


Abstract
Using an improved system for the functional identification of active antisense fragments, we have isolated antisense fragments which inactivate the p53 tumour suppressor gene. These antisense fragments map in two small regions between nt 350 and 700 and nt 800 and 950 of the coding sequence. These antisense fragments appear to act by inhibition of p53 mRNA translation both in vivo and in vitro. Expression of these antisense fragments overcame the p53-induced growth arrest in a cell line which expresses a thermolabile mutant of p53 and extended the in vitro lifespan of primary mouse embryonic fibroblasts. Continued expression of the p53 antisense fragment contributed to immortalisation of primary mouse fibroblasts. Subsequent elimination of the antisense fragment in these immortalised cells led to restoration of p53 expression and growth arrest, indicating that immortal cells continuously require inactivation of p53. Expression of MDM2 or SV40 large T antigen, but not E7 nor oncogenic ras, overcomes the arrest induced by restoration of p53 expression. Functional inactivation of both p21 and bax (by overexpression of Bcl2), but not either alone, allowed some bypass of p53-induced growth arrest, indicating that multiple transcriptional targets of p53 may mediate its antiproliferative action. The ability to conditionally inactivate and subsequently restore normal gene function may be extremely valuable for genetic analysis of genes for which loss-of-function is involved in specific phenotypes.
INTRODUCTION
Human tumours are caused by the progressive accumulation of activating mutations in oncogenes and loss of function mutations in tumour suppressor genes. A subset of tumour suppressor genes function to prevent tumour formation or growth by inducing cell cycle arrest or apoptosis in response to potentially oncogenic events. For example, tumour suppressor genes such as p53 and Rb encode proteins that are activated and restrain proliferation following expression of activated alleles of oncogenes, such as H-ras (1–3).
Indeed, p53 is the most commonly mutated gene in human cancer (reviewed in 4–7), underscoring its importance in the suppression of tumour formation. p53 functions as a transcription factor which plays an important role in the maintenance of genomic stability. Following genotoxic damage, p53 is induced and acts to restrain proliferation by inducing the expression of genes which lead to growth arrest (such as p21) or apoptosis (such as bax). By inhibiting proliferation following DNA damage, p53 action prevents the accumulation of potentially oncogenic mutations.
p53 also functions to suppress cellular immortalisation (8–11). Normal primary somatic cells are capable of undergoing a finite number of divisions in culture until they undergo cellular senescence, characterised by growth arrest, a large flat morphology and insensitivity to further mitogenic stimulation (reviewed in 12–14). In contrast, many tumour cells exhibit unlimited division potential, indicating that they have bypassed the barriers to immortalisation, such as cellular senescence. p53 appears to play a direct role in controlling the onset of cellular senescence. p53 transcriptional activity increases with ageing of the cells (15), wild-type p53 activity is necessary for growth arrest in senescence (11,16,17) and a high percentage of cells that escape from senescence have lost p53 activity (18–21).
Numerous antitumour therapeutic strategies have been proposed based on the ability to restore wild-type p53 function by refolding of mutant protein or relocalisation of mislocalised protein (22,23). However, while p53 is among the best studied tumour suppressor genes to date, it is not yet clear whether continual p53 inactivation is necessary for continued expression of the immortalisation phenotype, i.e. whether re-expression of wild-type levels of p53 is sufficient to halt the proliferation of immortalised cells with different genetic backgrounds. This must be true for such therapeutic strategies to be successful. The ability to conditionally inhibit tumour suppressor function and subsequently restore wild-type levels of gene expression under normal physiological control is essential to answer this question and can also be used to examine functional relationships between genes in mammalian cells. While a variety of techniques are available to inhibit gene function, such as genetic disruptions in mice and somatic cell gene disruptions in human cultured cells, these techniques are effectively not reversible. An alternative approach which is applicable to a wide variety of cell types and is potentially reversible is the delivery or expression of antisense constructs.
The most common approach to targeted gene inhibition in mammalian cells involves the intracellular expression of antisense RNA complementary to the mRNA of the target gene (24,25). Antisense RNA techniques offer a powerful tool for down-regulating the expression of specific genes both in vitro and in vivo (26–33). A number of molecular mechanisms have been proposed to explain the action of antisense RNAs (34). For example, sense–antisense RNA duplexes can form in the nucleus where they are rapidly degraded, improperly processed or not transported to the cytosol in cultured cells (30). In transient systems or when antisense RNA is injected into the cytoplasm, the antisense RNA may act by binding to the mRNA and inhibiting its translation (35).
Despite the ease of expressing antisense RNA for any cloned gene, many antisense RNA constructs have little or no biological effect (36–38). Some of the more effective antisense RNAs comprise only a part of the coding sequence and there are no clear rules to predict which portion of the mRNA will give rise to the most effective antisense inhibitor (38–41).
We have used a systematic approach to select antisense RNA fragments which are active in inhibiting the function of the p53 tumour suppressor gene. These antisense fragments are highly effective in repressing p53 protein expression and biological function. Reversible expression of these antisense fragments has been used to demonstrate the role of p53 in immortalisation and shed light on its functional relationship to other immortalisation genes. The ability to conditionally inactivate and subsequently restore normal gene function may be extremely valuable for the analysis of the function of a wide range of genes; this methodology could be more widely applied to provide insight into the function of other antiproliferative genes.
MATERIALS AND METHODS
Generation of the mouse p53 antisense library
mRNA was transcribed in vitro from pBluescript-p53 using T7 RNA polymerase (Stratagene). After RNA synthesis, the product was treated with RNase-free DNase (10 U/µg DNA) for 30 min at 37°C and phenol extracted. RNA was electrophoresed on a 4% acrylamide–7 M urea gel and p53 mRNA was purified. Aliquots of 2 µg of p53 mRNA were used in generating the library. Randomly primed cDNA fragments of the p53 gene were synthesised, size selected (50–500 nt) on a S400 column (Pharmacia) and cloned into the EcoRI and XhoI sites of pMARXIVpuro in the antisense orientation.
Cell culture and preparation of mouse embryo fibroblasts (MEFs)
MEFs were prepared from day 13.5 embryos derived from CD1 mice. The head and blood organs were removed, then the torso was minced and dispersed in 0.1% trypsin (45 min at 37°C). Cells were grown for two population doublings and then frozen. MEFs were subcultured 1:4 upon reaching confluence; each passage was considered to be two population doubling levels (PDs). p53–/– MEFs were obtained from embryos derived from crosses between p53+/– mice. To create a cell line expressing temperature-sensitive p53, immortalised p53–/– MEFs were infected with pWZL-p53Val135. Clones were isolated at the permissive temperature (39°C) and a clone was selected that showed highly efficient growth arrest following a temperature shift to the restrictive temperature (32°C). All cultures were maintained in DMEM (Gibco) plus 10% foetal bovine serum (Sigma). Where necessary, cells were selected when indicated with 75 µg/ml hygromycin (Calbiochem), 400 µg/ml G418 (Sigma) or 4 µg/ml puromycin (Fluka).
3T3 immortalisation protocols
MEFs were infected and selected as before. Every 3 days, cells were trypsinised, counted and 106 cells were plated per 10 cm plate.
Retroviral-mediated gene transfer
Samples of 5 × 106 LinXE ecotropic retrovirus producer cells were plated per 10 cm dish, incubated for 24 h and transfected by calcium phosphate precipitation using 20 µg of retroviral plasmid. After 48 h, the virus-containing medium was filtered (0.45 µm filter; Millipore) and supplemented with 8 µg/ml polybrene (Sigma) and an equal volume of fresh medium. One day before infection, target fibroblasts were plated at 8 × 105 cells/10 cm dish and incubated overnight. For infections, culture medium was replaced by the appropriate viral supernatant and the culture plates were centrifuged (1 h, 1500 r.p.m.) and incubated at 37°C for 16 h.
In vitro recovery of the proviruses
Total genomic DNA was extracted from cells from a confluent 10 cm plate. It was then treated with RNase A (50 µg/ml, 30 min) and proteinase K (100 µg/ml final concentration) and extracted twice with phenol/chloroform. Following ethanol precipitation, genomic DNA was washed extensively with 70% ethanol and dissolved in 200 µl of ultrapure water.
To excise the proviruses, 10 µg of genomic DNA was digested with CRE recombinase (DNA final concentration 0.1 µg/µl) for 3 h at 37°C, extracted with phenol/chloroform and ethanol precipitated. DNA was washed extensively with 70% ethanol and dissolved in 5 µl of water. Aliquots of 2–5 mg of total DNA were electroporated into DH10B-lac-trfA bacteria and proviruses recovered from zeocin-resistant bacterial colonies.
Immunoblot analysis
Cells were washed twice with ice-cold PBS and lysed in lysis buffer (150 mM NaCl, 1% NP-40, 50 mM Tris–HCl, pH 8.0, 1 mM PMSF, 1 µg/ml leupeptin, 25 µg/ml aprotinin, 1 mM EDTA). After 15 min on ice, lysates were vortexed (5 min at 4°C) and cleared by centrifugation. Aliquots of 100–150 µg of total protein (Bio-Rad protein assay) were separated by 10% SDS–PAGE and transferred to nitrocellulose membranes. Western blot analysis was carried out using standard procedures and detected using ECL (Amersham). Ab1-421 (Oncogene Research) was used to detect mouse p53; antibodies 240 and 246 (Santa Cruz) were used to detect mutant and wild-type p53 protein by immunoprecipitation. An affinity-purified rabbit polyclonal antibody was used for p21WAF, followed by detection with horseradish peroxidase-conjugated donkey anti-rabbit or sheep anti-mouse antibodies (Amersham).
Northern blot
Total RNA was isolated from subconfluent cultures (Trizol; Gibco BRL). Ten micrograms of RNA was resolved by electrophoresis, transferred to Hybond-N+ membranes and probed according to standard procedures. p53 mRNA was detected using a radiolabelled fragment containing the entire coding sequence of mouse p53.
In vitro translation
In vitro translation was performed in rabbit reticulocyte lysates (TNT T7-T3; Promega) following the vendor’s instructions. Mouse p53 was transcribed and translated from pBlueScript-p53 under control of the T7 promoter. Where necessary, p53 antisense fragments cloned into pBlueScript under the control of the T3 promoter were included in the reaction.
RESULTS
Isolation of p53 antisense fragments
We have tested our system against the p53 gene that has been used previously as the target in similar approaches (42,43). To obtain antisense fragments which inhibited p53 activity, we undertook a phenotype-based screen to directly identify fragments which inhibited p53 gene function. We first generated clones of immortalised fibroblasts derived from p53-disrupted mice which expressed a thermolabile p53 mutant (p53ts p53–/– MEF). Clones grew normally at a permissive temperature (39°C) at which p53 is inactive, but arrested when shifted to a restrictive temperature (32°C) where p53 adopts the wild-type conformation (44).
A random antisense fragment library directed against the coding sequence of a mouse p53 library was constructed and cloned into the MMLV-based retroviral vector pMARXIVpuro (45). This library contained ~104 independent clones. Library DNA was transfected into ecotropic retrovirus packaging cells and replication-deficient viruses were infected into exponentially growing p53ts p53–/– MEF cells. Following selection for puromycin resistance, ~105 cells/100 mm culture dish were plated and shifted to 32°C. After 3 weeks, approximately 30 colonies were formed at 32°C, indicating that they had overcome the growth inhibitory action of p53. These colonies were subcloned and grown at 32°C for an additional 2 weeks. After this second round of phenotypic selection, 17 colonies continued to grow.
Genomic DNA was extracted from positive colonies and proviruses containing the antisense fragment were excised. A total of 19 different constructs were recovered. The majority of the colonies carried a single provirus. Fourteen of the 19 constructs had a p53-derived fragment in the antisense orientation (Table 1).
Table 1. p53 antisense fragments recovered from a phenotypic screen in p53ts p53–/– MEF cells.
| Cell clone | Provirus recovered | Length | Sequence | Assigned number |
|---|---|---|---|---|
| 2 | p53 antisense | 141 | 706–565 | 15 |
| 3 | p53 antisense | 90 | 937–847 | 61 |
| 4 | p53 antisense | 114 | 920–806 | 2 |
| 5 | p53 antisense | 113 | 906–793 | 57 |
| p53 antisense | 120 | 923–803 | 55 | |
| 6 | p53 antisense | 87 | 923–836 | 71 |
| 7 | p53 antisense | 114 | 920–806 | 2 |
| 10 | p53 antisense | 273 | 670–357 | 76 |
| 11 | empty vector | |||
| 12 | Tamdem (sense) | 120 | 1065–1185 | 53 |
| 13 | p53 antisense | 114 | 920–806 | 2 |
| 14 | p53 antisense | 40 | 940–900 | 13 |
| p53 antisense | 114 | 920–806 | 2 | |
| 15 | Empty primers | |||
| 16 | p53 antisense | 114 | 920–806 | 2 |
| 17 | p53 antisense | 114 | 920–806 | 2 |
| 18 | Empty primers | |||
| 19 | p53 antisense | 121 | 911–790 | 17 |
| 21 | Empty vector |
The active antisense fragments localised to two small clusters between nt 350 and 700 (2/14) and between nt 800 and 950 (12/14). Among these antisense fragments, number 2 was recovered six times from independent colonies. Sequencing of representatives of the unselected library showed that the fragments were randomly distributed over the p53 coding sequence (data not shown), indicating that the selection for these specific regions was a property of the phenotypic screen.
Biological properties of p53 antisense fragments
The effectiveness of these antisense fragments was confirmed by reinfecting p53ts p53–/– MEF cells with positive proviruses. Following drug selection, cells were plated at 5 × 104 cells/100 mm dish and shifted to 32°C. Control cells with the vector alone did not grow at the restrictive temperature (Fig. 1) and exhibited a flat enlarged cellular morphology (data not shown). In contrast, cells expressing the p53 antisense fragments showed a high rate of colony formation at this temperature; fragments derived from the first region (number 15) showed less penetrance than those derived from the second region (numbers 55, 71, 12 and 2). Most of the cells expressing the p53 antisense fragments were small, refractive and did not cease proliferation at subconfluent densities (data not shown). Thus, selected p53 antisense fragments were effective in inhibiting the p53-induced arrest.
Figure 1.
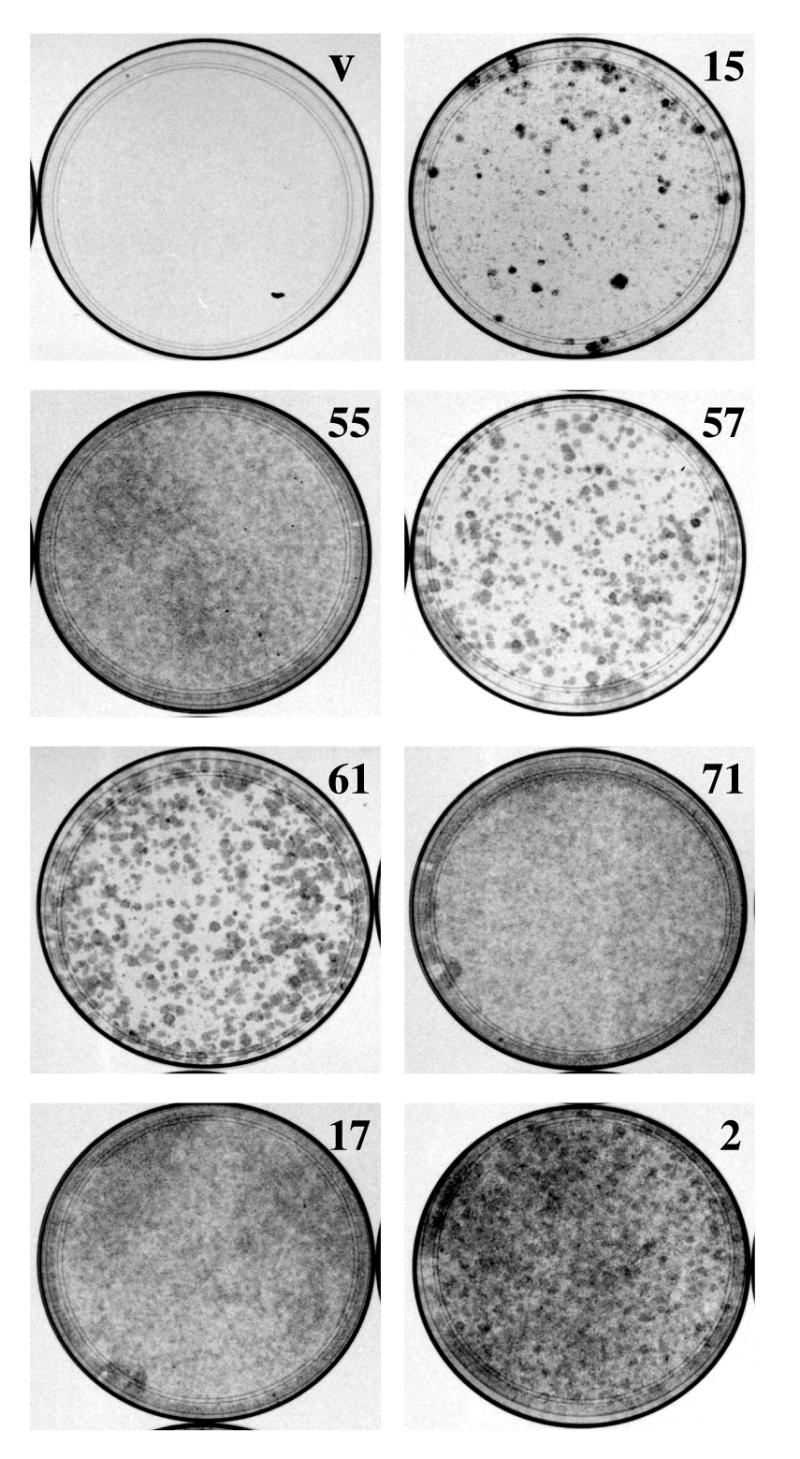
Expression of p53 antisense fragments overcomes p53-induced growth arrest. p53ts p53–/– MEF cells were infected with viruses that expressed p53 antisense fragments (numbers 15, 55, 57, 61, 71, 17 and 2) or empty vector (v). After drug selection for virally transduced cells, 5 × 104 cells were plated in 10 cm dishes and grown at restrictive temperature (32°C) for 15 days. Cells were then fixed and stained with crystal violet.
We next tested the effect of antisense expression on p53 protein levels. Following infection of p53ts p53–/– MEF cells and drug selection, cells were lysed and levels of p53 protein were analysed. While expression of each of the antisense fragments reduced p53 protein levels, antisense fragments derived from the second region were more effective (Fig. 2A).
Figure 2.
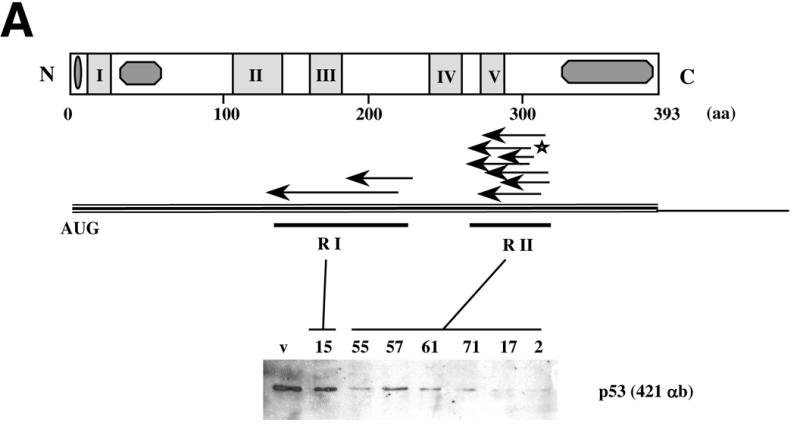
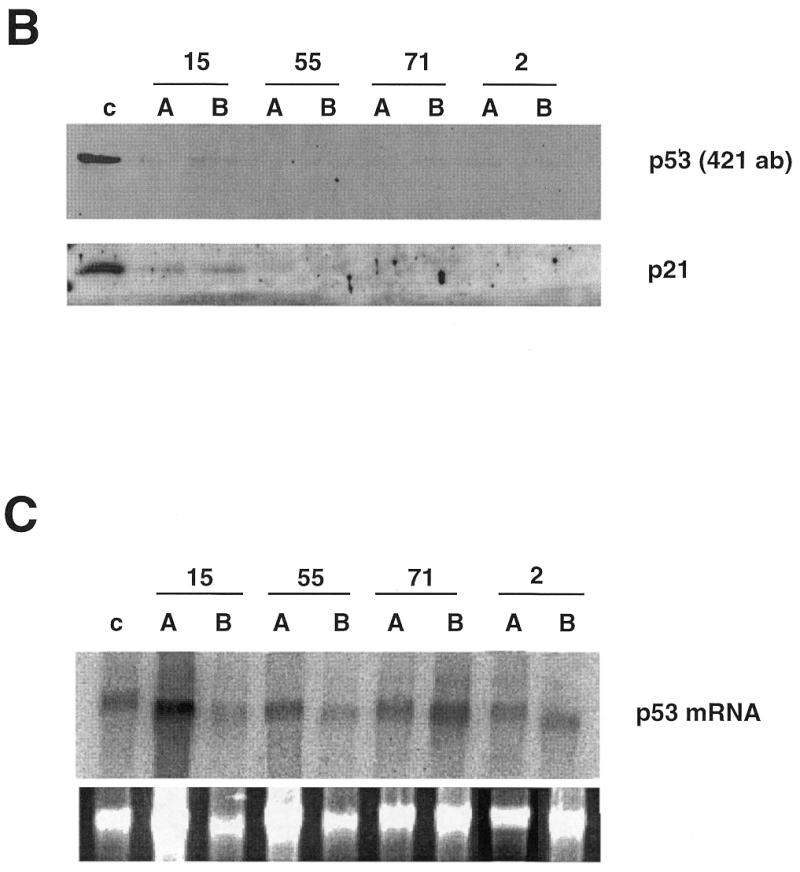
Localisation and activity of p53 antisense fragments. (A) p53 antisense fragments isolated following phenotypic selection mapped to two regions, RI and RII, of the p53 coding sequence. Expression of these antisense fragments reduces p53 protein levels in vivo. p53ts p53–/– MEF cells were infected with viruses expressing different antisense fragments (numbers 15, 55, 57, 61, 71, 17 and 2) or control vector (v). Infectants were selected, cells were lysed and p53 protein levels were assayed by western blot. (B) Expression of p53 antisense fragments reduces the expression of p21, a transcriptional target of p53. Two clones (A and B) of antisense-expressing p53ts p53–/– MEF cells (numbers 15, 55, 71 and 2) or vector alone (c) were selected and shifted to 32°C for induction of p53 activity. After 24 h, cells were lysed and levels of p53 or p21 protein were analysed by western blot. (C) Expression of p53 antisense fragments does not affect levels of p53 mRNA in vivo. Northern blot of p53 mRNA in colonies expressing p53 antisense fragments. Total RNA was prepared from antisense-expressing clones as above, separated by gel electrophoresis, transferred to Hybond membranes and probed with a radiolabelled p53-specific probe (Upper). (Lower) Ethidium bromide stained 28S band of the total RNA. The results of densitometric analysis of p53 versus 28S mRNA bands showed no significant variation with respect to control cells (not shown).
Variations in penetrance of individual antisense constructs within a population might result from differences in expression levels. We therefore infected p53ts p53–/– MEF cells with different antisense fragments, shifted cells to 32°C and after 3 weeks selected p53-resistant colonies. Two different clones expressing each of the antisense sequences numbers 15, 55, 71 and 2 were expanded and p53 levels were analysed (Fig. 2B). p53 protein was only detectable in colonies expressing antisense fragment number 15, consistent with its less penetrant ability to induce colony formation and inhibit p53 accumulation in mass culture. In each case, the level of p53 protein in the selected clones was lower than in the mass culture, indicating that there was selection for clones in which the antisense fragments were more active.
p53 functions via its activity as a transcription factor by inducing expression of growth arrest-inducing genes such as p21 (46). To test whether antisense fragment expression interfered with p53 activity we analysed levels of p21 protein in cells that had been infected with antisense-expressing or control vectors. Following a shift to 32°C control cells arrested and induced expression of p21, while colonies derived from p53 antisense selection were unable to induce p21 under similar conditions, indicating that reduced p53 activity was correlated with decreased protein expression following antisense expression (Fig. 2B).
To try to understand the mechanism by which the antisense fragments inhibited p53 expression, we next tested whether antisense fragment expression interfered with p53 mRNA expression. Antisense fragment expression had no detectable effect upon p53 mRNA expression (Fig. 2C), indicating that the p53 antisense fragments did not function via a mechanism involving mRNA degradation.
To further elaborate on the mechanism of antisense fragment action, we analysed the effect of RNA antisense expression in a cell-free system. Mouse p53 was transcribed using T7 polymerase and translated in reticulocyte lysates (Fig. 3). In the same reaction, antisense constructs or control vectors were expressed under control of the T3 promoter. Expression of p53 antisense fragments greatly reduced (number 15) or almost completely abolished (numbers 55, 71 and 2) p53 translation (Fig. 3A). Levels of inhibition in vitro correlate with those found in vivo (Fig. 2), indicating that the p53 antisense fragments might act by inhibiting translation of p53 protein. To assess whether the different effects of the antisense fragments might be due to differences in the efficiency of translational inhibition we translated p53 in the presence of increasing molar concentrations of antisense fragments derived from the first (number 15) and second (number 2) regions of p53 (Fig. 3D). In each case antisense number 2 is more efficient than number 15 in inhibiting translation, however, increasing levels of each improves their relative efficiency of inhibition (Fig. 3C and D).
Figure 3.
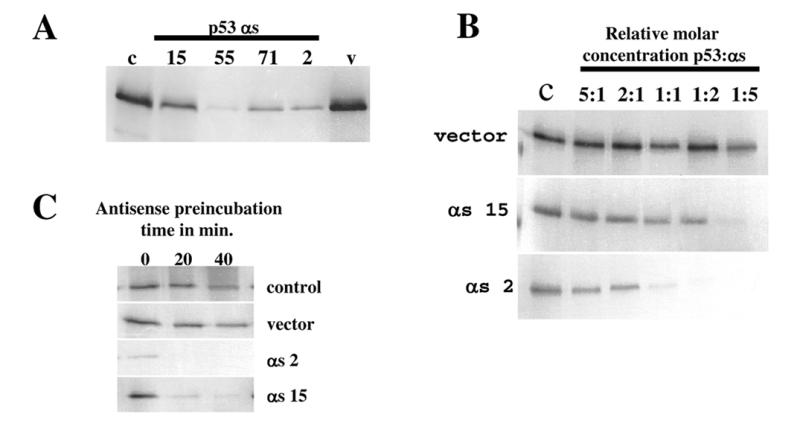
Inhibition of p53 translation by expression of p53 antisense fragments in vitro. (A) p53 protein was translated in vitro in the presence of p53 antisense fragment transcripts (numbers 15, 55, 71 and 2), control vector (v) or no additional vector (c). (B) In vitro translation of p53 protein in the presence of different molar ratios of p53 antisense transcripts (numbers 15 and 2) or control vector. (C) Effect of preincubation of antisense transcripts on translation of p53 protein.
These results are consistent with a mechanism in which the antisense fragments interfered with p53 protein synthesis rather than degradation of the RNA duplex or blocking of transport across the nuclear membrane, although this last mechanism cannot be excluded in vivo.
Biological effect of p53 antisense fragments in primary fibroblasts
We next tested the ability of the p53 antisense fragments to interfere with wild-type p53 function in primary cells. Specifically, we tested whether expression of p53 antisense fragments, like loss of p53 function, was capable of extending the in vitro lifespan of primary MEFs. MEFs in passage 3 were infected with viruses expressing p53 antisense fragments or control vector. Infected cells were selected and cultured until passage 5, the passage immediately prior to the passage at which wild-type MEFs undergo cellular senescence. Cells were plated at 3 × 105/100 mm dish and cultured for 15 days. Under these conditions wild-type cells do not form colonies, however, cells that display an extended cellular lifespan can continue to divide and can form colonies (16,47). MEFs infected with vector alone displayed a flat phenotype and grew very poorly. Surprisingly, the same phenotype was observed in cells expressing antisense fragment 15. In contrast, cells which expressed p53 antisense fragments derived from the second region were able to grow and form colonies (Fig. 4).
Figure 4.
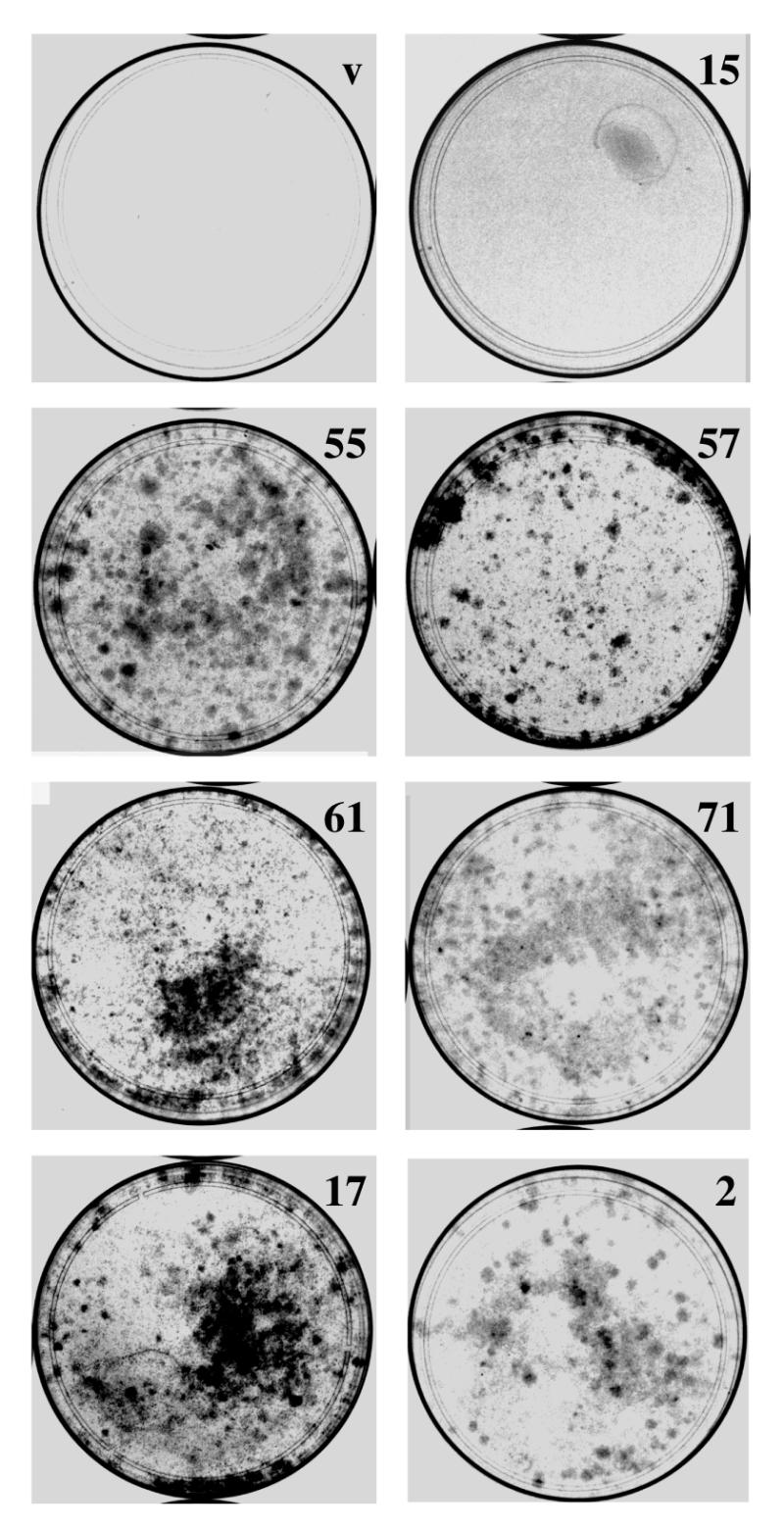
Expression of p53 antisense fragments induces an extended lifespan of primary MEFs. Primary MEFs were infected with antisense-expressing viruses (numbers 15, 55, 57, 61, 71, 17 and 2) or control vector (v) in PD 2. After selection, cells were passaged until PD 12, at which time 105 cells were plated in 10 cm dishes and grown for 15 days. Cells were then fixed and stained with crystal violet. Cells which exhibit an extended lifespan are capable of colony formation under these conditions.
We also carried out a similar functional screen to identify p53 antisense fragments which were capable of directly inducing an extended lifespan in primary MEFs. From this screen we isolated five antisense fragments, all of which mapped to region II of the p53 gene (data not shown), independently confirming the effectiveness of fragments derived from this region.
We next tested the relative efficiency of the p53 antisense fragments in producing an extended lifespan. Expression of antisense fragments showed a similar penetrance in inducing colony formation as expression of full-length antisense or dominant negative p53 (175H) or expression of HPV E7 (which inactivates members of the Rb family). However, they did not produce as extended a lifespan as that of MEFs derived from a p53-disrupted mouse (Table 2).
Table 2. Relative penetrance of p53 inactivation in inducing an extended lifespan of primary MEFs.
| MEF genotype | Relative penetrance |
|---|---|
| Wild type | 0 |
| p53–/– | 3.3 × 10–2 |
| p53αs | 4 × 10–3 |
| p53(175H) | 4 × 10–3 |
| E7 | 3 × 10–3 |
Primary MEFs were prepared from wild-type mouse embryos and infected with viruses expressing p53 antisense fragments, dominant negative p53 or E7. Murine fibroblasts were prepared from p53–/– mouse embryos for comparison. Cells were plated in PD 12, grown for 15 days, fixed and stained. Relative penetrance was measured as number of colonies formed per plated cell.
Recovery of physiological levels of p53 expression in immortalised MEFs
We next sought to test the effect of restoration of wild-type p53 levels in immortalised cells. For these experiments, we cloned the p53 antisense fragments into pMarxIV (45), a retroviral vector that contains a CRE recombinase target site (loxP site). Upon integration of the retrovirus into the genome, the loxP site is duplicated such that the genes carried by the virus are flanked on either side by loxP sites. Subsequent expression of CRE recombinase causes excision at these loxP sites, leading to removal from the genome and eventual loss of the construct. Therefore, gene expression in pMarxIV is effectively reversible. Thus, antisense constructs directed against p53 would be excised following CRE expression, leading to loss of the antisense fragment and restoration of physiological levels of p53 (Fig. 5A).
Figure 5.
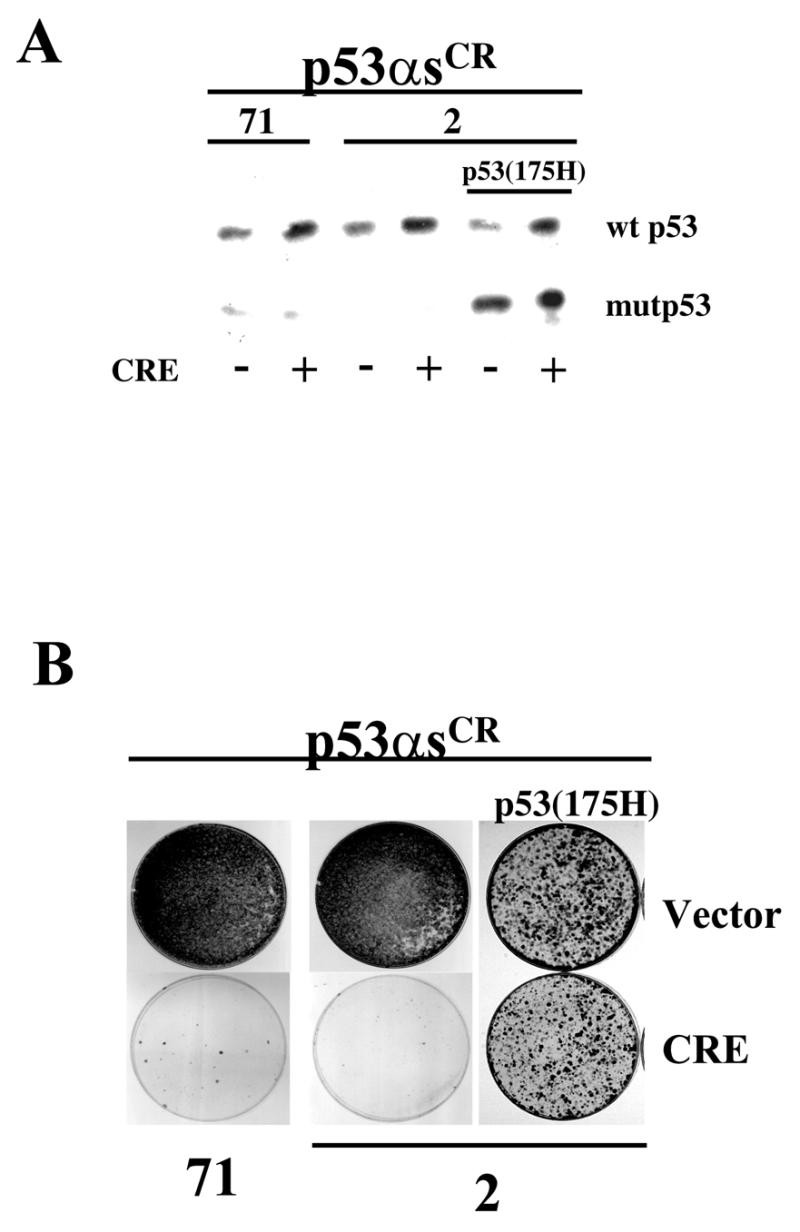
Recovery of p53 expression leads to growth arrest in immortalised cells. Presenescent MEFs were infected with viruses directing the expression of CRE-excisable pMarxIVp53αs71 or pMarxIVp53αs2 and immortalised using a 3T3 immortalisation protocol. Immortalised cells (p53αsCR) were infected with a control vector or pWZL-Hygro-CRE to excise the integrated p53 antisense construct leading to restoration of p53 expression. p53αsCR2 cells were infected with a virus expressing dominant negative p53, p53(175H), prior to CRE infection. (A) Levels of expression of wild-type p53 (wt p53) or mutant p53 (mutp53) in the p53αsCR cells after infection with CRE or vector alone. (B) Colony formation of p53αsCR cells infected with viruses carrying CRE or vector alone. Cells were plated at equal densities, cultured in the presence of hygromycin for 10–15 days, fixed and stained with crystal violet.
We took advantage of this feature to test whether continuous inactivation of p53 function was required for immortalisation. Presenescent MEFs were infected with two different p53αs (numbers 2 and 71) in pMarxIV and immortal cell lines, p53αsCR cells (CR for CRE-reversible) were generated using a 3T3 protocol. At PD 32, p53αsCR cells were infected with a CRE-expressing virus to ablate antisense expression. Cells in which p53 function had been restored failed to form colonies, while control cells continued to proliferate (Fig. 5). As an additional control, immortalised cell lines that had been generated following infection with non-excisable p53 antisense-expressing viruses did not arrest following CRE recombinase expression (data not shown). Finally, expression of dominant negative p53 from a non-excisable vector in the p53αsCR reversible cell line overcame the arrest induced by excision of the p53 antisense construct, indicating that the recovery of mortality was solely dependent on p53. Together these data indicate that continuous inactivation of p53 function is required for continued immortalisation.
Having a cell line which was reversibly dependent on loss of p53 function allowed us to test whether expression or inactivation of other genes could suppress the requirement for continued p53 inactivation. We therefore tested whether expression of MDM2 (a gene which targets p53 for degradation) (48,49), SV40 large T antigen (a viral oncogene which binds to and inactivates p53 and Rb) (50), HPV E7 (a protein which inactivates members of the pRB protein family) (51) or an allele of activated H-ras (a potent cytoplasmic oncogene) (52) could bypass the growth arrest induced by re-establishment of p53 expression in p53αsCR cells. In this system expression of MDM2 and large T antigen, but not E7 nor H-ras, inhibited p53-induced growth arrest.
p53 presumably inhibits immortalisation by directing expression of the relevant transcriptional targets. We therefore sought to test whether inactivation of known transcriptional targets of p53, such as the antiproliferative gene p21 and pro-apoptotic gene bax, was sufficient to overcome the arrest induced by re-expression of p53 in p53αsCR cells. Therefore, we generated a p53αsCR cell line in a p21–/– genetic background or expressed Bcl2 (a gene which binds to and inactivates bax) in p53αsCR cells. Overexpression of Bcl2 or inactivation of p21waf1 did not overcome the arrest induced by p53 recovery (Fig. 6). To test both genes simultaneously, we expressed Bcl2 in p21–/– p53αsCR cells. In this case, a partial bypass of the p53-dependent mortality was observed.
Figure 6.
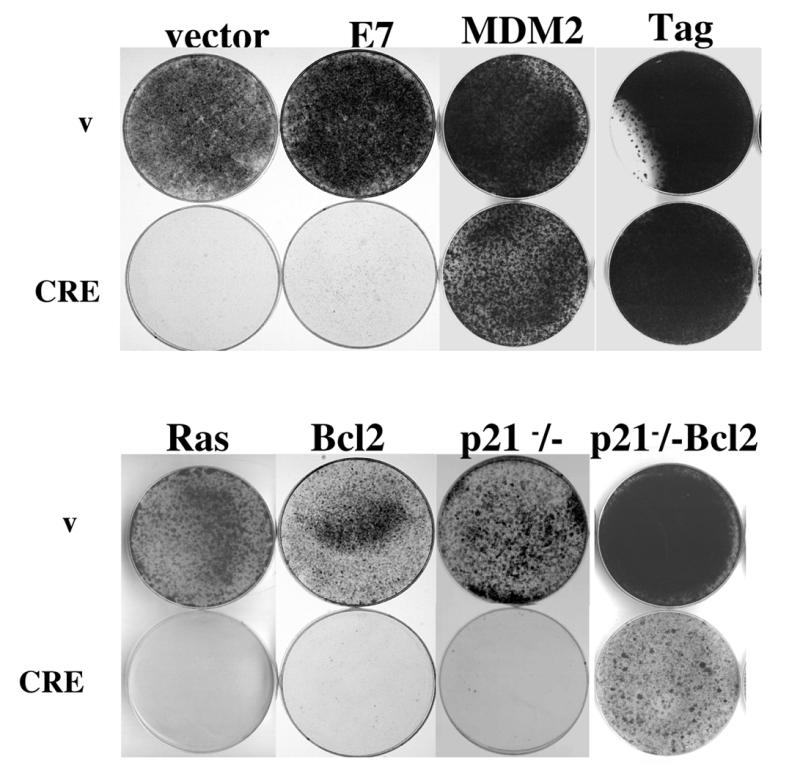
Bypass of growth arrest induced by restoration of p53 function. p53αsCR cells were infected with viruses directing the expression of various genes or a p53αsCR cell line was constructed in a p21–/– genetic background. Cells were then infected with a control or CRE-expressing virus, plated and grown for 10–15 days under selective conditions, fixed and stained with crystal violet.
DISCUSSION
Here, we have described a general strategy for identifying active antisense fragments and reversibly inactivating gene function. The identification of antisense sequences capable of suppressing target gene function has been a major goal in designing antisense vectors for gene therapy and for research use. In many cases, small antisense fragments have been more effective than full-length antisense constructs, spurring the development of methods to predict the most active antisense region. Although numerous reports describing methodologies to identify active antisense fragments exist, no consistently reliable method has yet been described. The identification of active antisense fragments by screening random fragments on the basis of their ability to produce a biological phenotype is a direct way to identify active antisense fragments. Although functional screens for small sense or antisense gene fragments have been previously described (42,43,53–55), the method described here is optimised to specifically identify small antisense fragments and provide a simple method to recover positive fragments.
We have used this methodology to identify antisense fragments that inhibit the function of the p53 tumour suppressor. The identified fragments mapped to two regions of the p53 gene and were at least as effective as full-length p53 antisense constructs in inhibiting translation of p53 mRNA (data not shown). A previous report (42) described the isolation of p53 antisense fragments that conferred cisplatin resistance to a human ovarian adenocarcinoma cell line. They found four small active antisense fragments that mapped between nt 360 and 700 of human p53, which overlaps with region I in our study. The identification of a second region for effective p53 antisense fragments in our study may be due to the use of p53 derived from different species or to the screens not being fully saturated.
Like inactivation of p53 by genetic disruption or expression of dominant interfering alleles, the p53 antisense fragments were capable of inducing an extended lifespan in primary MEFs, although not as efficiently as knockout. Experiments showing that expression of dominant negative p53 could functionally substitute for the p53 antisense fragments clearly demonstrated the specificity of the antisense action.
Expression of the p53 antisense fragments induced an extended lifespan and contributed to the immortalisation of primary MEFs. The observation that this immortalisation was reversible upon removal of the antisense construct provides a clear demonstration that continued inactivation of p53 is necessary for immortalisation. As well, it indicates that expression of the p53 antisense fragment was sufficient to prevent selection for mutations at the p53 genomic locus during immortalisation, again confirming the efficiency and specificity of action. Reversibly immortal cell lines have been constructed using conditional expression of SV40 large T antigen (56,57). While SV40 large T antigen can bind and inactivate p53, it can also inactivate members of the Rb family as well as other unrelated proteins (50), making it difficult to determine the role of each protein in suppressing immortalisation. The use of antisense RNA provides a more specific tool to analyse the function of individual proteins.
We have constructed conditionally immortalised cell lines that are dependent on the absence of p53 function. Thus, we have created a conditional cell line in which we can restore wild-type levels of p53. Such a cell line is extremely useful for identifying and probing pathways of p53 action. The observation that expression of MDM2 or SV40 large T antigen, but not E7 nor oncogenic ras, overcomes the arrest induced by restoration of p53 expression is consistent with a role of these proteins in functionally inactivating wild-type levels of p53 (48,49). In addition, this cell line might be extremely useful in identifying the relevant transcriptional targets of p53 in preventing immortalisation. Indeed, functional inactivation of both p21 and bax, known transcriptional targets of p53, did allow some bypass of p53-induced growth arrest, indicating that multiple transcriptional targets of p53 may mediate its antiproliferative action.
Reintroduction of missing tumour suppressors into tumours is being developed as a strategy of cancer therapy. We have shown that restoration of unaltered but inactive p53 leads to growth arrest depending on the nature of co-existing genetic alterations. p53 induces arrest in the presence of oncogenes such as ras and Bcl2, even in the absence of p21waf1. However, disturbing the p53 signal with binding oncogenes such as MDM2 and SV40 large T antigen inhibits this antitumorigenic effect, suggesting that inactivation of these proteins, when deregulated, will be more effective than reintroduction of wild-type p53.
The application of our system to a broader range of tumour suppressor genes may be useful in characterising their role in suppressing tumourigenic phenotypes.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by grants from the Leukaemia Research Fund (LRF 9728), Cancer Research Campaign (CRC no. SP 2366/0201 and CRC no. SP 2366/0101) and EMBO. D.H.B. was supported by the Hugh and Catherine Stevenson Fund.
REFERENCES
- 1.Lin A.W., Barradas,M., Stone,J.C., van Aelst,L., Serrano,M. and Lowe,S.W. (1998) Genes Dev., 12, 3008–3019. [DOI] [PMC free article] [PubMed]
- 2.Serrano M., Lin,A.W., McCurrach,M.E., Beach,D. and Lowe,S.W. (1997) Cell, 88, 593–602. [DOI] [PubMed]
- 3.Xu H.J., Zhou,Y., Ji,W., Perng,G.S., Kruzelock,R., Kong,C.T., Bast,R.C., Mills,G.B., Li,J. and Hu,S.X. (1997) Oncogene, 15, 2589–2596. [DOI] [PubMed]
- 4.Ko L.J. and Prives,C. (1996) Genes Dev., 10, 1054–1072. [DOI] [PubMed]
- 5.Lane D.P. (1994) Br. Med. Bull., 50, 582–599. [DOI] [PubMed]
- 6.Steele R.J., Thompson,A.M., Hall,P.A. and Lane,D.P. (1998) Br. J. Surg., 85, 1460–1467. [DOI] [PubMed]
- 7.Vogelstein B. and Kinzler,K.W. (1992) Cell, 70, 523–526. [DOI] [PubMed]
- 8.Foster B.A., Coffey,H.A., Morin,M.J. and Rastinejad,F. (1999) Science, 286, 2507–2510. [DOI] [PubMed]
- 9.Gao Q., Hauser,S.H., Liu,X.L., Wazer,D.E., Madoc-Jones,H. and Band,V. (1996) Cancer Res., 56, 3129–3133. [PubMed]
- 10.Sugrue M.M., Shin,D.Y., Lee,S.W. and Aaronson,S.A. (1997) Proc. Natl Acad. Sci. USA, 94, 9648–9653. [DOI] [PMC free article] [PubMed]
- 11.Wynford-Thomas D., Jones,C.J. and Wyllie,F.S. (1996) Biol. Signals, 5, 139–153. [DOI] [PubMed]
- 12.Barrett J.C., Annab,L.A. and Futreal,P.A. (1993) Adv. Exp. Med. Biol., 330, 27–43. [DOI] [PubMed]
- 13.Stein G.H. and Dulic,V. (1995) Bioessays, 17, 537–543. [DOI] [PubMed]
- 14.Wynford-Thomas D. (1999) J. Pathol., 187, 100–111. [DOI] [PubMed]
- 15.Bond J.A., Haughton,M., Blaydes,J., Gire,V., Wynford-Thomas,D. and Wyllie,F. (1996) Oncogene, 13, 2097–2104. [PubMed]
- 16.Bond J.A., Wyllie,F.S. and Wynford-Thomas,D. (1994) Oncogene, 9, 1885–1889. [PubMed]
- 17.Gollahon L.S. and Shay,J.W. (1996) Oncogene, 12, 715–725. [PubMed]
- 18.Dalal S., Gao,Q., Androphy,E.J. and Band,V. (1996) J. Virol., 70, 683–688. [DOI] [PMC free article] [PubMed]
- 19.Finlay C.A. (1992) Bioessays, 14, 557–560. [DOI] [PubMed]
- 20.Harvey D.M. and Levine,A.J. (1991) Genes Dev., 5, 2375–2385. [DOI] [PubMed]
- 21.Rogan E.M., Bryan,T.M., Hukku,B., Maclean,K., Chang,A.C., Moy,E.L., Englezou,A., Warneford,S.G., Dalla-Pozza,L. and Reddel,R.R. (1995) Mol. Cell. Biol., 15, 4745–4753. [DOI] [PMC free article] [PubMed]
- 22.Gallagher W.M. and Brown,R. (1999) Ann. Oncol., 10, 139–150. [DOI] [PubMed]
- 23.Komarova E.A. and Gudkov,A.V. (1998) Semin. Cancer Biol., 8, 389–400. [DOI] [PubMed]
- 24.Mukhopadhyay T. and Roth,J.A. (1995) Cancer J. Sci. Am., 1, 233–248. [PubMed]
- 25.Mukhopadhyay T. and Roth,J.A. (1996) Crit. Rev. Oncogen., 7, 151–190. [DOI] [PubMed]
- 26.Burfeind P., Chernicky,C.L., Rininsland,F. and Ilan,J. (1996) Proc. Natl Acad. Sci. USA, 93, 7263–7268. [DOI] [PMC free article] [PubMed]
- 27.Cheng J.Q., Ruggeri,B., Klein,W.M., Sonoda,G., Altomare,D.A., Watson,D.K. and Testa,J.R. (1996) Proc. Natl Acad. Sci. USA, 93, 3636–3641. [DOI] [PMC free article] [PubMed]
- 28.Holt J.T., Arteaga,C.B., Robertson,D. and Moses,H.L. (1996) Hum. Gene Ther., 7, 1367–1380. [DOI] [PubMed]
- 29.Izant J.G. and Weintraub,H. (1985) Science, 229, 345–352. [DOI] [PubMed]
- 30.Kim S.K. and Wold,B.J. (1985) Cell, 42, 129–138. [DOI] [PubMed]
- 31.Nellen W. and Sczakiel,G. (1996) Mol. Biotechnol., 6, 7–15. [DOI] [PubMed]
- 32.Sokol D.L. and Murray,J.D. (1996) Transgenic Res., 5, 363–371. [DOI] [PubMed]
- 33.Zhang Y., Mukhopadhyay,T., Donehower,L.A., Georges,R.N. and Roth,J.A. (1993) Hum. Gene Ther., 4, 451–460. [DOI] [PubMed]
- 34.Denhardt D.T. (1992) Ann. NY Acad. Sci., 660, 70–76. [DOI] [PubMed]
- 35.Melton D.A. (1985) Proc. Natl Acad. Sci. USA, 82, 144–148. [DOI] [PMC free article] [PubMed]
- 36.Kerr S.M., Stark,G.R. and Kerr,I.M. (1988) Eur. J. Biochem., 175, 65–73. [DOI] [PubMed]
- 37.Leiter J.M., Krystal,M. and Palese,P. (1989) Virus Res., 14, 141–159. [DOI] [PMC free article] [PubMed]
- 38.van der Krol A.R., Mol,J.N. and Stuitje,A.R. (1988) Gene, 72, 45–50. [DOI] [PubMed]
- 39.Daugherty B.L., Hotta,K., Kumar,C., Ahn,Y.H., Zhu,J.D. and Pestka,S. (1989) Gene Anal. Tech., 6, 1–16. [DOI] [PubMed]
- 40.Holzmayer T.A., Pestov,D.G. and Roninson,I.B. (1992) Nucleic Acids Res., 20, 711–717. [DOI] [PMC free article] [PubMed]
- 41.Rhodes A. and James,W. (1990) J. Gen. Virol., 71, 1965–1974. [DOI] [PubMed]
- 42.Gallagher W.M., Cairney,M., Schott,B., Roninson,I.B. and Brown,R. (1997) Oncogene, 14, 185–193. [DOI] [PubMed]
- 43.Ossovskaya V.S., Mazo,I.A., Chernov,M.V., Chernova,O.B., Strezoska,Z., Kondratov,R., Stark,G.R., Chumakov,P.M. and Gudkov,A.V. (1996) Proc. Natl Acad. Sci. USA, 93, 10309–10314. [DOI] [PMC free article] [PubMed]
- 44.Martinez J., Georgoff,I., Martinez,J. and Levine,A.J. (1991) Genes Dev., 5, 151–159. [DOI] [PubMed]
- 45.Hannon G.J., Sun,P., Carnero,A., Xie,L.Y., Maestro,R., Conklin,D.S. and Beach,D. (1999) Science, 283, 1129–1130. [DOI] [PubMed]
- 46.el-Deiry W.S., Tokino,T., Velculescu,V.E., Levy,D.B., Parsons,R., Trent,J.M., Lin,D., Mercer,W.E., Kinzler,K.W. and Vogelstein,B. (1993) Cell, 75, 817–825. [DOI] [PubMed]
- 47.Carnero A., Hudson,J.D., Price,C.M. and Beach,D.H. (2000) Nature Cell Biol., 2, 148–155. [DOI] [PubMed]
- 48.Haupt Y., Maya,R., Kazaz,A. and Oren,M. (1997) Nature, 387, 296–299. [DOI] [PubMed]
- 49.Kubbutat M.H., Jones,S.N. and Vousden,K.H. (1997) Nature, 387, 299–303. [DOI] [PubMed]
- 50.Pipas J.M. (1998) Dev. Biol. Stand., 94, 313–319. [PubMed]
- 51.Syrjanen S.M. and Syrjanen,K.J. (1999) Ann. Med., 31, 175–187. [DOI] [PubMed]
- 52.Barbacid M. (1987) Annu. Rev. Biochem., 56, 779–827. [DOI] [PubMed]
- 53.Gudkov A.V., Kazarov,A.R., Thimmapaya,R., Axenovich,S.A., Mazo,I.A. and Roninson,I.B. (1994) Proc. Natl Acad. Sci. USA, 91, 3744–3749. [DOI] [PMC free article] [PubMed]
- 54.Gudkov A.V., Zelnick,C.R., Kazarov,A.R., Thimmapaya,R., Suttle,D.P., Beck,W.T. and Roninson,I.B. (1993) Proc. Natl Acad. Sci. USA, 90, 3231–3235. [DOI] [PMC free article] [PubMed]
- 55.Roninson I.B., Gudkov,A.V., Holzmayer,T.A., Kirschling,D.J., Kazarov,A.R., Zelnick,C.R., Mazo,I.A., Axenovich,S. and Thimmapaya,R. (1995) Cancer Res., 55, 4023–4028. [PubMed]
- 56.Westerman K.A. and Leboulch,P. (1996) Proc. Natl Acad. Sci. USA, 93, 8971–8976. [DOI] [PMC free article] [PubMed]
- 57.Wright W.E. and Shay,J.W. (1992) Exp. Gerontol., 27, 383–389. [DOI] [PubMed]