input of 125pg cDNA, according to the manufacturer’s specifications. Subsequently, the libraries were multiplexed and sequenced at a depth of 20 million reads per sample (50bp SR) on a HiSeq4000.
Tissue RNA-seq
Whole tissue duodenal biopsies stored at -80C were thawed on ice and transferred to Starstedt tubes containing 350uL RLT Plus supplemented with 1% 2-mercaptoethanol and equal quantities of 1.0mm and 0.5mm zirconium oxide beads (Next Advance). Biopsies were bead beat 3 times for 1min at a setting of 9 on a Bullet Blender 24, with one minute of cooling on ice between each beating. Lysates were processed using the AllPrep DNA/RNA/miRNA Universal Kit (Qiagen). 500ng of purified RNA was used as input in the TruSeq Stranded mRNA Library Prep kit (Illumina) to generate sample libraries according to manufacturer’s specifications. Libraries were multiplexed and sequenced at a depth of 20 million reads per sample (50bp SR) on a HiSeq4000.
Mouse RNA-seq data processing to obtain raw counts
Mouse RNA-seq raw data were processed using a standard workflow based on the GENPIPES framework 73. Specifically, the “stringtie” type “rnaseq” pipeline was used. Reads were first trimmed using Trimmomatic software 74. Trimmed reads were aligned to the Mus_musculus.GRCm38 mouse reference genome using the STAR aligner 75 following a two-pass mapping protocol. Alignments were then sorted and filtered for duplicates using Picard(sort, markduplicates) (“Picard Toolkit” Broad Institute http://broadinstitute.github.io/picard/; Broad Institute). Gene-level read counts for downstream processing were calculated from spliced alignments using HTseq count 76.
Mouse RNA-seq data analysis: quality control filtering and normalization
All statistical analyses of the mouse RNA-seq data were performed using R (v.4.0.3). From the raw count matrices, genes expressed (i.e., having at least two counts) in fewer than two samples were removed. The resulting matrices will be referred to as the count matrices. Counts were normalized by applying the variance stabilizing transformation (i.e. vst(), default parameters) from the DESeq2 R package (v.1.30.1) 77. Batch effects were removed using the removeBatchEffect() function (batch = “sort_batch”) from the limma R package (v.3.46.0) 78.
Mouse RNA-seq data analysis: differential expression and gene set enrichment
Comparisons of gene expression between sample groups were made using DESeq2 to fit a negative binomial generalized linear model with a group variable. Wald statistics were used to determine the significance of the group coefficient, i.e., the log2-fold change (LFC) in expression between groups. We used the Benjamini-Hochberg method for controlling the false discovery rate (FDR). The P values reported are FDR adjusted. Genes with an adjusted P-value of at most 0.05 were considered differentially expressed (DE) between groups. The LFCs and FDR-adjusted P values were given as input to the fgsea() function from the fgsea R package (v.1.16.0) 79, which implements a preranked gene set enrichment analysis. The rankings of the genes were based on the FDR-adjusted P values. The mouse KEGG pathway database (mmuKegg) 80 and/or the Gene Ontology Biological Processes (GO-BP) database 81,82 were the gene sets used in the enrichment analyses. Enriched pathways (i.e., P <0.05) were collapsed to independent pathways to avoid repetitive terms, using the fgsea collapsePathways() function.
(i). Identifying region-specific GATA4-regulated genes
This set of DE genes was determined by grouping together “ileum-like” samples, i.e., WT ileum and GATA∆IEC jejunum samples, and comparing them with WT Jejunum samples. Comparisons were performed separately for the tissue and IECs. A threshold of >0.25 for the absolute value of the LFC was used to filter the very high number of DE genes in the tissue RNA-seq data, whereas no LFC threshold was used for the EC data.
(ii). Microbiota-dependent and -independent genes
We determined the influence of microbiota on the region-specific GATA4-regulated genes by systematically comparing jejunum tissue samples. For two analyses, we compared genotypes while maintaining a fixed microbiota status; for the third analysis, we jointly analyzed the effects of genotype and microbiota, including an interaction term (we were too underpowered to use only the latter approach). Thus, we identified three groups: (1) DE genes in GATA∆IEC SPF, relative to WT SPF, (2) DE genes in GATA∆IEC GF, relative to WT GF, and (3) genes with a significant interaction between genotype and microbiota.
Microbiota-dependent genes: Genes that were strongly DE (i.e., P <0.01, |LFC| > 0.6) in group 1 but not DE (i.e., P >0.2) in group 2, or vice versa, were deemed microbiota dependent. Furthermore, genes in group 3 that had a strong, significant interaction term (i.e., P <0.01, |LFC| > 0.9) were also considered microbiota dependent.
Microbiota-independent genes: Genes that were strongly DE (i.e., P <0.01, |LFC| > 0.6) in both groups (with LFC of the same sign) were deemed microbiota independent. Further, genes in group 3 that had a weak or insignificant interaction term (i.e., P >0.2 or |LFC| < 0.3) were included.
Both sets of genes, (a) and (b), were then intersected with the previously determined set of region-specific, GATA4-regulated genes, resulting in region-specific, GATA4-regulated genes that were either microbiota dependent or independent.
Mouse RNA-seq data analysis: data visualization
(i). Principal components plots
The principal components analysis (PCA) was done using the pca() function from the PCAtools R package (v.2.2.0.) 83. The top 500 genes selected by highest row variance in the centered (across rows) normalized count matrix were used to calculate the principal components. For the analysis of the immune genes (Figure 1C), the normalized counts matrix was first intersected with the genes in the immune module. Using the full set of genes to calculate the PCA shows the same patterns but with a larger spread within sample groups.
(ii). Heatmaps
Heatmaps were plotted using the Heatmap() function from the ComplexHeatmap R (v.2.6.2) package 84. The z-scored (i.e., across rows) normalized expression values were used to plot the heatmaps.
Annotation of GATA4-targeted genes
A previously published and publicly available table of annotated GATA4 ChIP-Seq peaks 9 was used to annotate GATA4 targets among the DE genes reported.
Generation of immune, IL-17, and IFNγ gene modules
We curated modules of immune genes, IL-17-associated genes, and IFNγ-associated genes. The immune module was created using two curated and publicly available databases: IRIS 7 and ImmPort 6. These human genes were then converted to mouse homologs. Genes that did not have homologs were discarded. The resulting module consists of 4,279 genes (Table S2.The IL-17 and IFNγ modules consist of known gene pathways from established databases, publications, and experimental data using IFNγ−/−, IFNγr−/−, IL17−/−, IL17r−/−, or IFNγ, IL17 treated cell lines or mice. Specifically, the IL-17 module consisted of genes encompassing the following pathways from mmuKEGG: 04657, 04659 and msigDB: m6335, m19422, m298, m300, m461, m460, m39560, m8578, m8581, m8579, m8927, m8928, and the following papers 14,85.
The IFNγ module consisted of genes encompassing the following pathways from msigDB: m22085, m5972, m5970, m4551, m9583 , m39363, m161, m6305, m6313, m6696, m6695, m6689, m6688, m6523, m6522, m6513, m6512, m1898, m2913, m8662, m8657, m5913, from the Gene Ontology database: GO:0034341, from the Reactome database: R- 913531, and the following papers 86,87.
Human RNA-seq data analysis
Adaptors and low-quality bases were trimmed using Trim Galore (v 0.4.4). Resulting reads were then aligned to the human reference sequence Ensembl GRCh38 release 87 using Kallisto 88. Next, the derived pseudo counts were normalized into log2 counts per million reads (CPM) using the voom function from the limma package (v3.46.0) 78. To evaluate the transcriptomic changes associated with GATA4 dysregulation in the small intestine and in the context of celiac disease we defined two contrast groups: “GATA4-lo”, to reflect loss of regionalization, and “GATA4-hi”, as a normal jejunum tissue. Using the normalized expression of GATA4 to rank all control, ACeD and GFD samples, we defined the “GATA4-hi” group as the samples in the top 30% of GATA4 expression (ACeD n =6, Control n= 18, GFD n= 18). The “GATA4-lo” group was defined by ACeD samples in the bottom 30% of GATA4 expression across only ACeD samples (n=15). Next, to define a universe set for the differential expression and enrichment analyses, we generated a set of 11,657 homologous genes expressed in the human and mouse cohorts. We then tested transcriptome-wide for significant differences in expression between the GATA4-hi and GATA4lo groups of samples, using a linear model that accounted for sex, age, batch, and technical covariates, and corrected for multiple testing. Over-enrichment analysis of gene ontologies biological processes were performed using the enrichGO function from the clusterProfiler (v3.0.4) 89. All statistical analyses within the human cohort and the overlaps with the mouse GATA4ΔIEC dataset were performed using R (v4.0.3).
Using the log2(CPM) expression values, we calculated single sample Gene Set Enrichment scores (ssGSEA) for the retinol pathway, IL17 downstream genes, and the MSigDB hallmark gene sets 90, using the R Bioconductor Package GSVA 91. These ssGSEA scores where then used to test association between GATA-hi vs GATA-lo contrasts groups, as well as association with presence or absence of bacteria. Associations were evaluated using a linear model accounting for sex, age and technical covariates.
Data and materials availability:
Raw and processed genomics data produced for this study will be deposited in the NCBI Gene Expression Omnibus (GEO), and the accession number will be included. Code used in this study will be made publicly available on Github.
Supplementary Material
Table S1: Mouse GATA4 transcriptomic analysis, related to Figure 1
Table S2: Immune gene modules, related to Figure 1B–D
Table S3: Differential expressed genes between GATA4 hi and GATA4 lo ACeD patients, related to Figure 5
Table S4: Detectable bacterial taxa by 16s in control or ACeD patients biopsies, related to Figure 5
Table S5: Association of biological pathways with specific bacteria in control or ACeD patients, related to Figure 5
Highlights:
GATA4 prevents small intestinal inflammation by restricting bacterial colonization
GATA4 regulates bacterial colonization in part by regulating IgA and retinol metabolism
GATA4-deficiency, SFB and C. rodentium synergize to drive immunopathology
TH7 immunity is found in celiac patients with GATA4-deficiency and Actinobacillus
Acknowledgements:
We would like to thank the patients and their family members, as well as the University of Chicago Celiac Disease Center, for supporting our research. We would like to thank Drs. Sonia Kupfer, Carol Semrad, Ritu Verma, Hilary Jericho, and Ian Wilson from the Celiac Disease Center for consenting and recruiting patients. We would like to thank Joaquín Sanz Remón for his help with the general transcriptional analysis of celiac disease and control patient biopsies. We thank Drs. Kenya Honda and Gabriel Nuñez for providing rat SFB and ΔEAE C. rodentium, respectively. We thank Ivaylo Ivanov for providing SFB TCR transgenic mice and for helpful discussions. We thank Steven Erickson from Dr. Albert Bendelac’s lab for generating IgA- and Jh-deficient mice using CRISPR-Cas technologies based on previously published mouse models (Chen et al., 1993; Harriman et al., 1999). We thank Betty Theriault, Kristin Kolar, and the entire Gnotobiotic Research Animal Facility at the University of Chicago for their help with the experiments involving gnotobiotic mice. We thank Yimei Chen and Tera Lavoie of the Advanced Electron Microscopy Core at University of Chicago for their help with transmission electron microscopy. We thank the Human Tissue Resource Center, the Integrated Light Microscopy Core Facility, Flow Cytometry Facility, and the Genomics Facility at the University of Chicago for their technical support. We thank Toufic Mayassi and Cezary Ciszewski for the thoughtful discussions and insightful comments. Finally, we thank Valerie Abadie for a critical reading of the manuscript and help with the graphical abstract. This work was supported by the National Institutes of Health via T32 AI007090 to Z.M.E., T32 GM007281 to D.G.S., U01AI109695 for A.M., U01 AI125250 to A.B., R01 AI144094 to A.B., R01DK103761 to N.S., R01 DK067180 to B.J., and the Digestive Diseases Research Core Center C-IID P30 DK42086 at the University of Chicago to B.J.
Footnotes
Declaration of interests:
The authors declare no competing interests.
Inclusion and diversity:
We support inclusive, diverse, and equitable conduct of research.
Supplemental Information
Supplemental information includes six figures and five tables.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mowat AM, and Agace WW (2014). Regional specialization within the intestinal immune system. Nature reviews. Immunology 14, 667–685. 10.1038/nri3738. [DOI] [PubMed] [Google Scholar]
- 2.Donaldson GP, Lee SM, and Mazmanian SK (2016). Gut biogeography of the bacterial microbiota. Nature reviews. Microbiology 14, 20–32. 10.1038/nrmicro3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esterházy D, Canesso MCC, Mesin L, Muller PA, de Castro TBR, Lockhart A, ElJalby M, Faria AMC, and Mucida D. (2019). Compartmentalized gut lymph node drainage dictates adaptive immune responses. Nature 569, 126–130. 10.1038/s41586-019-1125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bosse T, Piaseckyj CM, Burghard E, Fialkovich JJ, Rajagopal S, Pu WT, and Krasinski SD (2006). Gata4 is essential for the maintenance of jejunal-ileal identities in the adult mouse small intestine. Mol Cell Biol 26, 9060–9070. 10.1128/mcb.00124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Battle MA, Bondow BJ, Iverson MA, Adams SJ, Jandacek RJ, Tso P, and Duncan SA (2008). GATA4 is essential for jejunal function in mice. Gastroenterology 135, 1676–1686.e1671. 10.1053/j.gastro.2008.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhattacharya S, Dunn P, Thomas CG, Smith B, Schaefer H, Chen J, Hu Z, Zalocusky KA, Shankar RD, Shen-Orr SS, et al. (2018). ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci Data 5, 180015. 10.1038/sdata.2018.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelley J, de Bono B, and Trowsdale J. (2005). IRIS: a database surveying known human immune system genes. Genomics 85, 503–511. 10.1016/j.ygeno.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Lee JY, Hall JA, Kroehling L, Wu L, Najar T, Nguyen HH, Lin WY, Yeung ST, Silva HM, Li D, et al. (2020). Serum Amyloid A Proteins Induce Pathogenic Th17 Cells and Promote Inflammatory Disease. Cell 180, 79–91.e16. 10.1016/j.cell.2019.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson CA, Wojta K, Pulakanti K, Rao S, Dawson P, and Battle MA (2017). GATA4 Is Sufficient to Establish Jejunal Versus Ileal Identity in the Small Intestine. Cellular and molecular gastroenterology and hepatology 3, 422–446. 10.1016/j.jcmgh.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aronson BE, Rabello Aronson S, Berkhout RP, Chavoushi SF, He A, Pu WT, Verzi MP, and Krasinski SD (2014). GATA4 represses an ileal program of gene expression in the proximal small intestine by inhibiting the acetylation of histone H3, lysine 27. Biochimica et biophysica acta 1839, 1273–1282. 10.1016/j.bbagrm.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, Alonzo F, Ng C, Chen A, Lin X, et al. (2014). Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 510, 152–156. 10.1038/nature13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herp S, Durai Raj AC, Salvado Silva M, Woelfel S, and Stecher B. (2021). The human symbiont Mucispirillum schaedleri: causality in health and disease. Med Microbiol Immunol 210, 173–179. 10.1007/s00430-021-00702-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, Suda W, Imaoka A, Setoyama H, Nagamori T, et al. (2015). Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 163, 367–380. 10.1016/j.cell.2015.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prakash T, Oshima K, Morita H, Fukuda S, Imaoka A, Kumar N, Sharma VK, Kim SW, Takahashi M, Saitou N, et al. (2011). Complete genome sequences of rat and mouse segmented filamentous bacteria, a potent inducer of th17 cell differentiation. Cell host & microbe 10, 273–284. 10.1016/j.chom.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, and Núñez G. (2012). Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science (New York, N.Y.) 336, 1325–1329. 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collins JW, Keeney KM, Crepin VF, Rathinam VA, Fitzgerald KA, Finlay BB, and Frankel G. (2014). Citrobacter rodentium: infection, inflammation and the microbiota. Nature reviews. Microbiology 12, 612–623. 10.1038/nrmicro3315. [DOI] [PubMed] [Google Scholar]
- 18.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, and Said HM (2004). TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol 286, G367–376. 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 19.Turner JR (2006). Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am J Pathol 169, 1901–1909. 10.2353/ajpath.2006.060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, Geboes K, Ceuppens JL, and Rutgeerts P. (2002). Anti-tumor necrosis factor treatment restores the gut barrier in Crohn's disease. Am J Gastroenterol 97, 2000–2004. 10.1111/j.1572-0241.2002.05914.x. [DOI] [PubMed] [Google Scholar]
- 21.Simmons CP., Goncalves NS., Ghaem-Maghami M., Bajaj-Elliott M., Clare S., Neves B., Frankel G., Dougan G., and MacDonald TT. (2002). Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-gamma. Journal of immunology (Baltimore, Md. : 1950) 168, 1804–1812. 10.4049/jimmunol.168.4.1804. [DOI] [PubMed] [Google Scholar]
- 22.Matsunaga Y, Clark T, Wanek AG, Bitoun JP, Gong Q, Good M, and Kolls JK (2021). Intestinal IL-17R Signaling Controls Secretory IgA and Oxidase Balance in Citrobacter rodentium Infection. Journal of immunology (Baltimore, Md. : 1950) 206, 766–775. 10.4049/jimmunol.2000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shulzhenko N, Morgun A, Hsiao W, Battle M, Yao M, Gavrilova O, Orandle M, Mayer L, Macpherson AJ, McCoy KD, et al. (2011). Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nature medicine 17, 1585–1593. 10.1038/nm.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Trounstine M, Alt FW, Young F, Kurahara C, Loring JF, and Huszar D. (1993). Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JH locus. Int Immunol 5, 647–656. 10.1093/intimm/5.6.647. [DOI] [PubMed] [Google Scholar]
- 25.Bunker JJ, Erickson SA, Flynn TM, Henry C, Koval JC, Meisel M, Jabri B, Antonopoulos DA, Wilson PC, and Bendelac A. (2017). Natural polyreactive IgA antibodies coat the intestinal microbiota. Science (New York, N.Y.) 358. 10.1126/science.aan6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bunker JJ, Flynn TM, Koval JC, Shaw DG, Meisel M, McDonald BD, Ishizuka IE, Dent AL, Wilson PC, Jabri B, et al. (2015). Innate and Adaptive Humoral Responses Coat Distinct Commensal Bacteria with Immunoglobulin A. Immunity 43, 541–553. 10.1016/j.immuni.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. (2014). Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158, 1000–1010. 10.1016/j.cell.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donaldson GP, Ladinsky MS, Yu KB, Sanders JG, Yoo BB, Chou WC, Conner ME, Earl AM, Knight R, Bjorkman PJ, and Mazmanian SK (2018). Gut microbiota utilize immunoglobulin A for mucosal colonization. Science (New York, N.Y.) 360, 795–800. 10.1126/science.aaq0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamada N, Sakamoto K, Seo SU, Zeng MY, Kim YG, Cascalho M, Vallance BA, Puente JL, and Núñez G. (2015). Humoral Immunity in the Gut Selectively Targets Phenotypically Virulent Attaching-and-Effacing Bacteria for Intraluminal Elimination. Cell host & microbe 17, 617–627. 10.1016/j.chom.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goncalves A., Roi S., Nowicki M., Dhaussy A., Huertas A., Amiot MJ., and Reboul E. (2015). Fat-soluble vitamin intestinal absorption: absorption sites in the intestine and interactions for absorption. Food Chem 172, 155–160. 10.1016/j.foodchem.2014.09.021. [DOI] [PubMed] [Google Scholar]
- 31.Herr FM, Wardlaw SA, Kakkad B, Albrecht A, Quick TC, and Ong DE (1993). Intestinal vitamin A metabolism: coordinate distribution of enzymes and CRBP(II). Journal of lipid research 34, 1545–1554. [PubMed] [Google Scholar]
- 32.Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, Otipoby KL, Yokota A, Takeuchi H, Ricciardi-Castagnoli P, et al. (2006). Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science (New York, N.Y.) 314, 1157–1160. 10.1126/science.1132742. [DOI] [PubMed] [Google Scholar]
- 33.Jijon HB, Suarez-Lopez L, Diaz OE, Das S, De Calisto J, Parada-kusz M, Yaffe MB, Pittet MJ, Mora JR, Belkaid Y, et al. (2018). Intestinal epithelial cell-specific RARα depletion results in aberrant epithelial cell homeostasis and underdeveloped immune system. Mucosal Immunol 11, 703–715. 10.1038/mi.2017.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gattu S, Bang Y-J, Pendse M, Dende C, Chara AL, Harris TA, Wang Y, Ruhn KA, Kuang Z, Sockanathan S, and Hooper LV (2019). Epithelial retinoic acid receptor β regulates serum amyloid A expression and vitamin A-dependent intestinal immunity. Proceedings of the National Academy of Sciences 116, 10911. 10.1073/pnas.1812069116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johansen FE, and Kaetzel CS (2011). Regulation of the polymeric immunoglobulin receptor and IgA transport: new advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol 4, 598–602. 10.1038/mi.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jabri B, and Sollid LM (2009). Tissue-mediated control of immunopathology in coeliac disease. Nature reviews. Immunology 9, 858–870. 10.1038/nri2670. [DOI] [PubMed] [Google Scholar]
- 37.Monteleone I, Sarra M, Del Vecchio Blanco G, Paoluzi OA, Franzè E, Fina D, Fabrizi A, MacDonald TT, Pallone F, and Monteleone G. (2010). Characterization of IL-17A-producing cells in celiac disease mucosa. Journal of immunology (Baltimore, Md. : 1950) 184, 2211–2218. 10.4049/jimmunol.0901919. [DOI] [PubMed] [Google Scholar]
- 38.Castellanos-Rubio A, Santin I, Irastorza I, Castaño L, Carlos Vitoria J, and Ramon Bilbao J. (2009). TH17 (and TH1) signatures of intestinal biopsies of CD patients in response to gliadin. Autoimmunity 42, 69–73. 10.1080/08916930802350789. [DOI] [PubMed] [Google Scholar]
- 39.Bodd M, Ráki M, Tollefsen S, Fallang LE, Bergseng E, Lundin KE, and Sollid LM (2010). HLA-DQ2-restricted gluten-reactive T cells produce IL-21 but not IL-17 or IL-22. Mucosal Immunol 3, 594–601. 10.1038/mi.2010.36. [DOI] [PubMed] [Google Scholar]
- 40.Mayassi T, Ladell K, Gudjonson H, McLaren JE, Shaw DG, Tran MT, Rokicka JJ, Lawrence I, Grenier JC, van Unen V, et al. (2019). Chronic Inflammation Permanently Reshapes Tissue-Resident Immunity in Celiac Disease. Cell 176, 967–981.e919. 10.1016/j.cell.2018.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brar P, Kwon GY, Holleran S, Bai D, Tall AR, Ramakrishnan R, and Green PH (2006). Change in lipid profile in celiac disease: beneficial effect of gluten-free diet. Am J Med 119, 786–790. 10.1016/j.amjmed.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 42.Bledsoe AC, King KS, Larson JJ, Snyder M, Absah I, Choung RS, and Murray JA (2019). Micronutrient Deficiencies Are Common in Contemporary Celiac Disease Despite Lack of Overt Malabsorption Symptoms. Mayo Clin Proc 94, 1253–1260. 10.1016/j.mayocp.2018.11.036. [DOI] [PubMed] [Google Scholar]
- 43.Caminero A., McCarville JL., Galipeau HJ., Deraison C., Bernier SP., Constante M., Rolland C., Meisel M., Murray JA., Yu XB., et al. (2019). Duodenal bacterial proteolytic activity determines sensitivity to dietary antigen through protease-activated receptor-2. Nat Commun 10, 1198. 10.1038/s41467-019-09037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cenit MC, Olivares M, Codoñer-Franch P, and Sanz Y. (2015). Intestinal Microbiota and Celiac Disease: Cause, Consequence or Co-Evolution? Nutrients 7, 6900–6923. 10.3390/nu7085314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abadie V, Kim SM, Lejeune T, Palanski BA, Ernest JD, Tastet O, Voisine J, Discepolo V, Marietta EV, Hawash MBF, et al. (2020). IL-15, gluten and HLA-DQ8 drive tissue destruction in coeliac disease. Nature 578, 600–604. 10.1038/s41586-020-2003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taavela J, Viiri K, Popp A, Oittinen M, Dotsenko V, Peräaho M, Staff S, Sarin J, Leon F, Mäki M, and Isola J. (2019). Histological, immunohistochemical and mRNA gene expression responses in coeliac disease patients challenged with gluten using PAXgene fixed paraffin-embedded duodenal biopsies. BMC Gastroenterol 19, 189. 10.1186/s12876-019-1089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vuoristo M, Kesäniemi YA, Gylling H, and Miettinen TA (1993). Metabolism of cholesterol and apolipoprotein B in celiac disease. Metabolism 42, 1386–1391. 10.1016/0026-0495(93)90187-s. [DOI] [PubMed] [Google Scholar]
- 48.Kemppainen TA, Kosma VM, Janatuinen EK, Julkunen RJ, Pikkarainen PH, and Uusitupa MI (1998). Nutritional status of newly diagnosed celiac disease patients before and after the institution of a celiac disease diet--association with the grade of mucosal villous atrophy. Am J Clin Nutr 67, 482–487. 10.1093/ajcn/67.3.482. [DOI] [PubMed] [Google Scholar]
- 49.Xu M, Pokrovskii M, Ding Y, Yi R, Au C, Harrison OJ, Galan C, Belkaid Y, Bonneau R, and Littman DR (2018). c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 554, 373–377. 10.1038/nature25500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barlow JT, Bogatyrev SR, and Ismagilov RF (2020). A quantitative sequencing framework for absolute abundance measurements of mucosal and lumenal microbial communities. Nat Commun 11, 2590. 10.1038/s41467-020-16224-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.D'Argenio V, Casaburi G, Precone V, Pagliuca C, Colicchio R, Sarnataro D, Discepolo V, Kim SM, Russo I, Del Vecchio Blanco G, et al. (2016). Metagenomics Reveals Dysbiosis and a Potentially Pathogenic N. flavescens Strain in Duodenum of Adult Celiac Patients. Am J Gastroenterol 111, 879–890. 10.1038/ajg.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi Z, Zou J, Zhang Z, Zhao X, Noriega J, Zhang B, Zhao C, Ingle H, Bittinger K, Mattei LM, et al. (2019). Segmented Filamentous Bacteria Prevent and Cure Rotavirus Infection. Cell 179, 644–658.e613. 10.1016/j.cell.2019.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Palma G, Nadal I, Medina M, Donat E, Ribes-Koninckx C, Calabuig M, and Sanz Y. (2010). Intestinal dysbiosis and reduced immunoglobulin-coated bacteria associated with coeliac disease in children. BMC Microbiol 10, 63. 10.1186/1471-2180-10-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumar V, Jarzabek-Chorzelska M, Sulej J, Karnewska K, Farrell T, and Jablonska S. (2002). Celiac disease and immunoglobulin a deficiency: how effective are the serological methods of diagnosis? Clin Diagn Lab Immunol 9, 1295–1300. 10.1128/cdli.9.6.1295-1300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pickard JM., Maurice CF., Kinnebrew MA., Abt MC., Schenten D., Golovkina TV., Bogatyrev SR., Ismagilov RF., Pame EG., Turnbaugh PJ., and Chervonsky AV. (2014). Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature 514, 638–641. 10.1038/nature13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, et al. (2014). Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157, 340–356. 10.1016/j.cell.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 57.Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, Lee JY, Ziel JW, Miraldi ER, Domingos AI, et al. (2015). An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell 163, 381–393. 10.1016/j.cell.2015.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deloris Alexander A, Orcutt RP, Henry JC, Baker J Jr., Bissahoyo AC, and Threadgill DW (2006). Quantitative PCR assays for mouse enteric flora reveal strain-dependent differences in composition that are influenced by the microenvironment. Mamm Genome 17, 1093–1104. 10.1007/s00335-006-0063-1. [DOI] [PubMed] [Google Scholar]
- 59.Sagaidak S, Taibi A, Wen B, and Comelli EM (2016). Development of a real-time PCR assay for quantification of Citrobacter rodentium. J Microbiol Methods 126, 76–77. 10.1016/j.mimet.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 60.Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, and Stahl DA (1990). Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol 56, 1919–1925. 10.1128/aem.56.6.1919-1925.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsai PY, Zhang B, He WQ, Zha JM, Odenwald MA, Singh G, Tamura A, Shen L, Sailer A, Yeruva S, et al. (2017). IL-22 Upregulates Epithelial Claudin-2 to Drive Diarrhea and Enteric Pathogen Clearance. Cell host & microbe 21, 671–681.e674. 10.1016/j.chom.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37, 852–857. 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glöckner FO (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–596. 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harriman GR, Bogue M, Rogers P, Finegold M, Pacheco S, Bradley A, Zhang Y, and Mbawuike IN (1999). Targeted deletion of the IgA constant region in mice leads to IgA deficiency with alterations in expression of other Ig isotypes. Journal of immunology (Baltimore, Md. : 1950) 162, 2521–2529. [PubMed] [Google Scholar]
- 65.Husby S., Koletzko S., Korponay-Szabó IR., Mearin ML., Phillips A., Shamir R., Troncone R., Giersiepen K., Branski D., Catassi C., et al. (2012). European Society for Pediatric Gastroenterology, Hepatology, and Nutrition Guidelines for the Diagnosis of Coeliac Disease. Journal of Pediatric Gastroenterology and Nutrition 54. [DOI] [PubMed] [Google Scholar]
- 66.Molloy MJ, Grainger JR, Bouladoux N, Hand TW, Koo LY, Naik S, Quinones M, Dzutsev AK, Gao JL, Trinchieri G, et al. (2013). Intraluminal containment of commensal outgrowth in the gut during infection-induced dysbiosis. Cell host & microbe 14, 318–328. 10.1016/j.chom.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lefrançois L, and Lycke N. (2001). Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer's patch, and lamina propria cells. Curr Protoc Immunol Chapter 3, Unit 3.19. 10.1002/0471142735.im0319s17. [DOI] [PubMed] [Google Scholar]
- 68.Earley ZM, Akhtar S, Green SJ, Naqib A, Khan O, Cannon AR, Hammer AM, Morris NL, Li X, Eberhardt JM, et al. (2015). Burn Injury Alters the Intestinal Microbiome and Increases Gut Permeability and Bacterial Translocation. PloS one 10, e0129996. 10.1371/journal.pone.0129996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bogatyrev SR, and Ismagilov RF (2020). Quantitative microbiome profiling in lumenal and tissue samples with broad coverage and dynamic range via a single-step 16S rRNA gene DNA copy quantification and amplicon barcoding. bioRxiv, 2020.2001.2022.914705. 10.1101/2020.01.22.914705. [DOI] [Google Scholar]
- 70.Bogatyrev SR, Rolando JC, and Ismagilov RF (2020). Self-reinoculation with fecal flora changes microbiota density and composition leading to an altered bile-acid profile in the mouse small intestine. Microbiome 8, 19. 10.1186/s40168-020-0785-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, Lozupone C, Zaneveld JR, Vázquez-Baeza Y, Birmingham A, et al. (2017). Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5, 27. 10.1186/s40168-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barlow JT, Leite G, Romano AE, Sedighi R, Chang C, Celly S, Rezaie A, Mathur R, Pimentel M, and Ismagilov RF (2021). Quantitative sequencing clarifies the role of disruptor taxa, oral microbiota, and strict anaerobes in the human small-intestine microbiome. Microbiome 9, 214. 10.1186/s40168-021-01162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bourgey M, Dali R, Eveleigh R, Chen KC, Letourneau L, Fillon J, Michaud M, Caron M, Sandoval J, Lefebvre F, et al. (2019). GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience 8. 10.1093/gigascience/giz037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bolger AM, Lohse M, and Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Anders S, Pyl PT, and Huber W. (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A. (2021). Fast gene set enrichment analysis. bioRxiv, 060012. 10.1101/060012. [DOI] [Google Scholar]
- 80.Waterston RH., Lindblad-Toh K., Birney E., Rogers J., Abril JF., Agarwal P., Agarwala R., Ainscough R., Alexandersson M., A P., et al. (2002). Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562. 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 81.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.The Gene Ontology resource: enriching a GOld mine. (2021). Nucleic Acids Res 49, D325–d334. 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blighe K. (2021). PCAtools: everything Principal Component Analysis. https://github.com/kevinblighe/PCAtools. [Google Scholar]
- 84.Gu Z, Eils R, and Schlesner M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 85.Kumar P, Monin L, Castillo P, Elsegeiny W, Horne W, Eddens T, Vikram A, Good M, Schoenborn AA, Bibby K, et al. (2016). Intestinal Interleukin-17 Receptor Signaling Mediates Reciprocal Control of the Gut Microbiota and Autoimmune Inflammation. Immunity 44, 659–671. 10.1016/j.immuni.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu SY, Sanchez DJ, Aliyari R, Lu S, and Cheng G. (2012). Systematic identification of type I and type II interferon-induced antiviral factors. Proceedings of the National Academy of Sciences of the United States of America 109, 4239–4244. 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. (2016). Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 167, 1540–1554.e1512. 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bray NL, Pimentel H, Melsted P, and Pachter L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 89.Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov JP., and Tamayo P. (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hänzelmann S, Castelo R, and Guinney J. (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Mouse GATA4 transcriptomic analysis, related to Figure 1
Table S2: Immune gene modules, related to Figure 1B–D
Table S3: Differential expressed genes between GATA4 hi and GATA4 lo ACeD patients, related to Figure 5
Table S4: Detectable bacterial taxa by 16s in control or ACeD patients biopsies, related to Figure 5
Table S5: Association of biological pathways with specific bacteria in control or ACeD patients, related to Figure 5
Data Availability Statement
All the data supporting the findings of the article are available within the main text or supplementary information. The published article includes datasets generated during this study. Original RNA-seq data has been deposited in GEO: GSE205743. Original 16S rRNA sequencing datasets analyzed in this study are available at the NCBI BioProject: PRJNA797871. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. Original code for analyzing these datasets have been deposited in Zenodo and is publicly available. DOIs are listed in the key resources table.